

Abstract
The RNA field has been revolutionized by methods that allow genome-scale identification of RNA-protein interaction sites. Two reports now introduce more efficient approaches, opening the technology to wider adoption (Van Nostrand et al., 2016; Zarnegar et al., 2016).
Methods combining UV-crosslinking and RNA binding protein (RBP) immunoprecipitation with high-throughput sequencing (CLIP-Seq) promise to allow comprehensive high-resolution identification of all of the RNA binding sites occupied by any RBP of interest. In CLIP-Seq experiments, cells are irradiated with ultraviolet light to covalently trap RNA-protein interactions, followed by immunopurification of a specific RBP. RNAs covalently linked to the target RBP are partially digested to generate short sequence tags suitable for cDNA library preparation and high-throughput sequencing. The resulting datasets offer a transcriptome-wide picture of RBP binding sites at up to single nucleotide resolution. A series of variations on CLIP-Seq have been used to evaluate the functions and mechanisms of dozens of RNA binding proteins (Flynn et al., 2015; Hafner et al., 2010; König et al., 2010; Licatalosi et al., 2008), but technical barriers to their universal application persist.
CLIP-Seq methods are complex–protocols list 40 or more individual steps requiring several days to complete–and prone to failure. Despite methods calling for large numbers of input cells (typically tens of millions), it is often difficult to obtain sufficient material to generate high-complexity cDNA libraries for sequencing. To begin to address this problem, Zarnegar and colleagues first established a method to track RNA through a CLIP-Seq experiment more easily. Whereas previous CLIP-Seq protocols use 5′ radiolabeling to monitor RNAs through gel electrophoresis, adapter ligation, and RNA purification steps, their method, infrared-CLIP (irCLIP), uses an oligonucleotide labeled with an infrared fluorescent dye for 3′-adapter ligation (Zarnegar et al., 2016). The ligated product can then be quickly and sensitively detected at multiple points in the protocol. The authors use this system to optimize several aspects of the CLIP-Seq workflow, including improving the fragmentation of immunopurified RNA and streamlining or eliminating RNA precipitation and purification steps. The benefits accrued from multiple points of optimization result in much lower required starting cell numbers than other CLIP-Seq variants. While input requirements will ultimately depend on the abundance of the RBP of interest, its crosslinking efficiency to its cognate RNAs, and other factors, these improvements allowed productive sequencing of cDNA libraries from as few as 20,000 cells.
Among the most notable advances introduced in irCLIP is the use of thermostable group II intron reverse transcriptase (TGIRT) for cDNA synthesis. These enzymes exhibit a number of favorable properties when compared to widely used retroviral reverse transcriptases, including higher processivity, thermostability, and fidelity, as well as the ability to act on highly structured or modified RNA templates (Mohr et al., 2013). Simply adapting the library construction protocol to use the TGIRT enzyme led to an approximate eight-fold increase in cDNA production when compared to the optimized murine leukemia virus-derived Superscript III enzyme (Zarnegar et al., 2016). For difficult templates, this approach may yield even greater dividends.
Independently, van Nostrand and colleagues pursued a parallel path toward democratization of CLIP-based approaches. Their twist on the technique, “enhanced” CLIP (eCLIP), similarly promises tremendous gains in efficiency over previous methods. As with irCLIP, eCLIP involves streamlining of several steps of RNA and cDNA handling, all directed at minimizing loss of precious low-abundance material. Most importantly, eCLIP incorporates improved RNA-seq library preparation methods to vastly increase the efficiency of the adapter ligation steps required for reverse transcription and deep sequencing (Shishkin et al., 2015). Together, these enhancements lead to as much as a 1000x decrease in the PCR amplification required to generate high quality libraries for sequencing when compared to previous methods (Van Nostrand et al., 2016). As part of the ENCODE consortium, the authors have already used eCLIP to generate 102 datasets from 73 RBPs, illustrating its scalability and broad applicability.
Rather than the ideal of unbiased, comprehensive identification of RNA-protein interactions, current methods preferentially identify interactions between abundant RBPs and abundant RNAs. A major shortcoming in many existing CLIP-Seq methods is the lack of controls to monitor non-specific background or to account for differences in abundance of distinct cellular RNAs in the starting material. Combined with the inefficiencies addressed by irCLIP and eCLIP, a lack of standardized normalization and background subtraction methods for data analysis results in over-representation of highly abundant RNAs in CLIP-Seq datasets and can frequently lead to false-positives. To address this problem, the eCLIP pipeline includes controls for normalization to input RNA abundance to aid in accurate interpretation of CLIP-Seq data. Specifically, RNA from crude input extracts is fragmented and size-selected in parallel with immunopurified RNA. This input sample can then be used to test for significant enrichment of mRNA regions in CLIP-Seq experiments relative to input samples. This addition has the potential to reduce false-positives, aid in identifying interactions between RBPs and low-abundance RNAs, and improve reproducibility.
Notably, eCLIP and irCLIP take largely complementary approaches to CLIP-Seq optimization. In addition to the immediate benefits available to RNA biologists, this suggests that future gains could be made by combining the lessons learned in each study. Together, they open the door to more routine use of CLIP-Seq to study a wider range of RNA-protein interactions, in biological systems that go beyond the common transformed cell lines thus far used for most CLIP-Seq experiments.
Figure 1.
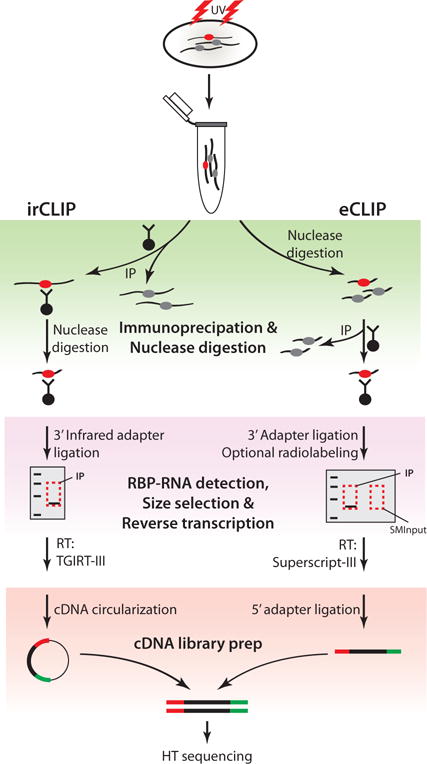
Schematic of CLIP-Seq workflow and major modifications introduced in irCLIP and eCLIP (Zarnegar et al. 2016; Van Nostrand et al. 2016). Top, cells are UV-irradiated to covalently link RNA-protein complexes, followed by lysis and RBP immunopurification. In-lysate nuclease digestion precedes RBP purification in eCLIP, while on-bead nuclease digestion is performed on immunopurified complexes in irCLIP. Ligation of an IR dye-labeled 3′ adapter in irCLIP allows rapid detection of RNA-protein complexes, rather than immunoblotting or radiolabeling as in eCLIP. Purified material is resolved by SDS-PAGE, transferred to nitrocellulose membranes, and size-selected in both methods, but eCLIP introduces purification and sequencing of size-matched RNA to allow normalization to input RNA levels (SMInput). In irCLIP, TGIRT-III reverse transcriptase is used to enhance cDNA synthesis, and a second adapter-ligation step is omitted by circularization of cDNA. In contrast, cDNA generated in eCLIP is ligated to a second adapter using optimized ligation methods. The products are then amplified by PCR and analyzed by high-throughput sequencing.
References
- Flynn RA, Martin L, Spitale RC, Do BT, Sagan SM, Zarnegar B, Qu K, Khavari PA, Quake SR, Sarnow P, Chang HY. Dissecting noncoding and pathogen RNA-protein interactomes. RNA. 2015;21:135–143. doi: 10.1261/rna.047803.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, A M, Jr, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Ghanem E, Smith W, Sheeter D, Qin Y, King O, Polioudakis D, Iyer VR, Hunicke-Smith S, Swamy S, Kuersten S, Lambowitz AM. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA. 2013;19:958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM, Novod N, Hung DT, Gnirke A, Garber M, Guttman M, Livny J. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat Methods. 2015;12:323–325. doi: 10.1038/nmeth.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, Stanton R, Rigo F, Guttman M, Yeo GW. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat Methods. 2016 doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, Khavari PA. irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods. 2016 doi: 10.1038/nmeth.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]