

Abstract
α-Klotho is highly expressed in the kidney, and its extracellular domain is cleaved and released into the circulation. Chronic kidney disease (CKD) is a state of α-Klotho deficiency, which exerts multiple negative systemic effects on numerous organs including the cardiovascular system. Since acute kidney injury (AKI) greatly escalates the risk of CKD development, we explored the effect of α-Klotho on prevention and treatment on post-AKI to CKD progression and cardiovascular disease. Therein, ischemia reperfusion injury-induced AKI was followed by early administration of recombinant α-Klotho or vehicle starting one day and continued for four days after kidney injury (CKD prevention protocol). A CKD model was generated by unilateral nephrectomy plus contralateral ischemia reperfusion injury. Late administration of α-Klotho in this model was started four weeks after injury and sustained for 12 weeks (CKD treatment protocol). The prevention protocol precluded AKI to CKD progression and protected the heart from cardiac remodeling in the post-AKI model. One important effect of exogenous α-Klotho therapy was the restoration of endogenous α-Klotho levels long after the cessation of exogenous α-Klotho therapy. The treatment protocol still effectively improved renal function and attenuated cardiac remodeling in CKD, although these parameters did not completely return to normal. In addition, α-Klotho administration also attenuated high phosphate diet-induced renal and cardiac fibrosis, and improved renal and cardiac function in the absence of pre-existing renal disease. Thus, recombinant α-Klotho protein is safe and efficacious, and might be a promising prophylactic or therapeutic option for prevention or retardation of AKI-to-CKD progression and uremic cardiomyopathy.
Keywords: acute kidney injury, cardiovascular disease, chronic kidney disease, ischemia reperfusion, phosphate
α-Klotho was originally identified when the serendipitous disruption of its locus led to premature multiorgan failure resembling aging.1 Conversely, ubiquitous overexpression of α-Klotho leads to extended life span.2 α-Klotho is a single-pass transmembrane protein highly expressed in the kidney.1 It is proteolytically cleaved (cleaved α-Klotho),3–5 and its transcript is proposed to be alternatively spliced to generate a soluble form (secreted α-Klotho).1 These extracellular polypeptides of α-Klotho constitute “soluble α-Klotho,” which functions as an endocrine substance with far-reaching effects.6 Soluble α-Klotho exerts multiple actions, including antioxidation, antisenescence, pro-autophagy, anti-apoptosis, antifibrosis, pro-stem cell, and anti-insulin actions.6–8 Numerous lines of evidence have shown that the majority of circulating α-Klotho is derived from the kidney.3,9 Chronic kidney disease (CKD) is a state of “pan-α-Klotho deficiency,” with drastic reduction in renal and circulating α-Klotho in rodents and humans.10–12 It has become clear that α-Klotho deficiency is not a mere biomarker but pathogenic for CKD, because α-Klotho repletion improves multiple renal and extrarenal parameters in both acute and chronic loss of renal function.11–14
The modalities of circulating α-Klotho augmentation in preclinical studies include whole body and hepatic transgenic overexpression, adeno-associated viral delivery, stimulation of endogenous α-Klotho production, or administration of recombinant α-Klotho, which has only been achieved in certain situations.14–16 However, transgenic or viral-based gene delivery are not easily translatable to human application, and the stimulation of endogenous α-Klotho production is modest and may be impossible when CKD is advanced. Recombinant α-Klotho therapy remains the most promising avenue for human application. The proof-of-concept for recombinant α-Klotho therapy was demonstrated in acute infusions where biologic effects such as acute renoprotection and modulation of renal transporters were demonstrated.14,17–19 However, treatment in CKD requires longterm administration of α-Klotho protein. In the present study, we tested parenteral recombinant α-Klotho therapy in both prophylactic and therapeutic capacities and demonstrated that it alleviates onset and delays progression of CKD, as well as ameliorates uremic cardiomyopathy, which is the grimmest complication of CKD.20,21 These preclinical data support the principle and usher in great promise on the horizon for the translational application of recombinant α-Klotho therapy in human CKD.
METHODS
Animal experiments
All animal experimental protocols were approved by the Institutional Animal Care and Use committee from University of Texas Southwestern Medical Center. The genetic background was 129sv, and equal numbers of male and female mice at 10 to 12 weeks of age were randomly assigned to sham, acute kidney injury (AKI), or CKD groups and received vehicle or α-Klotho treatment. AKI was induced by bilateral renal artery clamping for 35 minutes followed by reperfusion as described previously.11,18 Animals were fed normal rodent chow throughout the experimental period. CKD was induced by uninephrectomy plus contralateral 30-minute ischemia followed by reperfusion as described previously.11,18 Sham-treated animals underwent laparotomy and manual manipulation of the kidneys for the same surgical duration. Two weeks after surgery, animals were switched from normal rodent chow to 2.0% phosphate diet (Teklad 08020; Harlan, Madison, WI) for an additional 14 weeks.
Cardiac magnetic resonance imaging (MRI)
Cardiac function was evaluated by cardiac MRI using a 7 T small animal magnetic resonance scanner (Varian Inc., Palo Alto, CA) with a 38 mm birdcage radiofrequency coil as described previously.22 Under inhalational anesthesia (1.5%–2% isoflurane), MRI acquisitions were gated using both cardiac and respiratory triggering. Two-dimensional gradient echo images on 3 orthogonal planes (transverse, coronal, and sagittal) were acquired to determine the orientation (long axis) of the heart in each mouse. Axial images perpendicular to the long axis of the heart were chosen for cine imaging.11,23 Epicardial and endocardial borders were manually traced for calculation of left ventricular end systolic and end diastolic volume using ImageJ software. Total left ventricular volumes, stroke volume, cardiac output, and left ventricular ejection fraction were calculated as described previously.11 Left ventricle wall thickness was measured via midventricular short-axis images (just below the papillary muscle). Operators performing MRI acquisition and data analysis were blinded to the identity of the experimental groups.
Chemicals and biologics
The following antibodies were used: anti-α-actinin (Sigma-Aldrich, St. Louis, MO), anti-β-actin (Sigma-Aldrich), anti-Erk and anti-phospho-Erk (Cell Signaling Technology Inc., Danvers, MA), anti-α-Klotho Kl1 (KM2076) (Trans Genic Inc., Kobe, japan), and anti-α-smooth muscle actin (α-SMA) (Sigma-Aldrich). Wheat germ agglutinin (Molecular Probes Inc., Eugene, OR) was used to highlight the cardiac myocyte silhouettes. Secondary antibodies coupled to horseradish peroxidase for immunoblotting, or to FITC and Alexa Fluor for immunohistochemistry, were purchased from Molecular Probes/Invitrogen (Molecular Probes Inc.).
Soluble α-Klotho protein containing the ectodomain of mouse α-Klotho (amino acid number 31–982) with V5 and 6xHis tags at the C-terminus was generated as described previously.2 In the AKI model, α-Klotho protein in phosphate-buffered saline (0.01 mg/kg body weight/day) was injected i.p. for 4 consecutive days starting 24 hours after AKI induction. The same volume of phosphate-buffered saline served as control. In the CKD model, α-Klotho protein (0.3 mg/kg body weight/month) was administered i.p. using osmotic minipumps with phosphate-buffered saline as control (1004, Alzet Durect Corp., Cupertino, CA). Minipumps were replaced monthly, resulting in a total of 3 pumps per animal.
Kidney histology and histopathology
Kidneys were fixed in 4% paraformaldehyde at 4°C overnight followed by preparation of paraffin-embedded kidney section for hematoxylin and eosin or trichrome stain, respectively, which were photographed by a renal pathologist blinded to the experimental protocol using Zeiss light microscope (Carl Zeiss Micro-Imaging Inc. Thornwood, NY). A pathologist blinded to the experimental conditions evaluated the images using a semiquantitative pathological scoring system.18,24 The fibrotic area and intensity were quantified with Image) software using published protocols and presented as fibrotic area/total scanned area.11,18
Assessment of cardiac fibrosis and hypertrophy
Paraffin-embedded heart sections were stained with trichrome and photographed with a Zeiss light microscope and analyzed using Image) software.11,23 The scar ratio (percentage) was defined as trichrome-positive area divided by the scanned area of cardiac musculature. To measure cardiomyocyte surface areas, paraffin-embedded sections were labeled with Alexa Fluor 555–conjugated wheat germ agglutinin (Invitrogen, Carlsbad, CA) as described previously.11,23 Immunofluorescent images were obtained on a Zeiss laser scanning microscope (Carl Zeiss Micro-Imaging Inc.). ImageJ software was used to quantify cross-sectional cell surface area along the mid-chamber free wall based on wheat germ agglutinin–positive staining.11
Plasma and urine biochemistry and assay of plasma α-Klotho, and full-length as well as C-terminal FGF23
Plasma and urine creatinine concentrations were measured with a P/ACE MDQ Capillary Electrophoresis System and photodiode detector (Beckman-Coulter, Fullerton, CA) at 214 nm.24 Other biochemical parameters in plasma and urine were determined using established methods.24
Plasma α-Klotho was measured with an immunoprecipitation-immunoblot assay.10–12,23 Soluble α-Klotho protein containing the ectodomain of mouse α-Klotho (amino acid number 31–982; R&D, Minneapolis, MN) was used for calibration. Briefly, 100 μl of mouse plasma were subjected to immunoprecipitation with 1 μg of sb106 anti-α-Klotho antibody,10 and the immune complex was eluted from protein G beads with 2.5X Laemmli sample buffer after washing, fractionated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with rat anti-human α-Klotho monoclonal antibody (KM2076; Trans Genic Inc., Kobe, japan).
Intact FGF23 (iFGF23) was measured by intact FGF23 ELISA kit (Kainos, Japan) following manufacturer’s instructions, whereas C-terminal FGF23 (cFGF23) including full-length and C-terminal peptide of FGF23 was measured by mouse C-terminal ELISA kit (Immunotopics International, San Clemente, CA) according to manufacturer’s instructions. We measured both iFGF23 and cFGF23 to explore whether their levels change in parallel.
Immunohistochemistry and immunoblot in heart and kidney
Four pm sections of paraffin-embedded kidney and heart tissues were de-paraffined. After blocking, primary antibodies were incubated at 4°C overnight followed by secondary antibodies conjugated to fluorescein isothiocyanate or Alexa red. Rliodamine-phalloidin (1:50) (Molecular Probes Inc.) counterstain was used for detection of β-actin filaments. Sections were visualized using a Zeiss LSM-510 laser scanning microscope.
Cell lysates from heart and kidney were prepared as described previously.12,23 Thirty μg of protein were solubilized in Laemmli’s buffer, fractionated by SDS-PAGE, transferred to PVDF membrane, and blotted using specific primary antibodies and monoclonal mouse antibody for β-actin (1/5000 dilution). Primary antibodies were incubated overnight at 4°C. After washing, membranes were incubated with secondary antibodies conjugated with horseradish peroxidase (Amersham Life Sciences, Piscataway, NJ). Specific signal was visualized using ECL kit (Amersham Life Sciences).
Statistical analyses
Data are expressed as means ± SD (n = 4). As appropriate, statistical analysis was performed using Student’s unpaired t-test or 1-way analysis of variance (ANOVA) followed by ad hoc Student–Newman–Keuls test for multiple comparison between 2 groups when applicable. Sigma Plot software (12.5 v; Systat Software Inc. Chicago, IL) was used. A value of P ≤ 0.05 was considered statistically significant.
RESULTS
Early administration of recombinant α-Klotho protein prevents AKI-to-CKD progression
We previously showed that early administration of recombinant α-Klotho protein preserves renal function and histology 4 weeks after ischemic challenge.18 To explore more clinically relevant longer-term renal and extrarenal effects of therapy, we extended the study to 20 weeks after AKI to examine whether recombinant α-Klotho can reduce the risk of AKI-to-CKD progression (Supplementary Figure S1A). At 20 weeks after AKI, α-Klotho–treated mice had higher creatinine clearance (ClCr) and lower plasma phosphate than vehicle-treated mice (Supplementary Figure S1B and S1C). Interestingly, endogenous plasma α-Klotho levels in α-Klotho–treated AKI mice were higher than in vehicle-treated AKI mice 20 weeks after treatment, but still lower than that in sham-treated mice with or without α-Klotho administration (Figure 1a). α-Klotho–treated AKI mice had much less trichrome staining and lower abundance of the fibrosis marker (α-SMA) in the kidney (Supplementary Figure S2) than vehicle-treated AKI mice, suggesting that α-Klotho suppresses fibrosis in the kidney after AKI. Lower levels of α-Klotho protein in the kidney of vehicle-treated AKI mice (Figure 1b) might be responsible for the higher renal fibrosis and lower renal function in AKI mice without α-Klotho treatment. In support of this concept, early and short-term treatment for only 4 days with recombinant α-Klotho after AKI had a long-lasting (20 weeks) impact on phosphate metabolism as well as renal function and morphology (Supplementary Figures S1 and S2).
Figure 1. αKlotho administration after acute kidney injury (AKI) maintained higher plasma and renal αKlotho levels and improved cardiac function in ischemia-reperfusion injury (IRI)–induced AKI mice.
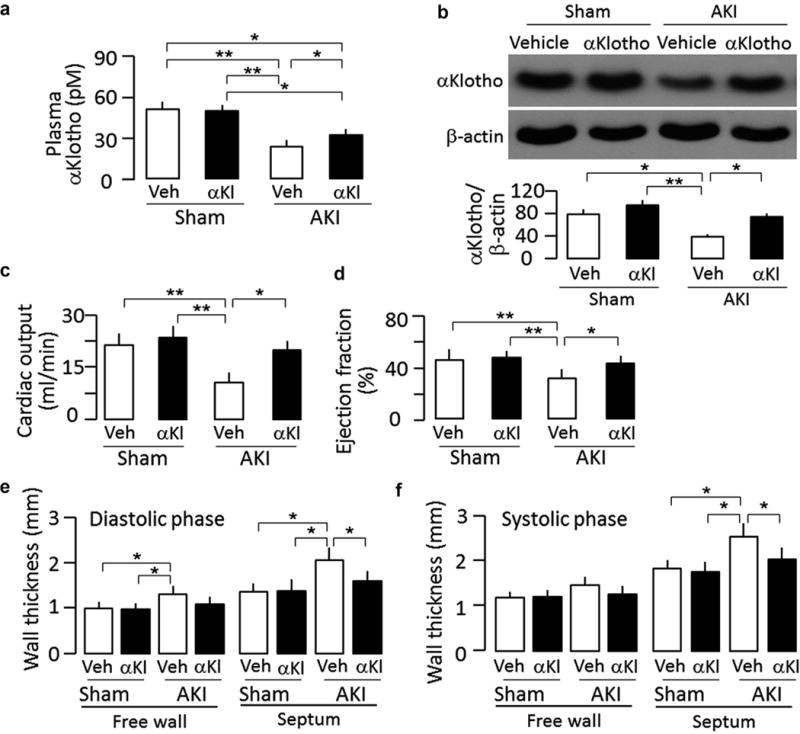
Sham-treated or IRI-induced AKI wild-type mice were Injected i.p. with αKlotho (αKl) protein (0.01 mg/kg) or vehicle (Veh) (phosphate-buffered saline) for 4 consecutive days starting 24 hours after surgery. Throughout the experimental period, all mice were fed normal rodent chow. At 20 weeks after surgery, animals were killed. (a) Plasma αKlotho. (b) αKlotho protein in the kidney. Upper panel shows representative immunoblots for αKlotho and (β-actin in the kidney. Bottom panel is a summary of normalized protein quantification from all examined immunoblots. (c) Cardiac output, (d) Left ventricular ejection fraction, (e) Left ventricular wall thickness at diastole, (f) Left ventricular wall thickness at systole. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test, and accepted when: *P < 0.05, **P < 0.01 between 2 groups.
Early administration of recombinant α-Klotho protein prevents uremic cardiomyopathy
AKI is an independent risk to cardiovascular disease after adjustment for other comorbidities.25 Remarkably, treatment with α-Klotho protein for only 4 days after AKI preserved cardiac function and reduced cardiac hypertrophy and cardiac fibrosis (Figure 1c–f and Figure 2a and b) 20 weeks after AKI. Development of cardiomyocyte hypertrophy (Figure 2c) and upregulation of α-actinin and α-SMA protein (Figure 2d) in vehicle-treated AKI mice were consistent with cardiac hypertrophy and fibrosis in cardiac histology (Figure 2a and b) and cardiac MRI results (Figure 1c–f). With this experimental design, it is difficult to conclude whether the observed amelioration of cardiac remodeling is a direct effect of α-Klotho on the heart or secondary to prevention of CKD, but regardless of the mechanism(s), the significance is that this severe extrarenal complication can be partially prevented by early α-Klotho treatment.
Figure 2. αKlotho administration attenuated cardiac remodeling after acute kidney injury (AKI).
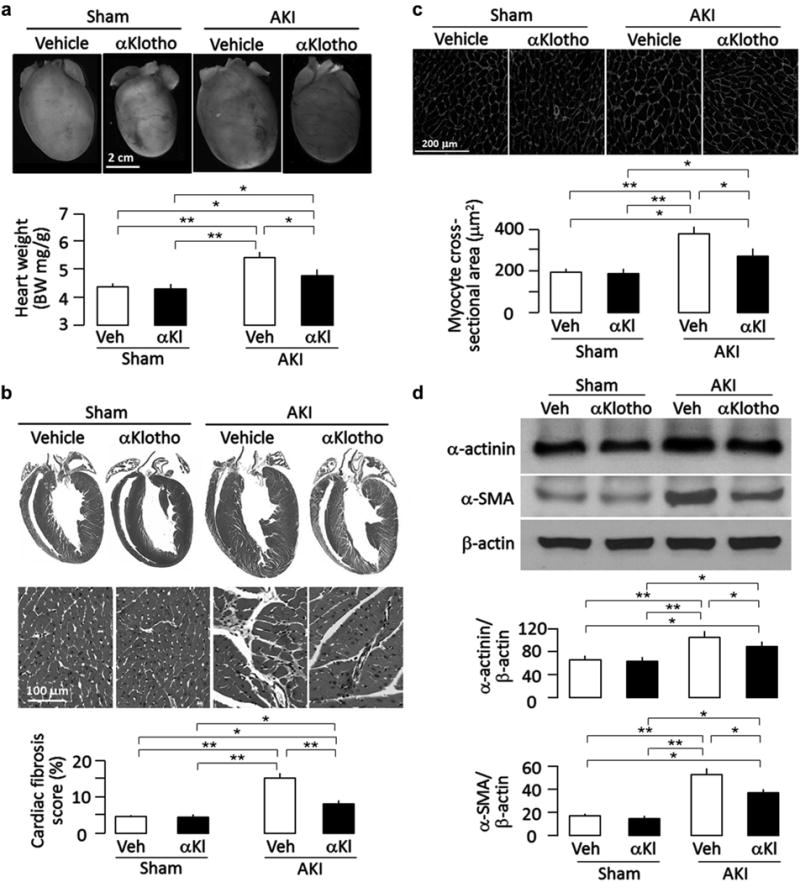
Wild-type mice subjected to ischemia-reperfusion injury (IRI)–induced AKI and sham surgery were treated with αKlotho (αKl) protein (described in Supplementary Figure S1A) or vehicle (Veh) and killed 20 weeks after surgery. All mice were fed normal rodent chow, (a) Cardiac hypertrophy in mice after AKI. Upper panel shows representative gross macrographs of hearts. Bottom panel is a summary of ratio of heart weight to body weight of examined mice. (b) Cardiac fibrosis in mice after AKI. Upper panel shows representative macrographs of sagittal sections (trichrome stain). Middle panel shows representative micrographs of left ventricular sections (trichrome stain). Bottom panel is a summary of semiquantification of trichrome-positive area to whole heart section calculated using ImageJ software. (c) Hypertrophic cardiomyocytes in post-AKI mice. Upper panel shows representative micrographs of heart sections stained with WGA. Bottom panel is a summary of cardiomyocyte size calculated using ImageJ software, (d) Hypertrophic and fibrotic markers in the heart. Upper panel shows representative immunoblots for α-actinin, α-smooth muscle actin (α-SMA), and (β-actin protein. Bottom panel shows a summary of normalized protein quantification from all examined immunoblots. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05, **P < 0.01 between 2 groups. WGA, wheat germ agglutinin.
Chronic administration of α-Klotho protein slows down CKD progression
For CKD subjects without a definite identifiable episode of AKI or those past this window of treatment opportunity, one needs to determine whether α-Klotho administration after the onset of CKD will still be efficacious. To define the effects of late administration of recombinant α-Klotho protein on CKD progression and cardiac complications, we started to treat mice with recombinant α-Klotho protein at 4 weeks after AKI, when CKD development has already initiated,18 and continued α-Klotho administration for 12 weeks (Figure 3a).
Figure 3. Effects of chronic administration of αKlotho (αKl) after established chronic kidney disease (CKD) on plasma phosphate, renal function, plasma FGF23, and plasma αKlotho in CKD mice.
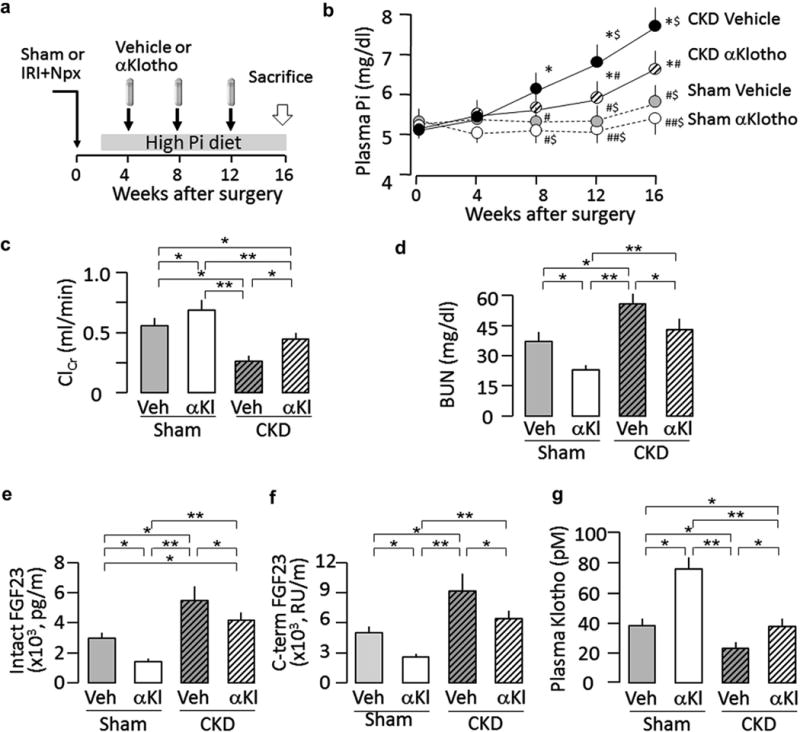
(a) Animal experimental design. Using osmotic minipump implants from week 4 to 16 after induction of CKD. The minipumps contained vehicle (Veh; phosphate-buffered saline) or αKlotho (0.3 μg/g body weight). All mice were fed normal rodent chow or high-phosphate (2%) experimental chow for 12 weeks starting 2 weeks after surgery. (b) Plasma phosphate 0,4,8,12, and 16 weeks after surgery. (c) Creatinine clearance (ClCr) at 16 weeks. (d) Blood urea nitrogen (BUN) at 16 weeks. (e) Plasma intact FGF23 at 16 weeks, (f) Plasma C-terminal FGF23 (C-term FGF23) at 16 weeks, (g) Plasma αKlotho at 16 weeks. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05 versus sham-vehicle; #P < 0.05, ##P < 0.01 versus CKD-vehicle; $P < 0.05 versus CKD-αKlotho at 0, 4, 8,12, and 16 weeks of surgery, respectively, for (b). *P < 0.05, **P < 0.01 between 2 groups for (c–g). IRI, ischemia-reperfusion injury; Npx, nephrectomy; Pi, phosphate.
At 4 weeks after AKI, CKD mice treated with vehicle had steady elevation of plasma phosphate, which was modestly but significantly blunted by α-Klotho treatment (Figure 3b) even though α-Klotho therapy was started 4 weeks after CKD induction surgery. Note that α-Klotho also reduced plasma phosphate in sham-treated mice fed a high-phosphate diet (Figure 3b), supporting α-Klotho’s role as a phosphaturic substance.17,26 Recombinant α-Klotho protein improved ClCr and reduced blood urea nitrogen in both CKD and sham-treated mice fed with high phosphate (Figure 3c and d), indicating that recombinant α-Klotho protein preserved renal function. Furthermore, α-Klotho–treated CKD mice and sham-treated mice fed a high-phosphate diet, respectively, both had lower plasma iFGF23 and cFGF23 and higher plasma α-Klotho than did vehicle-treated CKD mice and sham-treated mice fed a high-phosphate diet, respectively (Figure 3e–g). Lower plasma FGF23 levels (Figure 3e and f) can be secondary to higher plasma α-Klotho levels (Figure 3g) or simply related to amelioration of CKD (Figure 3c and d, and Figure 4a and b).
Figure 4. Effects of chronic administration of αKlotho after established chronic kidney disease (CKD) on kidney histology and renal αKlotho expression.
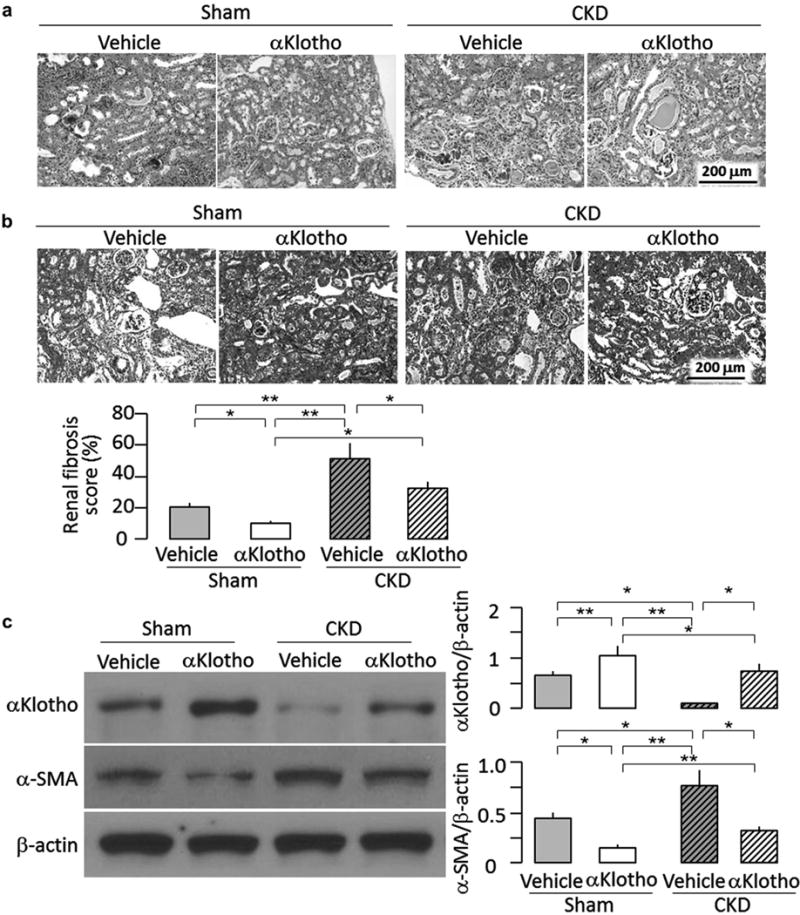
Animal experimental design was shown in Figure 3a. At 16 weeks, the kidneys were harvested for renal histology and immunoblot analysis, (a) Representative renal micrographs of hematoxylin-eosin stained kidney sections, (b) Renal fibrosis in mice at 16 weeks. Upper panel shows representative micrographs of trichrome-stained kidney sections. Bottom panel shows renal fibrosis scores from trichrome section calculated using ImageJ software, (c) αKlotho and fibrotic markers in the kidney. Left panel shows representative immunoblots for αKlotho, α-smooth muscle actin (α-SMA), and β-actin protein. Right panel shows a summary of normalized protein quantification from all examined immunoblots. Data are expressed as means ± SD of 4 mice from each group. Statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05, **P < 0.01 between 2 groups.
CKD mice had severe interstitial cell infiltration and fibrosis in the kidney, tubular atrophy and casts, and sclerotic glomeruli, all of which were dramatically attenuated by α-Klotho protein (Figure 4a and b). Sham-treated mice chronically fed a 2% phosphate diet also had tubulointerstitial infiltration and fibrosis, although much milder than did CKD mice (Figure 4a and b), indicating that high phosphate diet per se can induce kidney injury in normal mice when given for a sufficiently long duration. High phosphate-induced kidney damage in sham-treated mice was also alleviated by recombinant α-Klotho protein (Figure 4a and b), further supporting the concept that α-Klotho is renoprotective. Both CKD and sham-treated mice injected with α-Klotho had higher α-Klotho and lower α-SMA protein levels in the kidney (Figure 4c) compared to vehicle-treated counterparts, which is consistent with the observed higher levels of plasma α-Klotho (Figure 3g) and with the decrease in renal fibrosis (Figure 4a and b).
Chronic administration of α-Klotho protein protects against uremic cardiomyopathy
α-Klotho administration, even when started 4 weeks after CKD induction, still effectively ameliorated cardiac hypertrophy and fibrosis (Figure 5a and b). In addition, chronic α-Klotho treatment also alleviated the effects of highphosphate feeding on cardiac remodeling in sham-treated mice (Figure 5a–d), which is compatible with the previously published observation that cardiac remodeling in the setting of preserved renal function and normal phosphate homeostasis is much milder in transgenic α-Klotho overexpressors than in WT mice.11 It appears plausible that the cardioprotection of α-Klotho observed in CKD mice is at least partially due to its antiphosphotoxic effects, in part from its direct action on the heart, among other factors.
Figure 5. Effects of chronic administration of αKlotho on cardiac remodeling in chronic kidney disease (CKD) mice.
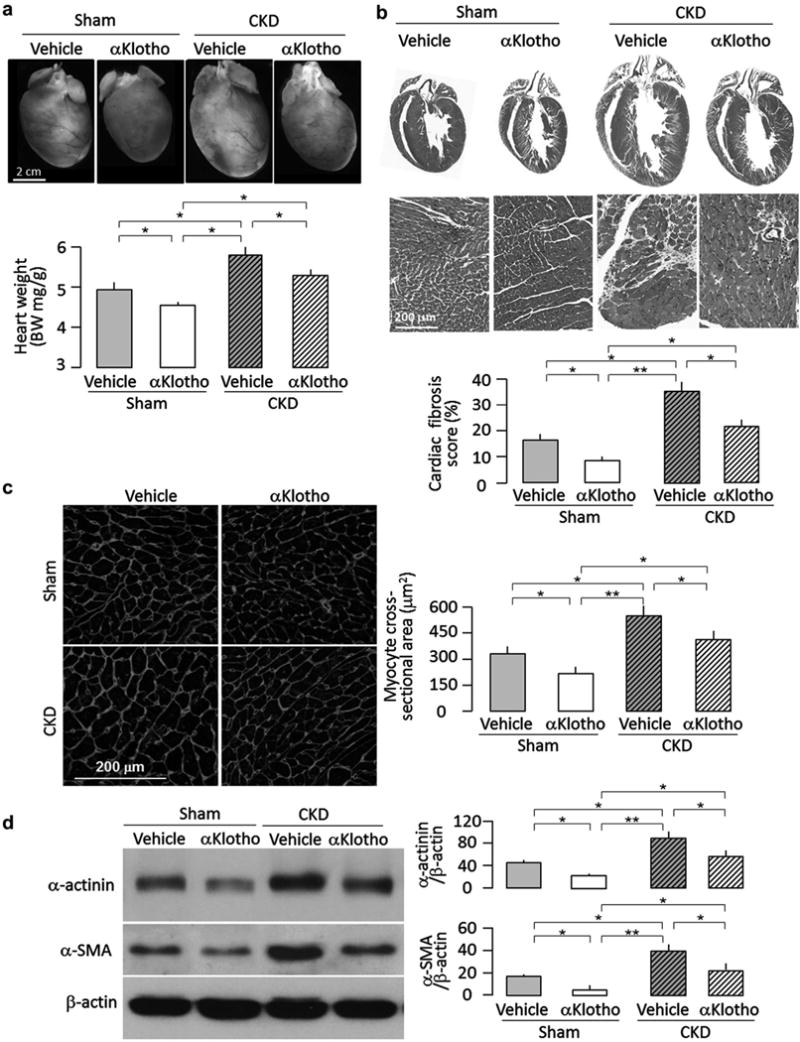
Animal experimental design was shown in Figure 3a. At 16 weeks after surgery, hearts were harvested for histology, immunoblot, and Immunohlstochemlstry. (a) Cardiac hypertrophy In CKD mice. Upper panel shows representative gross macrographs of hearts. Bottom panel shows a summary of ratio of heart weight to body weight for the examined mice, (b) Cardiac fibrosis in CKD mice. Upper panel shows representative macrographs of sagittal sections (trichrome stain). Middle panel shows representative micrographs of left ventricular sections (trichrome stain). Bottom panel is a summary of semiquantification of trichrome-positive area to whole heart section performed using ImageJ software. Data are expressed as means ± SD of 4 mice from each group, (c) Hypertrophic cardiomyocytes in CKD mice. Left panel shows representative micrographs of left ventricular sections stained with wheat germ agglutinin. Right panel is a summary of cardiomyocyte size calculated using ImageJ software, (d) Hypertrophic and fibrotic markers in the heart. Left panel shows representative immunoblotsfor α-actinin, α-smooth muscle actin (α-SMA), and β-actin protein. Right panel is a summary of normalized protein quantification from all examined immunoblots. Data are expressed as means ± SD of 4 mice from each group. Statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05, **P < 0.01 between 2 groups.
DISCUSSION
Several important conclusions can be drawn from this data set. First, the data further reinforce the notion that α-Klotho deficiency is not just a mere biomarker but a principal pathogenic factor in the development of CKD. Second, the data provide the proof-of-principle that recombinant α-Klotho protein replacement therapy in CKD is feasible, safe, and efficacious. Third, α-Klotho therapy can be prophylactic or therapeutic, and it has impacts on the kidney as well as on extrarenal organs such as the heart. Fourth, α-Klotho therapy is not just effective against CKD after ischemic AKI but also protects against phosphotoxic insults of the kidney.
Since the discovery of α-Klotho, a significant amount of efforts has been devoted to characterizing its role as an antiaging agent.1,2 The cytoprotective role of α-Klotho for cellular health is well-documented and undisputed.16,24,27,28 α-Klotho is 1 of the 3 members of the Klotho family (α, β, and γ) and is the only protein demonstrated to circulate in extracellular fluid including blood, urine, and cerebrospinal fluid.4 Studies using transgenic overexpression or viral-based gene delivery11,18,24,27,29 have shown that α-Klotho repletion in CKD models confers beneficial effects, strongly indicating that α-Klotho exerts a pathogenic role rather than being a mere biomarker.10,15,30 Several maneuvers have been predicted or shown to stimulate endogenous α-Klotho levels.31–33 For example, vitamin D analogues in rodents increased endogenous α-Klotho levels and decreased vascular calcification in a rodent CKD model.31 Regardless of whether there is adequate residual endogenous capacity to increase production, one can alternatively provide exogenous α-Klotho protein with the same rationale as for treatment with 25 and 1, 25-dihydroxyvitamin D and erythropoietin to correct deficiencies of these kidney-derived hormones in CKD. We previously demonstrated that intermittent i.p. injection of α-Klotho over days is feasible.14 In addition to studies using single-14,17,19 or multiple-dose injections over days with short follow-up,18 we now ascertained the long-term effects of recombinant α-Klotho therapy in this study. Ischemia-reperfusion injury (IRI)–induced AKI is followed by a period of α-Klotho deficiency, while replacement with exogenous α-Klotho after AKI hastens recovery of renal function and enhances return of endogenous renal and plasma α-Klotho levels.18 We now fully tested the hypothesis that α-Klotho protein administration is therapeutic and showed that this maneuver is effective in ameliorating onset and progression of CKD up to 20 weeks.
This finding has far-reaching clinical relevance. The consequences of AKI extend beyond its acute stage. Epidemiologic data have shown that subjects who have experienced an episode of AKI have an increased risk of CKD despite initial recovery.34 This poses a health care problem of colossal magnitude, and unfortunately there are no specific or efficacious countermeasures to prevent, delay, or reverse AKI-to-CKD progression. We previously showed that treatment of α-Klotho deficiency from post-AKI day 1 to 4 decreases renal fibrosis and increases creatinine clearance.18 We now extended the findings to a timeline compatible with human CKD after AKI. α-Klotho robustly decreased renal fibrosis, preserved renal function, and maintained phosphate homeostasis. A most encouraging result was that the grimmest extrarenal complication of CKD, pathologic cardiac remodeling, was appreciably attenuated.
Undoubtedly, the prospect of being able to prevent progression from AKI to CKD is highly significant. However, many patients present with established CKD, precluding the possibility of preventive therapy. In these subjects, 2 principal goals are to delay progression of CKD and prevent extrarenal complications of CKD that increase morbidity and mortality. We used a combination model of post-ischemic damage, renal mass reduction, and phosphate loading that fully exhibits the biochemical and histologic characteristics of human CKD.11,12 Contrary to the protocol in which α-Klotho was given immediately after IRI, we started α-Klotho therapy 4 weeks after IRI and contralateral nephrectomy, when endogenous α-Klotho deficiency and renal structural and functional lesions were already established. Three findings deserve comment: (1) the rate of progression of CKD was reduced; (2) extrarenal complications were ameliorated; and (3) the beneficial effect of α-Klotho was not limited to CKD but was also demonstrated in high-phosphate loading in the absence of CKD.
At 20 weeks after AKI induction, ClCr was more than 60% higher in the α-Klotho–treated mice compared to the vehicle-treated mice. This occurred even when α-Klotho treatment was started 4 weeks after induction, when renal damage was well under way. This potentially expands the therapeutic horizon of α-Klotho replacement to practically the entire CKD population. Although the effectiveness of α-Klotho in prevention or attenuation of kidney injury has been well-described in experimental models,12,14,18,24 the molecular mechanisms of renoprotection remain poorly defined. α-Klotho–mediated renoprotection and cytoprotection are proposed to be associated with, but not restricted to, suppression of apoptosis,12,24 cellular senescence,35,36 fibrosis,18 enhanced erythropoietin receptor signaling,37 antioxidation,27,38 and upregulation of autophagyflux.18 α-Klotho also improves phosphate homeostasis, which in turn is also associated with retardation of CKD progression.11,12,26 In addition, α-Klotho suppressed transient receptor potential channel 6in podocytes, ameliorated albuminuria by protecting the glomerular filter, and retarded CKD progression.39 α-Klotho–dependent tissue recovery after injury may be associated with preservation of stem cells.7 What is lacking for nearly 2 decades since the discovery of α-Klotho is the molecular target(s) of α-Klotho on the cell, such as the elusive “α-Klotho” receptor, if such an entity exists.
Cardiovascular complications are the major drivers of mortality in CKD.20,21 The mechanism of α-Klotho–mediated cardioprotection is complicated and may be associated with modulation of the transient receptor potential channels on the heart40 in addition to other mechanisms. Phosphotoxicity has received increasing attention as a pathogenic factor in the renal and extrarenai pathology in CKD.41,42 α-Klotho deficiency results in phosphate retention,11,17 and chronic high phosphate intake in rodents can compromise renal function even in the absence of CKD.18 For unclear reasons and via unknown mechanisms, high phosphate intake paradoxically suppresses endogenous phosphaturic α-Klotho.11,17,18 This study clearly shows that the detrimental renal and cardiac effects of chronic high-phosphate diet can be prevented by α-Klotho supplementation in the absence of CKD, which likely works at least partially via phosphaturia and subsequently via promotion of a negative systemic phosphate balance.
Epidemiologic data strongly support the association of FGF23 with cardiovascular disease and poor outcome in CKD.43–44 Faul et al. proposed a causal relationship in their finding of systemic injection of FGF23 leading to cardiac hypertrophy,23–45 possibly via activation of FGFR4.45 In contrast, other animal models dissociated FGF23 levels from cardiac hypertrophy.29 Using multiple animal models of high plasma FGF23, FGF23 levels can be correlated with cardiac hypertrophy and fibrosis only in the presence of α-Klotho deficiency,11 suggesting a multifactorial interactive model rather than single pathogenic factors. α-Klotho therapy abrogated chronic phosphate loading–induced pathologic cardiac remodeling and uremic cardiomyopathy with reduction of plasma iFGF23 and cFGF23. Whether the beneficial effects of α-Klotho on uremic cardiac remodeling are mediated by lowering of FGF23 actions remains to be explored.
In conclusion, early administration of α-Klotho prevented AKI-to-CKD progression and protected the heart from cardiac remodeling. Fate administration of α-Klotho still effectively improved renal function and attenuated cardiac remodeling, although renal function and morphology and cardiac remodeling were not returned to normal. Therefore, α-Klotho replacement might be a promising therapeutic strategy for retardation of AKI-to-CKD progression and improvement of uremic cardiomyopathy.
Supplementary Material
Figure S1. αKlotho administration after acute kidney injury (AKI) improved renal function and phosphate homeostasis, and maintained higher plasma αKlotho in ischemia-reperfusion injury (IRI)–induced AKI mice. (A) Animal experimental design. Sham-treated or IRI-induced AKI wild-type mice were injected i.p. with αKlotho protein (0.01 mg/kg) or vehicle (phosphate-buffered saline) for 4 consecutive days starting 24 hours after surgery. Throughout the experimental period, all mice were fed normal rodent chow. Blood and 24-hour urine were collected at 20 weeks after surgery for measurement of plasma as well as urine creatinine and plasma phosphate (Pi). (B) Creatinine clearance (ClCr). (C) Plasma Pi. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05, **P < 0.01 between 2 groups.
Figure S2. αKlotho administration after acute kidney injury (AKI) reduced renal fibrosis in ischemia-reperfusion injury (IRI)–induced AKI mice. Wild-type mice that underwent sham treatment or IRI-induced AKI were injected i.p. with αKlotho protein (0.01 mg/kg) or vehicle (phosphate-buffered saline) for 4 consecutive days starting 24 hours after surgery (depicted in Supplementary Figure S1A). Kidneys were collected at 20 weeks after surgery for kidney histology and molecular study. (A) Renal fibrosis in AKI versus sham mice at 20 weeks. Upper panel shows representative micrographs of trichrome-stained kidney sections. Bottom panel shows renal fibrosis scores calculated using ImageJ software. (B) α-Smooth muscle actin (α-SMA) protein expression in the kidney. Upper panel shows a representative immunoblotsfor α-SMA and β-actin protein in the kidney. Bottom panel shows a summary of normalized protein quantification from all examined immunoblots. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test, and accepted when: *P < 0.05, **P < 0.01 between 2 groups. Supplementary material is linked to the online version of the paper at www.kidney-international.org.
Acknowledgments
The investigators and the studies were supported by the National Institutes of Health (R01DK091392, R01-DK092461), the O’Brien Kidney Research Core (P30 DK-079328), and the Endowed Professor Collaborative Research Support Program of the Charles and Jane Pak Center for Mineral Metabolism and Clinical Research. The authors acknowledge Johanne Pastor, Han Cho, and Jean Paek for their valuable technical assistance.
Footnotes
DISCLOSURE
MK has a patent on Klotho peptides and antibodies; OM has consulted for AbbVie, Aliena, Ardelyx, Calico, Genzyme-Sanofi, and Takeda. All the other authors declared no competing interests.
References
- 1.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu MC, Shi M, Zhang J, et al. Renal production, uptake, and handling of circulating alphaKlotho. J Am Soc Nephrol. 2016;27:79–90. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CD, Tung TY, Liang J, et al. Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry. 2014;53:5579–5587. doi: 10.1021/bi500409n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Loon EP, Pulskens WP, van der Hagen EA, et al. Shedding of klotho by ADAMs in the kidney. Am J Physiol Renal Physiol. 2015;309:F359–F368. doi: 10.1152/ajprenal.00240.2014. [DOI] [PubMed] [Google Scholar]
- 6.Hu MC, Shiizaki K, Kuro-o M, et al. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–533. doi: 10.1146/annurev-physiol-030212-183727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bian A, Neyra JA, Zhan M, et al. Klotho, stem cells, and aging. Clin Interv Aging. 2015;10:1233–1243. doi: 10.2147/CIA.S84978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuro-o M. A potential link between phosphate and aging–lessons from Klotho-defident mice. Mech Ageing Dev. 2010;131:270–275. doi: 10.1016/j.mad.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindberg K, Amin R, Moe OW, et al. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169–2175. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker SL, Pastor J, Carranza D, et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015;30:223–233. doi: 10.1093/ndt/gfu291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu MC, Shi M, Cho HJ, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290–1302. doi: 10.1681/ASN.2014050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravikumar P, Li L, Ye J, et al. alphaKlotho deficiency in acute kidney injury contributes to lung damage. J Appl Physiol (1985) 2016;120:723–732. doi: 10.1152/japplphysiol.00792.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu MC, Shi M, Zhang J, et al. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010;78:1240–1251. doi: 10.1038/ki.2010.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu MC, Kuro-o M, Moe OW. Klotho and kidney disease. J Nephrol. 2010;23(Suppl 16):S136–S144. [PMC free article] [PubMed] [Google Scholar]
- 16.Chen TH, Kuro OM, Chen CH, et al. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. Eur J Pharmacol. 2013;698:67–73. doi: 10.1016/j.ejphar.2012.09.032. [DOI] [PubMed] [Google Scholar]
- 17.Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi M, Flores B, Gillings N, et al. αKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol. 2016;27:2331–2345. doi: 10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha SK, Hu MC, Kurosu H, et al. Regulation of renal outer medullary potassium channel and renal K(+) excretion by Klotho. Mol Pharmacol. 2009;76:38–46. doi: 10.1124/mol.109.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 2013;382(9889):339–352. doi: 10.1016/S0140-6736(13)60595-4. [DOI] [PubMed] [Google Scholar]
- 21.London GM. Left ventricular alterations and end-stage renal disease. Nephrol Dial Transplant. 2002;17(Suppl 1):29–36. doi: 10.1093/ndt/17.suppl_1.29. [DOI] [PubMed] [Google Scholar]
- 22.Aoyagi T, Kusakari Y, Xiao CY, et al. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2012;303:H75–H85. doi: 10.1152/ajpheart.00241.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panesso MC, Shi M, Cho HJ, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014;85:855–870. doi: 10.1038/ki.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82:516–524. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 26.Bian A, Xing C, Hu MC. Alpha Klotho and phosphate homeostasis. J Endocrinol Invest. 2014;37:1121–1126. doi: 10.1007/s40618-014-0158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravikumar P, Ye J, Zhang J, et al. alpha-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol. 2014;307:L566–L575. doi: 10.1152/ajplung.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikushima M, Rakugi H, Ishikawa K, et al. Anti-apoptotic and antisenescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun. 2006;339:827–832. doi: 10.1016/j.bbrc.2005.11.094. [DOI] [PubMed] [Google Scholar]
- 29.Xie J, Yoon J, An SW, et al. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. 2015;26:1150–1160. doi: 10.1681/ASN.2014040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu MC, Kuro-o M, Moe OW. The emerging role of Klotho in clinical nephrology. Nephrol Dial Transplant. 2012;27:2650–2657. doi: 10.1093/ndt/gfs160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau WL, Leaf EM, Hu MC, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–1270. doi: 10.1038/ki.2012.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narumiya H, Sasaki S, Kuwahara N, et al. HMG-CoA reductase inhibitors up-regulate anti-aging klotho mRNA via RhoA inactivation in IMCD3 cells. Cardiovasc Res. 2004;64:331–336. doi: 10.1016/j.cardiores.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Hu MC. Klotho connects intermedin1-53 to suppression of vascular calcification in chronic kidney disease. Kidney Int. 2016;89:534–537. doi: 10.1016/j.kint.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bucaloiu ID, Kirchner HL, Norfolk ER, et al. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012;81:477–485. doi: 10.1038/ki.2011.405. [DOI] [PubMed] [Google Scholar]
- 35.Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 2008;389:233–241. doi: 10.1515/BC.2008.028. [DOI] [PubMed] [Google Scholar]
- 36.Liu F, Wu S, Ren H, et al. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol. 2011;13:254–262. doi: 10.1038/ncb2167. [DOI] [PubMed] [Google Scholar]
- 37.Hu MC, Shi M, Cho HJ, et al. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013;84:468–481. doi: 10.1038/ki.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto M, Clark JD, Pastor JV, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280:38029–38034. doi: 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim JH, Xie J, Hwang KH, et al. Klotho may ameliorate proteinuria by targeting TRPC6 channels in podocytes [e-bup ahead of print] J Am Soc Nephrol. doi: 10.1681/ASN.2015080888. http://dx.doi.org/10.1681/ASN.2015080888. [DOI] [PMC free article] [PubMed]
- 40.Xie J, Cha SK, An SW, et al. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizuno M, Mitchell JH, Crawford S, et al. High dietary phosphate intake induces hypertension and augments exercise pressor reflex function in rats. Am J Physiol Regul Integr Comp Physiol. 2016;311:R39–R48. doi: 10.1152/ajpregu.00124.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selamet U, Tighiouart H, Sarnak MJ, et al. Relationship of dietary phosphate intake with risk of end-stage renal disease and mortality in chronic kidney disease stages 3–5: The Modification of Diet in Renal Disease Study. Kidney Int. 2016;89:176–184. doi: 10.1038/ki.2015.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez OM. Fibroblast growth factor 23, Klotho, and disordered mineral metabolism in chronic kidney disease: unraveling the intricate tapestry of events and implications for therapy. J Ren Nutr. 2013;23:250–254. doi: 10.1053/j.jrn.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf M, Molnar MZ, Amaral AP, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011;22:956–966. doi: 10.1681/ASN.2010080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grabner A, Amaral AP, Schramm K, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22:1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. αKlotho administration after acute kidney injury (AKI) improved renal function and phosphate homeostasis, and maintained higher plasma αKlotho in ischemia-reperfusion injury (IRI)–induced AKI mice. (A) Animal experimental design. Sham-treated or IRI-induced AKI wild-type mice were injected i.p. with αKlotho protein (0.01 mg/kg) or vehicle (phosphate-buffered saline) for 4 consecutive days starting 24 hours after surgery. Throughout the experimental period, all mice were fed normal rodent chow. Blood and 24-hour urine were collected at 20 weeks after surgery for measurement of plasma as well as urine creatinine and plasma phosphate (Pi). (B) Creatinine clearance (ClCr). (C) Plasma Pi. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test and accepted when: *P < 0.05, **P < 0.01 between 2 groups.
Figure S2. αKlotho administration after acute kidney injury (AKI) reduced renal fibrosis in ischemia-reperfusion injury (IRI)–induced AKI mice. Wild-type mice that underwent sham treatment or IRI-induced AKI were injected i.p. with αKlotho protein (0.01 mg/kg) or vehicle (phosphate-buffered saline) for 4 consecutive days starting 24 hours after surgery (depicted in Supplementary Figure S1A). Kidneys were collected at 20 weeks after surgery for kidney histology and molecular study. (A) Renal fibrosis in AKI versus sham mice at 20 weeks. Upper panel shows representative micrographs of trichrome-stained kidney sections. Bottom panel shows renal fibrosis scores calculated using ImageJ software. (B) α-Smooth muscle actin (α-SMA) protein expression in the kidney. Upper panel shows a representative immunoblotsfor α-SMA and β-actin protein in the kidney. Bottom panel shows a summary of normalized protein quantification from all examined immunoblots. Data are expressed as means ± SD of 4 mice from each group, and statistical significance was assessed by 1-way analysis of variance followed by Student-Newman-Keuls test, and accepted when: *P < 0.05, **P < 0.01 between 2 groups. Supplementary material is linked to the online version of the paper at www.kidney-international.org.