

Abstract
In this study, dual therapeutic-loaded GE11 peptide-conjugated liposomes were developed and applied to enhance therapeutic efficacies of standard-of-care regimens for the treatment of laryngeal cancer. The therapeutic strategy used here was a combination treatment with the chemotherapeutic docetaxel (DTX) and siRNA against the ABCG2 gene that regulates multidrug resistance in many tumor types. Liposome-encapsulated DTX/ABCG2–siRNA molecules were targeted to recognize tumor cells of squamous morphology by conjugation to the EGFR-targeting ligand, GE11. Targeted, drug-infused liposomes were nanosized and exhibited controlled release of DTX. Presence of GE11 peptides on liposomal surfaces enhanced the quantities of liposomal constructs taken up by Hep-2 laryngeal cancer cells. GE11 peptide-conjugated liposomes also enhanced cytotoxic effects against Hep-2 laryngeal cancer cells when compared to treatment with free DTX, thereby reducing IC50 values. Additionally, GE11 peptide-conjugated liposomes had significantly increased anti-tumor and apoptotic effects. Treatments with the GDSL nanoparticle formulation inhibited tumor growth in Hep-2 xenograft-bearing nude mouse models when compared to treatments with non-targeted NP constructs. Treatment of the mouse models with GE11 peptide-conjugated liposomes mitigated toxicities observed after treatment with free DTX. Taken together, liposomal encapsulation of DTX and ABCG2–siRNA improved the anti-tumor effects of treatment with free DTX in Hep-2 cell lines, and conjugation of GE11 peptides to liposomal constructs enhanced anti-tumor efficacies and specificities in laryngeal cancer cells.
Keywords: laryngeal cancer, docetaxel, ABCG2–siRNA, anti-tumor efficacy, liposomal chemotherapy carriers, GE11 peptide-targeted liposomes
Introduction
Cancer is one of the leading causes of death worldwide, with incidence rates increasing each year.1 Head and neck cancer is the sixth most common cause of cancer-related deaths worldwide.2,3 Laryngeal cancer, one of the most common subtypes of head and neck cancer, has an estimated annual mortality rate of ~100,000 persons and an annual incidence rate of 160,000 new cases. Treatment options for laryngeal cancers include surgery, radiotherapy, and chemotherapy.3,4 Surgery followed by concurrent chemotherapy is the standard clinical treatment modality.5
Docetaxel (DTX) is a mitotic chemotherapeutic agent widely indicated for the treatment of many malignancies. DTX acts by stabilizing the microtubule apparatus, thereby inhibiting cell division.6 DTX is a commonly used regimen in the treatment of laryngeal cancers. However, poor solubility and wide systemic side effects hamper the therapeutic potential of DTX.7 Systemic side effects of DTX include nephrotoxicity, neurotoxicity, and hypersensitivity reactions. Additionally, multidrug resistance (MDR) is a barrier of chemotherapeutic agents that reduces therapeutic efficacy rates and durability.8,9
Overexpressions of efflux pumps belonging to the ABC gene family are molecular mechanisms that contribute to MDR. Specifically, ABCG2 is overexpressed in many cancer cell types including laryngeal cancers.10 Previous data suggested that ABCG2 overexpression decreased cytotoxic effects of anticancer drugs. However, siRNA-mediated ABCG2 suppression inhibited cell proliferation.11,12 In this study, ABCG2–siRNA constructs (5′-CUGGAGAUGUUCUGAUAAA dTdT-3′) were employed to enhance the cytotoxic effects of DTX. In order to improve the physicochemical properties of DTX, to decrease the associated side effects, and to effectively deliver the ABCG2–siRNA, a novel therapeutic strategy was required.13
Among potential carrier molecules, liposomes are commonly investigated in clinical trials. Liposomes are composed of phospholipids arranged to create hydrophobic and hydrophilic cavities.14 Encapsulation of drugs in liposomes has increased the therapeutic efficacy of cargos and minimized associated side effects.15,16 Surface modifications of liposomes can reduce unwanted uptake by reticuloendothelial systems and can prolong blood circulation half-life times, thereby increasing therapeutic efficacies. Despite increased blood circulation times, lack of specificity in tumor tissues has hampered accumulation of chemotherapeutic agents in target tumor tissues.17,18 To enhance the accumulation of chemotherapeutic drugs in target tumor tissues, molecular markers that are overexpressed on the surfaces of laryngeal cancer cells need to be identified. EGFR is an important molecular marker that is overexpressed in squamous cell carcinomas, including laryngeal cancers.19,20 EGFR may serve as a suitable marker to target DTX and ABCG2–siRNA liposome-encapsulated molecules to laryngeal cancer cells. Previous studies have conjugated antibodies on the surface of nanoparticles (NPs) to increase the targeting specificity for cancer cells.21,22 Acharya et al and Tseng et al reported enhanced drug uptake in target cells when using EGFR-targeted drug delivery systems. A novel peptide, GE11 (YHWYGYT-PQNVI), was recently reported to possess targeting specificity against EGFR proteins. Enhanced intracellular deliveries and higher therapeutic efficacies of anti-tumor drugs were demonstrated using GE11 molecules as targeting ligands.23,24
The aim of this study was to increase the therapeutic efficacy of combined DTX and ABCG2–siRNA therapies in laryngeal cancer cells. Cationic liposomes were formulated and were loaded with DTX within the hydrophobic lipid bilayer. Additionally, ABCG2–siRNA polynucleotides were loaded on liposomal surfaces using electrostatic interactions. Liposomal surfaces were also conjugated with GE11 peptides as targeting ligands. The delivery system was tested to determine its physicochemical and biological characteristics. Additionally, a preclinical in vivo study was performed to assess the efficacy of the novel agent on Hep-2 laryngeal cancer cell-bearing nude mice.
Materials and methods
Materials
DTX was purchased from Sigma-Aldrich (St Louis, MO, USA). 1,2-Dioleoyl-sn-glycero-3-ethylphosphocholine (chloride salt) (EPC), cholesterol, (2,3-dioleoyloxy-propyl)-trimethylammonium (DOTAP), 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000)] (ammonium salt) (DSPE-PEG2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-poly(ethylene glycol)-maleimide (DSPE-PEG2000-MAL) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). GE11 (YHWYGYTPQNVIGGGGC, MW1872) was synthesized by GL Biochem Ltd. (Shanghai, China). All reagent-grade chemicals were used as received.
Preparation of drug-loaded liposomes and peptide conjugations
Combinations of EPC, DSPE-PEG2000, DSPE-PEG2000-MAL, DOTAP, and cholesterol were dissolved in an organic solvent in a round bottom flask and made into thin films using a rotary evaporator. Thin films were hydrated with phosphate-buffered saline (PBS) and extruded for 21 cycles using a mini-extruder (Avanti Polar Lipids, Inc.). To produce drug-loaded liposomes, DTX was dissolved with lipids during the thin film formation steps. To conjugate peptides to the liposomes, GE11 was dissolved in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, combined with liposomes containing DSPE-PEG2000-MAL (1:5 molar ratio) and incubated for 15 h in the dark. Unconjugated GE11 was removed using ultrafiltration tubes against distilled water. Cationic surface charges of NPs were employed to load ABCG2–siRNA onto the liposomes based on electrostatic interactions. The quantity of DTX loaded into the liposomes was evaluated using high-performance liquid chromatography (HPLC). ABCG2–siRNA conjugation rates were quantified using gel electrophoresis methods.
Particle size analyses
Hydrodynamic sizes and polydispersity indices (PDIs) of the particles were evaluated by dynamic light scattering methodologies using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. The formulations were suitably diluted and studied in triplicate.
Morphology analyses
The morphologies of individual NPs were studied using transmission electron microscopy (TEM; CM 200 UT; Philips, Amsterdam, the Netherlands). NPs were stained with 2% phosphotungstic acid, and a drop of each was placed on a carbon-coated copper grid. After 5 min, drops of NP solutions were cleared using tissue paper and dried under infrared radiation. The copper grid was then studied using TEM.
In vitro drug release assays
In vitro drug release assays of DTX from DTX-loaded liposome (DL) and GDSL were performed using dialysis methodologies. Briefly, DL and GDSL were dissolved in PBS (pH 7.4) and packed in dialysis tubes with molecular weight cutoffs of 3,000 Da. The tubes were then placed in a shaker water bath maintained at 100 rpm and 37°C. At predetermined time intervals, aliquots of sample were withdrawn and replaced with equal amounts of fresh PBS buffer. The amounts of drugs released into each PBS aliquot were evaluated using HPLC methods.
In vitro cell viability assays of Hep-2 cells
The Hep-2 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The effects of free DTX, ABCG2–siRNA, DL, DTX and siRNA-loaded liposome (DSL), and GDSL on Hep-2 cell viabilities were evaluated using MTT assays. In brief, Hep-2 cells were seeded onto 96-well plates at a seeding density of 1×104 cells/well. The cells were incubated for 24 h to allow attachment and growth. Cells were then treated with free DTX, DL, DSL, and GDSL in increasing concentrations and incubated for 24 h. Empty NP constructs were also applied to the Hep-2 cells and incubated for 24 h to ascertain toxic effects on the cells. After 24 h, cells were washed twice with PBS, MTT reagent aliquots (20 μL, 5 mg/mL) were added, and cells were incubated for an additional 24 h. Formazan crystals formed by the living cells were extracted by adding 100 μL of dimethyl sulfoxide (DMSO). Absorbance levels of the formazan crystals were evaluated at 570 nm using a microplate reader. Cell survival rates were normalized against untreated control cells. IC50 values were calculated using CompuSyn software (Version 1.0; ComboSyn Inc., Paramus, NJ, USA).
Apoptosis assays
Apoptosis rates of Hep-2 cancer cells were studied using an Annexin V/propidium iodide (PI) staining protocol. Briefly, cells were seeded at densities of 2×105 cells/well in a 12-well plate and incubated for 24 h. Media were refreshed with new aliquots containing free DTX, ABCG2–siRNA, DL, DSL, or GDSL, and then cells were incubated for 24 h. Following incubation, cells were washed twice with PBS and extracted. Cells were centrifuged, and pellets were reconstituted with binding buffer (100 μL). Cells were stained with 2 μL of Annexin and 2 μL of PI and incubated for 15 min. Cell volumes were brought up to 1 mL and studied using flow cytometric analyses (FACS Calibur flow cytometer; BD Biosciences, San Jose, CA, USA).
Hoechst 33382 staining assays
Apoptosis rates of Hep-2 cancer cells were studied using Hoechst 33382 staining methodologies. Briefly, cells were seeded at densities of 2×105 cells/well in 12-well plates and incubated for 24 h. Media were refreshed with new aliquots containing free DTX, ABCG2–siRNA, DL, DSL, or GDSL and then incubated for 24 h. Following incubation, cells were washed twice and fixed with 4% paraformaldehyde for 10 min. Cells were washed again and stained with Hoechst 33382 (10 μg/mL) for 10 min. The cells were washed and mounted, and cell nuclear morphologies were observed using fluorescence microscopy.
In vivo anti-tumor efficacy studies
Animal study protocols were approved by the Institutional Animal Ethics Committee of Shandong University, China and the handling and animal experiments were carried out as per the guidelines framed by the Animal Healthcare Committee, Shandong University, China. In vivo anti-tumor efficacies of the GE11-targeting liposomal DTX/ABCG2–siRNA constructs were performed in Hep-2-bearing tumor xenograft models. Briefly, 5×106 cells were subcutaneously injected into the right flank of nude mice and allowed to proliferate until masses reached a size of 80–100 mm3. Mice were randomly divided into 6 groups including controls, free DTX, ABCG2–siRNA, DL, DSL, and GDSL. Three fixed doses of DTX (5 mg/kg) were intravenously injected into mice every third day. Tumor volumes and body weights were measured at regular intervals. Tumor volumes were calculated by measuring lengths and widths of the tumors. The mice were humanely sacrificed at the study end point.
Statistical analyses
Analysis of variance (ANOVA) studies were performed on data gathered from all assays using one-way ANOVA methodologies. P-values <0.05 were considered statistically significant.
Results and discussion
Laryngeal cancer is a common subtype of head and neck cancer. Surgery followed by concurrent chemotherapy is the standard clinical treatment modality. DTX is a mitotic chemotherapeutic agent widely indicated for the treatment of many malignancies. However, systemic side effects and MDR of DTX reduce its therapeutic efficacy. The ABCG2 gene is overexpressed in laryngeal cancers and functions to remove DTX. In this study, ABCG2–siRNA constructs (5′-CUGGAGAUGUUCUGAUAAA dTdT-3′) were employed to enhance the cytotoxic effects of DTX. To target delivery of the chemotherapeutic drugs and the ABCG2–siRNA to laryngeal cancer cells, a stable liposome-based NP delivery system was designed. To target the system to laryngeal tumor cells, a recently described EGFR-targeting peptide (GE11; YHWYGYTPQNVI) was utilized on the liposomal constructs. Cationic liposomes were formulated in which DTX was loaded in the hydrophobic lipid bilayers of the liposomes, and ABCG2–siRNA molecules were loaded on liposomal surfaces using electrostatic interactions. Surfaces of the liposomes were also conjugated with GE11 peptides to target them to EGFR-expressing cells (Figure 1).
Figure 1.
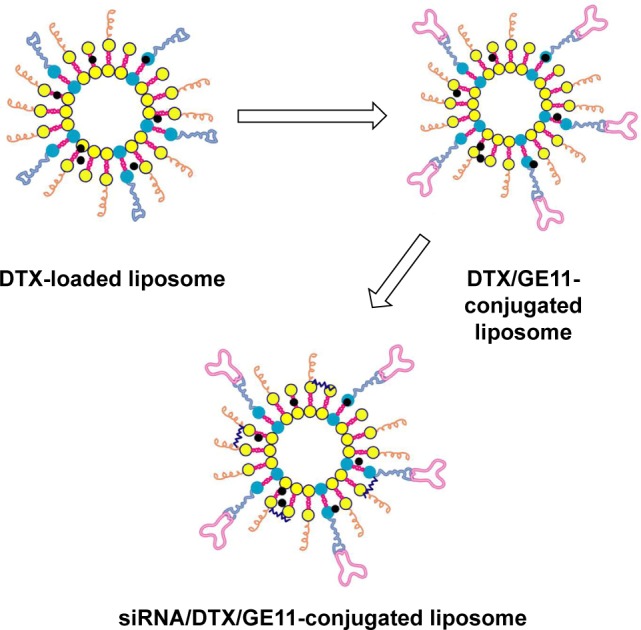
Schematic presentation of the steps for preparation of DTX and ABCG2–siRNA-loaded liposomes and conjugation of GE11 peptides on liposomal surfaces.
Abbreviation: DTX, docetaxel.
Physicochemical analyses of GE11 peptide-conjugated DTX/ABCG2–siRNA-loaded liposomes
DTX-loaded liposomes were prepared using thin film hydration methodologies. Liposomes were prepared in the presence of DSPE-PEG2000-MAL to promote conjugation of GE11 peptides to liposomal surfaces as active targeting ligands. The mean diameters of DTX/liposomes were ~110 nm with good dispersity indices. Conjugations of GE11 peptides increased NP sizes to approximately 150 nm, indicating the presence of targeting ligands on the surfaces of the DTX/liposomes (Figure 2A). Final DTX/ABCG2–siRNA-loaded NPs were of mean size ~180 nm with acceptable PDIs. Previous data have shown that liposomes that are smaller than 200 nm can be targeted to tumor cells and will accumulate in the tumors via enhanced permeation and retention effects.25 Moreover, the presence of polyethylene glycol on liposomal surfaces has been shown to enhance blood circulation rates of NPs in systemic environments. Particle sizes of the liposomal constructs were further confirmed by TEM imaging. DTX/liposomes were nanosized and uniformly distributed on TEM grids (Figure 2B). GE11-conjugated DTX/ABCG2–siRNA-loaded liposomes were slightly larger than DTX/liposomes and maintained spherical morphologies, indicating that surface modifications did not alter the shapes of the NPs.
Figure 2.

Physicochemical analyses of NPs.
Notes: (A) Particle size distributions of DL, DSL, and GDSL constructs. (B) TEM images of DL, DSL, and GDSL constructs; scale bars = 100 nm.
Abbreviations: DL, DTX-loaded liposome; DSL, DTX and siRNA-loaded liposome; DTX, docetaxel; NP, nanoparticle; TEM, transmission electron microscopy.
Drug-release studies
Concentrations of NPs used in the in vitro drug release assays were determined by dialysis methods in PBS (pH 7.4) at 37°C. Dialysis membranes separating molecules with molecular weights of 3,500 Da were chosen to retain the NP, while lower molecular weight unbound particles diffused into the external media. Sustained releases of DTX were observed from the NP constructs (Figure 3). Notably, relatively lower DTX release rates were observed from GDSL than DL (~68% and 80%, respectively). Slightly lower drug release rates were likely due to presence of the GE11 peptide on liposomal surfaces, which may extend path lengths for drug diffusions. A lack of unwanted liposome/DTX burst release events could minimize adverse events after chemotherapy treatments in systemic environments. Sustained DTX release times coupled with enhanced systemic circulation times of NP systems could be advantageous for cell-targeting downstream applications.26
Figure 3.

In vitro drug release assays measuring loss of DTX from DL and GDSL constructs.
Note: The release study was performed using dialysis methods at 37°C.
Abbreviations: DL, DTX-loaded liposome; DTX, docetaxel.
Cellular uptake analyses
Cellular uptake analyses of non-targeting and targeting liposomal formulations were first performed using flow cytometric methodologies. NPs were developed that encapsulated rhodamine-B as a fluorescent marker. Mean fluorescent intensity of GDSL constructs in Hep-2 cancer cells was significantly greater than mean fluorescent intensity of the non-targeted liposomal formulations (Figure 4). High fluorescence intensity measurements of GDSL constructs were attributed to preferential uptake of EGFR-bound GE11 peptide ligands present on the NP surfaces. Cellular uptake rates and mechanisms were further studied using confocal laser scanning microscopy (CLSM). Consistent with the flow cytometric analyses, CLSM analyses showed high intracellular fluorescence signals in Hep-2 cells treated with the GDSL formulation. Notably, strong rhodamine-B fluorescence intensities were observed in cytoplasmic regions, suggesting that NPs were internalized via endocytosis-mediated mechanisms. Previous literature suggested that entrapped dye would be released into acidic compartments before diffusing to active sites. Thus, we hypothesize that the rhodamine-B dye accumulated in the Hep-2 cells in acidic organelles such as endosomes or lysosomes. High cellular uptake of the NPs was expected to increase the DTX concentrations inside Hep-2 cells, thereby improving anti-tumor efficacies.
Figure 4.

In vitro cellular uptake assays of DSL and GDSL constructs into Hep-2 cancer cells.
Notes: (A) Flow cytometer analyses and (B) CLSM analyses. Scale bars = 20 μm.
Abbreviations: CLSM, confocal laser scanning microscopy; DAPI, 4′,6-diamidino-2-phenylindole, dihydrochloride; DSL, DTX and siRNA-loaded liposome; DTX, docetaxel; PE-A, flow cytometer fluorochrome conjugate.
In vitro cytotoxic effects of GE11-targeted DTX/ABCG2-liposomal NPs
The most critical therapeutic aspect of using NPs as delivery systems for tumor cells is improved safety of the reagents. Thus, cytotoxicity analyses were performed to assess safety of the NPs. As shown in Figure 5, NPs that were not loaded with DTX, ABCG2–siRNA, or GE11-targeting peptides did not affect viabilities of cancer cells, even when applied at high concentrations. Indeed, cell viability rates remained >90% at the 100 μg/mL concentration, indicating excellent biocompatibility of this nanoliposomal system (Figure 5A).
Figure 5.

In vitro cytotoxic analyses of (A) empty NPs and (B) DTX/ABCG2–siRNA-loaded NPs.
Notes: The cells were treated with a concentration gradient of each NP formulation and then evaluated by MTT assays. **P<0.001 is the statistical difference between DL and GDSL.
Abbreviations: DL, DTX-loaded liposome; DTX, docetaxel; DSL, DTX and siRNA-loaded liposome; DTX, docetaxel; NP, nanoparticle.
Next, in vitro cytotoxicities of free DTX, as well as DL-, DSL-, and GDSL-loaded NPs, were tested on Hep-2 laryngeal cancer cells. All formulations exhibited concentration-dependent cytotoxic effects. Drug-loaded liposome showed higher cytotoxicities compared to treatment with free DTX. The DL cytotoxic effects were further increased when combined with ABCG2–siRNA (Figure 5B), suggesting that inclusion of ABCG2–siRNA silenced MDR mechanisms and synergized with DTX. Furthermore, application of GE11-targeted DTX/ABCG2–siRNA liposomes had significantly higher cytotoxic effects than any of the other formulations. Additionally, IC50 values for GDSL particles were lower than with any other formulation. Enhanced cytotoxic effects of the GE11-targeted NPs may have resulted from enhanced cellular uptake rates in Hep-2 cancer cells owing to the presence of the targeting ligand binding to EGFR. This conclusion was consistent with a previous report showing that GE11 peptides mediated target-specific delivery of reporter genes to EGFR-overexpressing tumor cells in vitro.
Apoptosis assays
Anti-tumor efficacies of individual NP formulations were further evaluated using flow cytometry-based apoptosis assays. Hep-2 cells were treated with each NP formulation and stained with Annexin-V/PI staining kits (Figure 6). DTX-treated cells induced nearly 13% of cells to early apoptosis and 4% of cells to late apoptosis. Liposomes conjugated to ABCG2–siRNA induced 10% of cells to undergo apoptosis. DTX-loaded liposomes induced nearly 25% of cells to undergo apoptosis, indicating that the NP constructs effectively delivered the drug with anti-tumor effects. Importantly, application of the GDSL constructs induced significantly higher early (45%) and late (20%) apoptotic effects in the laryngeal cancer cells. Treatment with GDSL constructs caused an increased population of early and late apoptotic cancer cells making them the most effective means of delivering anti-tumor drugs tested here. The enhanced apoptotic effects were most likely due to enhanced cellular accumulation of GDSL NPs through EGFR-mediated endocytosis events. Furthermore, inclusion of MDR-inhibiting ABCG2–siRNA may have further enhanced DTX anti-tumor effects in this system.
Figure 6.

Annexin-V/PI-based apoptosis assays of Hep-2 cells.
Notes: Cells were treated with free DTX, ABCG2–siRNA, DL, DSL, or GDSL constructs. The apoptosis rates were evaluated using flow cytometric analyses.
Abbreviations: DL, DTX-loaded liposome; DTX, docetaxel; DSL, DTX and siRNA-loaded liposome; FITC, fluorescein isothiocyanate; LL, lower left; LR, lower right; PI, propidium iodide; UL, upper left; UR, upper right.
Quantifications of nuclear apoptosis
Nuclear apoptotic reactions of Hep-2 cancer cells were identified using Hoechst 33382 staining techniques. The control cell nuclei remained the same size and shape while nuclei treated with the NP formulations became irregular and distorted in shape (Figure 7). Apoptotic nuclear morphologies were observed after treatment with DTX and ABCG2–siRNA. After treatment with GDSL, apoptotic morphologies of the cells became more prominent, suggesting that the GDSL liposomes had enhanced anti-tumor effects. Targeting by the GE11 peptide may have enhanced cellular uptake rates of the NP constructs and induced greater apoptosis of the Hep-2 cancer cells. Prominent changes in the morphologies of cells treated with optimized NP formulations suggested that the final formulations would have promising anti-tumor effects in vivo.27
Figure 7.

Hoechst 33342-based apoptosis assays of Hep-2 cells.
Notes: Cells were treated with free DTX, ABCG2–siRNA, DL, DSL, or GDSL constructs. Nuclear morphologies of apoptotic cells were visualized using Hoechst staining.
Abbreviations: DL, DTX-loaded liposome; DSL, DTX and siRNA-loaded liposome; DTX, docetaxel.
In vivo anti-tumor efficacy assays
Xenograft nude mice were implanted with Hep-2 laryngeal cancer cells and were subsequently injected with each of the NP formulations. Anti-tumor efficacies of each formulation were evaluated by measuring tumor volumes after specified periods of time ranging from 0 to 25 days (Figure 8A). Treatment of the mouse models with free DTX, ABCG2–siRNA, DL, DSL, and GDSL inhibited tumor growth when compared with control mice (animals implanted with Hep-2 cells and not treated). Administration of free DTX or ABCG2–siRNA alone did not induce appreciable tumor inhibition. In contrast, nude mice treated with DL exhibited inhibition of tumor growth. DL-induced anti-tumor effects may be attributed to increased longer blood circulation times or better intracellular uptake of the constructs into Hep-2 xenografts. Similarly, animals treated with DSL constructs had inhibited tumor growth in the xenografts with lower tumor volumes when compared to animals treated with DL. Finally, animals treated with GDSL had significantly higher anti-tumor effects, with final tumor volumes of 450 mm3, rather than the 1,800 mm3 in mice implanted with untreated tumors. These results suggest that NP encapsulation of DTX and ABCG2–siRNA has anti-tumor efficacy in xenograft models implanted with Hep-2 cancer cells.
Figure 8.

In vivo anti-tumor efficacies of Hep-2 xenograft-implanted murine models.
Notes: Mice bearing Hep-2 tumors were treated with NP formulations, as listed, and (A) tumor volumes were measured on days 0–25. (B) Body weights were measured to determine toxicity of the treatments. *P<0.05; ***P<0.0001.
Abbreviations: DL, DTX-loaded liposome; DSL, DTX and siRNA-loaded liposome; DTX, docetaxel; NP, nanoparticle.
Clinical efficacies of DTX are hindered by concomitant systemic toxicities. Here, adverse effects of DTX treatment were measured by monitoring body weights of the subjects (Figure 8B). Treatment of xenograft-implanted mice with free DTX-induced toxicity resulted in a >20% mean body weight loss. In contrast, subjects treated with DL, DSL, or GDSL constructs did not experience losses of body weight, suggesting that treatment with NP constructs prevented some systemic side effects.
Combined treatment with ABCG2–siRNA and DTX effectively induced anti-tumor activities in Hep-2 cells. NP encapsulation of DTX resulted in higher therapeutic effects, possibly due to prolonged blood circulation times and enhanced accumulations of the carrier molecules in tumor tissues. The enhanced permeation and retention effects of the NPs may explain higher accumulations of these particles in tumor cells.28 In addition, surface conjugation of EGFR-targeting GE11 ligands resulted in increased anti-tumor effects of the NPs, presumably due to targeting of the particles to cancer cells. These findings were consistent with a previous report showing that the EGFR-targeting peptide GE11 mediated target-specific delivery of reporter genes to EGFR-overexpressing tumor cells in vitro.
Conclusion
The data presented here describe dual therapeutic-loaded GE11 peptide-conjugated liposomes that enhanced the combined therapeutic efficacies of DTX and ABCG2–siRNA in laryngeal cancers. The synthesized particles were nanosized and exhibited controlled release of DTX. GE11 surface peptides enhanced cellular uptake of the constructs in Hep-2 laryngeal cancer cells. The targeted liposomes had significantly higher cytotoxicities than free DTX and reduced the IC50 values. Consistently, GE11 peptide-conjugated liposomes enhanced anti-tumor and apoptotic effects compared to the non-targeted NPs. Additionally, treatment of Hep-2 xenograft-implanted nude mice with the GDSL NP formulation led to smaller tumors than those observed in animals receiving non-targeted NP constructs. Finally, animals receiving GDSL constructs exhibited reduced toxicity when compared to those receiving free DTX. Overall, treatment of laryngeal cancer cells with NP-encapsulated DTX and ABCG2–siRNA enhanced anti-tumor effects of DTX, and conjugation of the loaded liposomes with GE11 peptides targeted the constructs to laryngeal cancer cells.
Acknowledgments
Research presented here was supported by a grant from Shandong University, China.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Rousseau A, Badoual C. Head and neck: squamous cell carcinoma: an overview. Atlas Genet Cytogenet Oncol Haematol. 2012;16:145–155. [Google Scholar]
- 2.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Parkin D, Bray F. Chapter 2: the burden of HPV-related cancers. Vaccine. 2006;24(suppl 3):11–25. doi: 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 4.Marioni G, Marchese-Ragona R, Cartei G, Marchese F, Staffieri A. Current opinion in diagnosis and treatment of laryngeal carcinoma. Cancer Treat Rev. 2006;32(7):504–515. doi: 10.1016/j.ctrv.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Dietz A, Rudat V, Dreyhaupt J, et al. Induction chemotherapy with paclitaxel and cisplatin followed by radiotherapy for larynx organ preservation in advanced laryngeal and hypopharyngeal cancer offers moderate late toxicity outcome (DeLOS-I-trial) Eur Arch Otorhinolaryngol. 2009;266(8):1291–1300. doi: 10.1007/s00405-008-0846-y. [DOI] [PubMed] [Google Scholar]
- 6.Borrajo E, Abellan-Pose R, Soto A, et al. Docetaxel-loaded polyglutamic acid-PEG nanocapsules for the treatment of metastatic cancer. J Control Release. 2016;238:263–271. doi: 10.1016/j.jconrel.2016.07.048. [DOI] [PubMed] [Google Scholar]
- 7.Mirzaie ZH, Irani S, Mirfakhraie R, et al. Docetaxel-chitosan nanoparticles for breast cancer treatment: cell viability and gene expression study. Chem Biol Drug Des. 2016;88(6):850–858. doi: 10.1111/cbdd.12814. [DOI] [PubMed] [Google Scholar]
- 8.Dadras P, Atyabi F, Irani S, et al. Formulation and evaluation of targeted nanoparticles for breast cancer theranostic system. Eur J Pharm Sci. 2016;97:47–54. doi: 10.1016/j.ejps.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Ramasamy T, Ruttala HB, Kanu BG, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. doi: 10.1016/j.jconrel.2017.04.043. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Zhao JY, Qian XC, Cheng ZY, Liu Y, Wang Z. RASAL1 attenuates gastric carcinogenesis in nude mice by blocking RAS/ERK signaling. Asian Pac J Cancer Prev. 2015;16(3):1077–1082. doi: 10.7314/apjcp.2015.16.3.1077. [DOI] [PubMed] [Google Scholar]
- 11.de Jonge-Peeters SD, Kuipers F, de Vries EG, Vellenga E. ABC transporter expression in hematopoietic stem cells and the role in AML drug resistance. Crit Rev Oncol Hematol. 2007;62(3):214–226. doi: 10.1016/j.critrevonc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Priebsch A, Rompe F, Tonnies H, et al. Complete reversal of ABCG2-depending atypical multidrug resistance by RNA interference in human carcinoma cells. Oligonucleotides. 2006;16(3):263–274. doi: 10.1089/oli.2006.16.263. [DOI] [PubMed] [Google Scholar]
- 13.Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012;7(4):597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramasamy T, Kim JH, Choi JY, et al. pH sensitive polyelectrolyte complex micelles for highly effective combination chemotherapy. J Mater Chem B. 2014;2:6324. doi: 10.1039/c4tb00867g. [DOI] [PubMed] [Google Scholar]
- 15.Tran TH, Ramasamy T, Troung DH, Choi HG, Yong CS, Kim JO. Preparation and characterization of fenofibrate-loaded nanostructured lipid carriers for oral bioavailability enhancement. AAPS PharmSciTech. 2014;15(6):1509–1515. doi: 10.1208/s12249-014-0175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 17.Kim CS, Duncan B, Creran B, Rotello VM. Triggered nanoparticles as therapeutics. Nano Today. 2013;8(4):439–447. doi: 10.1016/j.nantod.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. doi: 10.1016/j.actbio.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Sheng Q, Liu J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br J Cancer. 2011;104(8):1241–1245. doi: 10.1038/bjc.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vidal F, de Araujo WM, Cruz AL, Tanaka MN, Viola JP, Morgado-Díaz JA. Lithium reduces tumorigenic potential in response to EGF signaling in human colorectal cancer cells. Int J Oncol. 2011;38(5):1365–1373. doi: 10.3892/ijo.2011.955. [DOI] [PubMed] [Google Scholar]
- 21.Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials. 2009;30(29):5737–5750. doi: 10.1016/j.biomaterials.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Tseng CL, Su WY, Yen KC, et al. The use of biotinylated-EGFmodified gelatin nanoparticle carrier to enhance cisplatin accumulation in cancerous lungs via inhalation. Biomaterials. 2009;30(20):3476–3485. doi: 10.1016/j.biomaterials.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19(14):1978–1985. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 24.Milane L, Duan Z, Amiji M. Therapeutic efficacy and safety of paclitaxel/lonidamine loaded EGFR-targeted nanoparticles for the treatment of multi-drug resistant cancer. PLoS One. 2011;6(9):24075–24086. doi: 10.1371/journal.pone.0024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming X, Carver K, Wu L. Albumin-based nanoconjugates for targeted delivery of therapeutic oligonucleotides. Biomaterials. 2013;34(32):7939–7949. doi: 10.1016/j.biomaterials.2013.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng XH, Wang Y, Huang D, et al. Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin-cisplatin nanoparticles. ACS Nano. 2011;5(12):9480–9493. doi: 10.1021/nn202410f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Master A, Malamas A, Solanki R, Clausen DM, Eiseman JL, Sen Gupta A. A cell-targeted photodynamic nanomedicine strategy for head and neck cancers. Mol Pharm. 2013;10(5):1988–1997. doi: 10.1021/mp400007k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48:131–143. doi: 10.1016/j.actbio.2016.10.034. [DOI] [PubMed] [Google Scholar]