

Abstract
Detection of diarrheagenic E. coli (DEC) typically depends on identification of virulence genes from stool cultures, not on stool itself. We developed a multiplex PCR assay that detects key DEC virulence genes (stx1, stx2, eae, bfpA, ipaH, LT, STh, aaiC, aatA). The assay involved a multiplex PCR reaction followed by detection of amplicon (s) using Luminex beads. The assay was evaluated on over 100 colony and broth specimens. We then evaluated the assay using DNA extracted from stool, colony pools and gram-negative broths, using stool spiked with known quantities of DEC. Performance of the assay on stool DNA was most quantitative, while stool broth DNA offered the lowest limit of detection. The assay was prospectively evaluated on clinical specimens in Tanzania. Stool DNA yielded higher sensitivity than colony pools compared with broth DNA as the standard. We propose using this assay to screen for DEC directly in stool or stool broths.
Introduction
Diarrhea is a major cause of global childhood mortality, leading to 1.5 million deaths each year or 15% of attributable mortality (Fischer Walker et al. 2010). The list of enteropathogens that can cause diarrhea is long and includes bacteria, viruses, protozoa, and helminths. DEC are major bacterial causes, however since E. coli are also a major component of normal microflora, identifying those with diarrheagenic potential requires cumbersome phenotypic or genotypic testing for virulence determinants. On the basis of these virulence determinants DEC are often categorized into Shiga-toxin producing (STEC), enteropathogenic (EPEC), enterotoxigenic (ETEC), enteroinvasive (EIEC), enteroaggregative (EAEC), and an EAEC subset termed Diffusely Adherent E. coli. A fatal outbreak of sprout-associated E. coli recently occurred in Germany, and this strain had both Shiga-toxin producing (stx2) and enteroaggregative features (Rasko et al. 2011).
Given the complexity of phenotypic assays to discriminate the DEC, PCR methods that amplify DEC virulence genes have proliferated, including singleplex PCR, multiplex PCR followed by gel-based analysis, and real-time PCR. The number of virulence targets that one needs to detect is often 8 or more, including chromosomal, plasmid, and phage DNA, thus the multiplex assays often need to be separated into multiple reactions. As examples, the assay of Fujioka et al. for stx1, stx2, eae, invE, astA, aggR, ST, and LT genes was separated into two amplifications (Fujioka et al. 2009). Aranda et al. also described two amplifications for eae, bfpA, CVD432, LT, ST, ipaH, and stx1 (Aranda et al. 2004). Gel electrophoresis is often used to distinguish amplicons, although band size discrimination can be difficult. Guion et al. described an 8-plex assay for DEC that used a real-time PCR platform and melt curves to discriminate products (Guion et al. 2008). In contrast to these methods, we describe a single multiplex PCR reaction that amplifies 9 virulence targets where amplicons are distinguished using sequence-specific probes on a Luminex platform. Advantages to this technique are the additional layer of specificity due to the probes and the ability to add additional targets given the multiplexing capability of the Luminex platform.
Most E. coli detection schemes utilize E. coli cultures. Specifically, stool is cultured on solid media such as MacConkey agar, a variable number of lactose fermenting colonies are picked to sample a diversity of E. coli strains, after which testing for virulence genes occurs. Both the culture and colony picking steps can be problematic. In many settings individuals with diarrhea are treated empirically with antibiotics, which hamper the sensitivity of culture. Picking colonies is tedious and creates an enormous amount of work for the laboratory. Most laboratories, therefore pool colonies for testing (Barletta et al. 2009), however the number of colonies to pool remains an open question. Galbadage et al. found that testing 20 colonies detected ETEC in twice as many individuals as testing 5 (Galbadage et al. 2009). This suggests potentially significant underestimation of DEC based on the colony picking process alone. For these reasons we investigated the performance of our assay on colonies versus broths, where theoretically all growth can be sampled, versus stool itself.
Materials and Methods
Strains, cultured specimens, and spiked materials
Positive control materials used in this study included reference E. coli strains EHEC 0157:H7, EPEC 2348/69, EIEC O124, ETEC H10407, and EAEC O42. Other materials included clinical DEC isolates from Armed Forces Research Institute of Medical Sciences (AFRIMS), Bangkok, Thailand and stool-containing broth specimens from the Virginia Division of Consolidated Laboratory Services (DCLS). Negative controls included E. coli strain ATCC 25922 (no virulence genes) and water. For sensitivity testing, individual reference strains of known colony forming units (cfu) were spiked into 200 mg aliquots of stool from a healthy DEC-negative donor and stored at −80°C until further processing or DNA isolation. Spiked stool specimens were plated onto MacConkey agar plates and incubated it at 37°C overnight, and inoculated a swab into 2ml of gram-negative (GN) broth followed by incubation for 16 hours in a shaking 37°C incubator.
Clinical stool specimens
Fifty-four fecal samples from inpatients with diarrhea were prospectively tested at Kilimanjaro Christian Medical Center in Moshi, Tanzania. All protocols were approved by the Ethics Committees of KCMC and the institutional review boards of the University of Virginia (UVA). Fresh stool samples were plated onto MacConkey agar using a sterile polyester swab and a GN broth (Becton Dickinson, Franklin Lakes, NJ) was inoculated with a pea-sized piece of stool. After overnight incubation 5 morphologically distinct lactose-fermentingcolonies were picked from the MacConkey agar and subcultured onto Trypticase Soy Agar with 5% Sheep blood (Becton Dickinson, Franklin Lakes, NJ).
DNA extraction
Lactose-fermentingcolonies were picked from the MacConkey agar, subcultured and suspended in 1 ml 0.05% Triton solution in a 2 ml screw top tube. The solution was briefly vortexed, heated at 100°C for 10 min, then centrifuged at 13000 rpm for 3 min, and 5 μl of supernatant was used for PCR. DNA was extracted from the GN broth by boiling or by using the QuickGene-810 and the QuickGene DNA tissue kit S (Fujifilm, Tokyo, Japan; http://www.fujifilm.com/products/life_science_systems/nucleic_acid_isolation/guides/pdf/common/D_Helicobacter_pylori_E.pdf). Stool DNA was isolated from 200 mg of stool using a slightly modified QIAmp Stool Mini Kit protocol (Qiagen Inc., Valencia, CA). Briefly, 1.4 ml of ASL buffer was added to the stool then pretreated by bead beating with 0.15-mm garnet beads (MO-BIO Laboratories, Carlsbad, CA) for 2 min followed by boiling for 7 min before continuing with the manufacturer’s extraction protocol. All DNA samples were stored at −80°C prior to use in PCR.
Luminex Multiplex PCR
Accepted virulence genes were targeted for DEC identification. Primer sequences for stx1 (Hidaka et al. 2009), stx2 (Paton and Paton 1998), ipaH (Vu et al. 2004), ial (Frankel et al. 1990), LT (Hidaka et al. 2009), aaiC (Boisen et al. 2008), and aatA (Schmidt et al. 1995) have been described previously (Table 1). Other primers were designed for targets that did not amplify efficiently in multiplex or when the amplicon was not suitable for Luminex detection. Multiple primer and probe combinations were evaluated before the final optimized multiplex PCR reaction. PCR amplification for the 9-plex reaction was performed in 25 μl volume with 12.5 μl of Qiagen Multiplex PCR Master Mix (Qiagen, Valencia, CA, USA) which contains dNTPs and 3 mmol/L MgCl2 final concentration, 0.2 μmol/L of each forward primer, 0.2 μmol/L of each reverse biotinylated (modified with biotin-TEG) primer, 5 μl Q-solution, 0.5–3.5 μl of nuclease free water (NFW) and 2–5 μl of DNA template. PCR was performed in a 9-plex at AFRIMS (ial target instead of ipaH target) and in a 6-plex at DCLS (targets included were stx1, stx2, LT, STh, eaeA, bfpA). PCR cycling condition consisted of an initial 15-min 95°C step followed by 40 cycles of 30 sec at 94°C, 30 sec at 60°C, 60 sec at 72°C, and a final 10-min extension 72°C step. Positive controls and negative controls (NFW and ATCC 25922 DNA template) were included in every run.
Table 1.
Primer and probe sequences
| Organism | Target | Reference | Forward, Reverse- biotinylated, Probe | Sequence (5′→3′) |
|---|---|---|---|---|
| EHEC | stx1 | (Hidaka et al. 2009) | EH132F | ACTTCTCGACTGCAAAGACGTATG |
| (Hidaka et al. 2009) | EH132Rbt | ACAAATTATCCCCTGAGCCACTATC | ||
| this work | EH132P | CTCTGCAATAGGTACTCCA | ||
|
| ||||
| EHEC | stx2 | (Paton and Paton 1998) | EH255F | GGCACTGTCTGAAACTGCTCC |
| (Paton and Paton 1998) | EH255Rbt | TCGCCAGTTATCTGACATTCTG | ||
| this work | EH255P | GGGGAGAATATCCTTTAATA | ||
|
| ||||
| EPEC/EHEC | eae | this work | EHEC179F | GTAAAGTCCGTTACCCCAACCTG |
| this work | EHEC179Rbt | CAAAGCGCACAAGACTACCA | ||
| this work | EHEC179P | GCACATAAGCAGGCAAAATAGC | ||
|
| ||||
| EPEC | bfpA | this work | EP300F | GGAAGTCAAATTCATGGGGG |
| this work | EP300Rbt | GGAATCAGACGCAGACTGGT | ||
| this work | EPEC300P | GCTGCAACCGTTACCGCAGG | ||
|
| ||||
| EIEC | ipaH | (Vu et al. 2004) | EI64F | CCTTTTCCGCGTTCCTTGA |
| (Vu et al. 2004) | EI64Rbt | CGGAATCCGGAGGTATTGC | ||
| (Vu et al. 2004) | EI64P | CGCCTTTCCGATACCGTCTCTGCA | ||
|
| ||||
| EIEC | ial | (Frankel et al. 1990) | EI320F | CTGGTAGGTATGGTGAGG |
| (Frankel et al. 1990) | EI320Rbt | GGAGGCCAACAATTATTTCC | ||
| (Frankel et al. 1990) | EI320P | CCATCTATTAGAATACCTGT | ||
|
| ||||
| ETEC | LT | (Hidaka et al. 2009) | ET62F | TTCCCACCGGATCACCAA |
| (Hidaka et al. 2009) | ET62Rbt | CAACCTTGTGGTGCATGATGA | ||
| (Hidaka et al. 2009) | ET62P | CTTGGAGAGAAGAACCCT | ||
|
| ||||
| ETEC | STh | this work | ET172F | TTCACCTTTCGCTCAGGATG |
| this work | ET172Rbt | AGCACCCGGTACAAGCAG | ||
| this work | ET172P | ATTACTGCTGTGAATTGTG | ||
|
| ||||
| EAEC | aaiC | (Boisen et al. 2008) | EA215F | ATTGTCCTCAGGCATTTCAC |
| (Boisen et al. 2008) | EA215Rbt | ACGACACCCCTGATAAACAA | ||
| this work | EA215P | GTAGTGCATACTCATCATTTAAG | ||
|
| ||||
| EAEC | aatA | (Schmidt et al. 1995) | EA237F | CTGGCGAAAGACTGTATCAT |
| this work | EA237Rbt | TTTTGCTTCATAAGCCGATAGA | ||
| this work | EA237P | TGGTTCTCATCTATTACAGACAGC | ||
Luminex detection
Luminex detection was performed using amplicon specific internal probes as previously described (Taniuchi et al. 2011a). Probes were amine modified at the 5′ terminus with 12 carbon spacers and covalently hybridized to the carboxylated Luminex beads using 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride. Previously published real-time probe sequences were utilized for detection of ipaH (Vu et al. 2004), ial (Frankel et al. 1990), and LT (Hidaka et al. 2009) amplicon. All other probes were designed for this study (Table 1). Hybridization of the amplicon to the beads was performed at 50°C for 35min using the Oligonucleotide Hybridization Protocol from Luminex (Luminex 2006). After addition of streptavidin PE to the bead-amplicon complex, detection was performed on either a Bioplex 200 (Bio-Rad, Hercules, CA, USA) or a Luminex 100 (Luminex Corporation, Austin, TX, USA) instrument.
Comparison Methods
For initial assay evaluation (Table 2), isolates or broths were identified by existing methods. The AFRIMS specimens were tested by DNA probe hybridization assay using digoxigenin labeled probes for LT (Sommerfelt et al. 1990), STh and STp (Sommerfelt et al. 1990), ial (Echeverria et al. 1990), stx1 and stx2 (Macario and Conway de Macario 1990), bfpA (Donnenberg et al. 1992; Giron et al. 1993), and eae (Jerse and Kaper 1991; Donnenberg et al. 1993). The DCLS broths were tested using in-house methods (PCR, Enzyme Immunoassay, and culture). Other strains (ATCC isolates 35401, 43893, and 11175, EDL 933, and O42 strains) and the clinical specimens in Tanzania were tested using an in-house multiplex PCR followed by gel electrophoresis (for primers and protocol see supplemental Table 1), which target the same genes as the PCR-Luminex assay (Luscher and Altwegg 1994; Cebula et al. 1995; Schmidt et al. 1995; Nguyen et al. 2005; Boisen et al. 2008). Briefly, the 9-plex PCR amplification was performed in 25 μl total volume consisting of 12.5 μl of Qiagen Multiplex PCR Master Mix (Qiagen, Valencia, CA, USA) which contains dNTPs and 3 mmol/L MgCl2 final concentration, 0.2 μmol/L of each forward primer, 0.2 μmol/L of each reverse primer, 5 μl Q-solution, 3.5 μl of nuclease free water (NFW) and 2 μl of DNA template. The cycling conditions were the same as the 9-plex amplification for the PCR-Luminex assay described earlier. Amplicons (range 147bp to 881bp) were detected by electrophoresis on the E-gel system (Life Technologies, Grand Island, NY, USA) using 2% agarose precast E-gels and E-gel Low Range Quantitative DNA Ladder (Life Technologies, Grand Island, NY, USA). Positive and negative controls (NFW and ATCC 25933 DNA template) were included with each run.
Table 2.
Performance of assay versus comparator methods on isolates and broths
| Target Gene | Isolates in-house+ | Isolates in-house− | Broth in-house+ | Broth in-house− | Total in-house+ | Total in-house− | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
|
Stx1 Luminex+ |
5 | 0 | 68 | 2 | 73 | 2 | 1.00 | 0.96 |
|
Stx1 Luminex− |
0 | 25 | 0 | 29 | 0 | 54 | ||
|
Stx2 Luminex+ |
4 | 0 | 33 | 0 | 37 | 0 | 1.00 | 1.00 |
|
Stx2 Luminex− |
0 | 21 | 0 | 66 | 0 | 87 | ||
|
eae Luminex+ |
10 | 0 | 76 | 3 | 86 | 3 | 1.00 | 0.94 |
|
eae Luminex− |
0 | 25 | 0 | 20 | 0 | 45 | ||
|
bfpA Luminex+ |
3 | 0 | 0 | 2 | 3 | 2 | 1.00 | 0.98 |
|
bfpA Luminex− |
0 | 26 | 0 | 97 | 0 | 123 | ||
|
ipaH Luminex+ |
1 | 0 | 1 | 0 | 1.00 | 1.00 | ||
|
ipaH Luminex− |
0 | 6 | 0 | 6 | ||||
|
ial Luminex+ |
4 | 0 | 4 | 0 | 1.00 | 1.00 | ||
|
ial Luminex− |
0 | 24 | 0 | 24 | ||||
|
LT Luminex+ |
6 | 0 | 0 | 1 | 6 | 1 | 1.00 | 0.99 |
|
LT Luminex− |
0 | 27 | 0 | 98 | 0 | 125 | ||
|
ST Luminex+ |
3 | 0 | 0 | 0 | 3 | 0 | 1.00 | 1.00 |
|
ST Luminex− |
0 | 27 | 0 | 99 | 0 | 126 | ||
|
aaiC Luminex+ |
3 | 0 | 3 | 0 | 1.00 | 1.00 | ||
|
aaiC Luminex− |
0 | 25 | 0 | 25 | ||||
|
aatA Luminex+ |
5 | 0 | 5 | 0 | 1.00 | 1.00 | ||
|
aatA Luminex− |
0 | 25 | 0 | 25 |
Statistical analysis
Luminex data was reported as median fluorescence intensity (MFI). We used a corrected MFI (cMFI = [MFI (sample) – MFI (background)]/MFI (background)] to accommodate for testing on different Luminex platforms under both high and low photomultiplier tube voltages and different softwares (Luminex 100 IS Software v. 2.3 in Thailand and Bioplex v. 5.0 at UVA and Tanzania). A cMFI cutoff of 3.0 was used at all sites except at DCLS which used 2.0. Where replicates or multiple cMFI are shown, data is reported as mean ± SEM. The cMFI values in clinical specimens were compared using Mann Whitney test (SPSS, Chicago, IL, USA). The correlation between cMFI and log cfu/200 mg of stool was calculated by Pearson correlation.
Results
We have previously developed PCR-based Luminex assays for enteric viruses, protozoa, helminths, and bacteria (Liu et al. 2011a; Liu et al. 2011b; Taniuchi et al. 2011a; Taniuchi et al. 2011b). In this work we adopted the same general principles of targeting conserved regions of virulence genes, designing short fragments to maximize PCR efficiency, and placing probes near one of the primer regions in order to shorten the distance from bead-probe to fluorophore-primer. We chose to target commonly accepted genes for EHEC (stx1, stx2, eae), EPEC (eae, bfpA), EIEC (ipaH or ial), ETEC (LT, STh), and EAEC (aaiC, aatA). We sourced primers from the literature where possible, however ultimately the assay required new primers for certain targets (eae, STh). Likewise, probes were designed new for many targets (stx1, stx2, aaiC, aatA) and most required significant refinement including 3 iterations for aaiC, stx1, and bfpA, and 2 for aatA, stx2, and STh.
We first tested the assay on DEC specimens of known pathotype confirmed with existing assays. This entailed use of laboratory strains, clinical isolates, and broths tested by DNA probes or in-house PCR assays targeting different regions of the genes (see Materials and Methods). The comparison of the Luminex protocol with the comparator methods revealed a sensitivity of 100% and specificity of 94–100% (Table 2). We further examined specificity of the assays by testing pure cultures or clinical specimens containing rotavirus, sapovirus, astrovirus, norovirus, Campylobacter, Vibrio, Salmonella, Shigella, Aeromonas, Yersinia, Entamoeba histolytica, E. dispar, Cryptosporidium, Cystoisospora, Cyclospora, Ascaris, Ancylostoma, Necator, Strongyloides, Trichuris, and found no detection on these materials with any of the assays (data not shown).
Since the assay was able to detect the virulence genes in cultures, we then evaluated how well the assay could detect the genes across specimen type. Many laboratories test for DEC from MacConkey colony pools, so we compared this material with DNA isolated from stool directly and from GN broths. To do so we spiked 101 – 108 cfu of EHEC O157:H7, EPEC 2348/69, EIEC O124, ETEC H10407, and EAEC O42 strain into aliquots of a single DEC PCR-negative healthy volunteer stool sample. These spiked stool samples were then cultured on MacConkey and 5 colonies picked or grown in GN broth overnight, as described in the Materials and Methods. DNA was extracted from these colony pools, broths, and the parental stool sample, and all extracts were tested using the multiplex PCR-Luminex assay in triplicate. This yielded several results (Figure 1). First, across all targets, there was no false positive detection of EHEC, EPEC, EIEC, ETEC, or EAEC with the other targets in the assay. Second, the stool itself yielded a quantitative correlation for most targets. Namely, a log-linear correlation between cfu/g of E. coli and Luminex cMFI was statistically significant for EHEC across stx1, stx2, eae, for EPEC across eae and bfpA, for EAEC for aatA, for ETEC across LT and STh, and EIEC. By contrast, the DNA extracted from broths and from colonies exhibited no statistically significant correlation between cfu/g of input stool and cMFI (P > 0.05). Third, using the standard cMFI cutoff for positivity of 3.0, broths yielded detection of 101 for most targets, however detection of ETEC was lost at 102 cfu/200 mg stool ( i.e., cMFI was < 3.0) and was low for STh. In contrast stool and colonies yielded detection at 103 cfu/200 mg stool (i.e., cMFI was > 3.0) for most targets. Fourth, there were clearly different levels of detection for the different targets; for instance, the STh assay offered lower cMFI than the LT assay for this STh/LT positive ETEC isolate in stools, broths, and isolates (P < 0.05 comparing mean cMFIs across all dilutions).
Figure 1.
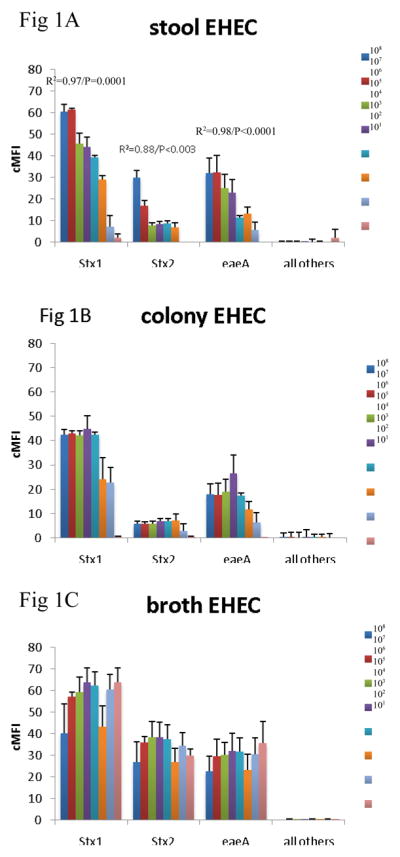
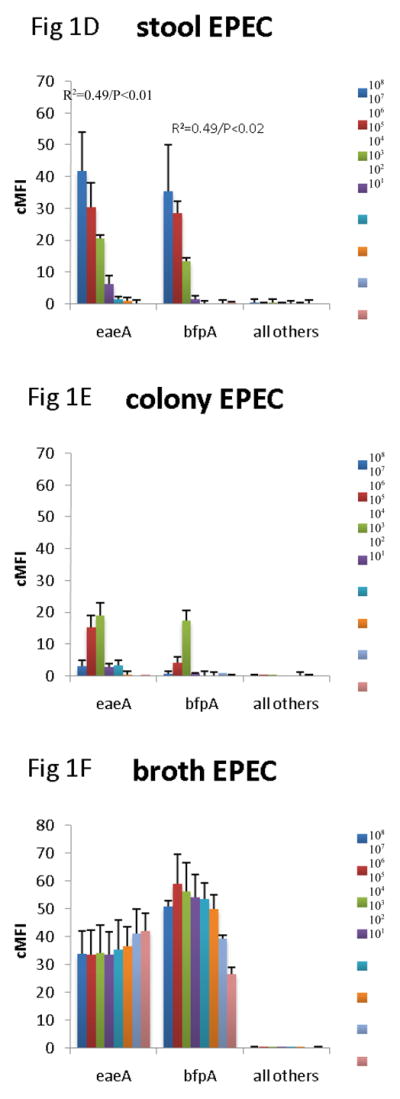


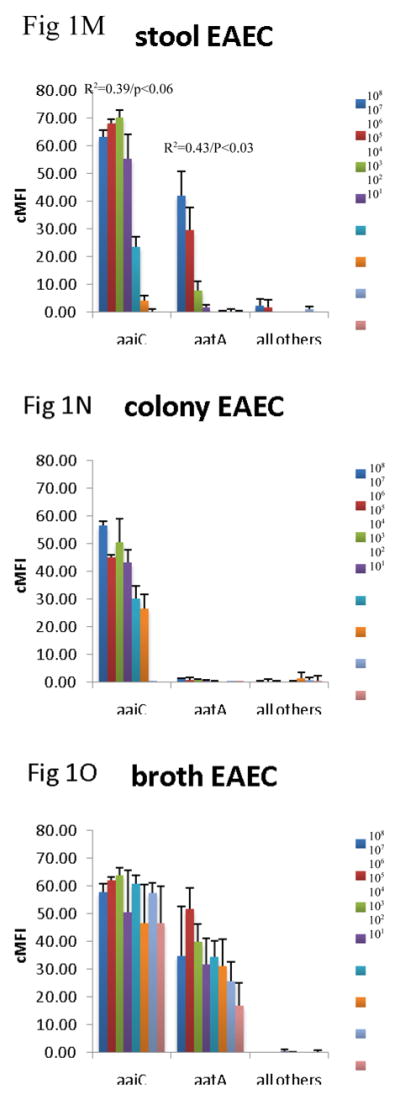
Comparison of stool DNA, broth DNA, and colony DNA in detection of DEC. Aliquots of a single stool sample were spiked with serial dilutions of EHEC 0157:H7 (AC), EPEC 2348/69 (D–F), EIEC O124 (G–I), ETEC H10407 (J–L), and EAEC O42 (M–O) as indicated. Spiked amounts are indicated in the legends and units are cfu per 200 mg of stool. Spiked samples were then processed by culture in GN broth and on MacConkey agar followed by picking 5 colonies. DNA was extracted from the direct stool versus colony pools versus GN broths and subjected to multiplex PCR in triplicate followed by Luminex detection. Data show cMFI values + SEM on the y axes for the targets indicated on the x axes. cMFI > 3.0 was considered positive. The y axes shown are different for each E. coli pathotype. Linear regression revealed R2 and P values as indicated.
We then prospectively evaluated the assay across the three specimen types in a field setting in Tanzania that experiences a diverse range of DEC. Given the previous finding that broths should be most sensitive, this specimen type was our standard. When stool was compared to broths we observed a sensitivity of 67–100% and specificity of 89%-100% across the assays (Table 3). Among discrepant samples, we frequently observed higher target fluorescence in specimens that were positive in both GN and stool versus specimens that were positive in only one specimen type, however differences were not statistically significant. The performance of the assay in colony DNA was also examined and this specimen type exhibited more discrepancies than stool DNA, with sensitivities of 33–50% and specificities of 89–100% (data not shown).
Table 3.
Evaluation of assay on clinical specimens
| Target Gene | GN+ | GN− | Sensitivity | Specificity |
|---|---|---|---|---|
|
Stx1 Stool+ |
7 (cMFIGN=9.7±2.4, cMFIStool=12.1±1.6) | 3 (cMFI=16.3±4.5) | 0.88 | 0.93 |
|
Stx1 Stool− |
1 (cMFI=3.3) | 43 | ||
|
Stx2 Stool+ |
0 | 0 | N/A | 1.00 |
|
Stx2 Stool− |
0 | 54 | ||
|
eae Stool+ |
11 (cMFIGN=11.6±3.2, cMFIStool=18.8±5.1) | 2 (cMFI=7.6±3.4) | 0.73 | 0.95 |
|
eae Stool− |
4 (cMFI=6.7±1.1) | 37 | ||
|
bfpA Stool+ |
0 | 4 | N/A | 0.93 |
|
bfpA Stool− |
0 | 50 | ||
|
ipaH Stool+ |
5 (cMFIGN=7.6±1.9, cMFIStool=10.5±1.9) | 5 (cMFI=9.5±2.8) | 0.71 | 0.89 |
|
ipaH Stool− |
2 (cMFI=3.3±0.0) | 42 | ||
|
LT Stool+ |
4 (cMFIGN=6.7±1.0, cMFIStool=14.4±6.2) | 0 | 0.67 | 1.00 |
|
LT Stool− |
2 (cMFI=3.2±0.0) | 48 | ||
|
ST Stool+ |
1 | 0 | 1.00 | 1.00 |
|
ST Stool− |
0 | 53 | ||
|
aaiC Stool+ |
7 (cMFIGN=19.4±5.6, cMFIStool=21.5±2.3) | 2 (cMFI=18.7±14.6) | 0.88 | 0.96 |
|
aaiC Stool− |
1 (cMFI=14.6) | 44 | ||
|
aatA Stool+ |
15 (cMFIGN=11.5±1.5, cMFIStool=10.7±1.3) | 4 (cMFI=9.6±2.7) | 0.83 | 0.89 |
|
aatA Stool− |
3 (cMFI=6.7±2.5) | 32 |
Discussion
This work describes an assay to detect diarrheagenic E. coli in stool or broths. We combined an accurate multiplex PCR method to detect characteristic DEC virulence genes, and evaluated the performance of the assay across specimen types – from stool to colonies to broths. We view this as important step towards the goal of directly identifying DEC in stool samples without the current limitations of colony picks or requiring phenotypic methods.
The genes targeted in this assay deserve discussion. Shiga-toxin producing E coli (STEC) are food-borne pathogens that have the potential to cause hemorrhagic colitis and hemolytic uremic syndrome, and are important to detect because antibiotics are not beneficial and may increase the risk of complications (Wong et al. 2000). STEC contain lysogenic bacteriophages that produce one or two shiga toxins, stx1 and stx2, the latter of which has several variants (De Baets et al. 2004). Our assay utilized published primers for both of the toxins that had been tested on 28 and 52 STEC strains, respectively, including known variants (Paton and Paton 1998; Hidaka et al. 2009). STEC are of important public health concern in developed countries, witness the recent Germany outbreak, and our assay exhibited 94–100% sensitivity/specificity on 73 stx1 and 37 stx2-positive stool broths submitted to the Virginia state laboratory and confirmed by in-house methods (Table 2). Currently the assay will be used for screening selected broth specimens for STEC at this laboratory.
A subset of STEC has the capacity to attach to and efface intestinal epithelial cells, a pathology mediated by the adhesin intimin encoded by eae. The same pathology and gene is also found within EPEC. Intimin sequence can be variable (Blanco et al. 2006) and indeed this target was relatively problematic in our hands, requiring multiple redesigns. Typical EPEC also contain a plasmid-borne E. coli adherence factor that contains bfpA, while “atypical” EPEC strains do not. Such atypical EPEC are more prevalent in many settings (Ochoa and Contreras 2011). Our spiking studies suggested that eae was more readily detected in the EHEC strain (detected 103) than EPEC (detected 105). This was surprising because it is a chromosomal gene, and was not due to sequence variations because we sequenced both eae amplicons and they were identical (data not shown).
Enteroinvasive E. coli were readily detected by targeting ipaH or ial, and in our hands the assays were interchangeable. These two genes are widely used for detection and are found on the invasion plasmid (Echeverria et al. 1992). The ipaH gene is also present on the chromosome thus we favored it for most of our work. The gene is exclusively found in Shigella and EIEC, and although there are sequence differences between these species, we targeted the conserved core region (bp 1065–1128). The assay yielded robust amplification and detected 102 cfu of EIEC bacteria per 200mg stool sample. We have also used this assay for detection of Shigella (Liu et al. 2011a), with similar sensitivity.
ETEC are common causes of diarrhea in children of the developing world. After adherence to the intestinal mucosa, ETEC produce one or both of two enterotoxins, heat-labile enterotoxin (LT) and heat-stable enterotoxin (ST). Globally it is estimated that 26% of strains contain LT, 45% contain ST, and the rest contain both (Isidean et al. 2011). The LT sequence is relatively conserved across human isolates. Two main subtypes of ST have been found in humans, STh (or STIb) and STp (or STIa). STh appears more common and has been reported to be the only diarrhea-associated subtype in some studies, while others have found both STh and STp to be associated with diarrhea (Steinsland et al. 2002; Bolin et al. 2006). ST was a difficult target given substantial sequence variation within GenBank and there appears to be some debate on published assays for ST. We first tried an assay based on the primers of Nguyen et al. (Nguyen et al. 2005), the comparator method of supplemental Table 1, which amplifies both STh and STp through wobble primer. This worked however Luminex signal was low and probe choices were limited. We thus redesigned the assay to enhance the signal, although this required preferential redesign for STh. For those interested in the rarer subtype STp we would recommend a singleplex test for this explicitly.
Although the HEp-2 adherence assay remains the defining feature of EAEC, we focused on the subset of EAEC strains that harbor the AggR regulon of virulence factors (the “typical” EAEC), as members of this group have been proven to be virulent in volunteer and epidemiologic studies. Typical EAEC carry key factors on a virulence plasmid and a chromosomal pathogenicity island, so we chose to detect one factor from each of these genetic elements (Czeczulin et al. 1999). aatA is a constituent of the EAEC virulence plasmid pAA2, corresponds to a widely used EAEC probe (Baudry et al. 1990), and has been found to be highly specific for EAEC versus other E. coli (Monteiro et al. 2009). aaiC encodes a secreted protein of the EAEC pathogenicity island AAI, which is coordinately regulated by the AggR activator, along with pAA plasmid-encoded factors (Dudley et al. 2006).
A major focus of this work was examining the performance of the assay in stool vs. broths vs. colonies, because ultimately we desired an assay that was workable on stool. Using spiked specimens we found detection of all targets in stool to be less sensitive than detection in broth, which is logical because broth allows for an additional round of amplification due to bacterial replication and the dilution of the inhibitory substances present in stool. However we found a substantial number of Tanzanian samples that were positive in stool only, not broth (for example, 9 of 54 specimens tested by ipaH). The significance of such stool+/culture- materials will need a significant amount of future study. Plausible explanations include a false positive result, low levels of DNA in stool of no significance, or prior antibiotic use rendering culture negative (essentially all individuals in Tanzania were on antimicrobials), the latter of which would be highly clinically relevant. The sensitivity of our assay in stool, with detection down to 103 cfu/200 mg for most targets, is similar to or better than previous reports that have tested stool DNA directly (Persson et al. 2007; de Boer et al. 2010; Barletta et al. 2011).
We also found DNA extracted directly from stool to be more quantitative than DNA extracted broth, again logical since the overnight broth amplification plateaus. The significance of pathogen quantitation in stool in predicting diarrhea is an area of active investigation, but there are emerging reports of its relevance in norovirus and EPEC infections (Phillips et al. 2009; Barletta et al. 2011). Evaluation of the role of quantitation in direct stool samples is warranted.
Our results reveal little benefit in picking colonies for E. coli, in that this technique was less sensitive than DEC detection from broths, no more sensitive than directly testing stool, and much more cumbersome. Thus we would propose using the assay to test on stool or broths for DEC as a screen, using broths if sensitivity is desired or stool itself if quantitation is desired, and if the screen is positive then one would have the option of continuing to culture the specimen in order to isolate the exact lactose fermenting bacterial strain for subsequent testing as necessary.
Supplementary Material
Supplemental Table 1. Primer and probe sequences of comparator method
Acknowledgments
This work was supported by U01 AI075396-01 from the National Institute of Health and Study of Risk Factors for Malnutrition using Molecular Genomic Tools Global Health Grant #51635 from the Bill and Melinda Gates Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aranda KR, Fagundes-Neto U, Scaletsky IC. Evaluation of multiplex PCRs for diagnosis of infection with diarrheagenic Escherichia coli and Shigella spp. J Clin Microbiol. 2004;42(12):5849–53. doi: 10.1128/JCM.42.12.5849-5853.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barletta F, Ochoa TJ, Ecker L, Gil AI, Lanata CF, Cleary TG. Validation of five-colony pool analysis using multiplex real-time PCR for detection of diarrheagenic Escherichia coli. J Clin Microbiol. 2009;47(6):1915–7. doi: 10.1128/JCM.00608-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barletta F, Ochoa TJ, Mercado E, Ruiz J, Ecker L, Lopez G, Mispireta M, Gil AI, Lanata CF, Cleary TG. Quantitative Real-time Polymerase Chain Reaction for Enteropathogenic Escherichia coli: A Tool for Investigation of Asymptomatic Versus Symptomatic Infections. Clin Infect Dis. 2011;53(12):1223–9. doi: 10.1093/cid/cir730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry B, Savarino SJ, Vial P, Kaper JB, Levine MM. A sensitive and specific DNA probe to identify enteroaggregative Escherichia coli, a recently discovered diarrheal pathogen. J Infect Dis. 1990;161(6):1249–51. doi: 10.1093/infdis/161.6.1249. [DOI] [PubMed] [Google Scholar]
- Blanco M, Blanco JE, Dahbi G, Mora A, Alonso MP, Varela G, Gadea MP, Schelotto F, Gonzalez EA, Blanco J. Typing of intimin (eae) genes from enteropathogenic Escherichia coli (EPEC) isolated from children with diarrhoea in Montevideo, Uruguay: identification of two novel intimin variants (muB and xiR/beta2B) J Med Microbiol. 2006;55(Pt 9):1165–74. doi: 10.1099/jmm.0.46518-0. [DOI] [PubMed] [Google Scholar]
- Boisen N, Struve C, Scheutz F, Krogfelt KA, Nataro JP. New adhesin of enteroaggregative Escherichia coli related to the Afa/Dr/AAF family. Infect Immun. 2008;76(7):3281–92. doi: 10.1128/IAI.01646-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin I, Wiklund G, Qadri F, Torres O, Bourgeois AL, Savarino S, Svennerholm AM. Enterotoxigenic Escherichia coli with STh and STp genotypes is associated with diarrhea both in children in areas of endemicity and in travelers. J Clin Microbiol. 2006;44(11):3872–7. doi: 10.1128/JCM.00790-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebula TA, Payne WL, Feng P. Simultaneous identification of strains of Escherichia coli serotype O157:H7 and their Shiga-like toxin type by mismatch amplification mutation assay-multiplex PCR. J Clin Microbiol. 1995;33(1):248–50. doi: 10.1128/jcm.33.1.248-250.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeczulin JR, Whittam TS, Henderson IR, Navarro-Garcia F, Nataro JP. Phylogenetic analysis of enteroaggregative and diffusely adherent Escherichia coli. Infect Immun. 1999;67(6):2692–9. doi: 10.1128/iai.67.6.2692-2699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baets L, Van der Taelen I, De Filette M, Pierard D, Allison L, De Greve H, Hernalsteens JP, Imberechts H. Genetic typing of shiga toxin 2 variants of Escherichia coli by PCR-restriction fragment length polymorphism analysis. Appl Environ Microbiol. 2004;70(10):6309–14. doi: 10.1128/AEM.70.10.6309-6314.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer RF, Ott A, Kesztyus B, Kooistra-Smid AM. Improved detection of five major gastrointestinal pathogens by use of a molecular screening approach. J Clin Microbiol. 2010;48(11):4140–6. doi: 10.1128/JCM.01124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Giron JA, Nataro JP, Kaper JB. A plasmid-encoded type IV fimbrial gene of enteropathogenic Escherichia coli associated with localized adherence. Mol Microbiol. 1992;6(22):3427–37. doi: 10.1111/j.1365-2958.1992.tb02210.x. [DOI] [PubMed] [Google Scholar]
- Donnenberg MS, Tacket CO, James SP, Losonsky G, Nataro JP, Wasserman SS, Kaper JB, Levine MM. Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest. 1993;92(3):1412–7. doi: 10.1172/JCI116717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley EG, Thomson NR, Parkhill J, Morin NP, Nataro JP. Proteomic and microarray characterization of the AggR regulon identifies a pheU pathogenicity island in enteroaggregative Escherichia coli. Mol Microbiol. 2006;61(5):1267–82. doi: 10.1111/j.1365-2958.2006.05281.x. [DOI] [PubMed] [Google Scholar]
- Echeverria P, Seriwatana J, Sethabutr O, Chatkaeomorakot A. Detection of Diarrheagenic Escherichia coli. In: Macario AJL, Conway de Macario E, editors. Gene probes for bacteria. San Diego: Academic Press; 1990. pp. 95–141. [Google Scholar]
- Echeverria P, Sethabutr O, Serichantalergs O, Lexomboon U, Tamura K. Shigella and enteroinvasive Escherichia coli infections in households of children with dysentery in Bangkok. J Infect Dis. 1992;165(1):144–7. doi: 10.1093/infdis/165.1.144. [DOI] [PubMed] [Google Scholar]
- Fischer Walker CL, Sack D, Black RE. Etiology of diarrhea in older children, adolescents and adults: a systematic review. PLoS Negl Trop Dis. 2010;4(8):e768. doi: 10.1371/journal.pntd.0000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel G, Riley L, Giron JA, Valmassoi J, Friedmann A, Strockbine N, Falkow S, Schoolnik GK. Detection of Shigella in feces using DNA amplification. J Infect Dis. 1990;161(6):1252–6. doi: 10.1093/infdis/161.6.1252. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Kasai K, Miura T, Sato T, Otomo Y. Rapid diagnostic method for the detection of diarrheagenic Escherichia coli by multiplex PCR. Jpn J Infect Dis. 2009;62 (6):476–80. [PubMed] [Google Scholar]
- Galbadage T, Jiang ZD, DuPont HL. Improvement in detection of enterotoxigenic Escherichia coli in patients with travelers’ diarrhea by increasing the number of E. coli colonies tested. Am J Trop Med Hyg. 2009;80(1):20–3. [PubMed] [Google Scholar]
- Giron JA, Donnenberg MS, Martin WC, Jarvis KG, Kaper JB. Distribution of the bundle-forming pilus structural gene (bfpA) among enteropathogenic Escherichia coli. J Infect Dis. 1993;168(4):1037–41. doi: 10.1093/infdis/168.4.1037. [DOI] [PubMed] [Google Scholar]
- Guion CE, Ochoa TJ, Walker CM, Barletta F, Cleary TG. Detection of diarrheagenic Escherichia coli by use of melting-curve analysis and real-time multiplex PCR. J Clin Microbiol. 2008;46(5):1752–7. doi: 10.1128/JCM.02341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka A, Hokyo T, Arikawa K, Fujihara S, Ogasawara J, Hase A, Hara-Kudo Y, Nishikawa Y. Multiplex real-time PCR for exhaustive detection of diarrhoeagenic Escherichia coli. J Appl Microbiol. 2009;106(2):410–20. doi: 10.1111/j.1365-2672.2008.04043.x. [DOI] [PubMed] [Google Scholar]
- Isidean SD, Riddle MS, Savarino SJ, Porter CK. A systematic review of ETEC epidemiology focusing on colonization factor and toxin expression. Vaccine. 2011;29(37):6167–78. doi: 10.1016/j.vaccine.2011.06.084. [DOI] [PubMed] [Google Scholar]
- Jerse AE, Kaper JB. The eae gene of enteropathogenic Escherichia coli encodes a 94-kilodalton membrane protein, the expression of which is influenced by the EAF plasmid. Infect Immun. 1991;59(12):4302–9. doi: 10.1128/iai.59.12.4302-4309.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gratz J, Maro A, Kumburu H, Kibiki G, Taniuchi M, Howlader AM, Sobuz SU, Haque R, Talukder KA, Qureshi S, Zaidi A, Haverstick DM, Houpt ER. Simultaneous detection of six diarrhea-causing bacterial pathogens with an in house PCR-Luminex Assay. J Clin Microbiol. 2011 doi: 10.1128/JCM.05416-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kibiki G, Maro V, Maro A, Kumburu H, Swai N, Taniuchi M, Gratz J, Toney D, Kang G, Houpt ER. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. J Clin Virol. 2011;50(4):308–13. doi: 10.1016/j.jcv.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luminex. Protocol. Luminex; Toronto, Canada: 2006. Sample Protocol for Oligonucleotide Hybridization. [Google Scholar]
- Luscher D, Altwegg M. Detection of shigellae, enteroinvasive and enterotoxigenic Escherichia coli using the polymerase chain reaction (PCR) in patients returning from tropical countries. Mol Cell Probes. 1994;8(4):285–90. doi: 10.1006/mcpr.1994.1040. [DOI] [PubMed] [Google Scholar]
- Monteiro BT, Campos LC, Sircili MP, Franzolin MR, Bevilacqua LF, Nataro JP, Elias WP. The dispersin-encoding gene (aap) is not restricted to enteroaggregative Escherichia coli. Diagn Microbiol Infect Dis. 2009;65(1):81–4. doi: 10.1016/j.diagmicrobio.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, Le Van P, Le Huy C, Gia KN, Weintraub A. Detection and characterization of diarrheagenic Escherichia coli from young children in Hanoi, Vietnam. J Clin Microbiol. 2005;43(2):755–60. doi: 10.1128/JCM.43.2.755-760.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa TJ, Contreras CA. Enteropathogenic Escherichia coli infection in children. Curr Opin Infect Dis. 2011;24(5):478–83. doi: 10.1097/QCO.0b013e32834a8b8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton AW, Paton JC. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J Clin Microbiol. 1998;36(2):598–602. doi: 10.1128/jcm.36.2.598-602.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson S, Olsen KE, Scheutz F, Krogfelt KA, Gerner-Smidt P. A method for fast and simple detection of major diarrhoeagenic Escherichia coli in the routine diagnostic laboratory. Clin Microbiol Infect. 2007;13(5):516–24. doi: 10.1111/j.1469-0691.2007.01692.x. [DOI] [PubMed] [Google Scholar]
- Phillips G, Lopman B, Tam CC, Iturriza-Gomara M, Brown D, Gray J. Diagnosing norovirus-associated infectious intestinal disease using viral load. BMC Infect Dis. 2009;9:63. doi: 10.1186/1471-2334-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Worsham PL, Abshire TG, Stanley ST, Bannan JD, Wilson MR, Langham RJ, Decker RS, Jiang L, Read TD, Phillippy AM, Salzberg SL, Pop M, Van Ert MN, Kenefic LJ, Keim PS, Fraser-Liggett CM, Ravel J. Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proc Natl Acad Sci U S A. 2011;108(12):5027–32. doi: 10.1073/pnas.1016657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Knop C, Franke S, Aleksic S, Heesemann J, Karch H. Development of PCR for screening of enteroaggregative Escherichia coli. J Clin Microbiol. 1995;33(3):701–5. doi: 10.1128/jcm.33.3.701-705.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerfelt H, Grewal HM, Bhan MK. Simplified and accurate nonradioactive polynucleotide gene probe assay for identification of enterotoxigenic Escherichia coli. J Clin Microbiol. 1990;28(1):49–54. doi: 10.1128/jcm.28.1.49-54.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinsland H, Valentiner-Branth P, Perch M, Dias F, Fischer TK, Aaby P, Molbak K, Sommerfelt H. Enterotoxigenic Escherichia coli infections and diarrhea in a cohort of young children in Guinea-Bissau. J Infect Dis. 2002;186(12):1740–7. doi: 10.1086/345817. [DOI] [PubMed] [Google Scholar]
- Taniuchi M, Verweij JJ, Noor Z, Sobuz SU, Lieshout L, Petri WA, Jr, Haque R, Houpt ER. High throughput multiplex PCR and probe-based detection with Luminex beads for seven intestinal parasites. Am J Trop Med Hyg. 2011;84(2):332–7. doi: 10.4269/ajtmh.2011.10-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi M, Verweij JJ, Sethabutr O, Bodhidatta L, Garcia L, Maro A, Kumburu H, Gratz J, Kibiki G, Houpt ER. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagn Microbiol Infect Dis. 2011;71(4):386–90. doi: 10.1016/j.diagmicrobio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu DT, Sethabutr O, Von Seidlein L, Tran VT, Do GC, Bui TC, Le HT, Lee H, Houng HS, Hale TL, Clemens JD, Mason C, Dang DT. Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J Clin Microbiol. 2004;42(5):2031–5. doi: 10.1128/JCM.42.5.2031-2035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930–6. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Primer and probe sequences of comparator method