

Abstract
Hemimegalencephaly (HME) is a heterogeneous cortical malformation characterized by enlargement of one cerebral hemisphere. Somatic variants in mammalian target of rapamycin (mTOR) regulatory genes have been implicated in some HME cases; however, ∼70% have no identified genetic etiology. Here, we screened two HME patients to identify disease-causing somatic variants. DNA from leukocytes, buccal swabs, and surgically resected brain tissue from two HME patients were screened for somatic variants using genome-wide genotyping arrays or sequencing of the protein-coding regions of the genome. Functional studies were performed to evaluate the molecular consequences of candidate disease-causing variants. Both HME patients evaluated were found to have likely disease-causing variants in DNA extracted from brain tissue but not in buccal swab or leukocyte DNA, consistent with a somatic mutational mechanism. In the first case, a previously identified disease-causing somatic single nucleotide in MTOR was identified. In the second case, we detected an overrepresentation of the alleles inherited from the mother on Chromosome 16 in brain tissue DNA only, indicative of somatic uniparental disomy (UPD) of the p-arm of Chromosome 16. Using methylation analyses, an imprinted locus on 16p spanning ZNF597 was identified, which results in increased expression of ZNF597 mRNA and protein in the brain tissue of the second case. Enhanced mTOR signaling was observed in tissue specimens from both patients. We speculate that overexpression of maternally expressed ZNF597 led to aberrant hemispheric development in the patient with somatic UPD of Chromosome 16p possibly through modulation of mTOR signaling.
Keywords: hemimegalencephaly, pachygyria
INTRODUCTION
There is growing recognition of an important role for de novo variants in specific syndromes (Barcia et al. 2012; Heinzen et al. 2012), as well as in more complex phenotypes like autism (Neale et al. 2012), intellectual disability (de Ligt et al. 2012), and epilepsy (Epi4K Consortium et al. 2013). Because these variants were detected in DNA from leukocytes of patients with a neurological disorder, it is presumed that these variants were newly acquired in parental gametes and present constitutionally in the zygote. However, disease-causing de novo somatic variants may arise postzygotically and be present only in a fraction of the cells in the brain. In fact, somatic variants have previously been reported in conditions associated with medically intractable epilepsy, including hypothalamic hamartoma (Hildebrand et al. 2016), focal cortical dysplasia (Lim et al. 2015; Nakashima et al. 2015), and hemimegalencephaly (HME) (HP:0007206), a severe malformation of cortical development characterized by enlargement of one cerebral hemisphere (Lee et al. 2012; Poduri et al. 2012; D'Gama et al. 2015). The mammalian target of rapamycin (mTOR) cascade has been implicated in a number of malformations of cortical development including HME (Crino 2011). Consistent with the known involvement of the mTOR-signaling pathway in HME, all identified somatic variants detected in surgically resected brain tissue occur in genes comprising the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR pathway and collectively are estimated to explain ∼30% of the cases of HME (Lee et al. 2012; Poduri et al. 2012; D'Gama et al. 2015). Previous studies have shown clearly that the mTOR cascade is activated in many cases of HME as evidenced by phosphoactivation of mTOR substrates such as p70S6kinase and ribosomal S6 protein (Ljungberg et al. 2006; Aronica et al. 2007; Boer et al. 2007). Most cases of HME are sporadic, with only one report of familial HME (Leventer et al. 2014), suggesting that new variants likely play an important role in the genetic architecture of this disease.
In this study, we sought to identify somatic variants in HME by genetically analyzing paired DNA samples from unaffected cells (leukocytes) and abnormal brain tissue from two patients to identify brain tissue–specific variants.
RESULTS
Clinical Presentation
HME1 is a female of European ancestry. She was developmentally delayed as an infant, and her first seizure (HP:0001250) occurred at 22 mo. Magnetic resonance imaging (MRI) at that time showed generalized overgrowth of the left frontotemporal lobes with regional cortical thickening and shallow sulci consistent with left-sided HME (Fig. 1A). Her seizures became progressively worse between 2 and 3 yr, occurring up to 50 times per day. At 36 mo, HME1 underwent left frontotemporoparietal craniotomy and functional hemispherectomy. Postoperatively, she made developmental gains and was seizure-free for nearly 24 mo. Seizures recurred 24 mo posthemispherectomy. MRI revealed residual fibers crossing the corpus callosum, and electroencephalography (EEG) was supportive of seizures secondarily generalizing from the left hemisphere. The patient then underwent a left-sided anatomic hemispherectomy with completion of the corpus callostomy 26 mo after her first surgery. Postoperatively, the patient has been free of overt seizures for 20 mo. However, EEG still shows brief bursts of spikes in the right hemisphere without clinical correlate. Neuropathologic analysis of a portion of the resected tissue revealed disorganized cerebral cortical cytoarchitecture with balloon cells (Fig. 1B) and persistent radial glial ladders (not shown). She was seen at 8 yr, and the examination showed that she was speaking in short sentences but with relatively severe dysarthria. She could read at the second-grade level. She also presented with right central facial and right upper extremity hemiplegia of the hand and hemiparesis of the arm and leg with the ability to walk and right-sided hyperreflexia and an upgoing toe on the right. She also had a right homonymous hemianopsia.
Figure 1.

Clinical and neuropathologic findings in HME1 and HME2. (A) T2-weighted magnetic resonance image (MRI) of HME1 showing left-sided hemimegalencephaly (HME). (B) Hematoxylin and eosin (H&E) staining of cortical brain tissue slices showing balloon cells (black arrows) in HME1 (left, 10× and right, 60×). (C) T2-weighted MRI of HME2 showing left-sided HME with pachygyria in the left parietooccipital region. (D) H&E (40×) or NeuN (60×) staining of cortical brain tissue slices from HME2 showing balloon cells (black arrow).
HME2 is a female of European ancestry. After an uncomplicated birth, HME2 began having complex partial seizures at 3 wk and drug-resistant infantile spasms shortly thereafter. MRI showed left HME with a broad area of parietooccipital pachygyria (Fig. 1C). She had a functional hemispherectomy at 6 mo. Postoperatively, subject continued to have seizures, and MRI showed some remaining connection at the corpus callosum and temporal lobe. At 6 mo after the initial surgery, the patient underwent completion of the functional hemispherectomy. The patient is currently seizure-free, 36 mo after the second surgery. Neuropathologic analysis of a portion of the resected tissue revealed markedly disorganized cerebral cortical cytoarchitecture with balloon cells (Fig. 1D). When seen at 5 yr, she was able to write her name with assistance. She knew three out of three colors and pointed to objects in the room but could not give definitions of other objects. She was able to count to 20. She was able to take up to 26 steps but needed someone to be behind her to make sure she did not fall. She presented with a right homonymous hemianopsia. She had right central facial and right upper extremity hemiplegia of the hand and hemiparesis of the arm and leg, as well as right-sided hyperreflexia and an upgoing toe on the right.
Genomic Analyses
The somatic variant calling pipeline from the paired exome sequence data of HME1 identified six candidate somatic variants predicted to be functional (possibly damaging or probably damaging by PolyPhen-2 missense, splice, or stop-gained single-nucleotide variants or frameshift indels) and absent from public exome databases and in-house sequenced controls (Supplemental Table 1). One of these candidate variants was a missense somatic single-nucleotide variant (sSNV) in MTOR (NM_004958.3:c.4447T>C;p.Cys1483Arg). Given the previous report of somatic variants in MTOR and genes comprising the PI3K–AKT–mTOR pathway in HME (Lee et al. 2012; D'Gama et al. 2015), we confirmed the presence and absence of the candidate somatic variant in MTOR in the brain and blood, respectively (Table 1). Both the sequencing data and the digital droplet polymerase chain reaction (PCR) analyses support that the variant is present in ∼6% of alleles in the brain (Fig. 2). We also confirmed that the variant was not detectable in DNA from the buccal swab from HME1, indicating the somatic variant occurred after the formation of the neuroectoderm.
Table 1.
Somatic variants identified in the brain tissue of two patients with hemimegalencephaly
| Patient | Gene | Chr. | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVar ID |
|---|---|---|---|---|---|---|---|
| HME1 | MTOR | 1 | NM_004958.3:c.4447T>C | p.Cys1483Arg | Substitution | SIFT: deleterious; PolyPhen: probably damaging | N/A |
| HME2 | ZNF597 | 16 | N/A | N/A | Maternal uniparental disomy of Chr. 16p | Hypomethylation of ZNF597 | N/A |
HGVS, Human Genome Variation Society; dbSNP, Database for Short Genetic Variations; dbVar, Database of Genomic Structural Variation.
Figure 2.

Genetic findings in HME1. (A) Aligned next-generation sequencing reads at the mammalian target of rapamycin (MTOR) variant marked by vertical gray lines (NM_004958.3:c.4447T>C;p.Cys1483Arg) with 5/85 (6%) supporting the variant allele in brain tissue DNA, compared with 0/57 (0%) in blood. The figure was generated using the Integrated Genomics Viewer. (B) Quantitative digital droplet polymerase chain reaction (PCR) of DNA from the brain, blood, and buccal scraping of HME1, compared with DNA from brain tissue and leukocytes of controls, indicating that the variant allele frequency (VAF%) in the brain tissue is ∼6%. Limit of detection of the assay is indicated by the dashed red line.
Using genome-wide genotyping of DNA from affected brain and blood for HME2, we identified somatic uniparental disomy (UPD) of the p-arm of Chromosome 16, as evidenced by an allelic imbalance of heterozygous variants along 16p in only the DNA from the abnormal brain tissue, despite no difference in the intensity of the overall genotyping signal (logRratio) (Fig. 3A; Table 1). A similar pattern was seen in DNA extracted for abnormal hippocampal tissue (Supplemental Fig. 1). Fluorescence in situ hybridization (FISH) confirmed the presence of a normal diploid Chromosome 16 (Supplemental Fig. 2) in temporal cortical brain tissue, confirming that the allelic imbalance was not due to duplication of 16p in some cells of the affected tissue. Similar to the somatic variant in HME1, we did not detect UPD in leukocyte DNA (Fig. 3A) or buccal DNA (Supplemental Fig. 1). Consistent with the maternal allele being overrepresented in the abnormal brain tissue, the ratio of the intensity of the genotyping probe measuring the alternate allele to the total intensity (b-allele frequency) for heterozygous variants in the brain tissue DNA of HME2 that are absent in the mother (i.e., those that were inherited from the father) is reduced compared with the blood. The opposite is true for heterozygous variants in HME2 brain absent in the father; that is, there is a greater representation of alleles that were inherited from the mother in the brain compared with the blood (Fig. 3B).
Figure 3.

Genetic findings in HME2. (A) Genome-wide genotyping data revealing somatic uniparental disomy (UPD) of the p-arm of Chromosome 16, as evidenced by an allelic imbalance exclusively in brain tissue based on the B-allele frequency for heterozygous variants deviating from 0.5 in DNA from brain tissue (red dots) compared DNA from leukocytes (blue dots), without changes in the overall signal intensity (logRratio). (B) Bar graph of the B-allele frequency of heterozygous variants detected in the leukocytes of probands broken out by the parental genotype.
Exome sequencing was performed with both brain and blood for HME2 to further scan for candidate disease-causing somatic variants. No somatic variants were identified from the paired exome sequence data that were predicted to be functional (possibly damaging or probably damaging by PolyPhen-2 missense, splice, or stop-gained SNVs or frameshift indels) and absent from public exome databases and in-house sequenced controls. The 16p UPD event was confirmed in the exome-sequencing data for the brain tissue as a somatic loss of heterozygosity by comparing the allele frequencies of the sequencing reads for the brain and leukocyte DNA. To ensure that no variants were overlooked because of a small percentage of the cells from the affected tissue, we performed targeted capture and sequencing of genes in the PI3K–AKT–mTOR pathway; the targeted regions were sequenced at an average sequencing coverage greater than 600-fold in both the brain and leukocyte DNA (Table 2). No candidate somatic variants were detected with in-depth analysis of 192 genes in the pathway, including DEPDC5, PIK3CA, MTOR, or AKT3.
Table 2.
Sequencing statistics for whole-exome sequencing and targeted resequencing (TRS)
| Family | Sequencing type | Individual | Tissue | Total reads | Percent of mapped reads | Average fold coverage across targeted regions | Percent of targeted region at 20× coverage |
|---|---|---|---|---|---|---|---|
| HME1 | Exome | Proband | Brain | 221,970,984 | 99.3 | 173 | 90.2 |
| HME1 | Exome | Proband | Blood | 146,470,259 | 99.3 | 114 | 85.1 |
| HME2 | Exome | Proband | Brain | 194,995,169 | 99.2 | 116 | 94.3 |
| HME2 | Exome | Proband | Blood | 244,128,539 | 99.3 | 140 | 95.4 |
| HME2 | TRS | Proband | Brain | 12,546,960 | 91.3 | 679 | 99.4 |
| HME2 | TRS | Proband | Blood | 17,601,695 | 92.8 | 975 | 99.5 |
| HME2 | Exome | Mother | Blood | 150,325,726 | 99.2 | 106 | 94.1 |
| HME2 | Exome | Father | Blood | 206,747,009 | 99.3 | 137 | 95.3 |
Because the number of copies of 16p genes is intact, UPD would be benign unless there is an imprinted gene or if a maternally inherited heterozygous variant was a deleterious homozygous variant in the subset of affected cells. To explore the latter hypothesis, we performed exome sequencing on leukocyte DNA from HME2 and from both unaffected parents. More than 94% of bases comprising the Consensus Coding DNA Sequence (CCDS) genes on the p-arm of Chromosome 16 were sequenced to at least 10-fold; one rare missense variant in PMM2 was transmitted to HME2 from the mother where the homozygous genotype at this site was not found in the unaffected parents or the databases of controls. However, this variant is predicted to be benign by PolyPhen-2 and therefore not a convincing candidate. We also used the exome data to look for rare de novo or newly homozygous variants that might be responsible for HME, and none were detected in the protein-coding regions of the genome in HME2.
We next compared the methylation profiles of temporal cortex tissue of HME2 with those of control brains, which included both autopsy brains and fresh frozen tissue from patients with focal cortical dysplasia documented not to have somatic UPD on the p-arm of Chromosome 16, to look for genomically imprinted loci along 16p. Statistical analyses comparing the methylation level of candidate hemimethylated sites in control tissue with that of HME2 identified two candidate imprinted loci, spanning ZNF597 and UMOD (multiplicity adjusted P-values of 0.012 and 0.032, respectively). ZNF597 has been previously reported to be imprinted (Nakabayashi et al. 2011; Barbaux et al. 2012).
Functional Analyses
To confirm imprinting of ZNF597 in HME2, we evaluated allelic expression of inherited heterozygous variants in the brain tissue of the proband from the RNA-seq data. We confirmed that ZNF597 is imprinted in the brain based on preferential mRNA expression of the maternal allele at a heterozygous polymorphism inherited from the mother (rs12737, HME2-leukocyte genotype: heterozygous (variant allele frequency in the sequencing reads [VAF] = 56%); HME2-brain genotype: heterozygous [VAF = 72%], HME2-mRNA genotype: homozygous [VAF = 98%]; maternal genotype: homozygous [VAF = 100%]; paternal genotype: heterozygous [VAF = 55%]). We also directly show that the UPD variant in the brain is associated with increased ZNF597 mRNA expression and protein expression in HME2 compared with control and HME1 levels, consistent with hypomethylation in HME2 (Fig. 4). UMOD was not expressed in the brain tissue, suggesting that this additional signal was a false positive. We directly show the UPD variant in the brain is associated with increased ZNF597 mRNA expression and protein expression, consistent with hypomethylation (Fig. 4). We observed no evidence of methylation differences in the six PI3K–AKT–mTOR pathway genes on 16p (Supplemental Fig. 3).
Figure 4.
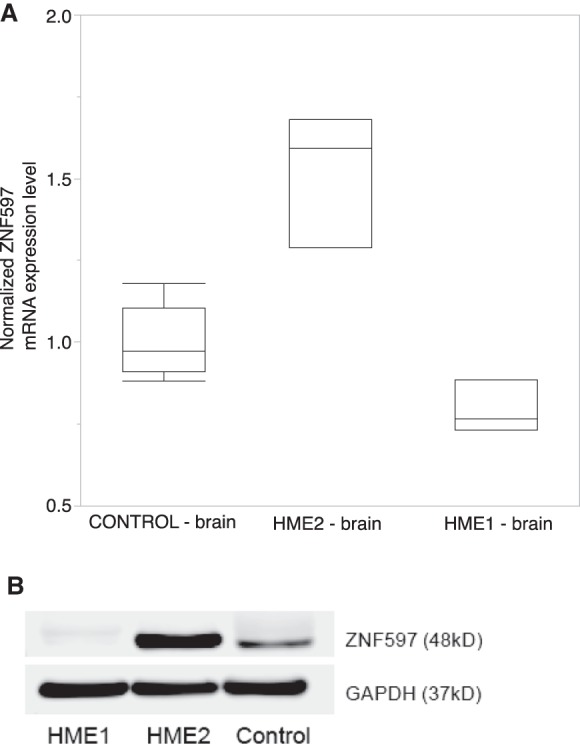
Increased (A) mRNA and (B) protein expression of ZNF597 in HME2 specimen lysates compared with HME1 and control brain tissue.
mTOR Hyperactivation
Because all previously identified somatic variants in HME have implicated the PI3K–AKT–mTOR pathway, we first assessed the level of mTOR expression in brain tissue from the two patients. Like the previously reported HME-associated somatic MTOR variants (Lee et al. 2012), immunolabeling with phospho-S6 antibodies reveals evidence for up-regulated mTOR signaling in HME1 brain tissue (Fig. 5). Similar to HME1, up-regulation of mTOR signaling was observed in brain tissue from HME2 (Fig. 5).
Figure 5.

mTOR activation in resected hemimegalencephaly (HME) tissues. (A) Phospho-S6 immunohistochemistry shows a few cytomegalic neurons and balloon cells in HME1. The inset shows a dysmorphic neuron and a balloon cell. (B) Phospho-S6 immunohistochemistry shows numerous cytomegalic neurons in HME2. Scale bar, 100 µm.
ZNF597 Expression
Because little is known about ZNF597 expression in normal human brain, we investigated the anatomic expression of ZNF597 in control brain specimens. There was modest expression of ZNF597 in control cortex and cerebellum (Supplemental Fig. 4). ZNF597 was detected in human fibroblasts, astrocytes, and U87 glioma cells (Supplemental Fig. 4). To better define interacting proteins, we used coimmunoprecipitation to define potential protein-binding partners of ZNF597. Previous work suggested an interaction between ZNF597 and ZDHHC17 (zinc finger, DHHC-type Containing 17), also known as Huntingtin Interacting Protein 14 (HIP14), which is a palmotyl acetyl transferase, known to interact with huntingtin, in yeast (Butland et al. 2014). We found that ZNF597 binds to ZDHHC17 (Fig. 6) when it is overexpressed in both 293T and U87 glioma cell lines.
Figure 6.
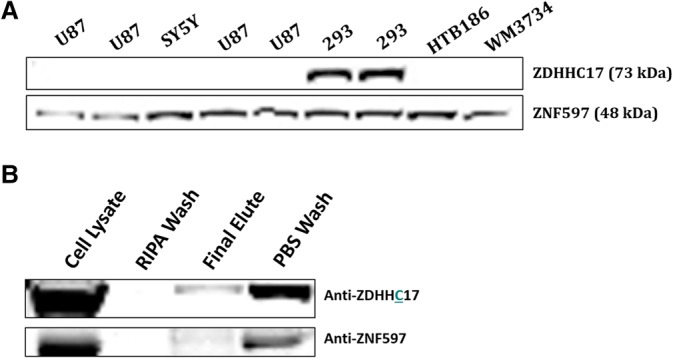
ZNF597 interacts with ZDHHC17. (A) Basal expression of ZNF597 and ZDHHC17 in several cell lines—for example, U87 glioma cells, SY5Y cells, 293FT cells, and human melanoma WM3734 cells. Note absent expression of ZDHHC17 in HTB186 cells. (B) (Top) Western assay demonstrates detection of ZDHHC17 protein following elution of bead-captured, Flag-tagged ZNF597 expressed in HTB186 cells (ZDHHC17 is not expressed by these cells under basal conditions). (Bottom) Detection of ZNF597 protein following elution of magnetic-bead-captured, Flag-tagged ZDHHC17 expressed in HTB186 cells. Some of the ZNF597 may be endogenous protein in HTB186 cells. Middle lanes depict minimal elution of bound proteins with either phosphate-buffered saline (PBS) or radio-immunoprecipitation assay (RIPA) buffer washes.
DISCUSSION
We identified and confirmed somatic variants in both patients evaluated in this study. In the first patient (HME1), we identified a somatic variant in MTOR (c.4447T>C;p.Cys1483Arg), located 1 bp away from the previously reported somatic HME variant (c.4448T>C; p.Cys1483Tyr) (D'Gama et al. 2015). A previous study identified the Cys1483Arg somatic variant in an individual with focal cortical dysplasia type IIb in 6% of alleles in the brain (Lim et al. 2015). In HME2, we found novel somatic UPD of the p-arm of Chromosome 16. UPD of the p-arm of Chromosome 16 of maternal origin in the germline is associated with abnormal intrauterine growth and a range of congenital malformations (Kalousek et al. 1993). Maternal UPD of Chromosome 16 has not been, to date, associated with malformations of cortical development (Kotzot and Utermann 2005); however, one case of germline maternal UPD 16 has been reported in an individual with likely Silver–Russell syndrome and relative macrocephaly (Azzi et al. 2015; Sachwitz et al. 2016). In this study, we show that the 16p UPD leads to hypomethylation and subsequent overexpression of ZNF597. A previous study also reported that maternal UPD of Chromosome 16 results in hypomethylation of ZNF597 (Nakabayashi et al. 2011). While overexpression of ZNF597 is one plausible explanation for the disease, we cannot rule out a maternally inherited heterozygous variant in a noncoding region that could be a deleterious homozygous variant in the cells with UPD or the presence of a noncoding de novo variant on the paternal chromosome that might be rescued by the somatic UPD event (Jongmans et al. 2012).
To date, somatic mutations identified in HME are thought to lead to the malformation of cortical development by altering mTOR signaling cascade (Lee et al. 2012; Poduri et al. 2012). Consistent with that observation, up-regulated mTOR signaling was found in the brain tissue of HME1 harboring a somatic MTOR mutation (Fig. 5). Interestingly, we also show evidence for up-regulated mTOR-signaling in HME2, despite the role of ZNF597 to the PI3K–AKT–mTOR pathway or in brain development more generally is not yet fully characterized. A previous study identified ZNF597 as a differentially spliced isoform of HIT-4 in mouse (Tanabe et al. 2010). Both HIT-4 and ZNF597 are zinc finger proteins that contain C2H2 domains, although their functions as putative transcription factors have not been confirmed. HIT-4/ZNF597 mRNA is widely expressed in rodent brain, and knockout of HIT-4/ZNF597 is lethal at embryonic day 7.5. Because the function of ZNF597 is unknown, we confirmed a previous finding that ZNF597 can bind to ZDHHC17, a Huntington interacting protein, thus providing some early insights into potential cell signaling pathways. Future studies are needed to determine whether hypomethylation of ZNF597 as a result of UPD of 16p can alter mTOR signaling and to more fully define the function of ZNF597 in cell growth and proliferation.
METHODS
Study Subjects
Two patients with HME were enrolled into the Genetics of Epilepsy study at Duke University. A blood sample was collected from the probands with HME and the unaffected parents. A buccal swab was also obtained from the probands. Both patients underwent resective surgery for the treatment of refractory seizures. Brain tissue specimens were collected and flash frozen from the temporal cortex of the first patient (HME1) and from the hippocampus and temporal cortex of the second patient (HME2). Formalin-fixed paraffin-embedded specimens were also collected as part of routine pathological analysis. Phenotypic information was compiled by interviewing the family, consulting with the treating neurologists and neurosurgeons, and by collecting data from the medical records.
The control brain tissue consisted of autopsy collected temporal cortex tissue obtained from the Kathleen Price Bryan Brain Bank or the Brain and Tissue Bank for Developmental Disorders (University of Maryland), as well as freshly collected brain tissue specimens from patients with focal cortical dysplasia. Control DNA samples from leukocytes were obtained from healthy controls enrolled in the Genetics of Memory Study at Duke University. All controls were confirmed not to have Chromosome 16p UPD or the MTOR variant identified in HME1.
All sample and phenotypic data collection were performed in accordance with the ethical standards set forth by the Duke University Institutional Review Board or the Maryland University Institutional Review Board and Committee on Human Research.
Genome-Wide Genotyping, Exome Sequencing, Targeted Resequencing, and RNA-seq
DNA samples extracted from leukocytes and brain tissue specimens collected from the HME probands were genotyped on the Illumina Human 610-Quad genome-wide genotyping array using standard protocols. DNA from buccal scrapings was genotyped on the Illumina HumanCore-12 genome-wide genotyping array. Copy-number variants were inferred using PennCNVsoftware (Wang et al. 2007). Exome sequencing and RNA-seq were performed within the Genomic Analysis Facility in the Center for Human Genome Variation (CHGV) at Duke University. Targeted resequencing was performed within the High Throughput Sequencing Facility in the Institute for Genomic Medicine (IGM) at Columbia University. For exome sequencing, sequencing libraries were prepared using the Illumina TruSeq or Kapa library preparation kit following the manufacturer's protocol, and targeted capture of the exome was performed with either Illumina TruSeq or Roche SeqCap EZ Exome v3.0 exome enrichment kits according to the manufacturer's protocol. For targeted resequencing, sequencing libraries were prepared using the Illumina TruSeq library preparation kit following the manufacturer's protocol, and targeted capture of the genes within the PI3K–AKT–mTOR pathway was performed with a custom designed enrichment kit (Roche SeqCap EZ) targeting 192 genes in the pathway, including the known HME genes (MTOR, AKT3, PIK3CA, and DEPDC5). Paired-end sequencing reads that were 125 bp in length were generated on either an Illumina HiSeq 2000 or an Illumina HiSeq 2500. Samples were exome-sequenced to an average coverage greater than 100-fold, and at least 95% of the CCDS genes were sequenced to at least 10-fold coverage on an Illumina HiSeq 2000 or 2500 sequencer. For RNA-seq, the Illumina TruSeq RNA kit was used to create sequencing libraries. Alignment and variant calling methods for exome-sequenced samples were described previously (Epi4K Consortium et al. 2013). Alignment of RNA-seq sequencing fragments was performed using TopHat software (Trapnell et al. 2009).
The minor allele frequency of identified variants on the p-arm of Chromosome 16 was assessed in internally available controls nonenriched for neuropsychiatric diseases and sequenced as part of other genetics studies (n = 2005), in approximately 6500 samples represented in the Exome Variant Server, National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS [August 2012]), and in approximately 65,000 samples present in the Exome Aggregation Consortium server (ExAC), Broad Institute, Cambridge, MA (http://exac.broadinstitute.org). sSNVs and somatic indels were called using Mutect2 (Cibulskis et al. 2013) and Varscan-2 software (Koboldt et al. 2009); loss of heterozygosity variants were called using Varscan-2. Qualifying variants were designated as those variants predicted to be deleterious (possibly damaging or probably damaging by PolyPhen-2 missense, splice, or stop-gained SNVs or frameshift indels) and missing from public databases and in-house controls, as well as from control brains that had been previously sequenced. Visual inspection of the alignment of the sequencing reads was performed with the Integrative Genomics Viewer (Robinson et al. 2011; Thorvaldsdóttir et al. 2013) to ensure the quality of the sequencing alignments and to rule out any variants with strand bias (i.e., variants for which the alternate allele only appeared on the plus or minus strand, which is indicative of a sequencing error).
Confirmation of Somatic MTOR Variant
The presence and absence of the MTOR variant in brain and blood/buccal DNA, respectively, was confirmed using digital droplet PCR (Bio-Rad QX200 Droplet Digital PCR System) beginning from 45 ng of DNA and using a standard digital droplet PCR protocol for a custom-designed TaqMan probe–based assay (F:CCATCATTCTAGGAAGCTCACCATT, R:CGACCCAGAGCTGATGCT, FAM-ATGCGCCGCCTCG, VIC-CATGCGCTGCCTCG).
Fluorescence In Situ Hybridization
Interphase fluorescence in situ hybridization (FISH) analysis was performed using the dual-color “break-apart” probe specific for the CBFB locus from Abbott Molecular. This probe set can detect rearrangement of the CBFB locus at 16q22, either by inversion 16 or translocation 16;16. In this context, it was used as a control for enumeration of the long arm (q) of Chromosome 16. FISH analysis was also performed using clone RP11-19H6 (BlueGnome) as a probe for enumeration of the short arm (p) of Chromosome 16. Localization of the probe to 16p13.3 was confirmed by inverted- DAPI banding.
Genome-Wide Methylation
Genome-wide methylation data were generated using the Infinium HumanMethylation450 BeadChip Kit. Unnormalized intensity data were normalized using wateRmelon methylation analysis package. Normalized data are shown in Supplemental Figure 5A. The distribution of the normalized intensity signal across 25 well-established imprinted regions (Pidsley et al. 2013) in control brain tissue samples is shown in Supplemental Figure 5B. Ninety percent of these known hemimethylated sites were located between normalized intensity values of 0.35 and 0.77. We then identified additional candidate imprinted sites along the p-arm of Chromosome 16 by filtering the average normalized intensity signals in control brain tissue also within this range. A total of 2635 probes along the 16p were within this range and annotated to a gene (446 genes). At each of these 2635 candidate imprinted loci, we calculated the sum of the squared t-statistic across each of the 446 genes, comparing the methylation signals for the two replicate analyses of DNA from the temporal cortex of HME2 with that for the DNA from 19 control temporal cortex specimens. We then calculated a marginal P-value for each gene by permutating the case and control status 10,000 times and comparing the observed sum of squared t-statistics to its permutation distribution. We also calculated multiplicity adjusted (for the 446 genes tested) P-values by comparing the sum of squared t-statistic for each gene with the permutation distribution of the max sum of squared t-statistics over all 446 genes.
ZNF597 Expression
ZNF597 mRNA expression was assessed using a commercially available TaqMan-based assay (ZNF597: Hs00331334_m1, Life Technologies). Quantitative real-time PCR was performed on RNA that was reverse-transcribed into cDNA by use of a High Capacity cDNA Synthesis Kit (Life Technologies). ZNF597 expression levels were normalized to mRNA expression of GAPDH (Life Technologies).
ZNF597 protein expression was assessed by western assay. Briefly, protein lysates were generated from HME1, HME2, and a single control cortex specimen using RIPA lysis buffer (50 mM Tris–HCl (pH 8.0); 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate, 0.1% SDS, protease and phosphatase inhibitors). Protein (30 µg) was separated on 4%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) Tris–Glycine gel (Bio-Rad), transferred onto polyvinylidene fluoride (PVDF) membranes, which were probed with ZNF antibodies overnight at 4°C and horseradish peroxidase (HRP)-conjugated secondary antibodies (GE Healthcare) for 1 h at room temperature (RT), and then visualized with ECL or ECL Plus (GE Healthcare). Membranes were probed with antibodies to GAPDH (Cell Signaling) to ensure equal protein loading. Additionally, cell lysates were taken from human fibroblasts (Coriel), human astrocytes (courtesy D. Kolson), and U87 glioma cells (courtesy D. O'Rorke) for analysis of ZNF597 expression.
Immunohistochemistry Assessing mTOR Signaling
Formalin-fixed, paraffin-embedded HME1 and HME2 brain specimens were microtome-sectioned at 7 µm and individually probed with antibodies recognizing the phosphorylated isoform of ribosomal S6 protein (phospho-S6; Ser 235/236; Cell Signaling, rabbit polyclonal, 1:200), native S6 protein (Cell Signaling, rabbit polyclonal, 1:200), or ZNF597 (AbCam, rabbit polyclonal, 1:200) overnight at 4°C. Sections were then probed with biotinylated secondary antibodies for 1 h at RT and visualized using avidin–biotin conjugation (Vectastain ABC Elite; Vector Labs) with 3,3′-diaminobenzidine. Dehydrated sections were mounted with coverslips (Permount). Light-microscopy images were acquired using a Leica DM4000 B microscope.
ZNF597 Protein–Protein Interactions
ZNF597 cDNA was obtained from OriGene (#RC206744) and cloned into pLD-puro-CcVA vector for high expression. The VA tag attached to the carboxyl terminus of ZNF597 consists of three Flag epitopes, a double tobacco etch virus (TEV) protease cleavage site, six His, and a StrepIII tag in series, ∼12 kDa in mass. ZDHHC17 (HIP14) cDNA was obtained from OriGene (#RC207982L1) containing both Myc and Flag tags at the carboxyl terminus. ZNF597 was expressed in 293FT cells, and ZDHHC17 was expressed in HTB186 cells (cerebellar medulloblastoma cells [ATCC]). Protein lysates were prepared with PBS-T (0.1% Tween-20) containing protease and phosphatase inhibitors. Anti-Flag Magnetic Beads (Sigma-Aldrich, A2220) were used for coimmunoprecipitation to trap ZNF597-Flag or ZDHHC17-Flag. The mixture was incubated at 4°C with end-to-end rotation overnight. Beads were washed with PBS and then RIPA buffer, and then were boiled in SDS-PAGE loading buffer. The released proteins were resolved on 4%–12% Bis–Tris protein gel (Invitrogen) and probed with corresponding antibody.
ADDITIONAL INFORMATION
Data Deposition and Access
The variants discovered in this study are listed in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) under accession numbers SCV000485071 (NM_004958.3:c.4447T>C) and SCV000485072 (uniparental disomy of Chromosome 16p). Patient consent was not obtained to deposit raw sequencing data.
Ethics Statement
All sample and phenotypic data collection was performed in accordance with the ethical standards set forth by the Duke University Institutional Review Board or the Maryland University Institutional Review Board and Committee on Human Research and with proper consent from study participants and their families. All necessary consent forms are on file at Duke University.
Acknowledgments
We graciously acknowledge the patients and their families for taking part in the research studies.
We acknowledge the following individuals or groups for the contributions of control samples: D. Daskalakis; R. Buckley; M. Hauser; J. Hoover-Fong, N.L. Sobreira, and D. Valle; A. Poduri; T. Young and K. Whisenhunt; Z. Farfel, D. Lancet, and E. Pras; R. Gbadegesin and M. Winn; K. Schmader, S. McDonald, H.K. White, and M. Yanamadala; R. Brown; S.H. Appel; E. Simpson; S. Halton, L. Lay; A. Holden; E. Behr; C. Moylan; A.M. Diehl and M. Abdelmalek; S. Palmer; G. Cavalleri; N. Delanty; G. Nestadt; D. Marchuk; V. Shashi; M. Carrington; R. Bedlack; M. Harms; T. Miller; A. Pestronk; R. Bedlack; R. Brown; N. Shneider; S. Gibson; J. Ravits; A. Gilter; J. Glass; F. Baas; E. Simpson; and G. Rouleau; The ALS Sequencing Consortium; The Murdock Study Community Registry and Biorepository; the Carol Woods and Crosdaile Retirement Communities; Washington University Neuromuscular Genetics Project; the Utah Foundation for Biomedical Research; and DUHS (Duke University Health System) Nonalcoholic Fatty Liver Disease Research Database and Specimen Repository.
The collection of control samples and data was funded in part by Biogen Idec.; Gilead Sciences, Inc.; New York-Presbyterian Hospital; The Columbia University College of Physicians and Surgeons; The Columbia University Medical Center; The Duke Chancellor's Discovery Program Research Fund 2014; Bill and Melinda Gates Foundation; The Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory; B57 SAIC-Fredrick Inc M11-074; The Ellison Medical Foundation New Scholar award AG-NS-0441-08; National Institute of Mental Health (K01MH098126, R01MH099216, and R01MH097993); National Institute of Allergy and Infectious Diseases (1R56AI098588-01A1); National Human Genome Research Institute (U01HG007672); National Institute of Neurological Disorders and Stroke (NINDS: U01-NS077303 and U01-NS053998); and National Institute of Allergy and Infectious Diseases Center (U19-AI067854 and UM1-AI100645).
Author Contributions
The study was conceived by N.G.G., E.L.H., and P.B.C. Patients were recruited and phenotyped by N.M.W., G.A.G., and M.A.M. Generation and analysis of the genome-wide genotyping data and exome-sequencing data were performed by N.G.G., K.D.C., and E.L.H. Protein and mRNA expression analyses were performed by N.G.G., K.D.C., P.B.C., and E.L.H. Protein–protein interaction analyses were conducted by P.B.C. Statistical analysis of the methylation status of 16p was performed by A.S.A., N.G.G., and E.L.H. FISH analyses were performed by H.G.L. and C.W.R. All authors participated in the writing and review of the manuscript.
Funding
This study was supported by NINDS R21NS078657 (E.L.H.), R01NS094596 (E.L.H. and P.B.C.), and R01NS082343-01 (P.B.C.) and Citizens United for Research in Epilepsy (CURE) (P.B.C.).
Competing Interest Statement
The authors have declared no competing interest.
Referees
Gholson Lyon
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
References
- Aronica E, Boer K, Baybis M, Yu J, Crino P. 2007. Co-expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol 114: 287–293. [DOI] [PubMed] [Google Scholar]
- Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, Netchine I, Harbison MD. 2015. A prospective study validating a clinical scoring system and demonstrating phenotypical–genotypical correlations in Silver–Russell syndrome. J Med Genet 52: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaux S, Gascoin-Lachambre G, Buffat C, Monnier P, Mondon F, Tonanny MB, Pinard A, Auer J, Bessières B, Barlier A, et al. 2012. A genome-wide approach reveals novel imprinted genes expressed in the human placenta. Epigenetics 7: 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, et al. 2012. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 44: 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer K, Troost D, Spliet WG, Redeker S, Crino PB, Aronica E. 2007. A neuropathological study of two autopsy cases of syndromic hemimegalencephaly. Neuropathol Appl Neurobiol 33: 455–470. [DOI] [PubMed] [Google Scholar]
- Butland SL, Sanders SS, Schmidt ME, Riechers S-P, Lin DTS, Martin DDO, Vaid K, Graham RK, Singaraja RR, Wanker EE, et al. 2014. The palmitoyl acyltransferase HIP14 shares a high proportion of interactors with huntingtin: implications for a role in the pathogenesis of Huntington's disease. Hum Mol Genet 23: 4142–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. 2011. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17: 734–742. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. 2012. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367: 1921–1929. [DOI] [PubMed] [Google Scholar]
- D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, Hossain A, Hatem NE, Barry BJ, Kwiatkowski DJ, et al. 2015. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol 77: 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium; Epilepsy Phenome/Genome Project; Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T. 2013. De novo mutations in epileptic encephalopathies. Nature 501: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, Tiziano FD, Fontaine B, Walley NM, Heavin S, et al. 2012. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 44: 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand MS, Griffin NG, Damiano JA, Cops EJ, Burgess R, Ozturk E, Jones NC, Leventer RJ, Freeman JL, Harvey AS, et al. 2016. Mutations of the sonic Hedgehog pathway underlie hypothalamic hamartoma with gelastic epilepsy. Am J Hum Genet 99: 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, Verwiel ET, Heijdra Y, Vulliamy T, Kamping EJ, Hehir-Kwa JY, Bongers EM, Pfundt R, van Emst L, van Leeuwen FN, et al. 2012. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet 90: 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalousek DK, Langlois S, Barrett I, Yam I, Wilson DR, Howard-Peebles PN, Johnson MP, Giorgiutti E. 1993. Uniparental disomy for Chromosome 16 in humans. Am J Hum Genet 52: 8–16. [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. 2009. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25: 2283–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzot D, Utermann G. 2005. Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A 136A: 287–305. [DOI] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, et al. 2012. De novo somatic mutations in components of the PI3K–AKT3–mTOR pathway cause hemimegalencephaly. Nat Genet 44: 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventer RJ, Jansen FE, Mandelstam SA, Ho A, Mohamed I, Sarnat HB, Kato M, Fukasawa T, Saitsu H, Matsumoto N, et al. 2014. Is focal cortical dysplasia sporadic? Family evidence for genetic susceptibility. Epilepsia 55: e22–e26. [DOI] [PubMed] [Google Scholar]
- Lim JS, Kim W-I, Kang H-C, Kim SH, Park AH, Park EK, Cho Y-W, Kim S, Kim HM, Kim JA, et al. 2015. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 21: 395–400. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D'Arcangelo G. 2006. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol 60: 420–429. [DOI] [PubMed] [Google Scholar]
- Nakabayashi K, Trujillo AM, Tayama C, Camprubi C, Yoshida W, Lapunzina P, Sanchez A, Soejima H, Aburatani H, Nagae G, et al. 2011. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum Mol Genet 20: 3188–3197. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, Shiina M, Shirozu H, Masuda H, Watanabe K, et al. 2015. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 78: 375–386. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, Wong CCY, Volta M, Lunnon K, Mill J, Schalkwyk LC. 2013. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, et al. 2012. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74: 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotech 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachwitz J, Strobl-Wildemann G, Fekete G, Ambrozaityte˙ L, Kucˇinskas V, Soellner L, Begemann M, Eggermann T. 2016. Examinations of maternal uniparental disomy and epimutations for Chromosomes 6, 14, 16 and 20 in Silver–Russell syndrome-like phenotypes. BMC Med Genet 17: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Hirano A, Iwasato T, Itohara S, Araki K, Yamaguchi T, Ichikawa T, Kumanishi T, Aizawa Y, Takahashi H, et al. 2010. Molecular characterization and gene disruption of a novel zinc-finger protein, HIT-4, expressed in rodent brain. J Neurochem 112: 1035–1044. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. 2007. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17: 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.