

Abstract
Metastatic triple-negative breast cancer comprises 12%–17% of breast cancers and carries a poor prognosis relative to other breast cancer subtypes. Treatment options in this disease are largely limited to systemic chemotherapy. A majority of clinical studies assessing efficacy of targeted therapeutics (e.g., the mammalian target of rapamycin [mTOR] inhibitor everolimus) in advanced breast cancer patients have not utilized predictive genomic biomarker-based selection and have reported only modest improvement in the clinical outcome relative to standard of care. However, recent reports have highlighted significant clinical responses of breast malignancies harboring alterations in genes involved in the phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling pathway to mTOR-inhibitor-involving regimens, underscoring the potential clinical benefit of treating subsets of breast cancer patients with molecularly matched targeted therapies. As the paradigm of cancer treatment shifts from chemotherapeutic regimens to more personalized approaches, the identification of additional reliable biomarkers is essential for identifying patients likely to derive maximum benefit from targeted therapies. Herein, we report a near-complete and ongoing 14-mo response to everolimus therapy of a heavily pretreated patient with biphenotypic, metastatic breast cancer. Genomic profiling of the metastatic triple-negative liver specimen identified a single reportable point mutation, STK11 F354L, that appears to have undergone loss of heterozygosity. No other alterations within the PI3K/mTOR pathway were observed. Published functional biochemical data on this variant are conflicting, and germline data, albeit with unclear zygosity status, are suggestive of a benign polymorphism role. Together with the preclinical data, this case suggests further investigation of this variant is warranted to better understand its role as a potential biomarker for mTOR inhibitor sensitivity in the appropriate clinical context.
Keywords: neoplasm of the breast
INTRODUCTION
Personalized medicine focuses on matching patients to appropriate therapeutic regimens based on the underlying genetic mechanisms driving their disease. Understanding the connection between genomic alterations and clinical outcomes promises to facilitate development of well-tolerated targeted therapies, aid in clinical trial selection, and ultimately improve cancer patient care. This approach has been exemplified in EGFR-mutant non-small-cell lung cancers and BRAF-mutant melanomas in which target inhibition with molecularly matched agents results in a dramatic clinical benefit (Arteaga and Baselga 2012).
Metastatic triple-negative breast cancer (TNBC) comprises 12%–17% of breast cancers (Carey et al. 2006; Foulkes et al. 2010) and carries a poor prognosis compared with other breast cancer subtypes (Dent et al. 2007; Liedtke et al. 2008). Treatment options for TNBC are largely limited to systemic chemotherapy because of a lack of efficacy for non-biomarker-directed targeted therapies (Singh et al. 2014). Utilizing platinum compounds as monotherapy in the neoadjuvant setting or in the context of combinational chemotherapy has demonstrated a mix of overall response rates (33%–80%), with the presence of a BRCA1/2 mutation being a key predictor of response (Silver et al. 2010; Maisano et al. 2011; Staudacher et al. 2011; Byrski et al. 2012). However, BRCA1/2 alterations are observed in 2%–5% of breast cancers, and predictive biomarkers of response to platinum regimens in the remaining patients remain unknown (The Cancer Genome Atlas Network 2012; Ciriello et al. 2015).
The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway is one of the most frequently deregulated pathways in human malignancies and has been implicated in breast cancer pathogenesis; 30%–35% of breast cancers harbor activating mutations in the PIK3CA oncogene or show loss of the PTEN tumor-suppressor gene via inactivating mutations or homozygous deletion (The Cancer Genome Atlas Network 2012). Although both mechanisms lead to constitutive activation of the downstream from AKT/mTOR signaling pathway, PTEN loss is enriched in TNBC (McAuliffe et al. 2010; Crown et al. 2012). Therefore, utilization of mTOR pathway inhibitors (e.g., everolimus and temsirolimus) was an attractive therapeutic strategy for the treatment of advanced breast cancers. However, in genomically unselected metastatic breast cancer patients, everolimus monotherapy demonstrated only modest clinical benefit with an overall response rate of 12% at a dose of 10 mg/day and 0% at a dose of 70 mg once weekly (Ellard et al. 2009). Moreover, a combination therapy of everolimus and the aromatase inhibitor exemestane resulted in a significant increase in median progression-free survival (6.9 mo) compared with exemestane alone (2.8 mo) in hormone receptor–positive (ER+/PR+), HER2-negative advanced breast cancer patients, although no significant increase in overall survival was reported (Baselga et al. 2012).
Clinical evidence demonstrating the efficacy of targeting the PI3K/AKT/mTOR pathway with mTOR inhibitors is mounting and suggests that subsets of patients may derive significant benefit from this approach. In one study of mesenchymal/metaplastic breast cancers treated with temsirolimus-based regimens, alterations in the PI3K/AKT/mTOR pathway were associated with therapeutic responses and prolonged stable disease (Moulder et al. 2015). A second study reported that six of eight patients with estrogen and/or progesterone receptor–positive gynecologic or breast malignancies featuring alterations of genes in the PI3K/AKT/mTOR pathway, including PIK3CA, AKT1, or PTEN, exhibited stable disease (SD), partial response (PR), or complete response (CR) following combined treatment with anastrozole and everolimus (Wheler et al. 2014). Recently, PIK3CA mutations and PTEN loss were identified as potential biomarkers for everolimus sensitivity in HER2+ breast cancer (André et al. 2016).
Here, we report a near-complete 14-mo response to everolimus therapy of a heavily pretreated patient with biphenotypic, metastatic breast cancer. Genomic profiling of her metastatic liver specimen identified a single reportable point mutation under loss of heterozygosity (LOH), STK11 F354L. The published literature suggests conflicting evidence supporting the role of this mutation in cancer. Although some data have predicted this variant to be a benign germline SNP, other data have demonstrated that this alteration can activate the PI3K/AKT/mTOR pathway. This case highlights the need for further studies aimed at assessing the role of this STK11 alteration in cancer progression and therapeutic response.
RESULTS
Clinical Presentation and Family History
The patient is a 49-year-old premenopausal woman who presented in 1997 with Grade III, T1c N1 M0, ER+/PR+ HER-2 intermediate invasive ductal carcinoma. The patient has no family history of breast cancer or evidence of BRCA1/2 mutations. The patient underwent a modified left mastectomy and received radiation therapy of the left chest wall and postoperative combination chemotherapy consisting of cyclophosphamide, methotrexate, and fluorouracil (CMF regimen), followed by docetaxel every 3 wk for six cycles and then tamoxifen for 5 yr (Fig. 1). Three years into treatment the patient underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy. Following completion of the 5-yr tamoxifen regimen, the aromatase inhibitor letrozole was administered for 7 yr through February 2009.
Figure 1.

Schematic of clinical course indicating treatment, duration, and disease recurrence. ER, estrogen receptor; PR, progesterone receptor; CMF, cyclophosphamide, methotrexate, and fluorouracil.
In late 2009, the patient experienced right upper quadrant pain and computed tomography (CT) imaging revealed extensive liver metastases. Liver biopsies confirmed metastatic undifferentiated carcinoma, which, unlike the original primary tumor, was ER−/PR−/HER2− (triple negative). The patient was treated with carboplatin and gemcitabine for 6 mo and a major response was observed on fused positron emission tomography (PET)–CT. Three months later, in November 2011, a follow-up PET–CT showed macronodular cirrhotic scarring but no sign of active disease in the liver. However, PET–CT imaging detected fluorodeoxyglucose (FDG)-avid spots in the spine at thoracic vertebrae T7 and T9. Biopsy specimens taken from the bone metastases were classified as poorly differentiated carcinoma, ER+/PR−/HER2−, consistent with the original breast cancer diagnosis. The patient received palliative radiation therapy and was started on anastrozole (1 mg tablet, daily) and fulvestrant (500 mg on days 1, 15, and 29 and once monthly thereafter) therapy. Three months later, in February 2012, PET–CT imaging revealed evidence of recurrence in the liver. Examination of a second biopsy taken from the liver was consistent with ER−/PR−/HER2− breast cancer, concordant with the immunohistochemistry (IHC) results from the initial liver metastasis. The patient continued to receive fulvestrant and anastrozole, with the addition of capecitabine chemotherapy (1000 mg/m2 b.i.d. [twice a day] 14 out of 21 days) from May to September 2012. Capecitabine was well tolerated except for mild-to-moderate hand–foot syndrome.
In September 2012, PET–CT scans indicated a complete metabolic response in all areas including both liver and spine, and cytotoxic chemotherapy was discontinued. The patient continued to receive fulvestrant and anastrozole for an additional 7 mo, at which time she presented with a palpable liver nodule with extensive left-sided musical rales involving the left chest and tumor-related symptoms of weight gain, bloating, poor endurance, and exertional dyspnea. A restaging PET–CT scan showed evidence of disease within the perihilar left upper lobe, and multiple additional foci of FDG avid metastases throughout both hepatic lobes. No additional FDG avid foci were observed in other locations, including the skeletal system, beyond sclerotic foci and photopenia within the thoracic spine in line with previously treated metastases. Results of the CT scans were consistent with response to combination hormonal therapy of the ER+ disease in the bone. However, significant disease progression of the ER−/PR−/HER2− liver metastases and a small degree of disease in the lung were observed. The patient was administered paclitaxel (80 mg/m2) weekly 3/4 (3–4 wk) from May to September 2013.
In January 2014, magnetic resonance imaging (MRI) and CT scans revealed extensive recurrent cancer throughout the liver (all lobes) and bone (including the axial and appendicular spine, T12, S1 vertebrae, the left iliac bone, and proximal left femur). An isolated left upper lobe lung nodule was also detected. In February 2014, the patient was started on megestrol acetate (40 mg twice daily) and 10 cycles of palliative chemotherapy with gemcitabine and carboplatin (days 1 and 8 out of a 21-d cycle) for 7 mo.
In June 2014, CT scans showed stable disease in terms of polyostotic bone metastases and liver metastases and slight regression of the dominant left hilar lung metastasis. At this time, with patient permission, the triple-negative liver specimen from the first recurrence (February 2012) was submitted for genomic profiling (Foundation Medicine, Cambridge, MA).
Genomic Analyses
Genomic profiling of the liver specimen from February 2012 identified a single reportable mutation: STK11 F354L (Tables 1 and 2) among the 236 cancer-related genes that were analyzed (Supplemental Table 1). STK11 encodes a serine/threonine kinase called LKB1 that activates AMPK and negatively regulates the mTOR pathway in response to changes in cellular energy levels (Shaw et al. 2004). Intriguingly, the STK11 mutation was the only detected alteration among several (19) genes involved in the PI3K/AKT/mTOR pathway (Table 3). We also identified four variants of unknown significance (Supplemental Table 2) whose role in cancer is currently unknown; however, none of these alterations suggested sensitivity to PI3K inhibitors. The STK11 F354L alteration was observed in 55% of sequencing reads spanning this region (Table 1). To investigate LOH at this locus, sequences were analyzed using a custom algorithm (see Methods) that incorporates mutant allele frequency, tumor purity, and copy-number changes at a given locus. Results from this analysis were consistent with the mutation being under LOH (Fig. 2). Therefore, this tumor lacked a wild-type copy of this gene.
Table 1.
Variant table
| Gene | Chr. | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP ID | Genotype | Mutant allele frequency (%) | Depth |
|---|---|---|---|---|---|---|---|---|---|
| STK11 | Chr 19 | c.1062C>G | p.F354L | Substitution | Deleterious | rs59912467 | het | 55 | 910 |
HGVS, Human Genome Variation Society; dbSNP, Single Nucleotide Polymorphism database; het, heterozygous.
Table 2.
Sequence coverage table
| Total reads | Total mapped reads | Target exon coverage (mean) | Target exon coverage at STK11 (mean) |
|---|---|---|---|
| 53,749,710 | 53,010,181 (98.6%) | 694.97 | 756.3 |
Table 3.
Summary of phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway genes analyzed
| Function | Gene | Alteration detected? (N/Y) |
|---|---|---|
| mTORC1/2 complex | MTOR | N |
| mTORC 1/2 complex | RPTOR | N |
| mTORC1/2 complex | RICTOR | N |
| mTORC-positive regulation | AKT1 | N |
| mTORC1-positive regulation | AKT2 | N |
| mTORC1-positive regulation | AKT3 | N |
| mTORC1-positive regulation | HRAS | N |
| mTORC1-positive regulation | IGF1 | N |
| PI3K pathway | PIK3C2G | N |
| PI3K pathway | PIK3C3 | N |
| mTORC1-positive regulation | PIK3CA | N |
| PI3K pathway | PIK3CG | N |
| mTORC1-negative regulation | PIK3R1 | N |
| mTORC1-negative regulation | PIK3R2 | N |
| mTORC1-negative regulation | PTEN | N |
| mTORC2-negative regulation | STK11 | Y |
| mTORC2-negative regulation | TSC1 | N |
| mTORC2-negative regulation | TSC2 | N |
| mTOR downstream regulator | TP53 | N |
Figure 2.

(A) Copy-number alteration plot of analyzed tumor sample sequenced. Chromosome numbers are annotated along the horizontal axis. Copy-number modeling from next-generation sequencing data was performed using established algorithms (see Methods). Log ratio values are noted on the left vertical axis; copy number (CN) for each region is noted on the right vertical axis. The region corresponding to STK11 is circled. (B) Chromosome 19 CN (left) and variant allele frequency (right) are enlarged. The region containing STK11 shows a CN of 1 with a corresponding drop in single-nucleotide polymorphism (SNP) allele frequencies, a signature of loss of heterozygosity.
Treatment Outcomes
Based on the presence of an STK11 alteration, in August 2014 the patient was started on exemestane (25 mg) and the mTOR inhibitor everolimus at 2.5 mg daily, with a dose increase of 2.5 mg every 2 wk until a target dose of 10 mg was achieved. The treatment was well tolerated with only a mild Grade I acneiform rash noted. The patient is currently approaching a complete response and treatment has been ongoing for 14 mo (Fig. 3). Consistent with this response, significant reductions in serum levels of alkaline phosphatase and cancer antigen 15-3 levels were observed within 3 mo of treatment initiation (Fig. 4). This response was far superior to median progression-free survival (6.9 mo) reported in the Phase III trial data (Baselga et al. 2012). Furthermore, the patient had failed multiple lines of chemotherapy and derived significant benefit from everolimus treatment in both the triple-negative and ER+ metastases.
Figure 3.

Representative positron emission tomography (PET)–computed tomography (CT) images (A) pre- and (B) posteverolimus treatment.
Figure 4.

Serum levels of (A) alkaline phosphatase (ALP) and (B) cancer antigen 15-3 (CA15-3) during clinical course of treatment.
DISCUSSION
Here we present a noteworthy response to everolimus in a heavily pretreated biphenotypic breast cancer patient whose tumor harbored an alteration in the mTOR-negative regulator gene, STK11 (Fig. 5). Although everolimus is Federal Drug Administration (FDA)-approved for breast cancer treatment, Phase III trial data in hormone-positive disease showed a response rate of 9.5% and a median overall survival of 6.9 mo (Baselga et al. 2012). In triple-negative disease, the response rate to everolimus with carboplatin was 28%, and progression-free survival was 3 mo (Singh et al. 2014). Previous studies have suggested that constitutive activation of PI3K/AKT/mTOR signaling via activating mutations in PIK3CA or inactivating events in PTEN may confer sensitivity to mTOR inhibitors in breast cancer (Wheler et al. 2014; Moulder et al. 2015).
Reliable biomarkers of mTOR-inhibitor sensitivity have yet to be firmly established. Anecdotal data from germline syndromes and sporadic cancers caused by mutations in this pathway suggest that aberrant activation of PI3K/AKT/mTOR signaling may confer sensitivity to these targeted agents. For example, germline mutations in TSC1/2, negative regulators of mTOR signaling, result in tuberous sclerosis and an increased risk of cancer. TSC1/2-driven cancers have demonstrated exquisite sensitivity to mTOR inhibitors, including everolimus (Wagner et al. 2010; Bissler et al. 2013; Franz et al. 2013; Mohammadieh et al. 2013). Somatic loss-of-function mutation in TSC1 and activating mutations in mTOR have been associated with exceptional responses to everolimus in bladder cancer and kidney cancer patients, respectively (Iyer et al. 2012; Voss et al. 2014; Wagle et al. 2014). Activating mutations in PIK3CA have been associated with higher response rates to PI3K/AKT/mTOR inhibitors across multiple cancer types in early-phase clinical trials (Janku et al. 2011, 2013). Finally, anecdotal evidence from a thymoma patient suggests that activating events in AKT1 (E17K) are also sensitive to these agents (Wheler et al. 2013).
Figure 5.
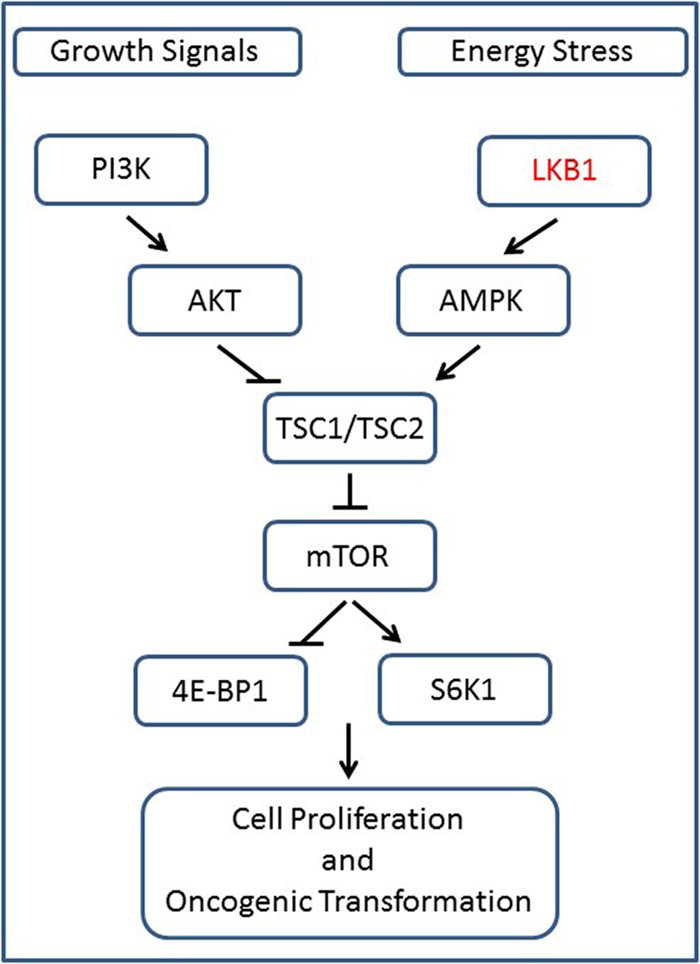
Schematic of the signaling pathway. The tumor-suppressor activity of LKB1 counteracts mitogen-induced phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling. Under energy-low cellular conditions, LKB1 activates AMPK, which, via activation of TSC1/TSC2, results in decreased mTOR activity.
Germline loss-of-function mutations in STK11 are responsible for Peutz–Jeghers syndrome (PJS), a rare autosomal dominant disorder (1 in 25,000–300,000) defined by polyps throughout the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk of cancer development (Beggs et al. 2010). One case report from a PJS patient with pancreatic adenocarcinoma describes a partial remission after everolimus treatment (Klumpen et al. 2011). However, the patient herein described has no evidence of PJS and was therefore not a candidate for germline testing. Somatic loss-of-function mutations in STK11 have been reported in lung adenocarcinoma (17%), intrahepatic cholangiocarcinoma (5%), and lung squamous cell carcinoma (1.7%) (The Cancer Genome Atlas Research Network 2012, 2014; Jiao et al. 2013). However, their association with sensitivity to targeted therapy remains largely unknown. Somatic mutations in STK11 are rare in breast cancers and have been reported in ∼0.2%–1.0% of invasive ductal carcinomas (MSKCC cBio portal, September 2015). Interestingly, the observed mutant allele frequency within the context of local copy-number loss suggests it is a germline alteration that has undergone LOH in this tumor. Collectively, these data suggest that the only copy of STK11 present in the tumor was the mutant allele.
The STK11 F354L alteration is located within the carboxyl terminus of LKB1. Preclinical data surrounding its functional effects are conflicting, and its role in cancer development is unclear. One study has shown that it impairs LKB1-mediated AMPK activation and leads to increased mTOR signaling (Forcet et al. 2005), whereas another study has shown that the mutation confers no change to LKB1 kinase activity (Launonen et al. 2000). Published reports have identified this alteration as both a germline variant in one family with PJS (Amos et al. 2004) and as a somatic mutation in cancer (Suzuki et al. 2012). These data are further complicated by the fact that this alteration has been identified in both cancer patients and normal controls at similar frequencies (6.3% and 5.6%, respectively) (Launonen et al. 2000). It is unclear how zygosity factors into a disease-specific function for this mutant. However, previous studies have shown that heterozygous germline truncation and missense polymorphisms are under somatic LOH-mediated selection in cancer (Ha et al. 2012; Lu et al. 2015). Thus, somatically acquired events may exploit susceptibilities in the germline genetic background of the patient. The fact that the mutation in this tumor appears to be under LOH is consistent with a loss-of-function role. Interestingly, in a study of Chinese patients with lung adenocarcinoma, 9/86 sequenced tumor samples harbored a germline STK11 F354L alteration, of which two exhibited LOH (Gao et al. 2010). Although heterozygous carriers may retain a functional copy of this gene, removal of the wild-type allele through LOH would result in a mutant-only form. In addition to a potential role in disease when under LOH, this alteration could also have a role in tumor maintenance and drug sensitivity. Other examples in the literature suggest that germline SNPs may modulate sensitivity to anticancer agents, regardless of their role in disease development (Huang et al. 2011; Ziliak et al. 2011; O'Donnell et al. 2012). Our data suggest that further research is warranted to better understand the role of this alteration in both tumorigenesis and mTOR inhibitor sensitivity.
The above data are provocative, but this study is not without limitations. Although STK11 was the sole gene with an identified alteration among the panel of PI3K/AKT/mTOR pathway genes sequenced, several alterative mechanisms to account for defects in this pathway remain possible. For example, changes to gene methylation or expression, microRNA expression, cellular metabolism, or other epigenetic modifications in the tumor sample were not assessed. It also remains plausible that other genomic events not captured by the current sequencing panel contributed to the patient's response. A further possibility is that additional alterations may have been acquired between the time of sequencing and treatment. However, given that significant therapeutic responses were observed across all metastatic tumor sites (liver, lung spin), this possibility seems less likely. Additional tissue for follow-up testing to investigate these possibilities was unavailable for this case.
As the paradigm of cancer treatment shifts from chemotherapeutic regimens to more personalized approaches, the discovery of reliable biomarkers is essential for identifying patients likely to derive maximum benefit from targeted therapies. Breast cancer treatment has already embraced this approach with the use of HER2-targeted therapies for HER2+ disease and antihormonal therapies for ER+/PR+ disease (Longo et al. 2007). Here, we present a case of a patient with biphenotypic breast cancer in whom multiple lines of chemotherapy had failed. The identification of an alteration in STK11 supported the initiation of therapy with the mTOR inhibitor everolimus. The patient has experienced an exceptional 14-mo response on this regimen. Despite controversy around the identified STK11 F354L variant, these data suggest a potential role of this variant in modulating mTOR inhibitor sensitivity when under LOH, regardless of its germline or somatic status. Additional research is warranted to better understand how this alteration affects the PI3K/AKT/mTOR signaling axis in the presence of mTOR inhibition.
METHODS
Local site permissions to use clinical samples were obtained for this work. Next-generation-sequencing-based genomic profiling was performed at a Clinical Laboratory Improvement Amendment (CLIA)-certified, College of American Pathologists (CAP)- and New York State (NYS)-accredited laboratory (Foundation Medicine). DNA was extracted from four formalin fixed paraffin embedded 10 micron sections. DNA was adaptor-ligated and capture was performed for all coding exons of 236 related genes and select introns from 19 genes frequently rearranged in cancer (Supplemental Table 1). Sequencing of captured libraries was performed using an Illumina HiSeq 2500 to a median exon coverage depth of 645×, and resultant sequences were analyzed for base substitutions, insertions, deletions, copy-number alterations (focal amplifications and homozygous deletions) and select gene fusions, as previously described (Frampton et al. 2013). Natural germline variants from The 1000 Genomes Project (dbSNP135) were removed, unless previously reported as somatic or characterized in the literature as pathogenic, and known driver alterations (COSMIC v62) were highlighted as biologically significant. All inactivating events (i.e., truncations and deletions) in known tumor-suppressor genes were also called as significant. To maximize mutation-detection accuracy (sensitivity and specificity) in impure clinical specimens, the test was previously optimized and validated to detect base substitutions at a ≥5% mutant allele frequency (MAF) and indels with a ≥10% MAF with ≥99% accuracy (Frampton et al. 2013). Copy-number modeling was performed by normalizing the tumor coverage profile to process-matched controls and performing a circular binary segmentation (CBS)-based segmentation to generate a genome-wide copy-number model. This model optimizes the copy-number profile with tumor ploidy and purity. Based on these optimized model parameters, expected tumor-derived minor allele frequency are made for each variant in each segment. Zygosity is then assigned as either homozygous or heterozygous based on the comparison between this expected minor allele frequency and the actual variant allele frequency. For example, in a region with a single-allele somatic deletion, LOH can be observed as a decrease CN in conjunction with a decrease in minor allele frequency (MAF) of germline polymorphisms. This decrease in allele frequency is proportional to the percentage of tumor cells within this sample. Thus, samples with higher tumor content will have a larger shift in germline polymorphism MAF. Segmentation calls are made based on the MAF. LOH calls are made based on the average MAF for a given segment in comparison to a model based on the tumor content of the sample and the copy-number profile.
ADDITIONAL INFORMATION
Data Deposition and Access
The ClinVar accession number for STK11 is SCV000299350 (https:www.ncbi.nlm.nih.gov.clinvar/).
Ethics Statement
The patient provided consent for genomic analysis of her sample and oral consent for the publication of this article. An institutional review board (IRB) determined that this case study was not human subject research and therefore the study did not require IRB approval.
Acknowledgments
We thank the patient for her willingness to share these data. We also thank Yuting He and James Sun (Foundation Medicine) for providing expertise and assistance with the LOH analysis.
Author Contributions
J.K. and J.C. contributed to conception and design. C.A.P., A.R., B.G., J.C., and J.K. contributed to acquisition of data. C.A.P., A.R., B.G., R.H., J.C., and J.K. contributed to analysis and interpretation of data. C.A.P., A.R., B.G., R.H., D.S., R.L.E., V.M., D.M., P.S., J.S.R., J.K., and J.C. contributed to writing, review, and/or revision of the manuscript. J.C. and J.K. contributed to study supervision.
Competing Interest Statement
C.A.P., A.R., B.G., R.H., D.S., R.L.E., V.A.M., D.M., P.S., J.S.R., and J.C. are employees of and have equity interest in Foundation Medicine, Inc.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Amos CI, Keitheri-Cheteri MB, Sabripour M, Wei C, McGarrity TJ, Seldin MF, Nations L, Lynch PM, Fidder HH, Friedman E, et al. 2004. Genotype–phenotype correlations in Peutz–Jeghers syndrome. J Med Genet 41: 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André F, Hurvitz S, Fasolo A, Tseng L-M, Jerusalem G, Wilks S, O'Regan R, Isaacs C, Toi M, Burris H, et al. 2016. Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2-overexpressing metastatic breast cancers: combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J Clin Oncol 34: 2115–2124. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Baselga J. 2012. Impact of genomics on personalized cancer medicine. Clin Cancer Res 18: 612–618. [DOI] [PubMed] [Google Scholar]
- Baselga J, Campone M, Piccart M, Burris HA III, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. 2012. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366: 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, Bertario L, Blanco I, Bülow S, Burn J, et al. 2010. Peutz–Jeghers syndrome: a systematic review and recommendations for management. Gut 59: 975–986. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ, et al. 2013. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 381: 817–824. [DOI] [PubMed] [Google Scholar]
- Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, Cybulski C, Marczyk E, Chrzan R, Eisen A, et al. 2012. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res 14: R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, et al. 2006. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295: 2492–2502. [DOI] [PubMed] [Google Scholar]
- Ciriello G, Gatza Michael L, Beck Andrew H, Wilkerson Matthew D, Rhie Suhn K, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. 2015. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown J, O'Shaughnessy J, Gullo G. 2012. Emerging targeted therapies in triple-negative breast cancer. Ann Oncol 23(Suppl 6): vi56–vi65. [DOI] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. 2007. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13: 4429–4434. [DOI] [PubMed] [Google Scholar]
- Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, Pritchard K, Eisen A, Vandenberg T, Taylor M, et al. 2009. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol 27: 4536–4541. [DOI] [PubMed] [Google Scholar]
- Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M. 2005. Functional analysis of Peutz–Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet 14: 1283–1292. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE, Reis-Filho JS. 2010. Triple-negative breast cancer. N Engl J Med 363: 1938–1948. [DOI] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. 2013. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, et al. 2013. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 381: 125–132. [DOI] [PubMed] [Google Scholar]
- Gao B, Sun Y, Zhang J, Ren Y, Fang R, Han X, Shen L, Liu X-y, Pao W, Chen H, et al. 2010. Spectrum of LKB1, EGFR, and KRAS mutations in Chinese lung adenocarcinomas. J Thorac Oncol 5: 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. 2012. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res 22: 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RS, Johnatty SE, Gamazon ER, Im HK, Ziliak D, Duan S, Zhang W, Kistner EO, Chen P, Beesley J, et al. 2011. Platinum sensitivity-related germline polymorphism discovered via a cell-based approach and analysis of its association with outcome in ovarian cancer patients. Clin Cancer Res 17: 5490–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, et al. 2012. Genome sequencing identifies a basis for everolimus sensitivity. Science 338: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, et al. 2011. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 10: 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, et al. 2013. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res 73: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. 2013. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 45: 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpen HJ, Queiroz KC, Spek CA, van Noesel CJ, Brink HC, de Leng WW, de Wilde RF, Mathus-Vliegen EM, Offerhaus GJ, Alleman MA, et al. 2011. mTOR inhibitor treatment of pancreatic cancer in a patient with Peutz–Jeghers syndrome. J Clin Oncol 29: e150–e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launonen V, Avizienyte E, Loukola A, Laiho P, Salovaara R, Järvinen H, Mecklin JP, Oku A, Shimane M, Kim HC, et al. 2000. No evidence of Peutz–Jeghers syndrome gene LKB1 involvement in left-sided colorectal carcinomas. Cancer Res 60: 546–548. [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, et al. 2008. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26: 1275–1281. [DOI] [PubMed] [Google Scholar]
- Longo R, Torino F, Gasparini G. 2007. Targeted therapy of breast cancer. Curr Pharm Des 13: 497–517. [DOI] [PubMed] [Google Scholar]
- Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MDM, Huang K-l, Wyczalkowski MA, Jayasinghe R, Banerjee T, et al. 2015. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun 6: e10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisano R, Zavettieri M, Azzarello D, Raffaele M, Maisano M, Bottari M, Nardi M. 2011. Carboplatin and gemcitabine combination in metastatic triple-negative anthracycline- and taxane-pretreated breast cancer patients: a phase II study. J Chemother 23: 40–43. [DOI] [PubMed] [Google Scholar]
- McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. 2010. Deciphering the role of PI3K/Akt/mTOR pathway in breast cancer biology and pathogenesis. Clin Breast Cancer 10Suppl 3: S59–S65. [DOI] [PubMed] [Google Scholar]
- Mohammadieh AM, Bowler SD, Silverstone EJ, Glanville AR, Yates DH. 2013. Everolimus treatment of abdominal lymphangioleiomyoma in five women with sporadic lymphangioleiomyomatosis. Med J Aust 199: 121–123. [DOI] [PubMed] [Google Scholar]
- Moulder S, Helgason T, Janku F, Wheler J, Moroney J, Booser D, Albarracin C, Morrow PK, Atkins J, Koenig K, et al. 2015. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann Oncol 26: 1346–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell PH, Stark AL, Gamazon ER, Wheeler HE, McIlwee BE, Gorsic L, Im HK, Huang RS, Cox NJ, Dolan ME. 2012. Identification of novel germline polymorphisms governing capecitabine sensitivity. Cancer 118: 4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. 2004. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6: 91–99. [DOI] [PubMed] [Google Scholar]
- Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, et al. 2010. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol 28: 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Novik Y, Stein S, Volm M, Meyers M, Smith J, Omene C, Speyer J, Schneider R, Jhaveri K, et al. 2014. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res 16: R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudacher L, Cottu PH, Diéras V, Vincent-Salomon A, Guilhaume MN, Escalup L, Dorval T, Beuzeboc P, Mignot L, Pierga JY. 2011. Platinum-based chemotherapy in metastatic triple-negative breast cancer: the Institut Curie experience. Ann Oncol 22: 848–856. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Oonishi T, Kudo T, Doi H. 2012. LKB1, TP16, EGFR, and KRAS somatic mutations in lung adenocarcinomas from a Chiba Prefecture, Japan cohort. Drug Discov Ther 6: 24–30. [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2012. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2014. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Hakimi AA, Pham CG, Brannon AR, Chen YB, Cunha LF, Akin O, Liu H, Takeda S, Scott SN, et al. 2014. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res 20: 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, Stewart M, Choueiri TK, Gandhi L, Cleary JM, et al. 2014. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov 4: 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N, Ligon AH, Antonescu CR, Ramaiya NH, Demetri GD, et al. 2010. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheler J, Hong D, Swisher SG, Falchook G, Tsimberidou AM, Helgason T, Naing A, Stephen B, Janku F, Stephens PJ, et al. 2013. Thymoma patients treated in a phase I clinic at MD Anderson Cancer Center: responses to mTOR inhibitors and molecular analyses. Oncotarget 4: 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheler JJ, Moulder SL, Naing A, Janku F, Piha-Paul SA, Falchook GS, Zinner R, Tsimberidou AM, Fu S, Hong DS, et al. 2014. Anastrozole and everolimus in advanced gynecologic and breast malignancies: activity and molecular alterations in the PI3K/AKT/mTOR pathway. Oncotarget 5: 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziliak D, O'Donnell PH, Im HK, Gamazon ER, Chen P, Delaney S, Shukla S, Das S, Cox NJ, Vokes EE, et al. 2011. Germline polymorphisms discovered via a cell-based, genome-wide approach predict platinum response in head and neck cancers. Transl Res 157: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.