

Abstract
Cisplatin is among the most widely used anticancer drugs and known to cause a dose‐limiting nephrotoxicity, which is partially dependent on the renal uptake carrier OCT2. We here report a previously unrecognized, OCT2‐independent pathway of cisplatin‐induced renal injury that is mediated by the organic anion transporters OAT1 and OAT3. Using transporter‐deficient mouse models, we found that this mechanism regulates renal uptake of a mercapturic acid metabolite of cisplatin that acts as a precursor of a potent nephrotoxin. The function of these two transport systems can be simultaneously inhibited by the tyrosine kinase inhibitor nilotinib through noncompetitive mechanisms, without compromising the anticancer properties of cisplatin. Collectively, our findings reveal a novel pathway that explains the fundamental basis of cisplatin‐induced nephrotoxicity, with potential implications for its therapeutic management.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Cisplatin is an important anticancer drug but it can cause severe, potentially life‐threatening injury to renal tubular cells, and the dogma is that this side effect is initiated by cisplatin uptake via the transporter OCT2.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ We have identified a new mechanism to import a nephrotoxic cisplatin metabolite into the kidney, namely, by the organic anion pathway transporters OAT1 and OAT3.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ Our study suggests that clinical exploration of OCT2 inhibitors as an adjunct to cisplatin therapy is unlikely to lead to complete kidney protection unless the identified organic anion pathway is also antagonized.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ Based on our results in mammalian models, certain FDA‐approved kinase inhibitors, including nilotinib, may be used to ameliorate cisplatin‐induced kidney injury in patients without affecting anticancer efficacy.
Cisplatin remains one of the most widely used agents for the treatment of multiple solid tumors in both children and adults. However, its clinical use is associated with dose‐limiting damage to renal tubular cells, cochlea, and peripheral nerves,1 and these complications may limit further treatment or even threaten life. There is no known treatment for cisplatin‐induced toxicities, and mechanistic details of these side effects remain poorly understood.2 The ability of cisplatin to cause damage to healthy tissues is dependent on the uptake carrier OCT2, which is highly expressed in renal proximal tubular cells, the cochlea, and dorsal root ganglia. Interestingly, cisplatin‐associated renal tubular damage is only partially diminished in mice that are knockout for the ortholog genes Oct1 (Slc22a1) and Oct2 (Slc22a2) (Oct1/2(–/–) mice),3, 4, 5 whereas the protection against platinum‐induced ototoxicity and neurotoxicity is complete.3, 6, 7 This suggests the existence of a secondary pathway that contributes, independently to Oct1/Oct2‐mediated uptake, to cisplatin‐induced renal damage.
As an initial step toward understanding the molecular mechanisms contributing to cisplatin‐induced nephrotoxicity in Oct1/2(–/–) mice, we previously reported that Oct1/Oct2‐mediated transport and p53 signaling are independently contributing to this side effect.8 The aim of the present study was to identify the mechanism that regulates cisplatin accumulation in renal tubular cells in the absence of Oct1/Oct2, and that acts upstream of p53. Our findings indicate that the Oct1/Oct2‐independent pathway is regulated by the transporters OAT1 (SLC22A6) and OAT3 (SLC22A8), which mediate accumulation of an anionic mercapturic acid metabolite formed in the γ‐glutamyl‐transpeptidase (GGT) pathway, and that access of cisplatin and this metabolite to tubular cells can be restricted by the tyrosine kinase inhibitor, nilotinib.
METHODS
Animal experiments
Male adult wildtype mice (8–12 weeks old), and sex‐ and age‐matched mice with a deficiency of Oct1 and Oct2 (Oct1/2(–/–)), all on an FVB background strain, were obtained from Taconic (Hudson, NY) and bred in‐house. C57BL/6 wildtype mice and corresponding Oat1(–/–) and Oat3(–/–) littermates were also bred in‐house. Animals were given a standard diet and water ad libitum, housed and handled in accordance with our Institutional Animal Care and Use Committee, and studies were performed in accordance with national animal protection laws. Gene expression was analyzed with RT2 Profiler PCR arrays (SABiosciences, Qiagen, Germantown, MD),4 and protein expression was performed as described.8
Evaluation of nephrotoxicity
All mice received a single i.p. dose of saline or cisplatin at 15 mg/kg (FVB mice) or 30 mg/kg (C57BL6 mice). Microscopic evaluation of nephrotoxicity by histology score was done by a pathologist who was blinded to the treatment of the animals.4 Ten random high‐power fields (10×) were selected per slide for scoring.5
Assessment of transporter function
Full‐length human OCT2, OAT1/OAT3, and mouse Oat1/Oat3 were obtained from Origene (Rockville, MD). Fusion‐flag fragments were amplified by high‐fidelity polymerase chain reaction (PCR). All engineered clones were validated by sequencing. Cells were maintained and uptake studies were done as described using dimethyl sulfoxide (DMSO)‐free and phenol‐red‐free conditions.5 Solutions of NAC‐1, a negatively charged N‐acetylcysteine S‐conjugate of cisplatin, were prepared fresh before each experiment by sequentially adding each compound until concentrations of 4 mM N‐acetylcysteine and 2 mM cisplatin were achieved. The structure and mass spectrum of the reaction product were previously confirmed using liquid chromatography‐tandem mass spectrometry.9
Isolation of mouse renal tubular cells
Primary murine renal tubular cells were isolated and cultured according to a modified protocol described previously.10 Briefly, renal cortical tissues were minced thoroughly and proximal tubular cells suspensions were obtained by phosphate‐buffered saline / ethylenediaminetetraacetic acid (PBS/EDTA) treatment and passage through cell strainers. After centrifugation at 2,000g for 10 min in Dulbecco's modified Eagle's medium (DMEM)/F‐12 medium, cells were plated in collagen‐coated flasks and cultured in DMEM/F‐12 medium supplemented with transferrin (5 μg/mL), insulin (5 μg/mL), hydrocortisone (0.05 μM), and vitamin C (50 μM). After about 1 week, confluent primary tubular cells were trypsinized and plated in 6‐well plates for uptake assays, as described above.
Evaluation of antitumor efficacy
Gene expression analyses in human tumors were obtained from the Pan Cancer gene expression data set, and extracted using the UCSC Xena browser. Accumulation of cisplatin and Pt‐DNA levels were determined as described.7 The cell growth inhibitory potential was evaluated with an methyl tetrazolium (MTT) assay using 48‐h continuous exposure. The influence of nilotinib (1 or 10 μM or vehicle; 15‐min preincubation) on the cytotoxicity of cisplatin (range, 0.01–100 μM) was evaluated in the replicating lung cancer cell lines A549, H23, H226, H322, H460, and H522 or the ovarian cancer cell lines IGROV‐1 and SKOV‐3. Nilotinib was dissolved in N‐methyl‐2‐pyrrolidone (NMP) and cisplatin stock solutions (10 mM) were prepared with the drug dissolved in cell culture medium.
Statistical analysis
Data are presented as mean values ± SD. Unpaired, two‐sided Student's t‐tests were calculated using Prism 5.0 (GraphPad, San Diego, CA), and P < 0.05 was considered statistically significant.
RESULTS
Phenotypic characterization of Oat‐deficient mice
Since tubular necrosis is not completely absent in Oct1/2(–/–) mice receiving cisplatin,3, 4, 5 we hypothesized that a secondary, organic anion transport mechanism is involved in the observed injury. To directly demonstrate a contribution of this organic anion system to cisplatin toxicity, we used a C57BL/6 mouse model with and without deletions of the Oat1 (Oat1(–/–) mice)11 and Oat3 genes (Oat3(–/–) mice),12 which are the main organic anion transporters localized to the basolateral membrane of renal tubular cells. The transporter‐deficient animals were phenotypically normal compared with wildtype mice as determined from a serum chemistry screen at baseline (Supplementary Table S1). Analysis of kidneys from Oat1(–/–) and Oat3(–/–) mice revealed that the expression of 84 ATP‐binding cassette (ABC) transporter and solute carrier (SLC) genes (Supplementary Table S2) was not substantially changed compared with tissues obtained from wildtype animals, with the exception of a downregulation of Slc22a6 and Slc22a8 “transcripts” (Figure 1 a,b). The renal protein expression of transporters with known or suspected relevance to cisplatin, such as Oct2,3, 4, 5 Abcc2 (Mrp2),13 and Ctr1 (Slc31a1)14 was unchanged relative to wildtype mice. However, Oat3 was virtually absent in the kidneys of Oat1(–/–) mice and reduced expression of Oat1 was observed in Oat3(–/–) mice (Figure 1 c,d), in line with published data.15
Figure 1.
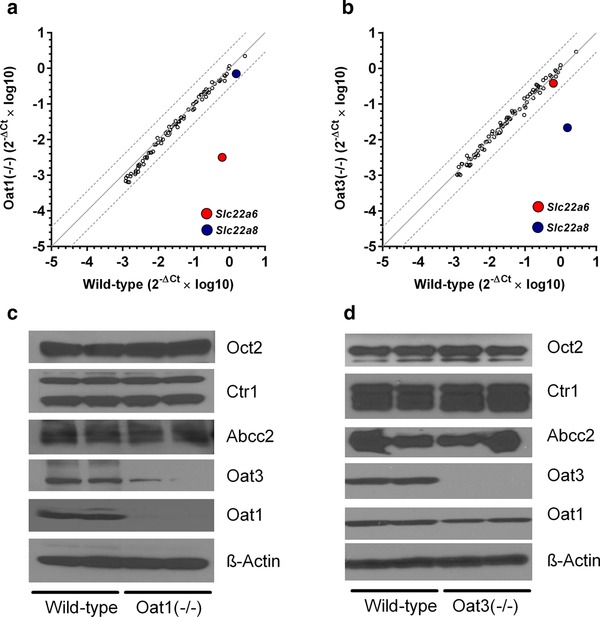
Phenotypic characterization of Oat1(–/–) and Oat3(–/–) mice. (a,b) Comparative expression of 84 transporter genes in kidneys at baseline of wildtype and Oat1(–/–) or Oat3(–/–) mice (n = 3 samples per group). Each symbol represents an average reading for a single gene, the solid line is the line of identity, and the dotted lines represent the 95% confidence intervals. The colored symbols represent the transporter genes for Oat1 (Slc22a6) and Oat3 (Slc22a8). (c,d) Comparative expression of five transporter proteins of known or suspected relevance to cisplatin in kidneys at baseline of wildtype and Oat1(–/–) or Oat3(–/–) mice.
Contribution of Oat1/Oat3 function to cisplatin toxicity
Next, we evaluated the comparative nephrotoxicity of cisplatin in Oat‐deficient mice. C57BL/6 mice, the strain for Oat1‐ or Oat3‐knockout, are relatively resistant to cisplatin‐induced nephrotoxicity compared with FVB mice, the strain used for Oct1/Oct2 knockout, presumably due to reduced renal expression of Oct1/Oct2 and impaired urinary excretion of cisplatin.8 A similar strain‐dependence has been reported for bromo‐dichloromethane‐induced nephrotoxicity, which is more severe in FVB mice than C57BL/6 mice.8 Therefore, cisplatin was administered to C57BL/6 mice at a relatively high single dose of 30 mg/kg. Under these conditions, we found that blood urea nitrogen (Figure 2 a) and creatinine (Figure 2 b), markers of acute renal tubular necrosis, were significantly increased within 1–3 days after administration in wildtype mice but not in Oat1(–/–) mice. The lesions observed in kidneys of cisplatin‐treated wildtype mice were characterized by dilated tubules filled with necrotic tubular epithelial cells, cellular debris, and proteinaceous casts, whereas the glomeruli, which do not express Oat1 or Oat3, were histologically normal (Figure 2 c,d). Similar observations were made in Oat3(–/–) mice (Figure 2 e–g), although the degree of cisplatin‐induced injury was higher than that observed in Oat1(–/–) mice (Figure 2 h). Because these mouse models do not unequivocally demonstrate that both Oat1 and Oat3 independently contribute to the observed injury, all related subsequent studies were done only in one of the models (the Oat1(–/–) mice).
Figure 2.
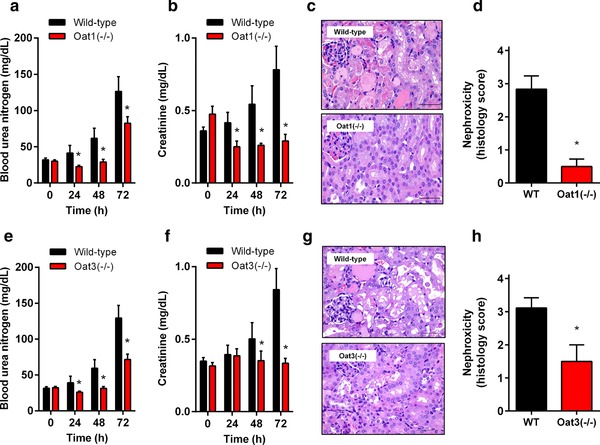
Influence of Oat1 or Oat3 loss on cisplatin‐induced nephrotoxicity. Levels of blood urea nitrogen (a) or serum creatinine (b) in wildtype or Oat1(–/–) mice (e,f for Oat3(–/–) mice) before and after cisplatin (30 mg/kg). (c) H&E staining of kidneys isolated from wildtype or Oat1(–/–) mice (g for Oat3(–/–) mice) 72 h after cisplatin. (d) Degree of nephrotoxicity based on histology scores observed in kidneys isolated from wildtype or Oat1(–/–) mice (h for Oat3(–/–) mice) 72 h after receiving cisplatin. Toxicity scores are based on percentage of damaged tubules: 0 (<10%; absent), 1 (11–25%; minimal), 2 (26–50%; mild), 3 (51–75%; moderate), and 4 (>75%; severe). All data are represented by mean values (bars) and SD (error bars), using n = 6 samples per group. The star (*) represents P < 0.05 vs. the corresponding wildtype group.
Transcriptional and functional profiling of Oat1(–/–) mouse kidney
As anticipated on the basis of the role of Oat1 as a xenobiotic uptake carrier in the kidney, we found that the rate and extent of the cumulative percentage of the cisplatin dose that was recovered in urine, expressed as the total of unchanged drug and metabolites, was significantly impaired in Oat1(–/–) mice compared with age‐matched wildtype mice (Figure 3 a). This finding is consistent with the contention that Oat1 transports cisplatin or a nephrotoxic metabolite into renal proximal tubular cells, and subsequently produces dose‐limiting renal toxicities. As a next step toward understanding the molecular mechanisms contributing to the lack of severe cisplatin‐induced nephrotoxicity in Oat1(–/–) mice, we performed tissue‐array analyses on kidney biopsies following cisplatin administration in vivo. Transcriptional profiling of 84 key genes implicated as potential biomarkers of drug‐induced proximal tubular damage (Supplementary Table S3) could clearly distinguish mouse genotype groups (Figure 3 b), and identified complex gene expression changes and a drug–response signature comprising of both upregulated and downregulated genes in wildtype mice that were largely absent in Oat1(–/–) mice. In particular, we found strong deregulation of multiple well‐characterized genes previously associated with cisplatin nephrotoxicity (Figure 3 c), including Havcr1 (KIM‐1), Noxa4 (NADPH oxidase 4), G6pc (glucose‐6‐phosphatase), and Calb1 (calbindin). None of the genes with altered expression in response to cisplatin differed among mouse genotypes in the absence of treatment. This suggests that cisplatin accumulation in renal tubular cells influences treatment sensitivity specifically in wildtype mice compared with Oat1(–/–) mice. This is consistent with the observation that pretreatment of wildtype mice with the Oat1/Oat3 inhibitor probenecid prevented cisplatin‐induced increases in blood urea nitrogen (Figure 3 d) and creatinine (Figure 3 e).
Figure 3.
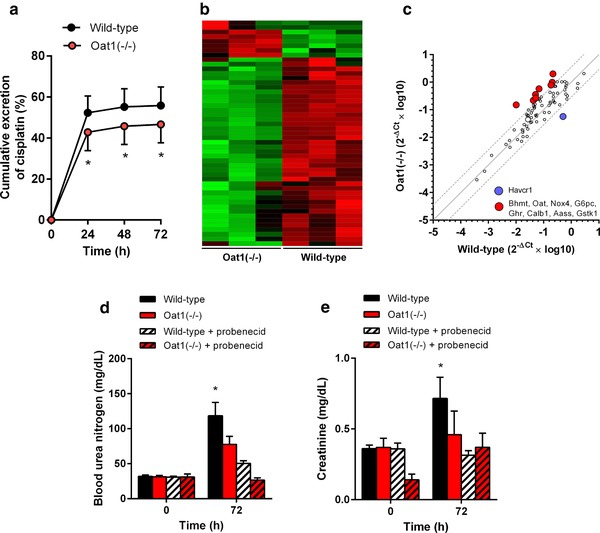
Cisplatin‐induced renal phenotypes in Oat1(–/–) mice. (a) Cumulative urinary excretion in wildtype and Oat1(–/–) mice within 72 h after cisplatin (30 mg/kg). (b,c) Comparative transcriptional profiling of 84 toxicity genes in kidney samples of wildtype and Oat1(–/–) mice after (30 mg/kg). Hierarchical clustering (b) was performed using Gapdh‐normalized data, and the color scale represents –1.5 SD (green) to 1.5 SD (red). In the correlation plot (c), each symbol represents a single gene, the solid line is the line of identity, and the dotted lines are the 95% confidence intervals. Levels of blood urea nitrogen (d) or serum creatinine (e) in wildtype or Oat1(–/–) mice are shown before and after cisplatin (30 mg/kg) with or without 3 probenecid administrations of 150 mg/kg (30 min before, 10 min after cisplatin, and 5 h after cisplatin). All data are represented by mean values (bars) and SD (error bars), using n = 3–6 per group. The star (*) represents P < 0.05 vs. the corresponding wildtype group.
OAT1/OAT3‐mediated transport of cisplatin metabolites
Previous studies have supported the existence of a mercapturic acid metabolite of cisplatin called NAC‐1 (Figure 4 a),9 a negatively charged N‐acetylcysteine S‐conjugate that acts as a precursor of a nephrotoxic and highly reactive thiol (Figure 4 b). To obtain evidence for a role of the organic anion system in the transport of this anionic cisplatin metabolite, we confirmed that NAC‐1, synthesized as described,9 preferentially accumulates in cells overexpressing OAT1 (Figure 4 c) or OAT3 (Figure 4 d) compared with control cells, and that this process is sensitive to inhibition by the OAT1/OAT3 inhibitor probenecid.
Figure 4.
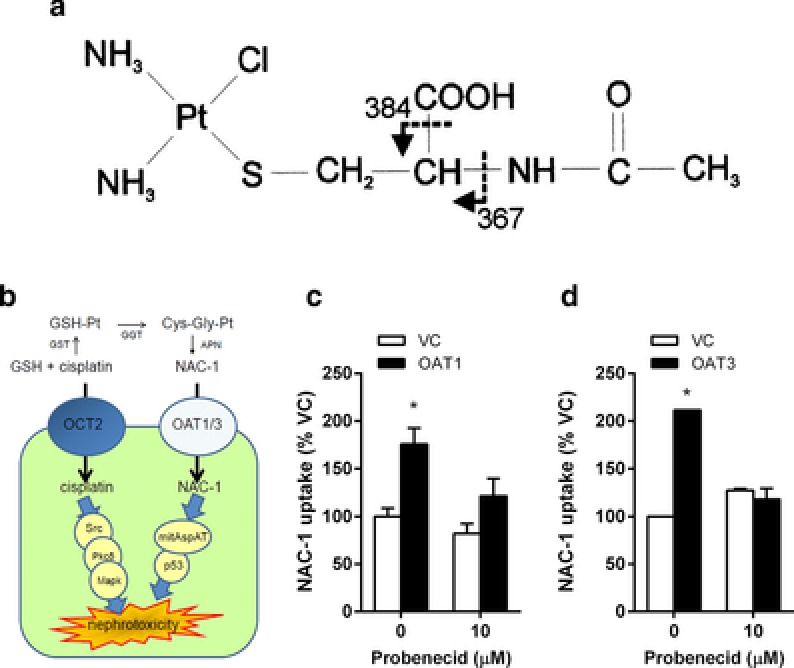
Contribution of OAT1 and OAT3 to renal transport of cisplatin metabolite NAC‐1. (a) Proposed OCT2‐dependent and ‐independent mechanisms of cisplatin‐induced nephrotoxicity. Involvement of OAT1 (b) and OAT3 (c) in the uptake of NAC‐1 (10 μM; 5‐min uptake) in the presence and absence of probenecid in transfected HEK293 cells. Data represent mean values (bars) and SD (error bars) using n = 6 samples per group, and were normalized to NAC‐1 uptake in the absence of probenecid in cells transfected with an empty vector (VC). The star (*) represents P < 0.05 vs. the VC group.
Identification of nilotinib as an OAT1/OAT3 inhibitor
Based on the ability of several tyrosine kinase inhibitors (TKIs) to potently inhibit both OCT2 function,16 as well as organic anion transporters such as OATP1B1,17 OATP1A2, and OATP2B1,18 we next evaluated the possibility that some of these same agents may also affect OAT1 and/or OAT3 function using the prototypical substrates p‐aminohippurate (PAH) and estrone‐3‐sulfate (E3S), respectively. Of 23 TKIs evaluated, we found that only nilotinib, an inhibitor of the Bcr‐Abl tyrosine kinase used for the treatment of patients with chronic myeloid leukemia, was able to potently inhibit OCT2 (Supplementary Figure S1), OAT1 (Figure 5 a), and OAT3 (Figure 5 b). Neither nilotinib (Figure 5 c) nor cisplatin (Figure 5 d) was identified as a transported substrate of OAT1 or OAT3, suggesting that the mechanism of inhibition by nilotinib is noncompetitive. We confirmed this observation further by showing that nilotinib can also inhibit the transport of 6‐carboxyfluorescein (6CF) in a model engineered to overexpress the murine orthologs Oat1 and Oat3 as well as proximal tubular cells isolated from wildtype mice (Figure 5 e). Interestingly, some of the TKIs evaluated in the screen caused an apparent increase in the activity of OAT1 or OAT3 by >1.5‐fold (Figure 5 a,b). The pharmacologic and therapeutic implications of this observation, however, remain to be investigated.
Figure 5.
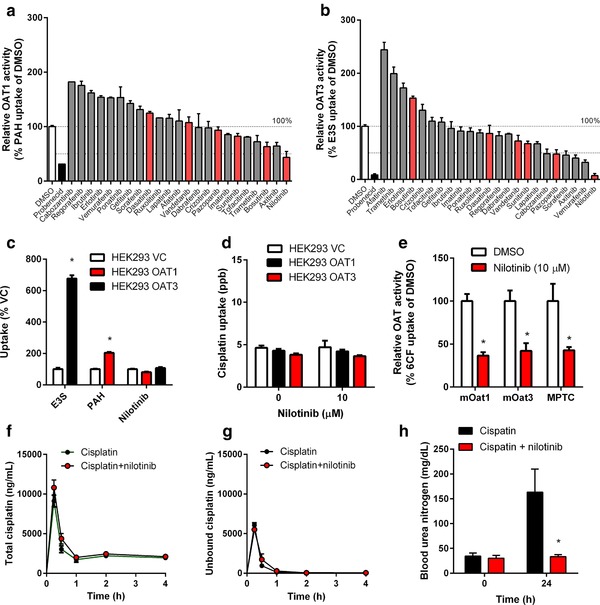
Inhibition of OAT1 and OAT3 function by nilotinib. Inhibition of OAT1 (a) and OAT3 (b) function by tyrosine‐kinase inhibitors (TKIs) in vitro (10 μM; 15‐min preincubation), using p‐aminohippurate (PAH) and estrone‐3‐sulfate (E3S) as OAT1 and OAT3 substrates in transfected HEK293 cells. Data (n = 6–9 per group) were normalized to substrate uptake in the absence of TKIs, and probenecid was used as a positive control inhibitor. TKIs with known OCT2‐inhibitory potential are shown in red. (c) Uptake of PAH (5 μM; 15‐min uptake) and E3S (1 μM; 15‐min uptake) by HEK293 cells overexpressing OAT1 and OAT3, respectively, and lack of uptake of nilotinib (1 μM; 15‐min uptake) by OAT1 or OAT3. Data (n = 6 per group) were normalized to substrate uptake in cells transfected with an empty vector (VC). (d) Lack of cisplatin uptake (500 μM; 60‐min uptake) by OAT1 or OAT3 in transfected HEK293 cells, with or without nilotinib (10 μM; 15‐min preincubation). (e) Inhibition of mouse Oat1 (mOat1) and mOat3 function in transfected HEK293 cells in vitro, and of organic anion transport function in mouse proximal tubular cells (MPTC) from FVB (wildtype mice) ex vivo by nilotinib (10 μM; 15‐min preincubation), using 6‐carboxyfluorescein (6CF) as a substrate. Data (n = 6 per group) were normalized to substrate uptake in the absence of nilotinib. (f, g) Plasma concentration–time curves for total cisplatin (f) or unbound cisplatin (g) after cisplatin (15 mg/kg) with or without pretreatment with nilotinib (150 mg/kg; p.o.) in DBA/lacJ (wildtype) mice. (h) Levels of blood urea nitrogen in Oct1/2(–/–) mice after cisplatin (15 mg/kg) with or without nilotinib pre‐treatment (15 mg/kg; p.o.). All data represent mean values (bars or symbols) and SD (error bars), and the star (*) indicates P < 0.05 vs. the respective control group.
Since the distribution of cisplatin into hepatocytes and its terminal elimination step into urine are transporter‐mediated processes involving Oatp1b2 (Slco1b2) and Mate1 (Slc47a1), respectively, which proteins are potentially sensitive to inhibition by nilotinib,17 we confirmed that the plasma levels of total cisplatin (Figure 5 f) and unbound cisplatin (Figure 5 g) were not affected by nilotinib due to an interaction at these sites (Supplementary Table S4). To explore nilotinib further as a dual Oat1/Oat3‐targeted agent for preventing cisplatin‐induced nephrotoxicity, it was administered concomitantly with cisplatin in Oct1/2(–/–) mice on an FVB strain. In line with our hypothesis, blood urea nitrogen levels were significantly reduced in Oct1/2(–/–) mice when nilotinib was administered in combination with cisplatin compared with cisplatin given alone (Figure 5 h).
Nilotinib as adjunct therapy during cisplatin treatment
Combining cisplatin with transporter‐inhibitors could possibly reduce the incidence and severity of cisplatin‐induced toxicity. However, it is important to establish that the anticancer efficacy of cisplatin is not compromised by nilotinib. The success of such a combination therapy would depend on two crucial factors: dosing/scheduling strategy and expression status of OCTs and OATs in cancer cells. To gain preliminary insights, we evaluated the expression profiles of OCT1/OCT2 and OAT1/OAT3 genes in 9,755 human tumor specimens using normalized RNAseq data from 31 individual cancer cohorts from The Cancer Genome Atlas (TCGA). This analysis indicated that the transporters are expressed at low levels in samples associated with the main cisplatin indications such as non‐small cell lung cancer (NSCLC) (Figure 6 a). As anticipated, the cellular uptake of cisplatin (Figure 6 b) and the nuclear Pt‐DNA adduct formation (Figure 6 c) in six different NSCLC cell lines were not substantially altered by nilotinib (Supplementary Figure S2). Most important, in vitro experiments where these cell lines were treated with the combination nilotinib‐cisplatin followed by MTT assays at 48 h indicated that nilotinib did not antagonize the cytotoxic effects of cisplatin under these conditions (Figure 6 d). Similar observations were made in ovarian cancer cell lines (Supplementary Figure S3), where nilotinib neither affected the uptake of cisplatin or NAC‐1 nor antagonized drug‐induced cytotoxicity.
Figure 6.
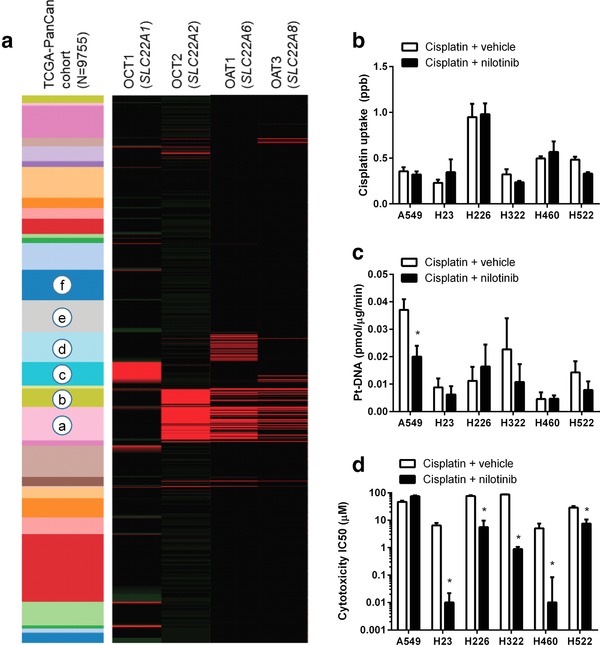
Nilotinib does not antagonize cisplatin‐mediated cell death. (a) Expression of the transporter genes SLC22A1 (OCT1), SLC22A2 (OCT2), SLC22A6 (OAT1), and SLC22A8 (OAT3) in 9,755 human tumor specimens using normalized RNAseq data from 31 individual pan‐cancer (PANCAN) cohorts from The Cancer Genome Atlas (TCGA). The expression values were normalized across cancer types, where the red color represents high gene expression values. The cohorts shown (top to bottom) include: thymoma, uterine carcinosarcoma, thyroid cancer, testicular cancer, sarcoma, rectal cancer, prostate cancer, pheochromocytoma, pancreatic cancer, ovarian cancer, ocular melanoma, mesothelioma, melanoma, lung cancer (squamous), lung cancer (adeno), glioma, liver cancer, large B‐cell lymphoma, kidney cancer (papillary), kidney cancer (clear cell), kidney chromophobe, head and neck cancer, glioblastoma, endometroid cancer, colon cancer, cervical cancer, breast cancer, bladder cancer, bile‐duct cancer, adrenocortical cancer, and acute myeloid leukemia. The highlighted cohorts include: a, kidney (clear cell); b, kidney (papillary); c, liver; d, glioma; e, non‐small cell lung cancer (NSCLC; adeno); and f, NSCLC (squamous). (b–d) Influence of nilotinib (10 μM; 15‐min preincubation) on the uptake (b), nuclear platination (Pt‐DNA) levels (c), and cytotoxicity (d) of cisplatin in the replicating NSCLC cell lines A549, H23, H226, H322, H460, and H522. Data (n = 4–16 per group) represent mean (bars) and SD (error bars), and the star (*) represents P < 0.05 vs. the respective control group.
DISCUSSION
The current study identified the organic anion transporters OAT1 and OAT3 as important, previously unrecognized contributors to cisplatin‐induced renal damage, which was shown to occur even in the absence of Oct1/Oct2‐dependent uptake of cisplatin into tubular cells. In particular, our results show that mice with a deficiency of the murine ortholog transporters Oat1 and Oat3 are partially protected from severe cisplatin‐induced nephrotoxicity. In addition, we found that nilotinib, a US Food and Drug Administration (FDA)‐approved TKI for the treatment of certain leukemias, is a potent inhibitor of organic anion and organic cation‐mediated transport, and that concomitant use of nilotinib with cisplatin abrogates renal tubular damage by simultaneously inhibiting both drug transport mechanisms.
In the precloning era, two carrier‐mediated systems for the renal secretion of organic compounds were known to exist, namely, an “organic cation system” and an “organic anion system”; classical substrates and inhibitors for the former include tetraethylammonium (TEA) and cimetidine, and PAH and probenecid, for the latter.19 In the late 1990s, the molecular entities responsible for the organic cation and organic anion systems in the kidney were finally identified as OCT2 and OAT1/OAT3, respectively.20 Although organic anions such as PAH and pyrazionate were originally reported to have no effect on the accumulation of cisplatin in renal cortex slices,21 subsequent in vivo investigation suggested involvement of the organic anion system in the development of cisplatin nephrotoxicity. In particular, a number of studies reported that probenecid can reduce the tubular secretion of total platinum after cisplatin administration in rats,22 rabbits,23 dogs,24 and humans,25 and can partially protect against cisplatin nephrotoxicity in mice.26, 27 Similar findings have been reported for furosemide,28 an agent now known, like probenecid, to be a potent inhibitor of OAT1 and OAT3.29 The present identification of the organic anion transporter pathway as a regulator of cisplatin‐induced nephrotoxicity is thus consistent with these prior observations.
Previous studies have indicated that rodent and human OAT1 and OAT3 can transport mercapturic acids,30 negatively charged N‐acetylcysteine S‐conjugates formed from the coupling of cysteine with endogenous or exogenous compounds. The existence of a mercapturic acid metabolite of cisplatin called NAC‐1,9 which acts as a precursor of a nephrotoxic and highly reactive thiol, led to our hypotheses that this conjugate is a substrate of OAT1 and/or OAT3 and is responsible for the OCT2‐independent mechanism leading to renal injury. The formation of NAC‐1 is initiated by a conjugation of cisplatin to glutathione either spontaneously or via glutathione‐S‐transferases, followed by cleavage first to a cysteinyl‐glycine‐conjugate and subsequently to NAC‐1 by GGT and aminopeptidases (APN), respectively, both of which are expressed on the surface of the proximal tubular cells. NAC‐1 is known to undergo transporter‐mediated uptake into LLC‐PK1 cells31 and rabbit proximal tubular cells32 by a mechanism that can be inhibited by PAH before undergoing final deconjugation by a pyridoxal 5′‐phosphate‐dependent enzyme, identified as mitochondrial aspartate aminotransferase (mitAspAT). The in vivo relevance of this pathway has been confirmed by the demonstration that genetic or pharmacological inhibition of GGT or mitAspAT33 offers partial protection against cisplatin‐induced nephrotoxicity. Our present findings show that the initiating event leading to NAC‐1‐mediated tubular injury is an uptake mechanism involving OAT1 and OAT3.
The demonstration that both OCT2 and OAT1/OAT3 play an important role in a clinically relevant cisplatin‐related toxicity provides a compelling rationale for the development of translational interventions for cisplatin‐containing regimens in patients involving the use of specific inhibitors of these carriers. Such agents would ideally have high potency, high specificity, low drug–drug interaction potential, intrinsic antitumor properties, favorable pharmaceutical properties, and nonoverlapping toxicity profiles. We hypothesized that the class of TKIs is of particular interest in this context, as these agents have many of the above features, and several members of the class have been found to potently inhibit various drug transporters, including OCT2.34 The ultimate identification of nilotinib as an inhibitor of OCT2, OAT1, and OAT3, without being itself a transported substrate, is consistent with a recent report suggesting that the in vitro inhibition constant of nilotinib for OAT3‐mediated transport of E3S is 0.41 μM,18 a concentration that is predicted to have potential in vivo significance.35 The mechanisms by which nilotinib affects OAT1 and OAT3 function requires further investigation.
Nilotinib was originally developed as an inhibitor of the Bcr‐Abl tyrosine kinase and has been used clinically as a once/twice daily oral tablet for the treatment of patients who are in the chronic and accelerated phases of Philadelphia‐chromosome‐positive chronic myeloid leukemia (CML). Compared with other TKIs, nilotinib has pharmaceutical and pharmacological features that suggest it might be an excellent modulator of cisplatin toxicities. These features include good oral absorption properties, a relatively slow systemic clearance, and a long half‐life of the terminal phase associated with relatively high and sustained plasma levels,36 thus ensuring sufficiently high and persistent local drug levels. Interestingly, high‐dose nilotinib pulse‐exposure is becoming an increasingly frequently applied concept in the treatment of CML,37 and the clinical experience with such a strategy will ultimately allow easy translation of our proposed concept to use nilotinib as a transporter inhibitor in conjunction with cisplatin‐based chemotherapy. It should be pointed out that, while most side effects associated with Bcr‐Abl inhibitors are mild, reversible, and easily managed, the nilotinib prescribing information carries a black box warning for QT prolongation, which could hinder immediate clinical implementation of the proposed intervention. In this context it is worth noting, however, that the median time from the start of nilotinib therapy using a conventional chronic regimen (i.e., once or twice daily dosing without interruption) to the onset of such events is >14 months (range, 2–68 months).38 Since in our studies we aim to interrogate the response of healthy tissues and cancer cells to the nilotinib–cisplatin combination following acute or intermittent exposure to the TKI, we anticipate that nilotinib will not be intrinsically toxic in the context of such studies.
The translational potential of a nilotinib‐based intervention strategy is further supported by our observation that the TKI does not antagonize the cytotoxic effects of cisplatin in preclinical models of the main cisplatin indications. These findings are in line with previously reported synergistic effects of nilotinib and cisplatin (or carboplatin) in leukemia39 and ovarian cancer,40 as well as the notion that other TKIs affecting Bcr‐Abl signaling can potentiate the in vitro and in vivo antitumor effects of cisplatin in models of lung cancer41 and several other tumor types.42 These results further suggest that cisplatin can be taken up into cancer cells by one or more distinct carriers independently of OCT1/OCT2 and OAT1/OAT3, and that these presently unknown carriers are insensitive to nilotinib‐mediated inhibition. Although in vivo confirmation is required, these initial observations indicate that combining cisplatin with TKIs such as nilotinib has the potential to reduce toxicities without compromising anticancer effects on tumor cells.
Collectively, we identified a previously unrecognized, OCT2‐independent pathway of cisplatin‐induced renal injury that is mediated by the organic anion transporters OAT1 and OAT3. The function of these transport systems can be simultaneously inhibited by nilotinib through noncompetitive mechanisms, without compromising the anticancer properties of cisplatin. These findings not only shed light on the etiology of cisplatin‐induced nephrotoxicity, but provide a rationale for the future development of new targeted interventions using transporter inhibitors to mitigate a debilitating side effect associated with cisplatin.
Supporting information
Supplementary Table S1. Serum chemistry at baseline in wildtype, Oat1(–/–), and Oat3(–/–) mice.
Supplementary Table S2. Genes included on the Mouse Transporter RT2 Profiles PCR array system.
Supplementary Table S3. Composition of the Mouse Nephrotoxicity RT2 Profiler PCR array.
Supplementary Table S4. Influence of nilotinib on the plasma pharmacokinetics of cisplatin in mice.
Supplementary Figure S1. Inhibition of OCT2 function by nilotinib.
Supplementary Figure S2. Cytotoxicity of nilotinib‐cisplatin in lung cancer models.
Supplementary Figure S3. Cytotoxicity of nilotinib‐cisplatin in ovarian cancer models.
Acknowledgments
We thank Guoqing Du and Erin Schuetz (St. Jude Children's Research Hospital, Memphis, TN) for technical assistance and helpful scientific discussion. The results shown are in part based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/). The project was supported in part by National Institutes of Health grants R01CA187176 and R01CA215802. These funding bodies had no role in the preparation of the article.
Author Contributions
A.S. and N.P. wrote the article; A.S., S.H., A.L., E.S., G.C., J.H.M.S., S.D.B., and N.P. designed the research; S.H., A.L., A.G., K.H., J.Y.K., L.L., A.V., J.S., D.S., E.S., and G.C. performed the research; A.S., S.H., A.L., A.G., K.H., J.Y.K., L.J.J., L.L., A.V., D.B.F., J.S., E.S., and G.C. analyzed the data; D.S. contributed new reagents/analytical tools.
Conflict of Interest/Disclosure
The authors declare no conflicts of interest.
References
- 1. de Jongh, F.E. et al Weekly high‐dose cisplatin is a feasible treatment option: analysis on prognostic factors for toxicity in 400 patients. Br. J. Cancer 88, 1199–1206 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yao, X. , Panichpisal, K. , Kurtzman, N. & Nugent, K. Cisplatin nephrotoxicity: a review. Am. J. Med. Sci. 334, 115–124 (2007). [DOI] [PubMed] [Google Scholar]
- 3. Ciarimboli, G. et al Organic cation transporter 2 mediates cisplatin‐induced oto‐ and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 176, 1169–1180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Filipski, K.K. , Mathijssen, R.H. , Mikkelsen, T.S. , Schinkel, A.H. & Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin‐induced nephrotoxicity. Clin. Pharmacol. Ther. 86, 396–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franke, R.M. et al Influence of Oct1/Oct2‐deficiency on cisplatin‐induced changes in urinary N‐acetyl‐beta‐D‐glucosaminidase. Clin. Cancer Res. 16, 4198–4206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lanvers‐Kaminsky, C. et al Human OCT2 variant c.808G>T confers protection effect against cisplatin‐induced ototoxicity. Pharmacogenomics 16, 323–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sprowl, J.A. et al Oxaliplatin‐induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. U. S. A. 110, 11199–11204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sprowl, J.A. et al Cisplatin‐induced renal injury is independently mediated by OCT2 and p53. Clin. Cancer Res. 20, 4026–4035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Townsend, D.M. , Marto, J.A. , Deng, M. , Macdonald, T.J. & Hanigan, M.H. High pressure liquid chromatography and mass spectrometry characterization of the nephrotoxic biotransformation products of Cisplatin. Drug Metab. Dispos. 31, 705–713 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pabla, N. et al Inhibition of PKCdelta reduces cisplatin‐induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Invest. 121, 2709–2722 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eraly, S.A. et al Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock‐out mice. J. Biol. Chem. 281, 5072–5083 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Sweet, D.H. et al Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (Oat3 (Slc22a8)) knockout mice. J. Biol. Chem. 277, 26934–26943 (2002). [DOI] [PubMed] [Google Scholar]
- 13. Wen, X. et al Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2‐null mice. Am. J. Pathol. 184, 1299–1308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pabla, N. & Dong, Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 73, 994–1007 (2008). [DOI] [PubMed] [Google Scholar]
- 15. Vallon, V. et al A role for the organic anion transporter OAT3 in renal creatinine secretion in mice. Am. J. Physiol. Renal Physiol. 302, F1293–1299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sprowl, J.A. et al A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 7, 10880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu, S. , Mathijssen, R. H. , de Bruijn, P. , Baker, S.D. & Sparreboom, A. Inhibition of OATP1B1 by tyrosine kinase inhibitors: in vitro‐in vivo correlations. Br. J. Cancer 110, 894–898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnston, R.A. , Rawling, T. , Chan, T. , Zhou, F. & Murray, M. Selective inhibition of human solute carrier transporters by multikinase inhibitors. Drug Metab. Dispos. 42, 1851–1857 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Hagenbuch, B. Drug uptake systems in liver and kidney: a historic perspective. Clin. Pharmacol. Ther. 87, 39–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. VanWert, A.L. , Gionfriddo, M.R. & Sweet, D.H. Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 31, 1–71 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Safirstein, R. , Miller, P. & Guttenplan, J.B. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 25, 753–758 (1984). [DOI] [PubMed] [Google Scholar]
- 22. Osman, N.M. & Litterst, C.L. Effect of probenecid and N′‐methylnicotinamide on renal handling of cis‐dichlorodiammineplatinum‐II in rats. Cancer Lett. 19, 107–111 (1983). [DOI] [PubMed] [Google Scholar]
- 23. Caterson, R. , Etheredge, S. , Snitch, P. & Duggin, G. Mechanisms of renal excretion of cisdichlorodiamine platinum. Res. Commun. Chem. Pathol. Pharmacol. 41, 255–264 (1983). [PubMed] [Google Scholar]
- 24. Klein, J. , Bentur, Y. , Cheung, D. , Moselhy, G. & Koren, G. Renal handling of cisplatin: interactions with organic anions and cations in the dog. Clin. Invest. Med. 14, 388–394 (1991). [PubMed] [Google Scholar]
- 25. Jacobs, C. , Coleman, C.N. , Rich, L. , Hirst, K. & Weiner, M.W. Inhibition of cis‐diamminedichloroplatinum secretion by the human kidney with probenecid. Cancer Res. 44, 3632–3635 (1984). [PubMed] [Google Scholar]
- 26. Ban, M. , Hettich, D. & Huguet, N. Nephrotoxicity mechanism of cis‐platinum (II) diamine dichloride in mice. Toxicol. Lett. 71, 161–168 (1994). [DOI] [PubMed] [Google Scholar]
- 27. Ross, D.A. & Gale, G.R. Reduction of the renal toxicity of cis‐dichlorodiammineplatinum(II) by probenecid. Cancer Treat. Rep. 63, 781–787 (1979). [PubMed] [Google Scholar]
- 28. Daley‐Yates, P.T. & McBrien, D.C. The renal fractional clearance of platinum antitumour compounds in relation to nephrotoxicity. Biochem. Pharmacol. 34, 1423–1428 (1985). [DOI] [PubMed] [Google Scholar]
- 29. Vallon, V. et al Overlapping in vitro and in vivo specificities of the organic anion transporters OAT1 and OAT3 for loop and thiazide diuretics. Am. J. Physiol. Renal Physiol. 294, F867–873 (2008). [DOI] [PubMed] [Google Scholar]
- 30. Bakhiya, N. et al Directing role of organic anion transporters in the excretion of mercapturic acids of alkylated polycyclic aromatic hydrocarbons. Drug Metab. Dispos. 35, 1824–1831 (2007). [DOI] [PubMed] [Google Scholar]
- 31. Townsend, D.M. , Deng, M. , Zhang, L. , Lapus, M.G. & Hanigan, M. H. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 14, 1–10 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolb, R.J. , Ghazi, A.M. & Barfuss, D.W. Inhibition of basolateral transport and cellular accumulation of cDDP and N‐acetyl‐L‐cysteine‐cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother. Pharmacol. 51, 132–138 (2003). [DOI] [PubMed] [Google Scholar]
- 33. Townsend, D.M. & Hanigan, M.H. Inhibition of gamma‐glutamyl transpeptidase or cysteine S‐conjugate beta‐lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 300, 142–148 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sprowl, J.A. , Mathijssen, R.H. & Sparreboom, A. Can erlotinib ameliorate cisplatin‐induced toxicities? J. Clin. Oncol. 31, 3442–3443, https://doi.org/10.1200/JCO.2013.50.8184 (2013). [DOI] [PubMed] [Google Scholar]
- 35. Giacomini, K.M. et al Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia, B. , Heimbach, T. , He, H. & Lin, T.H. Nilotinib preclinical pharmacokinetics and practical application toward clinical projections of oral absorption and systemic availability. Biopharm. Drug Dispos. 33, 536–549 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Lipka, D.B. et al Intracellular retention of ABL kinase inhibitors determines commitment to apoptosis in CML cells. PLoS One 7, e40853 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim, T.D. et al Clinical cardiac safety profile of nilotinib. Haematologica 97, 883–889 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang, F. et al Nilotinib enhances the efficacy of conventional chemotherapeutic drugs in CD34(+)CD38(‐) stem cells and ABC transporter overexpressing leukemia cells. Molecules 19, 3356–3375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weigel, M.T. et al Nilotinib in combination with Carboplatin and Paclitaxel is a candidate for ovarian cancer treatment. Oncology 87, 232–245 (2014). [DOI] [PubMed] [Google Scholar]
- 41. Zhang, K. , Wang, X. & Wang, H. Effect and mechanism of Src tyrosine kinase inhibitor sunitinib on the drug‐resistance reversal of human A549/DDP cisplatin‐resistant lung cancer cell line. Mol. Med. Rep. 10, 2065–2072 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Zhang, X. et al Imatinib sensitizes endometrial cancer cells to cisplatin by targeting CD117‐positive growth‐competent cells. Cancer Lett. 345, 106–114 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Serum chemistry at baseline in wildtype, Oat1(–/–), and Oat3(–/–) mice.
Supplementary Table S2. Genes included on the Mouse Transporter RT2 Profiles PCR array system.
Supplementary Table S3. Composition of the Mouse Nephrotoxicity RT2 Profiler PCR array.
Supplementary Table S4. Influence of nilotinib on the plasma pharmacokinetics of cisplatin in mice.
Supplementary Figure S1. Inhibition of OCT2 function by nilotinib.
Supplementary Figure S2. Cytotoxicity of nilotinib‐cisplatin in lung cancer models.
Supplementary Figure S3. Cytotoxicity of nilotinib‐cisplatin in ovarian cancer models.