

Abstract
Neoadjuvant androgen deprivation therapy (NADT) is one strategy for the treatment of early-stage prostate cancer; however, the long-term outcomes of NADT with radical prostatectomy including biochemical failure-free survival are not promising. One proposed mechanism is incomplete androgen ablation. In this study, we aimed to evaluate the efficiency of serum hydroxy-androgen suppression in patients with localized high-risk prostate cancer under NADT (leuprolide acetate plus abiraterone acetate and prednisone) and interrogate the primary sources of circulating hydroxy-androgens using our recently described stable isotope dilution liquid chromatography mass spectrometric method. For the first time, three androgen diols including 5-androstene-3β,17β-diol (5-adiol), 5α-androstane-3α,17β-diol (3α-adiol), 5α-androstane-3β,17β-diol (3β-adiol), the glucuronide or sulfate conjugate of 5-adiol and 3α-adiol were measured and observed to be dramatically reduced after NADT. By comparing patients that took leuprolide acetate alone vs leuprolide acetate plus abiraterone acetate and prednisone, we were able to distinguish the primary sources of these androgens and their conjugates as being of either testicular or adrenal in origin. We find that testosterone, 5α-dihydrotestosterone (DHT), 3α-adiol and 3β-adiol were predominately of testicular origin. By contrast, dehydroepiandrosterone (DHEA), epi-androsterone (epi-AST) and their conjugates, 5-adiol sulfate and glucuronide were predominately of adrenal origin. Our findings also show that NADT failed to completely suppress DHEA-sulfate levels and that two unappreciated sources of intratumoral androgens that were not suppressed by leuprolide acetate alone were 5-adiol-sulfate and epi-AST-sulfate of adrenal origin.
Keywords: androgen, localized high-risk prostate cancer, luteinizing hormone-releasing hormone agonist, abiraterone acetate, stable isotope dilution liquid chromatography mass spectrometry
Introduction
Prostate cancer (CaP) is the most commonly diagnosed and the third leading cause of cancer death in males of the United States (Siegel et al. 2017). Since the findings of Huggins and Hodges showed that castration could alleviate the symptoms of patients with advanced or metastatic CaP, the disease is recognized as being androgen dependent (Huggins & Hodges 2002, Perlmutter & Lepor 2007, Shafi et al. 2013, Wong et al. 2014). In males, the production of potent androgens: testosterone and 5α-dihydrotestosterone (DHT) occurs from two sources, Fig. 1. One is the testis that can directly produce testosterone and another is the adrenal, which produces precursors (e.g. dehydroepiandrosterone, DHEA; dehydroepiandrosterone sulfate, DHEA-S) for the formation of testosterone and DHT in peripheral tissues such as the prostate (Gomella et al. 2010, Cai & Balk 2011, Rege et al. 2013, Penning 2014, Sanchez-Guijo et al. 2016). Thus, androgen deprivation therapy (ADT) focusing on the suppression of testosterone and DHT has become the primary treatment for localized high-risk or metastatic CaP (Perlmutter & Lepor 2007, Shafi et al. 2013).
Figure 1.
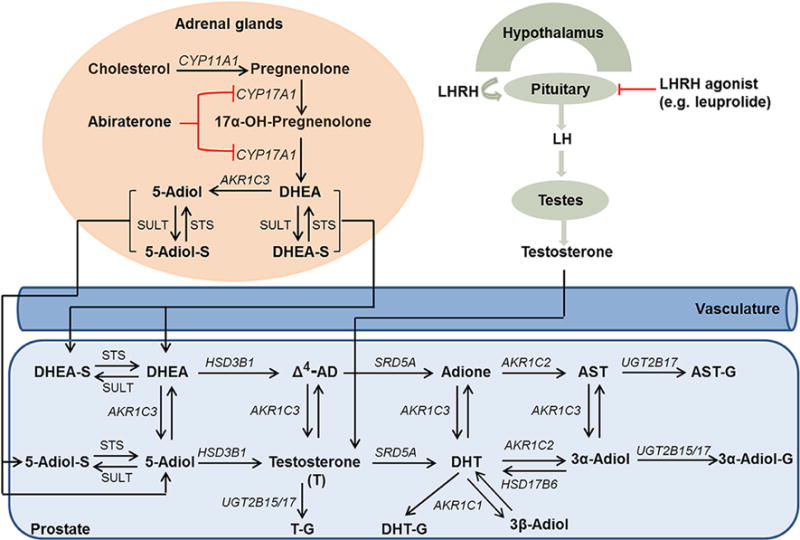
Androgen biosynthesis and metabolism in patients with prostate cancer. 3α-Adiol, 5α-androstane-3α,17β-diol; 3β-adiol, 5α-androstane-3β,17β-diol; 5-adiol, 5-androstene-3β,17β-diol; Adione, 5α-androstane-3,17-dione; Δ4-AD, Δ4-androstene-3,17-dione; AST, androsterone; DHEA, dehydroepiandrosterone; DHT, 5α-dihydrotestosterone; -G, glucuronide; -S, sulfate. Enzymes are identified by their gene names which are in italics. AKR1C1, 20α-hydroxysteroid dehydrogenase; AKR1C2, type 3 3α-hydroxysteroid dehydrogenase; AKR1C3, type 5 17β-hydroxysteroid dehydrogenase; CYP11A1, cytochrome P450 11A1; CYP17A1, cytochrome P450 17A1; HSD3B1, type 1 3β-hydroxysteroid dehydrogenase; HSD17B6, type 6 17β-hydroxysteroid dehydrogenase; SRD5A, 5α-reductase; STS, sulfatase; SULT, sulfotransferase.
Neoadjuvant androgen deprivation therapy (NADT) refers to ADT before patients undertake radical prostatectomy or radiation therapy (Kent & Hussain 2003). The aim of this approach is to downstage the malignant tumor, facilitate the complete surgical removal of the tumor and reduce the rate of cancer recurrence (Kent & Hussain 2003, Patel et al. 2016). NADT with radiotherapy has shown benefit in the improvement of clinical outcomes including progression-free survival and overall survival (Eom et al. 2014); however, the benefit of NADT followed by prostatectomy is still under investigation (Kent & Hussain 2003, Sonpavde et al. 2007). These early clinical trials used luteinizing hormone-releasing hormone agonists (LHRHa) or LHRHa in combination with antiandrogens (e.g. flutamide) to achieve the maximum androgen blockade for 3 months of treatment prior to prostatectomy (Fair et al. 1999, Klotz et al. 1999, Meyer et al. 1999, Debruyne & Witjes 2000, Schulman et al. 2000, Aus et al. 2002, Soloway et al. 2002, Yee et al. 2010). Although these trials significantly reduced the positive surgical margin, there was no difference in biochemical failure-free or progression-free survival rate in comparison to surgery alone during a long-term follow-up. To acquire more information on the efficacy of ADT and help clinicians explore the reasons for unimproved biochemical failure-free survival, a systematic analysis of the androgen metabolome in patients with NADT is necessary. However, few clinical trials of NADT have conducted a complete analysis of the effect of the treatment on the androgen metabolome due to the lack of analytical methods (Mostaghel et al. 2014, Taplin et al. 2014, Cho et al. 2015).
Recently, an NADT clinical trial using leuprolide acetate plus abiraterone acetate (AA) and prednisone in patients with localized high-risk prostate cancer was performed (Taplin et al. 2014). Leuprolide acetate (an LHRH agonist; LHRHa) can suppress the production of androgens (mainly testosterone) in testis, and AA inhibits the activities of cytochrome P450 17α hydroxylase/17,20-lyase (P45017A1) to block the conversion of pregnenolone to DHEA in the adrenal gland, which is the major source of precursors for testosterone and DHT in prostate and peripheral tissues in the absence of a functional testis (Fig. 1) (Gomella 2009, Goel & De 2011, Rege et al. 2013). One of the primary purposes of this clinical trial was to investigate if AA in addition to LHRHa could more effectively suppress the production of androgens in tissues in comparison to LHRHa alone. Patients on this clinical trial had serum levels of the keto-androgens including testosterone, DHT, DHEA, DHEA conjugates (sulfate and glucuronide), androsterone (AST) and Δ4-androstenedione (Δ4-AD), etc. measured by stable isotope dilution liquid chromatography-tandem mass spectrometry (SID-LC–MS/MS) following the derivatization of their ketone groups as Girard-T oximes (Taplin et al. 2014). However, androgen diols (e.g. 5-androstene-3β,17β-diol, 5-adiol; 5α-androstane-3α,17β-diol, 3α-adiol and 5α-androstane-3β,17β-diol, 3β-adiol) and their glucuronide or sulfate conjugates cannot be measured by this approach (Fig. 1). To make these measurements, we applied our recently reported SID-LC–MS/MS method coupled with picolinic acid derivatization to quantify nine hydroxy-androgens and their conjugates (glucuronide and sulfate) as picolinoyl esters in patients on this trial (Zang et al. 2017). Six of these androgens contain both a ketone group and hydroxyl group and can be called keto-androgens or hydroxy-androgens and can be determined by either Girard-T or picolinic acid derivatization (Tamae et al. 2013, Zang et al. 2017). By comparing patients that took leuprolide acetate alone vs leuprolide acetate plus AA and prednisone, we were able to distinguish the primary sources of these androgens and their conjugates as being of either testicular or adrenal in origin. Our findings also show that NADT failed to completely suppress DHEA-S levels and that 5-adiol-S and epi-AST-S of adrenal origin could not be reduced by leuprolide treatment alone.
Materials and methods
Serum from patients
Serum samples were collected from patients with localized high-risk CaP under a neoadjuvant randomized phase II trial of LHRHa (leuprolide acetate) plus AA with prednisone. Detailed information on this clinical trial (ClinicalTrials.gov Identifier: NCT00924469) has been reported elsewhere (Taplin et al. 2014). In brief, patients were randomly assigned into two groups. Patients in treatment group 1 (TX 1) received 12-week LHRHa followed by 12-week LHRHa plus AA and prednisone, and patients in treatment group 2 (TX 2) received a continuous 24-week LHRHa plus AA and prednisone treatment. Serum was collected at three time points (Day 1, Week 12 and Week 24). When 28 patients in each arm were compared, there were significant differences in serum androgen levels (Taplin et al. 2014). Based on the P values reported between the two groups, serum aliquots (250 μL) from 7 patients were selected randomly from each group as a convenience set and no preselection criteria were applied. Informed consent was obtained from all the patients. The trial was approved by institutional review boards at the enrollment sites. No patient enrollment occurred at the University of Pennsylvania where the studies were exempt from Human Subject IRB approval.
Materials for mass spectrometric analysis
Reagents were of ACS grade or higher (e.g. HPLC and Optima LC/MS) and were purchased from Thermo Fisher Scientific and used without further purification. Testosterone, epitestosterone (epi-T), DHEA, DHEA-S sodium salt, DHEA glucuronide (DHEA-G), AST, epi-AST, DHT, 5-adiol, 3α-adiol and 3β-adiol were purchased from Steraloids (Wilton, NH, USA). [2,3,4-13C3]-T ([13C3]-T) and [2,3,4-13C3]-DHT ([13C3]-DHT) were from C/D/N Isotopes (Point-Claire, Quebec, Canada) and Cambridge Isotopes (Andover, MA, USA), respectively. [2,3,4-13C3]-3α-adiol ([13C3]-3α-adiol) and [2,3,4-13C3]-3β-adiol ([13C3]-3β-adiol) were synthesized by an enzymatic method according to our published procedure (Zang et al. 2017). 4-Dimethylaminopyridine (DAP) 2-methyl-6-nitrobenzoic anhydride (MNBAn), picolinic acid (PA), triethylamine (TEA), anhydrous tetrahydrofuran (THF), β-glucuronidase from E. coli and sulfatase from Abalone entrails were from Sigma-Aldrich. Charcoal dextran stripped fetal bovine serum (CD-FBS) was from Atlanta Biologicals (Lawrenceville, GA, USA).
Preparation of serum samples
Serum samples were treated as previously described (Zang et al. 2017). In brief, the internal standard (IS) mixture of [13C3]-T, [13C3]-DHT, [13C3]-3α-adiol and [13C3]-3β-adiol (100 pg each) was spiked into 200 μL of serum. The samples were extracted with 2 mL of diethyl ether, and the organic layer was separated and evaporated. The dried residue was derivatized with 100 μL of picolinic acid reagent (50 mg PA, 40 mg MNBAn and 20 mg DAP in 1 mL THF) and 40 μL of TEA for 90 min at room temperature. After reaction, the picolinoyl ester derivatives were diluted by adding 1% aqueous acetic acid (1 mL) and purified using Strata C18-E SPE column (50 mg/mL) (Phenomenex, Torrance, CA, USA). The samples were dried and stored at −20°C.
Enzymatic hydrolysis was carried out for the analysis of androgen conjugates including glucuronide and sulfate by following the same procedure as reported before PA derivatization (Zang et al. 2017).
Calibration curves, quality control and mass spectrometric analysis
Calibration curves were prepared by a serial dilution of targeted androgen standard mixture in 200 μL of CD-FBS with a concentration range of 5–2500 pg/200 μL containing fixed amounts of ISs (100 pg each). Quality control (QC) samples were prepared with low, medium and high concentrations based on the range of the calibration curve from each targeted androgen. QC samples were analyzed along with serum samples. Androgen concentrations in serum were calculated based on the regression equations of calibration curves. Calibration curves were identical when performed in ethanol or CD-FBS indicating the absence of matrix effects and ion suppression. Precision and accuracy of the method varied by less than 20% of the coefficient of variation at the limit of quantitation (LOQ, data not shown).
A TSQ Quantum Ultra Triple Quadrupole mass spectrometer connected to Dionex UltiMate 3000 UHPLC system (Thermo Scientific) was employed for mass spectrometric analysis (Zang et al. 2017). The androgen picolinoyl derivatives were reconstituted in 100 μL of 60% acetonitrile in water (v/v) and 20 μL of the solution was injected to a Kinetex C18 column (100 mm × 2.1 mm, 2.6 μm, 100 Å; Phenomenex). The column was eluted with 0.05% (v/v) formic acid in water (buffer A) and 0.05% (v/v) formic acid in acetonitrile/methanol (40:60, v/v; buffer B). The gradient followed 20% B for 1 min, 20–60% B for 5 min, 60% B for 20 min, 60–95% B for 15 min and 95% B for 5 min. Data were analyzed by Xcalibur 3.0.63 software (Thermo Scientific). Concentrations of the analytes were calculated using the IS ratio method.
Statistical analysis
Concentrations of the targeted androgens were given as means with the ranges (Tables 1, 2 and 3). The comparison between TX 1 and TX 2 group at Week 12 was performed using Mann–Whitney U test in GraphPad Prism 6 (GraphPad Software) (Cho et al. 2015). P value less than 0.05 was considered as a statistically significant change.
Table 1.
Serum levels of unconjugated androgens.
| Androgens, unconjugated | Mean serum value (ng/dL)
|
P valuea | |||||
|---|---|---|---|---|---|---|---|
| TX 1: 24-week LHRHa/12-week AA + Prednisone (n = 7)
|
TX 2: 24-week LHRHa/24-week AA + Prednisone (n = 7)
|
||||||
| Day 1 (baseline) | Week 12 | Week 24 | Day 1 | Week 12 | Week 24 | ||
| T | 335.7 (213.9–691.2)b | 11.2 (4.7–18.5) | <1.0c | 313.0 (212.5–407.1) | <1.0 | <1.0 | 0.0006 |
| Epi-T | <1.0 | <1.0 | <1.0 | ((<1.0)–2.5)d | <1.0 | <1.0 | – |
| DHEA | 206.0 (125.6–391.6) | 227.0 (176.7–330.2) | 7.5 ((<1.0)–43.0) | 148.1 (65.9–251.1) | 1.8 ((<1.0)–4.1) | 2.4 ((<1.0)–4.9) | 0.0006 |
| DHT | 33.2 (18.7–71.1) | 3.3 ((<1.0)–10.4) | <1.0 | 33.0 (15.5–44.8) | <1.0 | <1.0 | 0.02 |
| AST | 17.6 (4.9–35.6) | 8.6 (2.7–22.0) | <1.0 | 11.5 (7.7–19.0) | <1.0 | <1.0 | 0.0006 |
| Epi-AST | 6.0 (2.5–15.9) | 5.3 (2.5–17.0) | <1.0 | 5.2 (2.8–6.2) | <1.0 | <1.0 | 0.0006 |
| 3α-Adiol | 9.9 (3.3–22.3) | <1.0 | <1.0 | 7.3 (4.2–10.6) | <1.0 | <1.0 | – |
| 3β-Adiol | 2.6 ((<1.0)–4.2) | <1.0 | <1.0 | 2.7 ((<1.0)–4.4) | <1.0 | <1.0 | – |
| 5-Adiol | 100.1 (35.9–191.2) | 44.2 (16.7–86.0) | <1.0 | 77.7 (34.0–121.3) | <1.0 | <1.0 | 0.0006 |
P values were calculated by comparing serum androgen levels at Week 12 between TX 1 and TX 2.
Range of serum levels from 7 patients in brackets.
Androgen level in each sample was less than LOD (limit of detection: 1.0 ng/dL for unconjugated androgen).
1.0 ng/dL (LOD) was used for the calculation of P values, when androgen levels were lower than LOD; and 2.5 ng/dL (LOQ) was used, when androgen levels were lower than LOQ.
3α-Adiol, 5α-androstane-3α,17β-diol; 3β-adiol, 5α-andorstane-3β,17β-diol; 5-adiol, 5-androstene-3β,17β-diol; AA, abiraterone acetate; AST, androsterone; DHEA, dehydroepiandrosterone; DHT, 5α-dihydrotestosterone; Epi-AST, epiandrosterone; Epi-T, epitestosterone; LHRHa, luteinizing hormone-releasing hormone agonist; T, testosterone; TX, treatment group; –, value is not available.
Table 2.
Serum levels of androgen glucuronides.
| Androgen glucuronide | Mean serum value (ng/dL)
|
P-valuea | |||||
|---|---|---|---|---|---|---|---|
| TX 1: 24-week LHRHa/12-week AA + Prednisone (n = 7)
|
TX 2: 24-week LHRHa/24-week AA + Prednisone (n = 7)
|
||||||
| Day 1 (baseline) | Week 12 | Week 24 | Day 1 | Week 12 | Week 24 | ||
| T-G | 99.8 (71.3–42.1)b | 19.7 (7.3–69.7) | <2.6c | 117.9 (65.0–216.9) | 4.2 (<2.6)–6.4)d | 3.7 ((<2.6)–6.4) | 0.0006 |
| Epi-T-G | <2.6 | <2.6 | <2.6 | ((<2.6)–13.1) | <2.6 | <2.6 | – |
| DHEA-G | 108.5 (45.0–188.1) | 82.2 (31.9–224.5) | <2.6 | 119.1 (7.4–514.4) | <2.6 | <2.6 | 0.0006 |
| DHT-G | 34.4 (15.3–68.8) | 10.9 ((<2.6)–27.8) | <2.6 | 56.7 (32.8–122.1) | <2.6 | <2.6 | 0.005 |
| AST-G | 2.5 × 103 (1666.1–3156.8) | 1.5 × 103 (1043.5–2934.4) | 18.4 ((<2.6)–62.4) | 2.5 × 103 (1223.7–4675.1) | 13.6 ((<2.6)–49.2) | 13.0 ((<2.6)–37.5) | 0.0006 |
| Epi-AST-G | 1.9 × 103 (931.4–3105.9) | 1.5 × 103 (614.2–3671.5) | 81.1 (11.1–337.2) | 2.7 × 103 (1002.9–7452.7) | 55.0 (16.4–117.7) | 71.9 (19.4–159.3) | 0.0006 |
| 3α-Adiol-G | 422.3 (223.1–776.9) | 87.7 (37.6–185.0) | <2.6 | 799.3 (74.3–2052.2) | <2.6 | <2.6 | 0.0006 |
| 3β-Adiol-G | – | – | – | – | – | – | – |
| 5-Adiol-G | 31.1 ((<2.6)–54.6) | 21.1 ((<2.6)–35.3) | <2.6 | 30.4 ((<2.6)–61.8) | <2.6 | <2.6 | 0.005 |
P values were calculated by comparing serum androgen levels at Week 12 between TX 1 and TX 2.
Range of serum levels for 7 patients in brackets.
Androgen glucuronide level in each sample was less than LOD (2.6 ng/dL for glucuronide conjugate).
2.6 ng/dL was used for the calculation of P values, when androgen glucuronide levels were lower than LOD; and 6.4 ng/dL (LOQ) was used, when androgen glucuronide levels were lower than LOQ.
3α-Adiol, 5α-androstane-3α,17β-diol; 3β-adiol, 5α-andorstane-3β,17β-diol; 5-adiol, 5-androstene-3β,17β-diol; AA, abiraterone acetate; AST, androsterone; DHEA, dehydroepiandrosterone; DHT, 5α-dihydrotestosterone; Epi-AST, epiandrosterone; Epi-T, epitestosterone; G, glucuronide; LHRHa, luteinizing hormone-releasing hormone agonist; T, testosterone; TX, treatment group; –, value is not available.
Table 3.
Serum levels of androgen sulfate conjugates.
| Androgen sulfate | Mean serum value (ng/dL)
|
P -valuea | |||||
|---|---|---|---|---|---|---|---|
| TX 1: 24-week LHRHa/12-week AA + Prednisone (n = 7)
|
TX 2: 24-week LHRHa/24-week AA + Prednisone (n = 7)
|
||||||
| Day 1 (baseline) | Week 12 | Week 24 | Day 1 | Week 12 | Week 24 | ||
| T-S | <204b | <204 | <204 | <204 | <204 | <204 | – |
| Epi-T-S | <204 | <204 | <204 | <204 | <204 | <204 | – |
| DHEA-S | 166 × 103 (121 × 103–259 × 103)c | 210 × 103 (115 × 103–375 × 103) | 3.1 × 103 (510–9750.3) | 96.7 × 103 (45.8 × 103–262 × 103) | 1.8 × 103 ((<204)–8004.5)d | 1.9 × 103 ((<204)–7557.6) | 0.0006 |
| DHT-S | <204 | <204 | <204 | <204 | <204 | <204 | – |
| AST-S | ((<204)–580.3) | <204 | <204 | ((<204)–3367.1) | <204 | <204 | – |
| Epi-AST-S | 4.0 × 103 (1980.0–7158.0) | 2.5 × 103 (510–6446.0) | <204 | 3.8 × 103 (571.5–7475.7) | <204 | <204 | 0.0006 |
| 3α-Adiol-S | <204 | <204 | <204 | <204 | <204 | <204 | – |
| 3β-Adiol-S | – | – | – | – | – | – | – |
| 5-Adiol-S | 13.6 × 103 (4774.2–33.6 × 103) | 11.9 × 103 (6330.7–29.0 × 103) | <204 | 8.0 × 103 (2199.3–18.8 × 103) | <204 | <204 | 0.0006 |
P values were calculated by comparing serum androgen levels at Week 12 between TX 1 and TX 2.
Androgen sulfate level in each sample was less than LOD (204 ng/dL for androgen sulfate conjugate).
Range of serum levels for 7 patients in brackets.
204 ng/dL (LOD) was used for the calculation of P-values, when androgen sulfate levels were lower than LOD; and 510 ng/dL (LOQ) was used, when androgen sulfate levels were lower than LOQ.
3α-Adiol, 5α-androstane-3α,17β-diol; 3β-adiol, 5α-andorstane-3β,17β-diol; 5-adiol, 5-androstene-3β,17β-diol; AA, abiraterone acetate; AST, androsterone; DHEA, dehydroepiandrosterone; DHT, 5α-dihydrotestosterone; Epi-AST, epiandrosterone; Epi-T, epitestosterone; LHRHa, luteinizing hormone-releasing hormone agonist; S, sulfate; T, testosterone; TX, treatment group; –, value is not available.
Results
Serum samples from seven patients in each group (TX 1 and TX 2) were included in this study. TX 1 is 12-week treatment of LHRHa followed by 12-week LHRHa plus AA and prednisone and TX 2 is 24-week treatment of LHRHa plus AA and prednisone. Androgen levels that decline after LHRHa treatment alone were considered of testicular origin, while androgen levels that decline only after LHRHa plus AA and prednisone treatment were considered to be of adrenal origin. The baseline characteristics and pathological outcomes from each patient were shown in Supplementary Table 1 (see section on supplementary data given at the end of this article). The representative LC–MS/MS chromatograms for nine hydroxy-androgens and four ISs as picolinoyl esters were shown in Supplementary Fig. 1.
Analysis of serum unconjugated androgens
In TX 1 baseline (Day 1), levels of testosterone (335.7 ng/dL), DHT (33.2 ng/dL), AST (17.6 ng/dL) and 5-adiol (100.1 ng/dL) were reduced by 97, 90, 51 and 56% at Week 12, respectively with leuprolide treatment alone (Table 1). Addition of AA and prednisone further reduced these levels to undetectable levels (<1.0 ng/dL: limit of detection) by Week 24. 3α-Adiol and 3β-adiol were reduced to undetectable levels at both Week 12 and Week 24. Serum levels of DHEA (206.0 ng/dL) and epi-AST (6.0 ng/dL) were not changed at Week 12 in comparison to baseline, but were reduced to 7.5 ng/dL (a 96% decrease) and to an undetectable level at Week 24, respectively when AA and prednisone were added to the leuprolide treatment. Epi-T was not detected (<1.0 ng/dL).
In TX 2 (Table 1), serum levels of T, DHEA, DHT, AST, epi-AST, 3α-adiol, 3β-adiol and 5-adiol were reduced to undetectable or lower levels at both Week 12 and Week 24 when leuprolide, AA and prednisone were given in combination, e.g. DHEA levels were reduced from 148.1 ng/dL at baseline to 1.8 ng/dL and 2.4 ng/dL at these respective times. Epi-T was only detected in one patient with a level of less than 2.5 ng/dL (LOQ) in TX 2 at baseline (Table 1). The individual patient variation of unconjugated androgens is shown in Figs 2, 3 and Supplementary Figs 2, 3, 4, 5, 6, 7.
Figure 2.
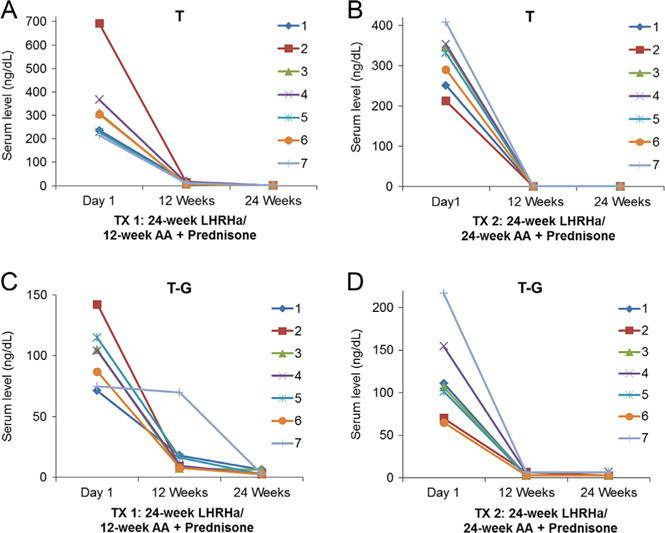
Serum levels of T and T-G from patients following neoadjuvant treatment. (A and C) 24-week LHRHa plus 12-week ‘AA + Prednisone’; (B and D) 24-week LHRHa plus 24-week ‘AA + Prednisone.’ AA, abiraterone acetate; G, glucuronide; LHRHa, luteinizing hormone-releasing hormone agonist; T, testosterone; TX, treatment. Sulfate conjugates were not detected in each group.
Figure 3.
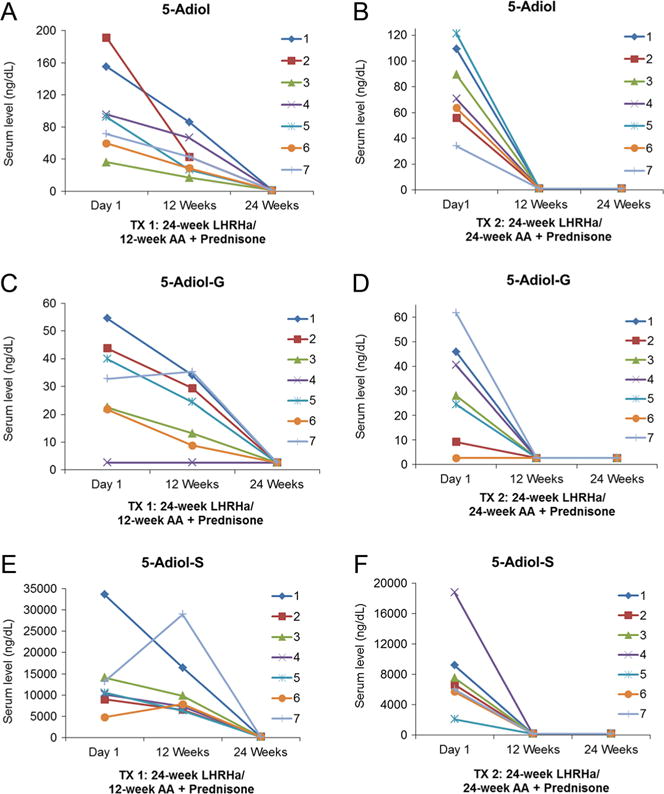
Serum levels of 5-adiol, 5-adiol-G and 5-adiol-S from patients following neoadjuvant treatment. (A, C and E) 24-week LHRHa plus 12-week ‘AA + Prednisone’; (B, D and F) 24-week LHRHa plus 24-week ‘AA + Prednisone.’ 5-Adiol, 5-androstene-3β, 17β-diol; AA, abiraterone acetate; G, glucuronide; LHRHa, luteinizing hormone-releasing hormone agonist; S, sulfate; TX, treatment.
Comparison of TX 1 and TX 2 at Week 12 (Table 1) showed that levels of testosterone (<1.0 ng/dL), DHEA (1.8 ng/dL), DHT (<1.0 ng/dL), AST (<1.0 ng/dL), epi-AST (<1.0 ng/dL) and 5-adiol (<1.0 ng/dL) in TX 2 were much lower than the levels achieved in TX 1 (P value <0.05). Epi-T, 3α-adiol and 3β-adiol were not detected (<1.0 ng/dL) in either group at Week 12 and were not compared.
Analysis of serum androgen glucuronides
In TX 1 baseline, serum levels of T-G (99.8 ng/dL), DHT-G (34.4 ng/dL), AST-G (2.5 × 103 ng/dL) and 3α-adiol-G (422.3 ng/dL) were decreased by 80, 68, 40 and 79% at Week 12, respectively by leuprolide treatment alone, Table 2. Addition of AA and prednisone decreased T-G, DHT-G and 3α-adiol-G to undetectable levels or reduced AST-G to 18.4 ng/dL resulting in a 99% decrease at Week 24. Serum levels of DHEA-G (108.5 ng/dL), epi-AST-G (1.9 × 103 ng/dL) and 5-adiol-G (31.1 ng/dL) were unchanged at Week 12 but were reduced to undetectable levels (DHEA-G and 5-adiol-G) or much lower levels (epi-AST-G: 81.1 ng/dL, a 96% decrease) at Week 24 after the addition of AA and prednisone to LHRHa. Epi-T-G was not detected. Levels of 3β-adiol-G were not quantified, because an undetermined compound co-eluting with 3β-adiol after glucuronidase digestion affected the quantitation of 3β-adiol-G, as reported before (Zang et al. 2017).
In TX 2 (Table 2), serum levels of DHEA-G, DHT-G, 3α-adiol-G and 5-adiol-G were observed to decrease to undetectable levels at both Week 12 and Week 24 when leuprolide, AA and prednisone were given in combination. T-G, AST-G and epi-AST-G were decreased to low levels (4.2, 13.6 and 55.0 ng/dL, respectively) at Week 12 and levels were not further changed at Week 24. Epi-T-G was only quantified in one patient in TX 2 at Day 1 and was not detected in the other patients. Levels of 3β-adiol-G were also not quantified for the reason described previously. The individual patient variation of androgen glucuronide levels was shown in Figs 2, 3 and Supplementary Figs 2, 3, 4, 5, 6.
Comparison of TX 1 and TX 2 at Week 12 (Table 2) showed that serum levels of T-G, DHEA-G, DHT-G, AST-G, epi-AST-G, 3α-adiol-G and 5-adiol-G were markedly reduced in TX 2 (P value <0.05). Glucuronide conjugates that were not detected (epi-T-G) or could not be quantified (3β-adiol-G) at Week 12 were not compared between two groups.
Analysis of serum androgen sulfates
In TX 1 baseline, no apparent change was observed for DHEA-S (166 × 103 ng/dL), epi-AST-S (4.0 × 103 ng/dL) and 5-adiol-S (13.6 × 103 ng/dL) at Week 12 by leuprolide treatment alone but a marked decrease of DHEA-S (3.1 × 103 ng/dL, a 98% decrease), epi-AST-S (undetectable level) and 5-adiol-S (undetectable level) was observed at Week 24 after the addition of AA and prednisone, Table 3.
In TX 2 (Table 3), a remarkable reduction of serum levels of DHEA-S (1.8 × 103 ng/dL and 1.9 × 103 ng/dL), epi-AST-S (undetectable level) and 5-adiol-S (undetectable level) at both Week 12 and Week 24 was observed. The individual patient variation of androgen sulfates is shown in Fig. 3 and Supplementary Figs 3, 5. For AST-S, only one patient in each group at baseline was quantified and also displayed a marked decrease to undetectable levels at Week 12 and Week 24. Levels of 3β-adiol-S were also not quantified for the reason described above. T-S, epi-T-S, DHT-S, AST-S and 3α-adiol-S were not detected in either treatment group.
Comparison of TX 1 and TX 2 at Week 12 showed that serum levels of DHEA-S, epi-AST-S and 5-adiol-S in TX 2 were decreased by 99% (DHEA-S) or to undetectable levels (epi-AST-S and 5-adiol-S) (P value <0.05). The other sulfate conjugates, which were not detected (T-S, epi-T-S, DHT-S, AST-S and 3α-adiol-S) or could not be quantified (3β-adiol-S) in both TX 1 and TX 2 at Week 12 were not compared.
Discussion
NADT is a treatment strategy for early-stage CaP (Kent & Hussain 2003, Gomella et al. 2010, Patel et al. 2016). Although NADT followed by radical prostatectomy improved the pathological outcomes at positive surgical margins, serum PSA levels and seminal vesicle invasion etc., there was no improvement in biochemical failure-free survival rate (Patel et al. 2016). Since one of the main purposes of NADT is to reduce the levels of androgens, a systematic analysis of the androgen metabolome is essential in order to gain information on the efficacy of drug treatment and investigate the reasons of unimproved biochemical failure-free survival rates. Here, we used a newly developed SID-LC–MS/MS method to determine nine hydroxy-androgens and their conjugates (glucuronide and sulfate) in patients with localized high-risk CaP under a randomized phase II NADT (LHRHa plus AA and prednisone) trial (Taplin et al. 2014, Zang et al. 2017). Three androgen diols (3α-adiol, 3β-adiol and 5-adiol) as well as the conjugates (3α-adiol-G, 5-adiol-G and 5-adiol-S) were determined simultaneously in this assay, which was not reported before. In addition, etiocholanolone (a 5β-isomer of AST) picolinate can be separated from the picolinoyl esters of AST, epi-AST and DHT and does not influence the determination of AST, epi-AST and DHT (see HPLC chromatograms in Supplementary Fig. 8). The serum levels of T, DHEA, DHT and AST from 7 patients in each group are comparable to the levels measured in a previous analysis using another LC-ESI-MS/MS method coupled with Girard-T derivatization for keto-androgen quantification (Supplementary Tables 2 and 3), which indicates the reliability of the current method (Taplin et al. 2014).
AA is an inhibitor of P45017A1, which can prevent the production of androgens (e.g. DHEA) from the adrenal gland, and LHRHa suppresses the production of androgenic steroids (mainly testosterone) from testis (Fig. 1) (Gomella 2009, Goel & De 2011). As such, the addition of AA to LHRHa could provide an intensive ADT (Taplin et al. 2014). In this assay, the comparison between TX 1 and TX 2 after 12-week treatment demonstrates that serum levels of testosterone, T-G, DHEA, DHEA-G, DHEA-S, DHT, DHT-G, AST, AST-G, epi-AST, epi-AST-G, epi-AST-S, 3α-adiol-G, 5-adiol, 5-adiol-G and 5-adiol-S can be further lowered by the administration of LHRHa plus AA and prednisone (Tables 1, 2 and 3).
Our study also distinguishes the primary sources of serum androgens and androgen conjugates in CaP patients as leuprolide targets testicular androgen biosynthesis and AA targets adrenal P45017A1 when given after leuprolide. It has been reported that testosterone, DHT, 3α-adiol and 3β-adiol are four androgenic steroids produced in testis in which testosterone is the most abundant androgen and the other three androgens are 1–10% of the amount of testosterone (Ruokonen et al. 1972, Tamm et al. 1987, Jarow & Zirkin 2005). The comparison between baseline and Week 12 in TX 1 showed that LHRHa alone remarkably reduced the serum levels of testosterone, DHT, 3α-adiol and 3β-adiol to either much lower or undetectable levels, which demonstrates that testis produces all four androgens and is the predominate source of these androgens in serum (Fig. 2 and Supplementary Figs 2, 6, 7). 5-Adiol is secreted from the adrenal gland and testis as a downstream metabolite of DHEA. By contrast, AST is secreted from both tissues as a product of the 17,20-lyase action of P45017A1 using 5α-pregnane precursors (Wieland et al. 1965, Ruokonen et al. 1972, Cai & Balk 2011, Nakamura et al. 2012). We find that serum 5-adiol and AST displayed a ~50% reduction in TX 1 at Week 12 and were reduced to undetectable levels at Week 24, which suggests that 5-adiol and AST are derived from both testis and the adrenal gland (Fig. 3, Table 1 and Supplementary Fig. 4). Serum DHEA and DHEA-S are predominantly formed in the adrenal gland (Abraham 1974, Turcu et al. 2014). Levels of DHEA and epi-AST were not changed at Week 12 in TX 1 but decreased to undetectable levels at Week 24 after the addition of AA and prednisone, which indicates that epi-AST is also predominately of adrenal origin (Supplementary Figs 3 and 5).
In humans, uridine diphosphoglucuronsyltansferase 2B (e.g. UGT 2B15/17) enzymes catalyze the conversion of unconjugated hydroxy-androgens to androgen glucuronides and are expressed in the liver, prostate, breast, testis and adrenal gland (Hum et al. 1999, Barbier et al. 2000, Nakamura et al. 2008). Based on our data (Table 2), serum levels of T-G, DHT-G and 3α-adiol-G in TX 1 at Week 12 displayed 80, 68 and 79% reduction in comparison to baseline, respectively, and were further reduced to undetectable levels at Week 24 after AA and prednisone addition, which indicates that the contributions to serum T-G, DHT-G and 3α-adiol-G could be from testis and the adrenal gland (Fig. 2 and Supplementary Figs 2, 6). AST-G was reduced by 40% at Week 12 in TX 1 and was further decreased to a much lower level at Week 24 (99% decrease), which suggests that serum AST-G is also derived from both testis and the adrenal gland (Supplementary Fig. 4). No apparent change was observed from DHEA-G, epi-AST-G and 5-aidol-G in TX 1 at Week 12 with LHRHa alone but they dramatically decreased after the addition of AA and prednisone, which implies that serum DHEA-G, Eip-AST-G and 5-adiol-G come mainly from the adrenal gland (Fig. 3 and Supplementary Figs 3, 5).
Comparison of serum testosterone, T-G, DHT and DHT-G showed that these levels were significantly different at Week 12 between the two treatment groups. This raises the issue of whether the residual testosterone and DHT come from the testis or the adrenal. LH-independent testis production of testosterone may not occur in the patients since the levels of serum testosterone in TX 1 at Week 12 were very similar to those achieved in patients who have undergone bilateral orchiectomy (Gomella 2009). By contrast, LC–MS/MS measurements of C19 steroids in the adrenal vein before and after ACTH stimulation showed elevated testosterone production (Rege et al. 2013).
For androgen sulfate conjugates (Table 3), serum levels of DHEA-S, epi-AST-S and 5-adiol-S at Week 12 in TX 1 did not display a distinct change in comparison to baseline but could be reduced to lower or undetectable levels at Week 24, which indicates that serum DHEA-S, epi-AST-S and 5-adiol-S are predominately produced in adrenal gland. Our studies also show that epi-AST-S and 5-adiol-S are unappreciated sources of intratumoral androgens in the absence of a P45017A1 inhibitor (Fig. 3 and Supplementary Figs 3, 5). These studies confirm that high levels of sulfotransferase exist in adrenal gland, which is consistent with previous reports on the expression of hydroxysteroid sulfotransferase (SULT 2A1) in the gland which catalyzes the formation of DHEA-S and 5-adiol-S (Lindsay et al. 2008, Rege et al. 2016).
During this NADT trial, although a large reduction was observed in serum androgens and their conjugates, a follow-up study reported that resistance to drug therapy still occurred early in these patients (Taplin et al. 2014). One proposed mechanism is that serum DHEA-S is retained at an adequate level for the conversion of potent androgens in prostatic tissue after ADT (Tamae et al. 2015). Based on the current data, serum levels of DHEA-S were still maintained at 3.1 × 103 or 1.9 × 103 ng/dL in TX 1 and TX 2 after 24 weeks, respectively (Table 3), which is three orders of magnitude higher than the other androgens and is consistent with the recent NADT study using LHRHa plus AA and prednisone (Cho et al. 2015). Thus, the current data support the role of serum DHEA-S as a reservoir for tissue androgen synthesis and the progression of CaP after ADT (Taplin et al. 2014, Cho et al. 2015, Tamae et al. 2015).
As serum DHEA-S, 5-adiol-S and epi-AST-S could be a depot for androgen synthesis after leuprolide treatment alone, steroid sulfatase inhibitors could be added to this regimen. As DHEA-S remains at a relatively high level compared to other androgens even after treatment with leuprolide plus AA and prednisone, a steroid sulfatase inhibitor could be added to this regimen as well. In addition, as shown in Fig. 1, AKR1C3 is a pivotal enzyme for the synthesis of androgens (5-adiol, testosterone, DHT and 3α-adiol) from DHEA, Δ4-AD, adione and AST in the adrenal and prostate (Nakamura et al. 2012, Penning 2014). Recent studies also show that AKR1C3 is a coactivator of the androgen receptor and the inhibition of AKR1C3 could overcome the resistance to both AA and enzalutamide, so AKR1C3 inhibitors (e.g. indomethacin) could be combined with LHRHa, AA/prednisone or antiandrogens to attain the optimal blockade of androgen signaling (Yepuru et al. 2013, Liu et al. 2015, 2017). Furthermore, type 1 3β-hydroxysteroid dehydrogenase (3BHSD1) is a key enzyme that converts DHEA or 5-adiol to Δ4-AD and testosterone, respectively, in the prostate and other peripheral tissues (Martz 2013). Its missense mutant (N367T) is a gain-in-stability mutation and increases androgen biosynthesis in the prostate and may contribute to abiraterone drug resistance. Carriers of this mutation could be targeted for NADT using a 3BHSD1 inhibitor (Chang et al. 2013, Li et al. 2015).
In summary, a newly developed LC-ESI-MS/MS method was applied to the systematic analysis of the androgen metabolome in CaP patients under an NADT clinical trial. The analysis demonstrates that LHRHa plus AA and prednisone can achieve more intense androgen suppression than LHRHa alone. In addition, the data help differentiate the primary sources of serum androgens and androgen conjugates in CaP patients. Furthermore, the data support the important role of serum DHEA-S in CaP progression after ADT and identifies 5-adiol-S and epi-AST-S as adrenal androgens as important precursors for intraprostatic androgen biosynthesis. Our findings suggest that more intense NADT to improve the long-term outcomes might be achieved by combining other inhibitors to existing regimens, e.g. steroid sulfatase, AKR1C3 or 3BHSD1 inhibitors.
Supplementary Material
Acknowledgments
The authors thank Jessica Murray in Prof. Trevor M Penning’s lab for her assistance in data analysis and thank Dominic Ciccimaro in Prof. Ian A Blair’s lab for his assistance in mass spectrometry analysis.
Consultancies: Mary-Ellen Taplin, Janssen Pharmaceuticals; Steven P Balk, Johnson & Johnson, Astellas Pharma; Trevor M Penning, Tokai Pharmaceuticals, SAGE Therapeutics. Grants: Mary-Ellen Taplin, Janssen Pharmaceuticals. Honoraria: Elahe A Mostaghel, Jassen Pharmaceuticals. Ownership of companies: Trevor M Penning is the founder of Penzymes, LLC. Board membership: None. Royalties and Patents: Trevor M Penning is an inventor on patents WO2012/142208A1; US2014/0107085A1; WO2013/059245A1; US2016/0303082A1; and WO2017/070448A1.
Funding
This work was supported by a NCI Grant 1P01-CA16 3227-04 and P30-ES013508 awarded to T M P. The original COU-AA-201 study was supported by Janssen Research & Development.
Footnotes
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/ERC-17-0107.
Author contribution statement
Experimental design: T Zang, T M Penning and D Tamae. Pathological data collection: M Taplin and W Xie. Data analysis and interpretation: T Zang, T M Penning, M Taplin and C Mesaros. Manuscript writing: All authors.
References
- Abraham GE. Ovarian and adrenal contribution to peripheral androgens during the menstrual cycle. Journal of Clinical Endocrinology and Metabolism. 1974;39:340–346. doi: 10.1210/jcem-39-2-340. [DOI] [PubMed] [Google Scholar]
- Aus G, Abrahamsson PA, Ahlgren G, Hugosson J, Lundberg S, Schain M, Schelin S, Pedersen K. Three-month neoadjuvant hormonal therapy before radical prostatectomy: a 7-year follow-up of a randomized controlled trial. BJU International. 2002;90:561–566. doi: 10.1046/j.1464-410X.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- Barbier O, Lapointe H, El Alfy M, Hum DW, Belanger A. Cellular localization of uridine diphosphoglucuronosyltransferase 2B enzymes in the human prostate by in situ hybridization and immunohistochemistry. Journal of Clinical Endocrinology and Metabolism. 2000;85:4819–4826. doi: 10.1210/jc.85.12.4819. [DOI] [PubMed] [Google Scholar]
- Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocrine-Related Cancer. 2011;18:R175–R182. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Mostaghel EA, Russell KJ, Liao JJ, Konodi MA, Kurland BF, Marck BT, Matsumoto AM, Dalkin BL, Montgomery RB. External beam radiation therapy and abiraterone in men with localized prostate cancer: safety and effect on tissue androgens. International Journal of Radiation Oncology, Biology, Physics. 2015;92:236–243. doi: 10.1016/j.ijrobp.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne FM, Witjes WP. Neoadjuvant hormonal therapy prior to radical prostatectomy: the European experience. Molecular Urology. 2000;4:251–256. discussion 257. [PubMed] [Google Scholar]
- Eom KY, Ha SW, Lee E, Kwak C, Lee SE. Is neoadjuvant androgen deprivation therapy beneficial in prostate cancer treated with definitive radiotherapy? Radiation Oncology Journal. 2014;32:247–255. doi: 10.3857/roj.2014.32.4.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair WR, Rabbani F, Bastar A, Betancourt J. Neoadjuvant hormone therapy before radical prostatectomy: update on the memorial sloan-kettering cancer center trials. Molecular Urology. 1999;3:253–260. [PubMed] [Google Scholar]
- Goel AK, De S. Abiraterone acetate, an inhibitor of adrenal androgen synthesis in ‘hormone refractory prostate cancer’. Indian Journal of Medical and Paediatric Oncology. 2011;32:43–45. doi: 10.4103/0971-5851.81890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomella LG. Effective testosterone suppression for prostate cancer: is there a best castration therapy? Reviews in Urology. 2009;11:52–60. [PMC free article] [PubMed] [Google Scholar]
- Gomella LG, Singh J, Lallas C, Trabulsi EJ. Hormone therapy in the management of prostate cancer: evidence-based approaches. Therapeutic Advances in Urology. 2010;2:171–181. doi: 10.1177/1756287210375270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. Journal of Urology. 2002;168:9–12. doi: 10.1016/S0022-5347(05)64820-3. [DOI] [PubMed] [Google Scholar]
- Hum DW, Belanger A, Levesque E, Barbier O, Beaulieu M, Albert C, Vallee M, Guillemette C, Tchernof A, Turgeon D, et al. Characterization of UDP-glucuronosyltransferases active on steroid hormones. Journal of Steroid Biochemistry and Molecular Biology. 1999;69:413–423. doi: 10.1016/S0960-0760(99)00061-8. [DOI] [PubMed] [Google Scholar]
- Jarow JP, Zirkin BR. The androgen microenvironment of the human testis and hormonal control of spermatogenesis. Annals of the New York Academy of Sciences. 2005;1061:208–220. doi: 10.1196/annals.1336.023. [DOI] [PubMed] [Google Scholar]
- Kent EC, Hussain MH. Neoadjuvant therapy for prostate cancer: an oncologist’s perspective. Reviews in Urology. 2003;5(Supplement 3):S28–S37. [PMC free article] [PubMed] [Google Scholar]
- Klotz LH, Goldenberg SL, Jewett M, Barkin J, Chetner M, Fradet Y, Chin J, Laplante S. CUOG randomized trial of neoadjuvant androgen ablation before radical prostatectomy: 36-month post-treatment PSA results. Canadian Urologic Oncology Group. Urology. 1999;53:757–763. doi: 10.1016/S0090-4295(98)00616-5. [DOI] [PubMed] [Google Scholar]
- Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay J, Wang LL, Li Y, Zhou SF. Structure, function and polymorphism of human cytosolic sulfotransferases. Current Drug Metabolism. 2008;9:99–105. doi: 10.2174/138920008783571819. [DOI] [PubMed] [Google Scholar]
- Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Research. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Molecular Cancer Therapeutics. 2017;16:35–44. doi: 10.1158/1535-7163.MCT-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz L. Escaping abiraterone. Science-Business eXchange. 2013;38:1–13. doi: 10.1038/scibx.2013.1049. [DOI] [Google Scholar]
- Meyer F, Moore L, Bairati I, Lacombe L, Tetu B, Fradet Y. Neoadjuvant hormonal therapy before radical prostatectomy and risk of prostate specific antigen failure. Journal of Urology. 1999;162:2024–2028. doi: 10.1016/S0022-5347(05)68092-5. [DOI] [PubMed] [Google Scholar]
- Mostaghel EA, Nelson PS, Lange P, Lin DW, Taplin ME, Balk S, Ellis W, Kantoff P, Marck B, Tamae D, et al. Targeted androgen pathway suppression in localized prostate cancer: a pilot study. Journal of Clinical Oncology. 2014;32:229–237. doi: 10.1200/JCO.2012.48.6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Nakajima M, Yamanaka H, Fujiwara R, Yokoi T. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metabolism and Disposition. 2008;36:1461–1464. doi: 10.1124/dmd.108.021428. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Rege J, Satoh F, Morimoto R, Kennedy MR, Ahlem CN, Honma S, Sasano H, Rainey WE. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein corticosteroids before and after adrenocorticotropic hormone stimulation. Clinical Endocrinology. 2012;76:778–784. doi: 10.1111/j.1365-2265.2011.04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VR, Leveillee RJ, Shah AD, Coelho RF, Kim ED. Medscape. New York, NY, USA: WebMD LLC; 2016. Neoadjuvant androgen deprivation therapy in prostate cancer. available at: http://emedicine.medscape.com/article/455994-overview. [Google Scholar]
- Penning TM. Androgen biosynthesis in castration-resistant prostate cancer. Endocrine-Related Cancer. 2014;21:T67–T78. doi: 10.1530/ERC-14-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter MA, Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Reviews in Urology. 2007;9(Supplement 1):S3–S8. [PMC free article] [PubMed] [Google Scholar]
- Rege J, Nakamura Y, Satoh F, Morimoto R, Kennedy MR, Layman LC, Honma S, Sasano H, Rainey WE. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. Journal of Clinical Endocrinology and Metabolism. 2013;98:1182–1188. doi: 10.1210/jc.2012-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rege J, Karashima S, Lerario AM, Smith JM, Auchus RJ, Kasa-Vubu JZ, Sasano H, Nakamura Y, White PC, Rainey WE. Age-dependent increases in adrenal cytochrome b5 and serum 5-androstenediol-3-sulfate. Journal of Clinical Endocrinology and Metabolism. 2016;101:4585–4593. doi: 10.1210/jc.2016-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruokonen A, Laatikainen T, Laitinen EA, Vihko R. Free and sulfate-conjugated neutral steroids in human testis tissue. Biochemistry. 1972;11:1411–1416. doi: 10.1021/bi00758a013. [DOI] [PubMed] [Google Scholar]
- Sanchez-Guijo A, Neunzig J, Gerber A, Oji V, Hartmann MF, Schuppe HC, Traupe H, Bernhardt R, Wudy SA. Role of steroid sulfatase in steroid homeostasis and characterization of the sulfated steroid pathway: evidence from steroid sulfatase deficiency. Molecular and Cellular Endocrinology. 2016;437:142–153. doi: 10.1016/j.mce.2016.08.019. [DOI] [PubMed] [Google Scholar]
- Schulman CC, Debruyne FM, Forster G, Selvaggi FP, Zlotta AR, Witjes WP. 4-Year follow-up results of a European prospective randomized study on neoadjuvant hormonal therapy prior to radical prostatectomy in T2-3N0M0 prostate cancer. European Study Group on Neoadjuvant Treatment of Prostate Cancer. European Urology. 2000;38:706–713. doi: 10.1159/000020366. [DOI] [PubMed] [Google Scholar]
- Shafi AA, Yen AE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacology and Therapeutics. 2013;140:223–238. doi: 10.1016/j.pharmthera.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA: A Cancer Journal for Clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Soloway MS, Pareek K, Sharifi R, Wajsman Z, McLeod D, Wood DP, Jr, Puras-Baez A, Lupron Depot Neoadjuvant Prostate Cancer Study Group Neoadjuvant androgen ablation before radical prostatectomy in cT2bNxMo prostate cancer: 5-year results. Journal of Urology. 2002;167:112–116. doi: 10.1016/S0022-5347(05)65393-1. [DOI] [PubMed] [Google Scholar]
- Sonpavde G, Chi KN, Powles T, Sweeney CJ, Hahn N, Hutson TE, Galsky MD, Berry WR, Kadmon D. Neoadjuvant therapy followed by prostatectomy for clinically localized prostate cancer. Cancer. 2007;110:2628–2639. doi: 10.1002/cncr.23085. [DOI] [PubMed] [Google Scholar]
- Tamae D, Byrns M, Marck B, Mostaghel EA, Nelson PS, Lange P, Lin D, Taplin ME, Balk S, Ellis W, et al. Development, validation and application of a stable isotope dilution liquid chromatography electrospray ionization/selected reaction monitoring/mass spectrometry (SID-LC/ESI/SRM/MS) method for quantification of keto-androgens in human serum. Journal of Steroid Biochemistry and Molecular Biology. 2013;138:281–289. doi: 10.1016/j.jsbmb.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP, Kantoff PW, Taplin ME, Penning TM. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chemico-Biological Interactions. 2015;234:332–338. doi: 10.1016/j.cbi.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm J, Volkwein U, Kurniawan E, Becker H. Concentrations of unconjugated 5 alpha-androstane-3 alpha, 17 beta-diol and 5 alpha-androstane-3 beta, 17 beta-diol and their precursor in human testicular tissue. Comparison with testosterone, 5 alpha-dihydrotestosterone, estradiol-17 beta, and with steroid concentrations in human epididymis. Journal of Steroid Biochemistry. 1987;26:345–348. doi: 10.1016/0022-4731(87)90099-9. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, Sanda MG, Davis JW, Loda M, True LD, et al. Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. Journal of Clinical Oncology. 2014;32:3705–3715. doi: 10.1200/JCO.2013.53.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcu A, Smith JM, Auchus R, Rainey WE. Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Comprehensive Physiology. 2014;4:1369–1381. doi: 10.1002/cphy.c140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland RG, Decourcy C, Levy RP, Zala AP, Hirschmann H. C-19-O-2 steroids and some of their precursors in blood from normal human adrenals. Journal of Clinical Investigation. 1965;44:159–168. doi: 10.1172/JCI105122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nature Reviews Clinical Oncology. 2014;11:365–376. doi: 10.1038/nrclinonc.2014.72. [DOI] [PubMed] [Google Scholar]
- Yee DS, Lowrance WT, Eastham JA, Maschino AC, Cronin AM, Rabbani F. Long-term follow-up of 3-month neoadjuvant hormone therapy before radical prostatectomy in a randomized trial. BJU International. 2010;105:185–190. doi: 10.1111/j.1464-410X.2009.08698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, Narayanan R. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clinical Cancer Research. 2013;19:5613–5625. doi: 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- Zang T, Tamae D, Mesaros C, Wang Q, Huang M, Blair IA, Penning TM. Simultaneous quantitation of nine hydroxy-androgens and their conjugates in human serum by stable isotope dilution liquid chromatography electrospray ionization tandem mass spectrometry. Journal of Steroid Biochemistry and Molecular Biology. 2017;165:342–355. doi: 10.1016/j.jsbmb.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.