


Abstract
PPM serine/threonine protein phosphatases function in signaling pathways and require millimolar concentrations of Mn2+ or Mg2+ ions for activity. Whereas the crystal structure of human PP2Cα displayed two tightly bound Mn2+ ions in the active site, recent investigations of PPM phosphatases have characterized the binding of a third, catalytically essential metal ion. The binding of the third Mg2+ to PP2Cα was reported to have millimolar affinity and to be entropically driven, suggesting it may be structurally and catalytically important. Here, we report the use of hydrogen/deuterium exchange–mass spectrometry and molecular dynamics to characterize conformational changes in PP2Cα between the active and inactive states. In the presence of millimolar concentrations of Mg2+, metal-coordinating residues in the PP2Cα active site are maintained in a more rigid state over the catalytically relevant time scale of 30–300 s. Submillimolar Mg2+ concentrations or introduction of the D146A mutation increased the conformational mobility in the Flap subdomain and in buttressing helices α1 and α2. Residues 192–200, located in the Flap subdomain, exhibited the greatest interplay between effects of Mg2+ concentration and the D146A mutation. Molecular dynamics simulations suggest that the presence of the third metal ion and the D146A mutation each produce distinct conformational realignments in the Flap subdomain. These observations suggest that the binding of Mg2+ to the D146/D239 binding site stabilizes the conformation of the active site and the Flap subdomain.
Graphical abstract
Metal ions are essential cofactors for approximately one-third of enzymes.1 In addition to an essential role in the catalytic step, metal ions also may stabilize the active state of the enzyme or contribute to substrate recognition.2 The metal-dependent protein phosphatases (PPM, formerly PP2C), one of four unrelated families of protein phosphatases, dephosphorylate phosphoserine and phosphothreonine residues in proteins.3,4 The PPM are characterized by their distinctive fold, the preponderance of highly conserved aspartate residues in the active site, and a requirement for supplementation with millimolar levels of Mg2+ or Mn2+ to support detectable activity with purified proteins.5,6 Human PP2Cα (PPM1A) exerts tumor suppressor activity through its activity on direct and indirect targets, such as p53, p38 MAPK, RelA, and ERK,7–9 and was the first member of the PP2C family to be structurally characterized.10 The PP2C fold consists of two antiparallel β-sheets surrounded by two sets of α-helices, with the active site located along one edge of the β-sheets.10,11 The two β-sheets are connected by one loop across the bottom and three loops across the top. The third loop, termed the Flap, is folded into a unique structure and is thought to be important for substrate recognition.12 In crystals formed at low pH, the active site contains two tightly bound Mn2+ ions and superficially resembles the active sites of other, evolutionarily unrelated serine/threonine protein phosphatases.10,11 However, neither the structures nor the enzyme kinetics suggested a molecular explanation for the requirement of divalent ion supplementation.10,11,13
The presence of a third divalent metal ion in the PP2C active site was first observed in structures of “eukaryotic-like” bacterial PP2C phosphatases,12,14–17 and subsequently in several plant PP2C phosphatases.18–21 Notably, the aspartic acid residues that coordinate the third metal ion are highly conserved among “eukaryotic-like” bacterial PP2Cs and eukaryotic PP2Cs.5,6 In human PP2Cα, the motif 8 aspartic acid, which also coordinates one of the tightly bound metal ions, and the motif 5 aspartic acid are identified as D239 and D146, respectively. Mutation of PP2Cα D239 or D146 results in a loss of activity.13,22,23 Recently, we used isothermal titration calorimetry (ITC) to characterize the binding of Mg2+ to essentially metal-free human PP2Cα.23 Binding of the first 2 equiv of Mg2+ to PP2Cα was exothermic and had micromolar-level affinities, whereas binding of the third Mg2+ equivalent was endothermic and had a millimolar-level affinity.23 The finding that the binding of the third metal ion is entropically rather than enthalpically driven suggests that it may have a structural role as well as a catalytic role.
Hydrogen/deuterium exchange–mass spectrometry (HDX–MS) is a powerful tool for characterizing protein structure.24,25 In continuous labeling HDX–MS experiments, a protein is diluted in a D2O buffer for a chosen interval, ti, allowing exchangeable hydrogens to be replaced by deuterium from the solvent. Using MS, the average mass increase of the protein (or individual peptides) is measured after a specified ti. Quenching of exchange reactions and rapid sample workup prior to MS permit the exchange rates of backbone amides in the native protein to be monitored. The rate of backbone amide exchange is dependent on solution pH, temperature, solvent accessibility, and protein structure, particularly the secondary structures that involve backbone amide hydrogen bond formation.24,26,27 The observed extent of amide deuterium uptake provides information about the conformational mobility of different regions of the protein.
In this work, we used HDX–MS techniques to characterize the conformational mobility of active and inactive states of wild-type (Wt) PP2Cα and a catalytically inactive mutant (PP2Cα D146A, Mt) that has decreased binding affinity for the third Mg2+ ion.23 We found that the active state of the enzyme was characterized by increased rigidity in the active site. The Mg2+ concentration and the D146A mutation separately, and in combination, exerted effects on conformational mobility in the active site, in the Flap subdomain, and in one set of buttressing α-helices. We further used molecular dynamics simulations to investigate changes in structure and hydrogen bond occupancy.
METHODS
Protein Expression, Purification, and Activity Assays
The purified PP2Cα and PP2Cα D146A proteins correspond to PPM1A isoform 1 (GenBank entry NP_066283.1) and the corresponding D146A mutant, each with the addition of amino acids SGGT at the N-terminus. Residue numbering refers to the canonical protein sequence numbering. The proteins were expressed from plasmid pET45b(+)-His-TEV-PP2Cα or pET45b(+)-His-TEV-PP2Cα D146A in Escherichia coli BL21-(DE3) cells and purified as described previously,23 with minor modifications. Purified proteins were at least 95% pure. Proteins were dialyzed against a buffer consisting of 50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 2 mmol/L MgCl2, 2 mmol/L β-mercaptoethanol (β-ME), 10% (v/v) glycerol, and 0.1 mmol/L EGTA or in the same buffer, except with 0.1 mmol/L MgCl2. Phosphatase activity assays were performed as described previously, with minor modifications.23 Initial rates of phosphatase activity were determined by incubating Wt or mutant PP2Cα with various concentrations of the p38α (175–185, 180pT) peptide in high-Mg2+ (HB-H) or low-Mg2+ (HB-L) buffer [10 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 2 mmol/L β-ME, 0.1 mmol/L EGTA, 10% (v/v) glycerol, and 2 or 0.1 mmol/L MgCl2] at 25 °C for 15 min. Phosphate was assessed using the Biomol green assay (Enzo Life Science), following the manufacturer’s protocol. To determine Km and kcat values, the initial rates were fitted to the Michaelis-Menten equation: v = Vmax[S]/(Km + [S]), where [S] is the concentration of the phosphopeptide substrate. The value of kcat was calculated from the equation Vmax = kcat[E]o and the concentration of the enzyme, [E]o.
HDX–MS
High- and low-Mg2+ buffers (HB-H and HB-L, respectively) were prepared in H2O for dilution and equilibration of proteins, or in D2O (99.99%) for H/D exchange experiments. pDcorrected values were measured with a glass electrode and corrected for deuterium isotope effects.25 HDX–MS experiments utilized a LEAP HDX-PAL (Leap Technologies) robotic system for sample handling. For each set of measurements, D2O exchange times (ti) of 30, 60, 300, 900, 3600, and 14400 s were selected, where the uncertainty of each immersion time is approximately twice the latency of the LEAP scheduler (Δt ≈ 3.5 s). The latency of each solution manipulation is measured and recorded in the system log, allowing explicit revision of ti when necessary.
Protein samples were diluted to 3 µmol/L in HB-H or HB-L and equilibrated at 1 °C for 15 min prior to experiments. Exchange was initiated by diluting 5 µL of protein (Wt or Mt) into 25 µL of exchange buffer at 25 °C. After immersion of each sample in D2O for ti, the exchange reaction was quenched by diluting 25 µL of exchanged protein into 35 µL of quench buffer [0.1 mol/L phosphate (pH 2.5) with 3 mol/L urea] at 1 °C. Quenched samples were immediately injected into an immobilized pepsin column (Water’s Enzymate BEH Pepsin Column) and digested for 3 min using a flow rate of 50 µL/min and a mobile phase of water with 0.1% formic acid. The resulting peptides were trapped on a C18 guard cartridge (10 mm × 1 mm, 5 µm particles, Grace Davison Discovery Science) and washed with water with 0.1% formic acid for 30 s to remove additives present in the equilibration, exchange, and quench buffers. Trapped peptides were eluted onto a C18 analytical column (5 cm × 1 mm, 1.9 µm particles, Hypersil GOLD aQ Analytical HPLC Column, Thermo Scientific) via a Dionex Ultimate 3000 UPLC system pumping at 50 µL/min with a 9.5 min binary gradient of water with 0.1% formic acid (solvent A) and an 80:20:0.1 (v/v/v) acetonitrile/water/formic acid mixture (solvent B). The following gradient was used. The level of B was increased from 5 to 35% over the first 3 min, increased from 35 to 70% over 5 min, increased from 70 to 100% over 0.5 min, held at 100% for 0.5 min, and decreased back to 5% over 0.5 min. All LC columns, values, and tubing were kept at 2 °C. Peptides were analyzed on a Thermo LTQ Orbitrap Elite instrument with the following settings: spray voltage, 3.7 kV; sheath gas flow rate, 25 (arbitrary units); capillary temperature, 270 °C; Orbitrap resolution, 60000. All exchange time points and control (0 s exchange) samples were analyzed in triplicate. A reduction step was omitted because PP2Ca does not contain disulfide bonds. Between HDX-MS analyses of protein samples, the pepsin column was washed by injecting 50 µL of 1.5 mol/L guanidine hydrochloride (Gdn-HCl) in water with 0.1% (v/v) formic acid. The analytical and trap columns were washed between protein samples by injecting 50 µL of an 80:20:0.1 (v/v/v) acetonitrile/water/formic acid mixture.
Peptide Identification
Peptide maps were generated for Wt and Mt proteins in each HEPES buffer. Proteins were diluted to 3 µmol/L and analyzed using the LEAP HDX-PAL as described above except that protein samples were diluted into H2O-based buffers prior to reactions being quenched. Peptides were identified by exact mass and tandem MS (MS/MS) on a Thermo LTQ Orbitrap Elite instrument. Each full mass spectral scan (mass range of m/z 300–2000) resulted in data-dependent selection of the six most abundant precursor ions for fragmentation by collision-induced dissociation. Dynamic exclusion was used with the following settings: repeat count, 1; repeat duration, 10 s; exclusion duration, 15 s. Thermo RAW data files were converted to mascot generic files using a mass matrix converter (Matrix Science, Oxford, U.K.). Peptide identification utilized MASCOT (Matrix Science) with the following parameters: enzyme, none; peptide tolerance, 20 ppm; MS/MS tolerance, 0.6 Da; maximal number of 13C atoms, 1; peptide charges, +2, +3, and +4.
HDX–MS Data Analysis and Visualization
Relative deuterium uptake was assessed using HDX Workbench,28 using the values of 83% deuterium in the exchange buffer and 80% recovery of the deuterium label. The area between exchange curves, Abec, a functional data analysis concept,29,30 was used as a measure of differences in hydrogen/deuterium exchange dynamics.31 We used a trapezoidal approximation to estimate Abec values comparing proteins in two states, a and b
| (1) |
where xa,i and xb,i are the remaining hydrogen fractions for the two states and i is the index over the six time points, ti. When the numbers of replicates for the two states are the same, the degrees of freedom is equal to 2nobs − 2 and the pooled standard deviation of Abec is given by
| (2) |
The exchange-competent fraction, ρ, is the inverse of the protection factor.24,27 Abec values can be related to the average log ratio of ρ values in the two states as
| (3) |
where and are the fractions of the jth of n detectable amide hydrogens in the exchange-competent state in protein states a and b, respectively, and the angle brackets indicate the average over the detectable amide hydrogens in the peptide.24,27 Under common conditions for HDX–MS experiments, amide hydrogens in the first two positions of a peptide exchange rapidly during sample workup, so the extent of deuterium incorporation in the native protein is not recovered for those positions.24 For small values of ρ, Abec is approximately equal to 〈ΔΔG〉/2.303RT, where 〈ΔΔG〉 is the average difference between protein states a and b of the free energy difference between the peptide in the native protein structure and the peptide fully exposed to solvent, R is the gas constant, and T is the absolute temperature.27,31 Wt and Mt PP2Cα peptides containing residue 146 differ in sequence and intrinsic exchange rate constants. The loss of hydrogen resulting from intrinsic exchange for fully exposed peptides was calculated as
| (4) |
where j is the index over n detectable amide hydrogens and is the pH-, temperature-, and sequence-dependent exchange rate constant.24,27,32
The statistical significance of deuterium uptake at specific time points or as summarized by Abec values was assessed using a two-tailed Student’s t test with the indicated level of significance. Results from repeated measurements are reported as means ± standard deviations (1σ). Analysis and visualization of time series data for peptide-level deuterium uptake were performed in the R computing environment.33 Structural visualizations were performed using the UCSF Chimera package, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institute of General Medical Sciences Grant P41-GM103311).34 Advice for colors was obtained from www.ColorBrewer2.org.
Molecular Dynamics Simulations
The dynamics of the protein were studied in three different states: PP2Cα at “high” [Wt(3Mg)] and “low” [Wt(2Mg)] Mg2+ ion concentrations and the D146A mutant [D146A(2Mg)] at a “low” Mg2+ ion concentration. The starting coordinates for Wt(2Mg) were obtained from Protein Data Bank (PDB) entry 1A6Q,10 with the conversion of the two manganese ions to Mg2+ ions. For the Wt(3Mg) simulation, a third Mg2+ ion was introduced between the side-chain carboxyl groups of D146 and D239. The starting coordinates for D146A(2Mg) were obtained by applying the D146A coordinate mutation to the Wt(2Mg) structure. Simulations were performed with the NAMD 2.9 software package,35 using CHARMM22 All-Hydrogen Topologies and Parameters.36 Each protein was centered and solvated with 11852 TIP3 model water molecules in a periodic image cell with initial dimensions of 95 Å × 77 Å × 57 Å. Electrostatic interactions were reduced to zero at 12.0 Å, using “switchdist” and “pairlistdist” settings of 10.0 and 14.0 Å, respectively. The Langevin method was used to propagate the dynamics with a target temperature of 300 K, and the Langevin-Piston method was used to maintain a target pressure of 1 atm. All bonds were kept rigid to allow for a time step of 2 fs. After appropriate minimization and equilibration for 50 ns, each system was run for 200 ns, saving coordinate snapshots every 1 ps for analysis. Ensemble representations of the three states were generated by aligning the α-carbons of core residues 56–60, 129–132, 140–143, 148–151, 234–237, and 284–288 for 19 snapshots evenly spaced over the simulation interval.
RESULTS
PP2Cα Is Inactive toward Phosphopeptide Substrates in the Presence of Low Mg2+ Concentrations
Earlier work has shown that Wt PP2Cα requires millimolar concentrations of divalent Mg2+ or Mn2+ ions to exhibit measurable activity and that the D146A mutant is enzymatically inactive.13,22,23 To investigate the conformational mobility of inactive and active states of PP2Cα, we chose solutions similar to those used previously to measure the binding of the third metal by ITC.23 PP2Cα was highly active toward a phosphopeptide substrate in the high-Mg2+ concentration buffer (2 mmol/L Mg2+) at 25 °C, exhibiting a kcat of 2.3 ± 0.3 s−1 and a Km of 84 ± 18 µmol/L. Note that all uncertainties reported herein represent one standard deviation, 1σ. PP2Cα did not exhibit measurable phosphatase activity in the low-Mg2+ buffer (0.1 mmol/L Mg2+), and PP2Cα D146A did not exhibit measurable activity in either buffer, as expected.22,23 As we reported previously on the basis of ITC experiments (Table 2 of ref 23), the association constant for binding of Mg2+ to PP2Cα at 25 °C was 1620 ± 325 L/mol, corresponding to a site occupancy of 0.76 ± 0.04 in the high-Mg2+ buffer. In addition, the association constant for binding of Mg2+ to PP2Cα D146A under the same conditions was 610 ± 14 L/mol, corresponding to a site occupancy of 0.55 ± 0.01 in the high-Mg2+ buffer, and binding of Mg2+ to the PP2Cα D146A/D243A double mutant was undetectable. We interpreted these results as indicating that the binding pocket for the third metal in PP2Cα contains two mutually exclusive subsites: the subsite involving D146 and D239, which is required for catalytic activity, and the subsite involving D239 and D243, which contributes to but is not required for catalytic activity.23
Hydrogen/Deuterium Exchange Dynamics of PP2Cα and PP2Cα D146A in the Presence of Low or High Concentrations of Divalent Mg2+ Ions
We used HDX–MS methods to investigate the conformational mobility of active and inactive states of PP2Cα and of the inactive mutant, PP2Cα D146A. A statistical summary (Table 1) and time-dependent deuterium uptake values for PP2Cα and PP2Cα D146A in the two buffer conditions are provided (Table S1). For PP2Cα in 2 mmol/L Mg2+ (WtH), peptide-level, average deuterium uptake values after exchange for 300 s have been overlaid as shaded hues onto a representation of the three-dimensional structure of PP2Cα (Figure 1A). The peptide containing residues 184–191, which forms a protruding point in the Flap region, exhibited the greatest extent of deuterium incorporation, while peptides from helix α6, which is protected from direct exposure to the solvent by the C-terminal α-helices, exhibited the lowest extent of deuterium incorporation. A higher level of deuterium incorporation was also detected in the β2–β3 loop at the lower edge of the active site, the β3–β4 loop, the lower portion of helices α1 and α2, most of the Flap region, the α7–α8 loop, and residues near the N- and C-termini of the protein. Peptide maps showing time-dependent deuterium uptake for PP2Cα in the two buffers are provided (Figures S1 and S2). After exchange for 300 s, significant differences in deuterium uptake between inactive and active states of the wild-type enzyme were limited to a few specific regions [WtL – WtH (Figure 1B)]. The strongest effect of a low Mg2+ concentration was an increase in the level of deuterium incorporation of nearly 20% in peptides containing residues 192–200. This portion of the Flap lies close to the active site and was relatively protected from deuterium exchange in the presence of 2 mmol/L MgCl2. Smaller increases in the level of deuterium incorporation were detected in the β2–β3 loop at the lower edge of the active site, in segments of helices α1 and α2, in the other segments of the Flap, and in the β6–β7 loop that contains D146, which is important for binding of the third Mg2+ ion. Helix α8, which is in the PP2C C-terminal extension, exhibited a significantly lower level of deuterium uptake in low-Mg2+ buffer than in high-Mg2+ buffer.
Table 1.
Statistical Summary of HDX–MS Analysis of PP2Cα and PP2Cα D146A in the Presence of High (H) or Low (L) Concentrations of MgCl2a
| WtH | WtL | MtH | MtL | |
|---|---|---|---|---|
| proteinb | PP2Cα | PP2Cα | D146A | D146A |
| exchange buffer [MgCl2] (mmol/L) | 2.0 | 0.1 | 2.0 | 0.1 |
| sequence coverage (%) | 91.7 | 91.7 | 91.7 | 91.7 |
| no. of unique peptide/charge states | 111 | 111 | 110 | 110 |
| no. of unique peptides | 85 | 85 | 84 | 84 |
| no. of peptides as a single charge state | 60 | 60 | 59 | 59 |
| no. of peptides as multiple charge states | 25 | 25 | 25 | 25 |
| mean correlation among multiple charge states | 0.997 | 0.983 | 0.998 | 0.993 |
| mean no. of replicates/peptide | 2.98 | 2.98 | 2.94 | 2.97 |
| mean no. of time points/peptide | 6.97 | 6.98 | 6.94 | 6.99 |
Exchange reactions occurred at 25.0 °C in pDcorrected = 7.5 buffer with 83.3% D2O.
Human PP2Cα(1–382) and PP2Cα D146A(1–382) were expressed in E. coli and retained four additional residues at the N-terminus (SGGT) after purification.
Figure 1.

Deuterium uptake in PP2Cα and PP2Cα D146A in the presence of high or low concentrations of magnesium ions. Peptide-level average deuterium uptake values, or differences in values, after exchange for 300 s at 25 °C are indicated by colored hues overlaid onto a representation of the three-dimensional structure of PP2Cα (PDB entry 1A6Q), with the addition of a third Mg2+ ion and selected annotations. (A) Deuterium uptake in PP2Cα in the presence of 2 mmol/L MgCl2 (WtH). (B) Differences in deuterium uptake in PP2Cα in the presence of 0.1 mmol/L MgCl2 compared with 2 mmol/L MgCl2 (WtL – WtH). (C) Differences in deuterium uptake in PP2Cα D146A compared with PP2Cα in the presence of 2 mmol/L MgCl2 (MtH – WtH). (D) Differences in deuterium uptake in PP2Cα D146A in the presence of 0.1 mmol/L MgCl2 compared with 2 mmol/L MgCl2 (MtL – MtH). When multiple peptides were detected for a region of the protein, the chosen peptide was representative. Deuterium uptake values represent the average of three observations. Differences in deuterium uptake between two conditions were considered significant on the basis of a two-tailed Student’s t test with p < 0.05.
Most regions of PP2Cα D146A exhibited rates of deuterium uptake higher than that of Wt PP2Cα under the same conditions. Peptide maps showing time-dependent deuterium uptake for PP2Cα D146A in the two buffers are provided (Figures S3 and S4). After exchange for 300 s in the high-Mg2+ buffer, many segments of PP2Cα D146A, especially within the catalytic domain, exhibited rates of deuterium uptake greater than that of the Wt protein (Figure 1C). Differences in deuterium uptake in PP2Cα D146A between low and high Mg2+ concentrations are depicted in Figure 1D (MtL – MtH). The strongest effect of a low Mg2+ concentration in the mutant was an increased rate of deuterium uptake in the middle portion of helix α2, with smaller increases evident in many portions of the protein. Notably, the Flap subdomain exhibited few differences between low and high Mg2+ concentrations, unlike Wt PP2Cα.
Effects of Mg2+ Concentration and the D146A Mutation on Deuterium Uptake Manifest in Specific Regions Distributed across the PP2Cα Catalytic Domain
To gain a global perspective on the effects of Mg2+ concentration and the D146A mutation on the dynamics of deuterium uptake, we adapted functional data analysis (FDA) methods, an approach that has been used to analyze short time series data in a variety of fields.29,30,37 We used the area between exchange curves, Abec, as a measure of dissimilarity in deuterium uptake dynamics between the two conditions.31 Under EX2 conditions, which usually apply to globular proteins in HDX experiments, Abec values can be related to the log ratio of the exchange-competent fractions in the two states.26,27,38 The largest effects on conformational stability exerted by the Mg2+ concentration or the D146A mutation were localized to specific regions within the catalytic domain, with relatively small effects localized in the C-terminal extension (Table S2 and Figure 2A). Generally, the destabilizing effects of the mutation were larger than the destabilizing effects of a low Mg2+ concentration. For most peptides, effects of the mutation in high- and low-Mg2+ buffers were highly correlated, with an R2 of 0.95 (Figure 2B, left panel). Four peptides from the Flap region (amino acids 192–201) deviated from the general trend, suggesting a selective stabilization of these peptides in Wt compared with Mt PP2Cα in high-Mg2+ buffers. In contrast, effects of altering the Mg2+ concentration on conformational dynamics in the Wt protein correlated less well with the effects in the Mt protein, with an R2 of 0.63 (Figure 2B, right panel). The same four peptides of amino acids 192–201 deviated from the general trend, suggesting a reduced effect of Mg2+ concentration in the Mt compared with that in the Wt protein in these peptides. Overall, only 5% of the variance in the effects of the mutation remains unexplained by a linear relationship between the effect of the mutation in high-versus low-Mg2+ buffers, whereas 37% of the variance in the effects of the magnesium ion concentration remains unexplained by a linear relationship between the effects in the Wt and the Mt proteins.
Figure 2.

Global effects of Mg2+ concentration and the D146A mutation on deuterium uptake. (A) Values of the area between exchange curves metric (Abec) for the indicated comparisons were calculated as the area between the remaining mean peptide-level hydrogen fractions vs log t curves, where t is the exchange time in seconds, and are plotted vs peptide midpoints. Symbol characteristics indicate statistical significance based on the two-tailed Student’s t test: empty symbols, not significant; lightly shaded symbols, p < 0.01; darkly shaded symbols, p < 0.001. Selected annotations are included. (B) Interactions between magnesium ion concentration effects and mutation effects. The peptide sequence position is indicated by light blue (N) to dark purple (C) symbol shading. The left panel shows mutation effects in 2 and 0.1 mmol/L MgCl2 buffers, and the right panel shows Mg2+ concentration effects in Wt compared with Mt proteins.
Amide Hydrogen Exchange Behaviors of Peptides Containing Metal-Coordinating Residues Differ between Active and Inactive Conformations of PP2Cα
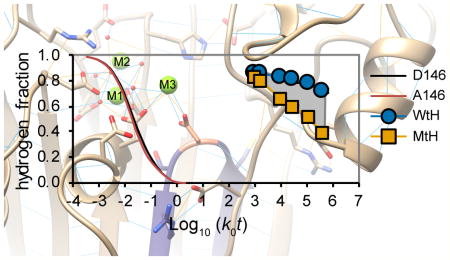
The PP2Cα active site (Figure 3A) contains a cluster of highly conserved, metal-coordinating residues that vary little in their structural arrangement among related PP2C phosphatases.12,14,15,19,22 The third metal ion, when detected in structures, is usually coordinated by residues homologous to D146 and D239.12,14,15,19,22 The D146 residue is located at the apex of a buried loop that crosses the active site just below the Flap subdomain. Under both buffer conditions, the rate of deuterium uptake in peptides containing the D146 residue of PP2Cα was significantly lower than in the corresponding PP2Cα A146 peptides for each exchange interval [p < 0.002 (Figure 3B, bottom left panel)]. For the Wt protein in high-Mg2+ buffer, the average rate of deuterium uptake after exchange for 300 s for peptides containing the D146 residue was 16.5 ± 0.5% (7th percentile), whereas the average rate of deuterium uptake for the mutant peptides was 34.7 ± 0.2% (33nd percentile), indicating a specific increase in the rate of deuterium uptake in the mutant protein for these peptides. In contrast, changes in Mg2+ concentration had only small effects on deuterium incorporation in these peptides for both Wt and Mt proteins. Peptides containing other metal-coordinating residues exhibited smaller effects of the mutation and/or the Mg2+ concentration. Deuterium uptake in peptides containing E37 was affected by both the mutation and the buffer conditions (Figure 3B, top left panel). Residues E37 and D38 indirectly coordinate M2 through metal-bound water molecules and are located in the β2–β3 loop at the bottom of the active site. For the Wt protein in high-Mg2+ buffer, deuterium uptake approached saturation after exchange for 4 h, while for the Mt protein in low-Mg2+ buffer, deuterium uptake approached saturation after exchange for 900 s. The rate of deuterium uptake in peptides containing D60 and G61, which directly coordinate the tightly bound metal ions M1 and M2, differed significantly between the Wt and Mt proteins [p < 0.01 for MtH-WtH, all time points (Figure 3B, top right panel)], but the magnitude of the effect was smaller than for residue 146-containing peptides. Peptides containing D282, which coordinates M1, exhibited smaller effects of the mutation and the Mg2+ concentration on exchange rates (Figure 3B, bottom right panel). For this peptide, deuterium uptake values generally increased in the following order: WtH < WtL ≈ MtH < MtL. This suggests that high concentrations of Mg2+ may compensate for some conformational alterations induced by the D146A mutation.
Figure 3.

Effects of Mg2+ concentration and the D146A mutation on deuterium uptake by peptides containing metal-coordinating residues. (A) Ribbon diagram depicting the active site of PP2Cα (based on PDB entry 1A6Q with an added third Mg2+). Metal-coordinating residues and residues R33 and D243 are labeled. Selected peptides containing metal-coordinating residues are colored light blue. The visible portion of the Flap subdomain is colored orange. Regions not detected in the exchange experiments are colored dark gray. Predicted backbone–backbone hydrogen bonds meeting strict (blue) or relaxed (orange) criteria are indicated. (B) Time course of deuterium uptake by selected peptides containing metal-coordinating residues of PP2Cα (Wt) and PP2Cα D146A (Mt) exposed to exchange buffers containing 2 mmol/L (H) or 0.1 mmol/L (L) MgCl2. Exchange times, in seconds, are plotted on a log scale. Values plotted on the graph are the means, generally computed from three replicate experiments, and bars (often smaller than the symbol) indicate the measurement precision (±1σ). (C) Abec for peptides containing metal-coordinating residues for the indicated comparisons. Bars indicate the measurement uncertainty (±1σ) propagated from panel B. Significance values, based on a two-tailed Student’s t test, are indicated: *p < 0.01; **p < 0.001; ***p < 0.0001.
Peptides containing D146 and A146 differ in their intrinsic exchange rate constants because of the difference in primary sequences.24,32 The observed differences in the incorporation of deuterium by these peptides in the native conformations of the Wt and Mt proteins produced Abec values that are approximately 10 times larger than the area between the exchange curves for the fully exposed Wt and Mt peptides (Figure S5). Thus, Abec values estimate the ratio of the exchange-competent fraction in the two states for peptides that do not contain the mutated residue, as the reference states are identical, and also for peptides containing the mutated residue, as the differences between the reference states are small compared with the magnitudes of the observed effects in the native state.
We calculated Abec values to estimate effects of Mg2+ concentration and the D146A mutation on the conformational stability of the active site (Figure 3C). For residue 146-containing peptides, the large magnitude of Abec values for comparison of the behaviors of the Mt and Wt proteins in either buffer suggests that for the D146A mutant, the average fraction of amide hydrogens in the exposed state is increased by a factor of ~3, indicating that the native state was apparently destabilized by at least 2.8 kJ/mol (0.7 kcal/mol) per amino acid residue compared with the Wt protein (eq 3). Abec values for β2–β3 loop peptides were significantly positive for three of the direct comparisons (p < 0.001), although the effects of the magnesium concentration were small for the Wt and Mt proteins. Abec values for peptides containing D60 and G61 suggest that the D146A mutation substantially affected the conformational stability of the active site under both buffer conditions, whereas altering the Mg2+ concentration had minimal effects.
Flap Subdomain Segments Are Differentially Affected by the Mg2+ Concentration and the D146A Mutation
The importance of the Flap subdomain for catalysis and substrate recognition by PP2C phosphatases remains incompletely understood. The PP2Cα Flap subdomain comprises numerous short secondary structural elements and contains a high density of conserved residues (Figure 4A).5,10,39 Backbone and side-chain hydrogen bonds stabilize the Flap subdomain (Figure 4B), but structural studies suggest that the PP2C Flap subdomain is flexible.15,16,40,41 The dynamic behavior of deuterium uptake varies considerably for different segments of the Flap subdomain (Figure 4C). The deuterium uptake by peptides of amino acids 160–183 that connect the main αββα sandwich with the Flap subdomain exhibited substantial effects of the D146A mutation across all exchange intervals for both buffer conditions (p < 0.01) and small effects of the Mg2+ concentration during the shorter exchange intervals (Figure 4C, left panel). The largest feature of recognizable secondary structure in the PP2Cα Flap subdomain is the nine-residue α3 helix (amino acids 169–177). As information about the deuterium content of the initial two residues of a peptide is usually lost during HDX-MS sample processing,24,32 the peptide of amino acids 160–183, which contains two proline residues, reports the average exchange behavior of 20 amide hydrogen atoms. For the Wt peptide, a limiting deuterium uptake value near 60% is consistent with the presence of a stable α-helix containing seven or eight amide protons. The limiting value of ≈75% for the mutant peptide suggests that only five or six amide hydrogens are protected from exchange by a stable secondary structure, suggesting destabilization of two residues of helix α3. The peptide of amino acids 184–191, which forms a protruding point of the Flap, exhibited high rates of exchange for both the Wt and Mt proteins under both buffer conditions (Figure 1 and Figures S1–S4). Deuterium uptake by the peptide of amino acids 192–200, which is derived from a part of the Flap that lies near the active site, was substantially affected by both the Mg2+ concentration and the D146A mutation (Figure 4C, middle panel). Generally, deuterium incorporation rates in this peptide increased in the following order: WtH < WtL < MtH < MtL. Given the presence of seven detectable amide protons in the peptide of amino acids 192–200, limiting values of deuterium uptake of ≈73 and ≈87% for the Wt and Mt proteins, respectively, are consistent with two protected amide protons and one protected amide proton for the Wt and Mt proteins, respectively. The peptide of amino acids 214–225, which connects the Flap subdomain to β9 in the main αββα sandwich, exhibited only small effects of the Mg2+ concentration, but substantial effects of the mutation for all exchange intervals (Figure 4C, right panel, p < 0.05). To compare the effects of Mg2+ concentration and the D146A mutation on the conformational stability of the Flap subdomain, we calculated Abec values (Figure 4D). For the peptide of amino acids 160–183, buffer conditions did not significantly affect the conformational stability of either protein, but the D146A mutant was substantially destabilized in both high- and low-Mg2+ buffers, compared with the Wt protein. Abec values of ~0.3 correspond to a 2-fold increase in the exchangeready fraction in the less stable state. Abec values for the peptide of amino acids 192–200 suggest distinct effects on conformational dynamics exerted by the Mg2+ concentration and the D146A mutation, separately and in combination. The peptide of amino acids 192–200 in the Wt protein was substantially stabilized by the high-Mg2+ buffer, whereas only a slight stabilization was observed for the Mt protein. Compared with the Wt protein, this segment was substantially destabilized in the Mt protein in high-Mg2+ buffer, while the effects of the mutation were weaker in low-Mg2+ buffer. A similar pattern was displayed for the peptide of amino acids 214–225, although the effects were weaker.
Figure 4.

Magnesium concentration and the D146A mutation exert complex effects on deuterium uptake by peptides in the Flap region. (A) Schematic diagram showing the PP2Cα primary sequence encompassing the Flap subdomain (H164–E218, dotted line), with icons representing defined secondary structure elements and lines representing selected peptides. (B) Ribbon diagram representing the three-dimensional structure of the PP2Cα Flap structure (based on PDB entry 1A6Q). Predicted backbone–backbone hydrogen bonds meeting strict (blue) or relaxed (orange) criteria are indicated. (C) Time course of deuterium uptake by selected Flap subdomain peptides of PP2Cα (Wt) and PP2Cα D146A (Mt) exposed to exchange buffers containing 2 mmol/L (H) or 0.1 mmol/L MgCl2 (L). Values plotted on the graph are the means, generally computed from three replicate experiments, and bars (often smaller than the symbol) indicate the measurement precision (±1σ). (D) Area between exchange curves metrics (Abec) plotted for selected Flap peptides for the indicated comparisons. Significance values, based on a two-tailed Student’s t test, are indicated: *p < 0.01; **p < 0.001; ***p < 0.0001. Bars indicate the measurement uncertainty (±1σ) propagated from panel C.
The Mg2+ Ion Concentration and the D146A Mutation Exert Strong Effects on the Conformational Mobility of Buttressing Helices α1 and α2
Some of the largest effects of the Mg2+ concentration and the D146A mutation on deuterium exchange rates were observed in peptides derived from portions of helices α1 and α2 (Figure 5A). After exchange for 30 s, deuterium incorporation levels in the upper portions of α1 (amino acids 67–73) were similar for all four conditions but diverged over longer exchange intervals. The rate of deuterium incorporation in the Wt protein in high-Mg2+ buffer increased slowly over the course of the experiment and reached 42.3 ± 2.8% after exchange for 4 h; exchange was more rapid in low-Mg2+ buffer, and the rate of deuterium incorporation reached 64.9 ± 0.8% after exchange for 4 h. Compared with the Wt protein in each buffer condition, the rate of deuterium incorporation was substantially and significantly higher in the Mt protein for exchange intervals of ≥300 s (p < 0.0001) and reached levels of 87.1 ± 1.0 and 89.3 ± 0.8% for high and low Mg2+ concentrations, respectively, after exchange for 4 h. Similar patterns were observed in peptides derived from amino acids 96–110. The portions of helices α1 and α2 that displayed marked effects of low Mg2+ concentrations and the D146A mutation did not exhibit unusual intrinsic flexibility, as suggested by crystallographic B factors. On the basis of the crystal structure (PDB entry 1A6Q), the average B factor for amino acids 67–73 and 96–110 is 18.4 ± 2.6 (15 residues), whereas the average for all α-helical residues is 21.0 ± 6.0 (130 residues).
Figure 5.

Hydrogen bond/electrostatic interaction networks link the β6-β7 loop, the Flap, and metal-coordinating residues. (A) Time course of deuterium uptake for selected α1/α2 peptides of PP2Cα and D146A PP2Cα exposed to exchange buffers containing 2 mmol/L (H) or 0.1 mmol/L (L) MgCl2. Values plotted on the graph are the means, generally computed from three replicate experiments, and bars (often smaller than the symbol) indicate the measurement precision (±1σ). (B) Area between exchange curves metrics (Abec) plotted for selected Flap peptides for the indicated comparisons. Significance values, based on a two-tailed Student’s t test, are indicated: *p < 0.01; **p < 0.001; ***p < 0.0001. Bars indicate the measurement uncertainty (±1σ) propagated from panel A. (C) Ribbon diagram showing a network of hydrogen bond/electrostatic interactions linking strongly affected regions of helices α1 and α2 (sandy brown) in the main domain (tan) to the Flap subdomain (sky blue).
The calculated Abec values for these α1 and α2 peptides were significantly positive for each of the four direct comparisons [p < 0.01 (Figure 5B)]. Calculated Abec values ranged from ~0.2 to ~0.8. These Abec values were calculated over the experimentally accessible exchange intervals but do not contain information about exchange occurring on time scales of <30 s or >4 h. As such, the calculated Abec values can be interpreted as a lower bound of the difference in the free energy of local unfolding between the compared states. As both deuterium uptake curves for the Mt protein are nearly complete, the calculated Abec for the MtH-MtL comparison provides an accurate estimate of the free energy difference; the three remaining comparisons should be interpreted as lower bounds.
The PP2Cα crystal structure (PDB entry 1A6Q) provides detailed structural information about PP2Cα with two Mn2+ ions bound, an inactive conformation. The structure suggests a network of side-chain hydrogen bond and electrostatic interactions linking helices α1 and α2 and residues in the Flap subdomain (Figure 5C). The Flap subdomain and the bridge connecting the metal-coordinating residues D60 and G61 with the top of helix α1 are linked by side-chain interactions between the highly conserved residues H62 and D199. Moreover, side-chain interactions between R102 and E218, side-chain interactions between D109 and K203, and an interaction between the side chain and backbone of R113 and K203 suggest a link between the Flap subdomain and the region of α2 that was highly affected by the low Mg2+ concentration and the D146A mutation. Furthermore, the structure suggests an extended hydrogen bond/electrostatic interaction network linking helix α2 in the main αββα sandwich to α3 in the Flap subdomain. Following strict criteria, hydrogen bonds can link the R102 and E218 side chains and the E218 backbone with the R195 side chain. The distances between the R195 NE and E171 OE1 atoms and between the R195 NH1 and E171 OE2 atoms (3.309 and 3.385 Å, respectively) support the formation of hydrogen bonds under relaxed criteria. Interestingly, the PP2Cα crystal structure also supports linkage of the lower portion of the active site, helices α1 and α2, and the Flap subdomain through formation of a small antiparallel β-strand-like structure involving backbone-backbone hydrogen bonds. Specifically, H62 and G126 can form two backbone-backbone hydrogen bonds, connecting the β4–α1 bridge with the α2–β5 bridge; the S127–L197, L197–S127, and D199–S125 residue pairs can form three backbone-backbone hydrogen bonds connecting the α2–β5 bridge with the Flap subdomain. Our results, in combination with a known structure for PP2Cα, suggest that mechanical linkages among the metal-coordinating residues, the Flap subdomain, and buttressing α-helices α1 and α2 differ between active and inactive conformations of PP2Cα.
Molecular Dynamics Simulations of PP2Cα and PP2Cα D146A Suggest Alterations in Structure and Hydrogen Bond Occupancy
Analysis of hydrogen/deuterium exchange rates suggests that millimolar concentrations of Mg2+ ions and the presence of an aspartic acid residue at position 146, both of which are required for catalytic activity, synergize to bring the PP2Cα catalytic domain into a tighter conformation. Although the effects in some regions of the protein are dominated by either the Mg2+ ion concentration or the mutation at position 146, many regions are affected by both factors. To gain additional understanding of these effects, we performed molecular dynamics (MD) calculations to produce 200 ns of simulation for Wt PP2Cα with two Mg2+ ions bound [Wt(2Mg)], Wt PP2Cα with three Mg2+ ions bound [Wt(3Mg)], and PP2Cα D146A with two Mg2+ ions bound [D146A(2Mg)]. An overlay of representative structures, spaced across the MD simulation interval, identifies conformational differences and regions of conformational variability between the Wt(2Mg) and Wt(3Mg) structures (Figure 6A). Compared with the Wt(2Mg) structures (tan traces), those from the Wt(3Mg) simulation (light blue traces) show a shift of the flap subdomain away from the active site and toward the helices α1 and α2, which in turn are shifted toward the β2–β3 loop at the bottom of the active site. Within the active site, M3 exhibits a larger variation in its location than M1 or M2, consistent with its lower binding affinity.23 In Wt(3Mg) structures, M1 is shifted toward and M2 is shifted away from the β2–β3 loop at the bottom of the active site. In the absence of M3, the D146 side chain partially interacts with the R186 side chain, contributing to the wider range of conformations exhibited by the R186 side chain. In Wt(3Mg) structures, the D146 side chain is fully engaged in coordinating M3 and the R186 side chain exhibits a smaller range of conformations. Similarly, in the absence of M3, R33 exhibits greater conformational variability by interacting with either E37 or D282, while in the Wt(3Mg) structures, the R33 side chain interacts exclusively with the E37 side chain. Heat maps representing differences [Wt(2Mg) – Wt(3Mg)] in all pairs of Cα–Cα average distances are consistent with the variations exhibited by the representative structures (Figure S6). Notably, in the Wt(2Mg)–Wt(3Mg) comparison (Figure S6), the broad stripe of reddish regions from position 165 to 220 indicates that, in the presence of the third metal ion, average distances are increased between Flap subdomain Cα atoms and those of many regions of the remainder of the catalytic domain, including the β2–β3 loop, the loop at the top of helix α2, the loop between β6 and β7 that contains D146, the region containing D239 and D243, and the region surrounding Asp282. On average, D282 is closer (blue shadings) to several regions, including the M2-coordinating residues E37 and D38, the M1- and M2-coordinating residue D60, strands β3 and β4, and the upper portion of helix α2. Of interest is the suggestion that in the Wt(3Mg) simulation compared with the Wt(2Mg) simulation, helix α3 is closer to D195. Thus, the MD simulations suggest that the presence of a third metal ion causes widespread adjustments in the structure of the PP2Cα catalytic domain, principally in a more distant position of the Flap combined with a tighter arrangement of metal-coordinating residues in the active site.
Figure 6.

Molecular dynamics (MD) simulations suggest conformational effects of the binding of the third magnesium ion or the D146A mutation. (A) Ribbon diagram overlay of 19 MD snapshots for PP2Cα with three (blue traces, sea green Mg2+ ions) compared with two Mg2+ ions bound (tan traces, olive Mg2+ ions). Selected side chains are shown as sticks. (B) Ribbon diagram overlay of 19 MD snapshots for PP2Cα D146A (plum traces) compared with PP2Cα (tan traces), both with two Mg2+ ions bound (chartreuse and olive, respectively). Selected side chains are shown as sticks. (C) Differences in hydrogen bond occupancy for PP2Cα with three compared with two Mg2+ ions bound based on 200 ns of molecular dynamics simulations. Increased (top left diagonal, red symbols) or decreased (bottom right diagonal, blue symbols) hydrogen bond occupancies for the comparison of Wt(3Mg) vs Wt(2Mg) are indicated for donor/acceptor pairs. (D) Differences in hydrogen bond occupancy for PP2Cα D146A compared with PP2Cα, both with two Mg2+ ions bound, based on 200 ns of molecular dynamics simulations. Increased (top left diagonal, brown symbols) or decreased (bottom right diagonal, blue-green symbols) hydrogen bond occupancies for the comparison of D146A(2Mg) vs Wt(2Mg) are indicated for donor/acceptor pairs.
An overlay of representative structures from the Wt(2Mg) (tan traces) and D146A(2Mg) (plum traces) simulations shows that the N-terminal lobe of the Flap subdomain is positioned closer to the active site in the mutant (Figure 6B). Substitution of alanine at position 146 eliminates possible interactions between the R186 side chain and aspartate side chain, resulting in a smaller range of positions of the R186 side chain, but with an increased level of exposure to the solvent. Similarly, in the D146A(2Mg) structures, the R33 side chain interacts exclusively with the D282 side chain, in contrast to the Wt(2Mg) structures, in which the R33 side chain interacts with the D282 or E37 side chains. Heat maps representing differences [Wt(2Mg) - D146A(2Mg)] in all pairs of Cα-Cα average distances from 200 ns of MD simulations show a predominance of reduced average Cα–Cα distances in the D146A(2Mg) structures (blue) over much of the catalytic domain (Figure S7). The simulations suggest substantial alterations in the conformations of the loop connecting helix α1a to helix α2 and the bridge connecting α2 to β5. Interestingly, the Flap (amino acids 165–220) exhibits a pattern of alternating increased (red) and decreased (blue) Cα-Cα average distances in D146A(2Mg) compared with Wt(2Mg) structures, suggesting a structural reorganization.
We analyzed differences in hydrogen bond occupancy for PP2Cα with three compared with two Mg2+ ions bound and for PP2Cα compared with PP2Cα D146A, both with two Mg2+ ions bound (Figure 6C). The MD simulations suggest that the presence of a third bound metal ion increased the levels of H62–D199 and R102–E218 hydrogen bonding while decreasing the level of R124–D199 hydrogen bonding. We also analyzed differences in hydrogen bond occupancy for PP2Cα compared with PP2Cα D146A, both with two Mg2+ ions bound (Figure 6D). Mutation to an alanine at position 146 increased the level of hydrogen bonding between the R33 and E37 side chains while decreasing the level of hydrogen bonding between H62 and G126 backbones.
DISCUSSION
The PP2C phosphatases are characterized by their requirement for supplementation with millimolar concentrations of Mg2+ or Mn2+ ions to demonstrate measurable catalytic activity in vitro. Recent work has associated this requirement with the binding of a weakly associated third metal ion in the active site.12,15,16,22,23,41 The D146A mutation in PP2Cα or the corresponding mutation in related PP2C phosphatases abolished or severely reduced catalytic activity.22,23 However, details of the involvement of the third metal ion in either the catalytic mechanism or stabilization of the native structure of PP2C phosphatases remain incompletely understood. Here, we have used HDX–MS techniques to show that the two requirements for activity, millimolar concentrations of Mg2+ ions and the presence of an aspartic acid residue at position 146, combine to restrict the conformational mobility of the PP2Cα active site on a catalytically relevant time scale. The increased conformational mobility in the Flap subdomain and in portions of helices α1 and α2 that result from low Mg2+ ion concentrations or the loss of the aspartic acid side chain at position 146 suggests that structural bracing in these regions contributes to rigidity in the active state of the enzyme.
PP2Cα activity toward phosphopeptide or phosphoprotein substrates is abolished by mutation of metal-coordinating active site residues, but also by mutation of specific residues in the Flap subdomain or that link the Flap subdomain to the active site (Figure 7). Mutation of active site residues that directly coordinate M1 or M3 abolished (D239A and D146A) or sharply reduced (D60N, D239N, and D282N) phosphatase activity.22,23,42 Mutation of R33, which binds the phosphorylated substrate, mutation of residues indirectly coordinating M2 (E37 and D38), or mutation of D243, which is part of an alternative binding site for M3, moderately reduced PP2Cα activity.11,23,42 Mutation of residues lying just outside the active site (H40, T128, and S190) had little effect on activity.23,42 Interestingly, residues that are critical for the activity of PP2Cα toward physiological substrates form an arc running through the Flap alongside the active site. The R174A, R195A, and D199A mutants are functionally inactive in cell-based assays.9,43,44 Mutation of R186, N188, or R195 resulted in a loss of activity toward phosphopeptide substrates, but the activities toward the small molecule substrate p-nitrophenyl phosphate (pNPP) were only moderately affected.23 Other Flap subdomain residues residing outside the arc (H164, K165, and Q185) exhibited only moderately reduced activity toward either pNPP or phosphopeptide substrates.7,23
Figure 7.

Ribbon diagram of the PP2Cα active site and Flap subdomain with functional classification of the effects of mutations on phosphatase activity. The structural representation is based on PDB entry 1A6Q, with the addition of a third Mg2+ ion. Mutated residues are colored brown for little or no effect (≥65% of Wt activity, H40, T128, and S190), light blue for reduced activity (5–50% of Wt activity, R33, E37, D38, H62, H164, K165, Q185, and D243), orchid for a loss of activity (0–5% of Wt activity, D60, D146, D239, and D282), teal for altered specificity (inactive toward phosphopeptides but active toward pNPP, R186, N188, and R195), and pink for functionally inactive (no detected activity toward physiological substrates in cell-based assays, D199 and R174). For specific references, see the text.
The importance of specific Flap subdomain residues for the function of PP2Cα is supported by the effects of mutation of the corresponding residues in related PP2C phosphatases. PP2Cα R195 is one of eight residues within a 30-residue region that are highly conserved among eukaryotic PP2C phosphatases.3,39 Mutation to alanine of the corresponding residue in the Caenorhabditis elegans phosphatase FEM-2 led to a loss of phosphatase activity in vitro and a lack of functional activity in vivo.39 Similarly, mutation of the corresponding residue in the human homologue hFEM-2 (PPM1F) resulted in a loss of phosphatase activity toward physiological substrates in vitro and loss of pro-apoptotic activity upon overexpression in human cells.45 The PP2Cα residues G198 and D199 are also highly conserved.39 C. elegans FEM-2 G339 aligns with human PP2Cα G198; the G339R mutation exhibited a functional loss of FEM-2 activity in vivo and severely reduced phosphatase activity toward a phosphoprotein in vitro.46,47 Residue D193 of the Drosophila melanogaster PP2C phosphatase Alphabet aligns with human PP2Cα D199; the D193V mutation exhibited substantially reduced in vitro activity toward a phosphopeptide and functional loss of activity in vivo.43 Thus, residues aligning with R195, G198, and D199 are critical for the functional activity of PP2C phosphatases. These residues are located within the peptides of amino acids 192–200 or 192–201 that exhibited the strongest interplay between the effects of the Mg2+ concentration and the D146A mutation (Figure 2). We found that the peptides containing these residues exhibited distinct profiles of deuterium incorporation in each combination of Wt or Mt protein with high or low Mg2+ concentrations (Figure 4). In addition, R195 is positioned to participate in a side-chain-based hydrogen bonding/salt bridge network that may contribute to the increased conformational mobility in portions of α1 and α2 observed in inactive states of PP2Cα (Figure 5). Furthermore, in crystal structures of PP2Cα, the side chain of D199 is positioned to form a hydrogen bond with the side chain of H62, a residue that has been shown to be important for efficient catalytic activity.10,11,42
Arabidopsis thaliana phosphatase HAB1 is involved in regulating the response to environmental stress; complexes of Hab1 with a signaling hormone-bound receptor or with a substrate kinase have been characterized by X-ray crystallography and HDX–MS.19,21,48 The HAB1 Flap subdomain is critical for specific interaction with the regulatory receptor and the protein substrate SnRK2.6.19,21 An HDX–MS study of HAB1 in complex with SnRK2.6 found a reduced rate of deuterium uptake in the loop at the bottom of the active site, in the bridge to α1, and in the portion of the Flap subdomain corresponding to PP2Cα R195–D199.48 These regions correspond to the regions of PP2Cα affected by the Mg2+ concentration and the D146A mutation. A. thaliana phosphatase PPH1 is involved in regulating the response to varying levels of light.20 Crystal structures of PPH1 and of PPH1 D180E (an inactive mutant) in complex with a phosphopeptide show that a region of the Flap subdomain is important for specific interactions with the substrate.20 This region of PPH1 aligns to the protruding point of the Flap subdomain of PP2Cα. These structures of PP2C phosphatases in complex with model substrates illustrate the importance of the Flap subdomain for interaction with physiological substrates.
Crystal structures of various PP2C phosphatases have revealed one, two, or three metal ions in the active site.21,49,50 Moreover, structures for the same enzyme in different conformations or crystallized under different conditions may exhibit two or three metal ions in the active site.14–16,41 General mechanisms for PP2C catalysis involving two or three metal ions have been proposed.10,11,22,41,42 Metal ions can be involved in the catalytic process through activation of the attacking moiety, binding of the substrate, or stabilization of intermediates or transition states.1,51 The distributed effects of magnesium binding and of the D146A mutation throughout the active site and across the Flap subdomain suggest that substrate recognition and catalytic activity are coupled in PP2C phosphatases to achieve catalytic specificity. Additionally, the binding of Mg2+ to PP2Cα detected by ITC was weakened but not eliminated by the D146A or D243A mutation, suggesting two alternative binding sites.23 Among the crystal structures of PP2C phosphatases exhibiting a third metal in the active site, positions of the tightly bound metal ions and their coordinating residues can be superimposed with small variance, but the position of the third metal ion is considerably more variable. These observations are consistent with a mechanism that involves a shift in the position of the third metal ion during catalysis. In the work presented here, we detected specific stabilization of PP2C D146A in the presence of 2 mmol/L Mg2+ compared with 0.1 mmol/L Mg2+, suggesting that the magnesium occupancy of the alternate site involving D243 stabilizes the folded state, even in the absence of an aspartate at position 146.
In this work, we observed that changing the occupancy of the third metal binding site(s) or mutation of D146 affected the conformational mobility in the active site, portions of the Flap subdomain, and specific segments of helices α1 and α2. MD calculations support increased rigidity of the active site in the active state of the enzyme through an increased number of hydrogen bond interactions within the active site. Moreover, the importance of the Flap and buttressing helices α1 and α2 is supported by functionally characterized mutants of PP2Cα and related PP2C phosphatases. These observations will be useful in future investigations of the mechanism of PP2C phosphatase activity toward physiological substrates and in understanding how the activity can be activated or inhibited.
Supplementary Material
Acknowledgments
Funding
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and the Institute for Bioscience and Biotechnology Research/National Institute of Standards and Technology. E.S.G. and K.W.A. were supported by National Academy of Science-National Research Council postdoctoral fellowships. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
ABBREVIATIONS
- Abec
area between exchange curves
- β-ME
β-mercaptoethanol
- EX2
exchange conditions class 2
- HDX-MS
hydrogen/deuterium exchange-mass spectrometry
- FDA
functional data analysis
- HB-H
HEPES buffer with 2 mmol/L MgCl2
- HB-L
HEPES buffer with 0.1 mmol/L MgCl2
- MD
molecular dynamics
- Mt
mutant
- MtH
mutant enzyme in 2 mmol/L MgCl2
- MtL
mutant enzyme in 0.1 mmol/L MgCl2
- pNPP
p-nitrophenyl phosphate
- PPM
metal-dependent protein phosphatases
- RT
gas constant × absolute temperature
- Wt
wild type
- WtH
wild-type enzyme in 2 mmol/L MgCl2
- WtL
wild-type enzyme in 0.1 mmol/L MgCl2
Footnotes
ASSOCIATED CONTENT
Peptide-level mean deuterium uptake by PP2Ca or PP2Cα D146A in 2.0 or 0.1 mmol/L Mg2+ at 25 °C (XLSX)Peptide-level area between exchange curves, standard deviations, and p values (XLSX)Peptide map showing time-dependent deuterium uptake by human PP2Cα in the presence of 2 mmol/L MgCl2 (Figure S1), peptide map showing time-dependent deuterium uptake by human PP2Cα in the presence of 0.1 mmol/L MgCl2 (Figure S2), peptide map showing time-dependent deuterium uptake by human PP2Ca D146A in the presence of 2 mmol/L MgCl2 (Figure S3), peptide map showing time-dependent deuterium uptake by human PP2Ca D146A in the presence of 0.1 mmol/L MgCl2 (Figure S4), calculated and observed fractional remaining hydrogen for PP2Cα peptides containing D146 or A146 (Figure S5), differences in average Cα–Cα distances of PP2Ca between Wt(2Mg) and Wt-(3Mg) over 200 ns of molecular dynamics simulations (Figure S6), and differences in average Cα–Cα distances between Wt(2Mg) and D146A(2Mg) over 200 ns of molecular dynamics simulations of PP2Ca and PP2Ca D146A (Figure S7) (PDF)
Author Contributions
S.D. purified proteins and determined enzymatic activities. E.S.G., S.J.M. and L.M.M.J. developed the initial experimental design. E.S.G. conducted initial HDX experiments and K.W.A. final HDX experiments. S.J.M. and E.S.G. analyzed HDX data and wrote the draft. S.R. D. performed MD calculations. S.J.M. performed functional data analysis calculations and prepared figures. E.A. and J.W.H. provided facilities and material support. S.J.M. and E.S.G. contributed equally to this work.
Certain commercial materials and equipment were identified to adequately specify the experimental procedure. Such identification neither implies recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the material or equipment identified is the best available for the purpose.
The authors declare no competing financial interest.
References
- 1.Andreini C, Bertini I, Cavallaro G, Holliday GL, Thornton JM. Metal ions in biological catalysis: from enzyme databases to general principles. J. Biol. Inorg. Chem. 2008;13:1205–1218. doi: 10.1007/s00775-008-0404-5. [DOI] [PubMed] [Google Scholar]
- 2.Sissi C, Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37:702–711. doi: 10.1093/nar/gkp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moorhead GB, De Wever V, Templeton G, Kerk D. Evolution of protein phosphatases in plants and animals. Biochem. J. 2009;417:401–409. doi: 10.1042/BJ20081986. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Bork P, Brown NP, Hegyi H, Schultz J. The protein phosphatase 2C (PP2C) superfamily: detection of bacterial homologues. Protein Sci. 1996;5:1421–1425. doi: 10.1002/pro.5560050720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerk D, Silver D, Uhrig RG, Moorhead GB. ″PP2C7s″, Genes Most Highly Elaborated in Photosynthetic Organisms, Reveal the Bacterial Origin and Stepwise Evolution of PPM/PP2C Protein Phosphatases. PLoS One. 2015;10:e0132863. doi: 10.1371/journal.pone.0132863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li R, Gong Z, Pan C, Xie DD, Tang JY, Cui M, Xu YF, Yao W, Pang Q, Xu ZG, Li MY, Yu X, Sun JP. Metal-dependent protein phosphatase 1A functions as an extracellular signal-regulated kinase phosphatase. FEBS J. 2013;280:2700–2711. doi: 10.1111/febs.12275. [DOI] [PubMed] [Google Scholar]
- 8.Lu X, An H, Jin R, Zou M, Guo Y, Su PF, Liu D, Shyr Y, Yarbrough WG. PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene. 2014;33:2918–2927. doi: 10.1038/onc.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takekawa M, Maeda T, Saito H. Protein phosphatase 2Calpha inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J. 1998;17:4744–4752. doi: 10.1093/emboj/17.16.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- 11.Pan C, Tang JY, Xu YF, Xiao P, Liu HD, Wang HA, Wang WB, Meng FG, Yu X, Sun JP. The catalytic role of the M2 metal ion in PP2Calpha. Sci. Rep. 2015;5:8560. doi: 10.1038/srep08560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlicker C, Fokina O, Kloft N, Grune T, Becker S, Sheldrick GM, Forchhammer K. Structural analysis of the PP2C phosphatase tPphA from Thermosynechococcus elongatus: a flexible flap subdomain controls access to the catalytic site. J. Mol. Biol. 2008;376:570–581. doi: 10.1016/j.jmb.2007.11.097. [DOI] [PubMed] [Google Scholar]
- 13.Fjeld CC, Denu JM. Kinetic analysis of human serine/threonine protein phosphatase 2Calpha. J. Biol. Chem. 1999;274:20336–20343. doi: 10.1074/jbc.274.29.20336. [DOI] [PubMed] [Google Scholar]
- 14.Bellinzoni M, Wehenkel A, Shepard W, Alzari PM. Insights into the catalytic mechanism of PPM Ser/Thr phosphatases from the atomic resolution structures of a mycobacterial enzyme. Structure. 2007;15:863–872. doi: 10.1016/j.str.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Pullen KE, Ng HL, Sung PY, Good MC, Smith SM, Alber T. An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C–Family Ser/Thr protein phosphatase. Structure. 2004;12:1947–1954. doi: 10.1016/j.str.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Rantanen MK, Lehtio L, Rajagopal L, Rubens CE, Goldman A. Structure of Streptococcus agalactiae serine/threonine phosphatase. The subdomain conformation is coupled to the binding of a third metal ion. FEBS J. 2007;274:3128–3137. doi: 10.1111/j.1742-4658.2007.05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira SF, Goss L, Dworkin J. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 2011;75:192–212. doi: 10.1128/MMBR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dupeux F, Antoni R, Betz K, Santiago J, Gonzalez-Guzman M, Rodriguez L, Rubio S, Park SY, Cutler SR, Rodriguez PL, Marquez JA. Modulation of abscisic acid signaling in vivo by an engineered receptor-insensitive protein phosphatase type 2C allele. Plant Physiol. 2011;156:106–116. doi: 10.1104/pp.110.170894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melcher K, Ng LM, Zhou XE, Soon FF, Xu Y, Suino-Powell KM, Park SY, Weiner JJ, Fujii H, Chinnusamy V, Kovach A, Li J, Wang Y, Li J, Peterson FC, Jensen DR, Yong EL, Volkman BF, Cutler SR, Zhu JK, Xu HE. A gate-latch-lock mechanism for hormone signalling by abscisic acid receptors. Nature. 2009;462:602–608. doi: 10.1038/nature08613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei X, Guo J, Li M, Liu Z. Structural Mechanism Underlying the Specific Recognition between the Arabidopsis State-Transition Phosphatase TAP38/PPH1 and Phosphorylated Light-Harvesting Complex Protein Lhcb1. Plant Cell. 2015;27:1113–1127. doi: 10.1105/tpc.15.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soon FF, Ng LM, Zhou XE, West GM, Kovach A, Tan MH, Suino-Powell KM, He Y, Xu Y, Chalmers MJ, Brunzelle JS, Zhang H, Yang H, Jiang H, Li J, Yong EL, Cutler S, Zhu JK, Griffin PR, Melcher K, Xu HE. Molecular mimicry regulates ABA signaling by SnRK2 kinases and PP2C phosphatases. Science. 2012;335:85–88. doi: 10.1126/science.1215106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su J, Schlicker C, Forchhammer K. A third metal is required for catalytic activity of the signal-transducing protein phosphatase M tPphA. J. Biol. Chem. 2011;286:13481–13488. doi: 10.1074/jbc.M109.036467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanoue K, Miller Jenkins LM, Durell SR, Debnath S, Sakai H, Tagad HD, Ishida K, Appella E, Mazur SJ. Binding of a third metal ion by the human phosphatases PP2Calpha and Wip1 is required for phosphatase activity. Biochemistry. 2013;52:5830–5843. doi: 10.1021/bi4005649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engen JR, Wales TE. Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu. Rev. Anal. Chem. 2015;8:127–148. doi: 10.1146/annurev-anchem-062011-143113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallagher ES, Hudgens JW. Mapping Protein-Ligand Interactions with Proteolytic Fragmentation, Hydrogen/Deuterium Exchange-Mass Spectrometry. Methods Enzymol. 2016;566:357–404. doi: 10.1016/bs.mie.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Englander JJ, Del Mar C, Li W, Englander SW, Kim JS, Stranz DD, Hamuro Y, Woods VL., 3rd Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7057–7062. doi: 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hvidt A, Wallevik K. Conformational changes in human serum albumin as revealed by hydrogen-deuterium exchange studies. J. Biol. Chem. 1972;247:1530–1535. [PubMed] [Google Scholar]
- 28.Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffey N, Hinde J. Analyzing Time-Course Microarray Data Using Functional Data Analysis - A Review. Stat. Appl. Genet. Mol. Biol. 2011;10:23. doi: 10.2202/1544-6115.1671. [DOI] [Google Scholar]
- 30.Minas C, Waddell SJ, Montana G. Distance-based differential analysis of gene curves. Bioinformatics. 2011;27:3135–3141. doi: 10.1093/bioinformatics/btr528. [DOI] [PubMed] [Google Scholar]
- 31.Mazur SJ, Weber DP. The Area Between Exchange Curves as a Measure of Conformational Differences in Hydrogen-Deuterium Exchange Mass Spectrometry Studies. J. Am. Soc. Mass Spectrom. 2017;28:978–981. doi: 10.1007/s13361-017-1615-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins: Struct., Funct., Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna: 2015. [Google Scholar]
- 34.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackerell AD, 3rd, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 37.Montana G, Berk M, Ebbels T. Modelling short time series in metabolomics: a functional data analysis approach. Adv. Exp. Med. Biol. 2011;696:307–315. doi: 10.1007/978-1-4419-7046-6_31. [DOI] [PubMed] [Google Scholar]
- 38.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21:996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin-Sang ID, Spence AM. Caenorhabditis elegans sex-determining protein FEM-2 is a protein phosphatase that promotes male development and interacts directly with FEM-3. Genes Dev. 1996;10:2314–2325. doi: 10.1101/gad.10.18.2314. [DOI] [PubMed] [Google Scholar]
- 40.Su J, Forchhammer K. Determinants for substrate specificity of the bacterial PP2C protein phosphatase tPphA from Thermosynechococcus elongatus. FEBS J. 2013;280:694–707. doi: 10.1111/j.1742-4658.2011.08466.x. [DOI] [PubMed] [Google Scholar]
- 41.Wehenkel A, Bellinzoni M, Schaeffer F, Villarino A, Alzari PM. Structural and binding studies of the three-metal center in two mycobacterial PPM Ser/Thr protein phosphatases. J. Mol. Biol. 2007;374:890–898. doi: 10.1016/j.jmb.2007.09.076. [DOI] [PubMed] [Google Scholar]
- 42.Jackson MD, Fjeld CC, Denu JM. Probing the function of conserved residues in the serine/threonine phosphatase PP2Calpha. Biochemistry. 2003;42:8513–8521. doi: 10.1021/bi034074+. [DOI] [PubMed] [Google Scholar]
- 43.Baril C, Therrien M. Alphabet, a Ser/Thr phosphatase of the protein phosphatase 2C family, negatively regulates RAS/MAPK signaling in Drosophila. Dev. Biol. 2006;294:232–245. doi: 10.1016/j.ydbio.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 44.Sun W, Yu Y, Dotti G, Shen T, Tan X, Savoldo B, Pass AK, Chu M, Zhang D, Lu X, Fu S, Lin X, Yang J. PPM1A and PPM1B act as IKKbeta phosphatases to terminate TNFalpha-induced IKKbeta-NF-kappaB activation. Cell. Signalling. 2009;21:95–102. doi: 10.1016/j.cellsig.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan KM, Chan SL, Tan KO, Yu VC. The Caenorhabditis elegans sex-determining protein FEM-2 and its human homologue, hFEM-2, are Ca2+/calmodulin-dependent protein kinase phosphatases that promote apoptosis. J. Biol. Chem. 2001;276:44193–44202. doi: 10.1074/jbc.M105880200. [DOI] [PubMed] [Google Scholar]
- 46.Hansen D, Pilgrim D. Molecular evolution of a sex determination protein. FEM-2 (pp2c) in Caenorhabditis. Genetics. 1998;149:1353–1362. doi: 10.1093/genetics/149.3.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pilgrim D, McGregor A, Jackle P, Johnson T, Hansen D. The C. elegans sex-determining gene fem-2 encodes a putative protein phosphatase. Mol. Biol. Cell. 6:1159–1171. doi: 10.1091/mbc.6.9.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.West GM, Pascal BD, Ng LM, Soon FF, Melcher K, Xu HE, Chalmers MJ, Griffin PR. Protein conformation ensembles monitored by HDX reveal a structural rationale for abscisic acid signaling protein affinities and activities. Structure. 2013;21:229–235. doi: 10.1016/j.str.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teh AH, Makino M, Hoshino T, Baba S, Shimizu N, Yamamoto M, Kumasaka T. Structure of the RsbX phosphatase involved in the general stress response of Bacillus subtilis. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015;71:1392–1399. doi: 10.1107/S1399004715007166. [DOI] [PubMed] [Google Scholar]
- 50.Wynn RM, Li J, Brautigam CA, Chuang JL, Chuang DT. Structural and biochemical characterization of human mitochondrial branched-chain alpha-ketoacid dehydrogenase phosphatase. J. Biol. Chem. 2012;287:9178–9192. doi: 10.1074/jbc.M111.314963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.