

Abstract
Histone deacetylase inhibitors (HDACis) are a potent class of tumor-suppressive agents traditionally believed to exert their effects through loosening tightly-wound chromatin resulting in de-inhibition of various tumor suppressive genes. Recent literature however has shown altered intratumoral hypoxia signaling with HDACi administration not attributable to changes in chromatin structure. We sought to determine the precise mechanism of HDACi-mediated hypoxia signaling attenuation using vorinostat (SAHA), an FDA-approved class I/IIb/IV HDACi. Through an in-vitro and in-vivo approach utilizing cell lines for hepatocellular carcinoma (HCC), osteosarcoma (OS), and glioblastoma (GBM), we demonstrate that SAHA potently inhibits HIF-a nuclear translocation via direct acetylation of its associated chaperone, heat shock protein 90 (Hsp90). In the presence of SAHA we found elevated levels of acetyl-Hsp90, decreased interaction between acetyl-Hsp90 and HIF-a, decreased nuclear/cytoplasmic HIF-α expression, absent HIF-α association with its nuclear karyopharyin Importin, and markedly decreased HIF-a transcriptional activity. These changes were associated with downregulation of downstream hypoxia molecules such as endothelin 1, erythropoietin, glucose transporter 1, and vascular endothelial growth factor. Findings were replicated in an in-vivo Hep3B HRE-Luc expressing xenograft, and were associated with significant decreases in xenograft tumor size. Altogether, this study highlights a novel mechanism of action of an important class of chemotherapeutic.
Keywords: HDACi, Hsp90, SAHA, HIF, hypoxia
INTRODUCTION
Over the last decade the field of targeted chemotherapy has yielded significant improvements in the prognosis of patients with incurable malignancies. Despite the enthusiasm, there still exists a need for an effective therapy that interferes with components of the hypoxia signaling pathway. Transcriptional activity induced by the heterodimeric transcription factors hypoxia-inducible factor 1α and 2α (HIF-1α, HIF-2α) encourages neoplastic transformation to an aggressive phenotype through upregulation of genes involved with angiogenesis, tissue invasion, and metastasis [1–3]. Resulting intracellular metabolic changes facilitate tolerance to elevated levels of reactive oxygen species typically encountered after chemo- and/or radiotherapy [4–7]. Elevated intratumoral HIF levels carry a negative prognosis, and are an independent risk factor for decreased patient survival [8–10]. To this end, a variety of therapies inhibiting HIF and/or components of the hypoxia signaling cascade have been identified [11, 12]. However, agents that only target specific components of the hypoxia signaling pathway generally fail to produce an enduring clinical response [13–16]. The vast majority of HIF inhibitors utilized for pre-clinical and clinical investigation are non-specific for the hypoxia pathway [11].
Histone deacetylase inhibitors (HDACis) are a group of small-molecule compounds that have shown potent tumor-suppressive activity both in vitro and in clinical studies [17–21]. Their success led to FDA approval of two agents, Vorinostat (Suberoylanilide Hydroxamic Acid (SAHA), and Romidepsin (FK228), for treatment of cutaneous T cell lymphoma (CTCL) [22, 23]. There are currently over 120 clinical studies evaluating HDACi efficacy in other tumor subtypes [24]. The effects of HDACis are traditionally described as mediated through de-inhibition of genetic repression through loosening tightly wound chromatin [25–27]. HDACis prevent the removal of acetyl groups from the lysine residue on histone proteins, keeping the negatively charged acetyl molecule in close proximity to the negatively charged DNA phosphate backbone [27]. The mutual repulsion allows transcriptional access to genes generally suppressed by malignant cells, including tumor suppressors such as p21 and Rb [28–31]. Several recent studies have modified this view, clarifying class I HDACs (HDAC 1, 2, 3, 8) as the main class capable of acting on histones as their primary substrate [24]. Class I HDACs have shown the ability to promote cell proliferation and survival [29, 30], as well as endothelial sprouting and vascular branching [32]. However, it is the other classes of HDACs held largely accountable for angiogenesis initiation and propagation largely through the modification of non-histone proteins [32–40].
Knockout studies have demonstrated the vital importance of class IIa HDACs (HDAC4, 5, 7, 9), in particular HDAC7, for the development of the immature vasculature through cytoplasmic control of transcription factors [41]. Class IIb HDACs (HDAC6, 10) can act through HDAC6 to permit cytoplasmic accumulation of HIF through deacetylation of Hsp90 and ubiquitin [42]. Class III HDACs (SIRT1-7) act through Sirtuin 1 to deacetylate Foxo1, a transcription factor critical to blood vessel development [43–46]. Class IV HDACs consist solely of HDAC11, which has no known direct anti-angiogenic functions, rather functioning to regulate the immune system through control of cytokine expression [47, 48].
SAHA is a class I/IIb/IV HDACi that has potent inhibitory effects on the hypoxia signaling pathway. It's mechanism of hypoxia attenuation involves a high level of complexity due at least in part to its influence on multiple HDACs. SAHA has been shown to decrease levels of HIF-1α and VEGF in various tumor cell lines without a proportional change in the levels of HIF-1α mRNA [17, 32, 34]. Some authors have suggested this due to class II activity inducing direct HIFα acetylation, targeting HIF for destruction in a von Hippel-Lindau ubiquitin ligase (pVHL) or p53-dependent manner [38, 49, 50]. However, mechanistic studies have not reliably observed direct HIF-1α acetylation by N-acetyltransferase, and confirmatory studies have not seen increased pVHL activity after SAHA on a consistent basis [51–53]. Other groups have proposed a class I/II HDACi-mediated interaction with the Hsp70/90 chaperone axis causing increased ubiquitin-independent proteosomal degradation [54]. Even others suggest HIF translational inhibition through indirect interference with eukaryotic initiation factor-G3 (eIFG3) [34]. Clearly, a tremendous amount of interest has yielded multiple hypotheses for this complex mechanism. More evidence is needed to better understand SAHA-mediated hypoxia signaling suppression.
In the present study, we provide evidence suggesting that SAHA interferes with HIF signaling through direct acetylation of its associated chaperone, heat shock protein 90 (Hsp90). We propose this causes decreased HIF nuclear translocation mediated through decreased interaction of HIF with its nuclear karyopherin protein, Importin. Our findings were replicated across multiple tumor cells lines, and recapitulated in an in-vivo Hep3B HRE-Luc expressing xenograft. These data provide insight into the mechanism of action of the FDA-approved HDACi, SAHA, as well as identify Importin as a potential therapeutic target for treatment of hepatocellular carcinoma, and possibly other cancer sub-types characterized by aggressive hypoxia signaling.
RESULTS
Effects of SAHA on HIF-1 response to hypoxia
The effect of the type I/IIb/IV HDAC inhibitor SAHA on HIF expression was determined in multiple tumor-derived cell lines. In the hepatocellular carcinoma Hep3B cell line, exposure to low-dose (0.5 μM) and moderate-dose SAHA (1 μM) caused a decrease in the quantity of HIF-1α and HIF-2α under hypoxic conditions (Figure 1A). We evaluated the effect of SAHA on HIF-1/2α-associated transcriptional activation through a luciferase assay based on Hep3B cells carrying a stably transfected Hypoxia Responsive Element (HRE) luciferase reporter (Hep3B HRE-Luc). SAHA significantly reduced the transcriptional activity of HIF-1/2α under hypoxic conditions (p < 0.0001), while exerting minimal effect on HRE transcription under normal oxygen conditions (Figure 1B). In accordance with the reduction in the amount of HIF-1/2α HRE-reporter signaling, SAHA resulted in significant inhibition of hypoxia-responsive gene expression with downregulation of endothelin 1 (EDN1) (p < 0.0001), erythropoietin (EPO) (p < 0.001), glucose transporter 1 (GLUT1) (p < 0.0001), and vascular endothelial growth factor (VEGFA) (p < 0.0001). No significant change was observed in the mRNA expression levels of HIF1A or HIF2A in the same assay (Figure 1C). We confirmed the inhibitory effect of SAHA on the expression of HIF-1α and HIF-2α in glioblastoma (U87 MG) and osteosarcoma (U2OS and MG-63). The reduction of HIF-1/2α expression was similar among the conditions of low oxygen levels (1% O2) (Figure 1D), and in the presence of the hypoxia mimetic CoCl2 and oxoglutarate analog dimethyloxalylglycine (data not shown). This data suggests that SAHA suppresses HIF downstream transcriptional activity independent of a reduction in HIF mRNA levels.
Figure 1. SAHA suppresses HIF-1α and HIF-2α induction in response to hypoxia.
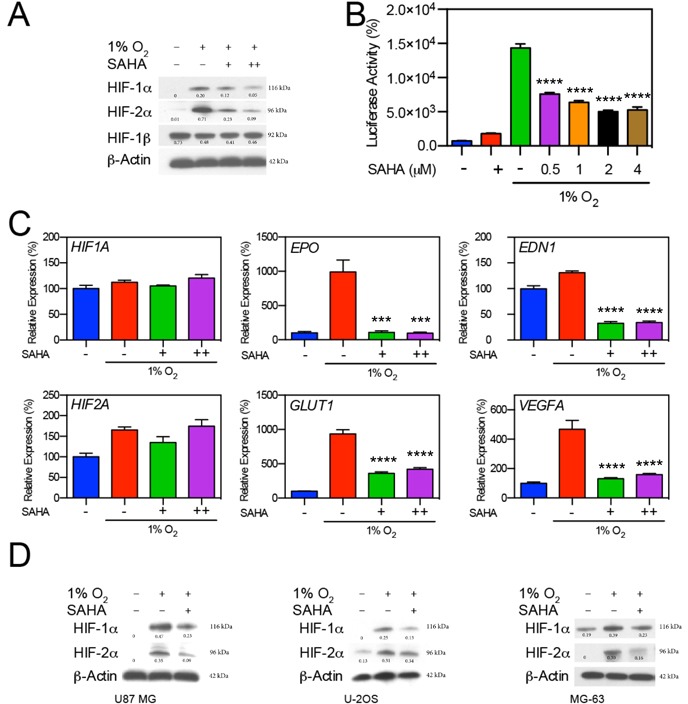
A. Hep3B cells were exposed to hypoxic conditions (1% O2) for 16 hr in the presence of 0.5 or 1 μM of SAHA (+, and ++, respectively). Representative western blot with normalized densitometric values (protein/actin loading control) show decreases in HIF-1α and HIF-2α expression upon exposure to SAHA. B. Luciferase reporter assay demonstrates significant decreases (p < 0.0001) in HRE-associated luciferase activity in response to SAHA under hypoxic conditions. C. Effects of SAHA on hypoxia related gene expression in Hep3B cells exposed to SAHA for 16 hr under conditions of 21% or 1% O2 analyzed by qRT-PCR, showing significant suppression of EDN1, EPO, GLUT1, and VEGFA in response to both 0.5 and 1 μM SAHA with no significant change in HIF1A or HIF2A expression. D. Tumor cell lines U87 MG, U2OS, and MG63 were exposed to 0.5 μM SAHA for 16 hr under 21% or 1% O2, with resulting HIF-1α and HIF-2α suppression similar to that observed in Hep3B cells. Representative western blot with normalized densitometric values (protein/actin loading control) are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
SAHA interferes with HIF-1/2α nuclear localization
To further investigate the underlying mechanisms of SAHA-mediated repression of HIF activity, we assessed HIF subcellular localization after incubation with SAHA. Notably, very low-dose SAHA treatment (0.1 mM) resulted in a slight decrease in total cellular HIF-1α levels in Hep3B cells (Figure 2A). A larger decrease in HIF-1α and HIF-2α concentrations were noted in the nuclear compartment after SAHA treatment (Figure 2B). Changes in HIF-expression were confirmed by luciferase assay demonstrating a significant reduction in HIF-associated signaling at very-low, moderate, and high SAHA concentrations (p < 0.01, p < 0.0001, p < 0.0001, respectively) (Figure 2C). Taken together, these findings suggest a SAHA-mediated hypoxia signaling interference contributing to out of proportion decreases in HIF nuclear concentrations and signaling relative to total cellular HIF levels.
Figure 2. SAHA interferes with nuclear translocation of HIF-1α and HIF-2α.
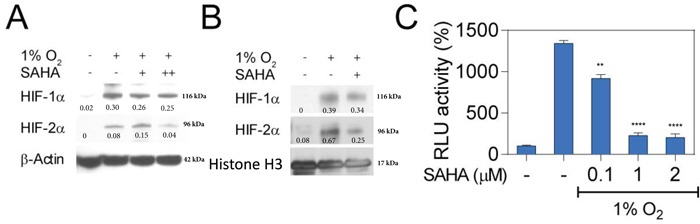
A. Hep3B cells were cultured under normoxic or hypoxic conditions for 16 hr. Representative western blot with normalized densitometric values (protein/actin loading control) show no change in total cellular HIF-1/2α expression at low concentrations of SAHA (0.1 μM, +), and moderate decrease at 2 μM (++). B. Representative western blot with normalized densitometric values (protein/histone loading control) show nuclear proteins isolated from Hep3B cells exposed to 0.1 μM (+) SAHA for 16 hr under indicated conditions with decreased nucleated HIF-1α and HIF-2α. C. Luciferase reporter assay demonstrates a significant decrease in HRE-associated luciferase activity in response to SAHA under hypoxic conditions. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Suppression of HIF by both HDACi and Hsp90 inhibition
The broad spectrum class I/IIb/IV HDAC inhibitor LB-205 [55, 56], and the Hsp90 inhibitor geldanamycin (GA) were tested and compared to SAHA in the setting of hypoxia to determine whether SAHA leads to HIF downregulation at least partly through inactivation of Hsp90, and/or whether SAHA mediates HIF downregulation in a manner conserved across class I/IIb/IV HDAC inhibitors. We found that SAHA, LB-205, and GA each showed varying but elevated suppression of HIF-1α and HIF-2α (Figure 3A). Each agent was also associated with a significant reduction in HIF-α-associated transcriptional activity (Figure 3B). The inhibitory HIF-signaling effects of LB-205 and GA were further evidenced by qRT-PCR demonstrating a significant suppression of downstream hypoxia signaling transcripts EDN1, VEGFA, GLUT1, and EPO (Figure 3C). The observed downstream inhibition was comparable to that seen after SAHA exposure (Figure 1C).
Figure 3. Effect of HDACi and Hsp90 inhibitor on HIFs expression.
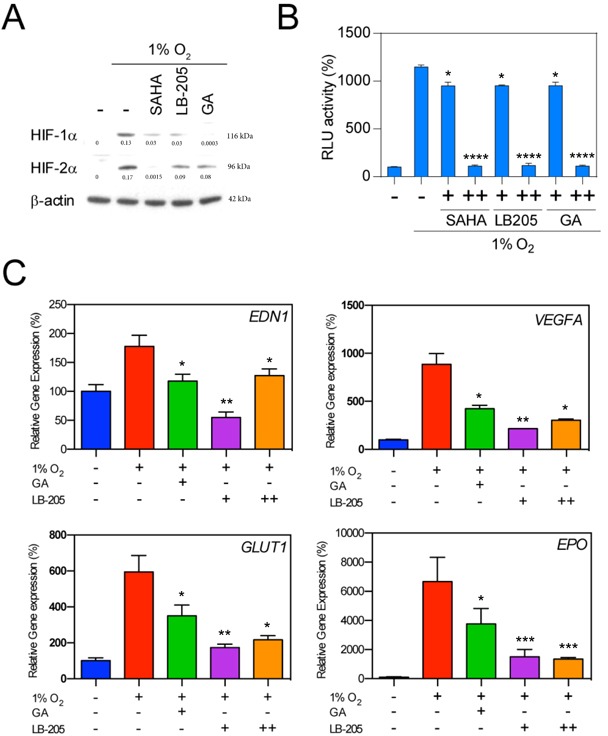
A. Hep3B cells were exposed to the HDACis SAHA (1 μM), LB205 (2 μM), and the Hsp90 inhibitor geldanamycin (GA, 2 μM) under hypoxic culture conditions. Representative western blot with normalized densitometric values (protein/actin loading control) demonstrate a decrease in HIF-1α and HIF-2α levels in all treatment conditions. B. Luciferase reporter assay demonstrates a significant decrease in HRE-associated luciferase activity in response to 0.1 and 2 μM SAHA (+ and ++, respectively), 1 and 5 μM LB-205 (+ and ++, respectively), and 1 μM and 2 μM geldanamycin (+ and ++, respectively). C. Hypoxia related genes were suppressed by 2 μM GA, and with 1 and 5 μM of LB-205 (+ and ++, respectively). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
SAHA decreases HIF-Hsp90 binding affinity
Hsp90 is a chaperone that functions to assist protein folding and stabilization. Minet et al., demonstrated that Hsp90 interacts with HIF-1α in normoxic and hypoxic conditions, contributing to HIF-1α activation and turnover [57]. Hsp90 activity is modulated therapeutically by HDACis through acetylation of its middle domain lysine residue [58]. Acetylated Hsp90 likely exhibits less affinity to its client protein, making it less able to function in protein folding and other chaperone relevant functions. Targeting Hsp90 through inhibition of HDAC6 results in accumulation of acetylated Hsp90 [59, 60]. We demonstrated through co-immunoprecipitation assay that moderate-dose SAHA decreases the affinity between HIF-1/2α and Hsp90 (Figure 4A and 4B), with an associated concomitant increase in the amount of acetylated Hsp90 (Figure 4C). To assess whether Hsp90 acetylation affects the quantity of HIF-1α and HIF-2α, we measured HIF-1/2α expression under hypoxic conditions in the presence of the Hsp90b recombinants K286Q, K286A, and D88N. These recombinants have shown in prior experiments the ability to simulate the activity of acetylated Hsp90 [61, 62]. We found that the quantity of HIF-1α and HIF-2α was decreased in the presence of the Hsp90β recombinants (Figure 4D). An HRE-luciferase assay was performed that confirmed a significant reduction in HIF-α associated downstream activity in the recombinant groups (p < 0.0001) (Supplementary Figure S1). We investigated the effect of SAHA on HIF-α nuclear translocation by studying the interaction between HIF-α and Importin, a karyopherin located on the nuclear membrane that facilitates HIF's nuclear transport. We found that in hypoxic conditions, HIF-1α and HIF-2α associate with Importin in Hep3B cells (Figure 4E and 4F). In the presence of SAHA, no interaction occurs between the two proteins (Figure 4E and 4F). This suggests that SAHA-mediated acetylation of Hsp90 leads to an attenuation of HIF-1/2α interaction with Importin (Figure 4E and 4F). We repeated the experiment in the presence and absence of the Hsp90 inhibitor GA, and similarly found that Hsp90 inactivation led to absent interaction between HIF-1/2α and Importin (Figure 4E and 4F). Together, these findings suggest that SAHA-mediated acetylation of Hsp90 affects Hsp90 function, contributing to decreased Hsp90-binding with HIF, and decreased/absent HIF binding to Importin. Given the function of Importin as a nuclear karyopherin, this suggests that SAHA interferes with HIF signaling at least partly by interfering with HIF nuclear translocation.
Figure 4. SAHA reduces the interaction between HIF-1/2α and Hsp90.
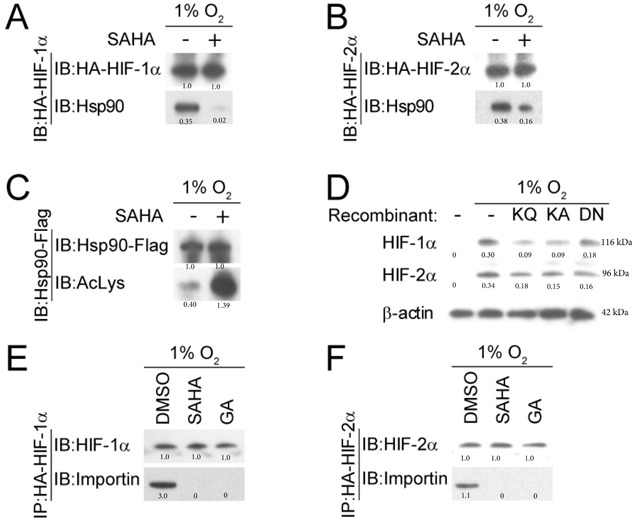
A., B. Representative western blot with normalized densitometric values (protein/HA-HIF loading control) after co-immunoprecipitation demonstrate diminished Hsp90 association with HIF-1α and HIF-2α in the presence of SAHA (1 μM). C. Hsp90 acetylation is increased by SAHA (1 μM), seen by immunoprecipitation with anti-Flag antibodies. Normalized densitometric values (protein/Hsp90-Flag loading control) are shown. D. Hsp90β recombinants that mimic acetylated (K286Q and K286A) and dominant-negative (D88N) forms of Hsp90β were cultured in Hep3B cells under hypoxic conditions, with resultant suppressed HIF-1α and HIF-2α expression compared to the WT Hsp90 (- recombinant) condition. Normalized densitometric values (protein/actin loading control) are shown. E., F. Co-immunoprecipitation and representative western blot shows reduced HIF-α-importin interaction in the presence of SAHA (1 μM) or GA (1 μM). Normalized densitometric values (protein/HIF loading control) are shown.
SAHA enhances HIF-1/2α ubiquitination and degradation
Under normoxic conditions, HIF-α is efficiently removed from the cytoplasm in a VHL-mediated E3 ubiquitin-ligase pathway involving the 26S proteasome [63]. We investigated HIF-1α and HIF-2α levels after giving SAHA in the presence/absence of MG-132, a 26S proteosome inhibitor that reverses pVHL ubiquitin-dependent degradation [63–67]. Consistent with our prior observations, the quantity of HIF-1/2α is reduced after moderate dose-SAHA therapy in hypoxic conditions. We found that HIF-1α and HIF-2α degradation was partly rescued by MG132, reaffirming the role of the 26S proteasome in SAHA-mediated HIF-1/2α degradation (Figure 5A). An immunoprecipitation assay was performed in the presence of SAHA to verify that SAHA increases HIF's association with ubiquitin, given ubiquitin's close functional role with the 26S proteasome, and given SAHA's ability to reverse 26S proteasome-mediated degradation of HIF. We found that HIF-ubiquitin association is enhanced in the presence of SAHA. To determine whether the observed effects were specific to SAHA or conserved across a similar class HDAC inhibitor, we replicated the experiment with LB-205. We found that LB-205 increases HIF ubiquitination to a similar degree as SAHA. These findings suggest that SAHA increases HIF degradation at least partly through a ubiquitin-dependent mechanism, in a manner likely conserved across I/IIb/IV HDAC inhibitors (Figure 5B-5C).
Figure 5. SAHA-induced degradation of HIF-1/2α is mediated by the ubiquitin and proteasome pathway.
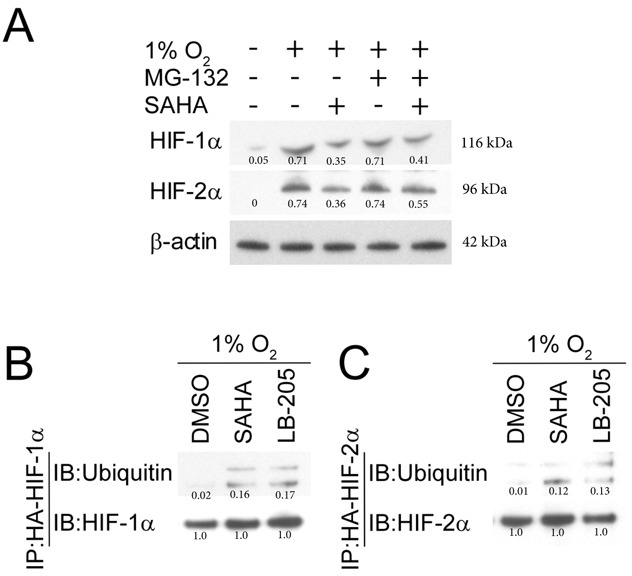
A. The proteasome inhibitor MG132 (5 μM) reverses SAHA (1 μM) mediated HIF-1α and HIF-2α downregulation in Hep3B cells exposed to hypoxic conditions for 8 hours. Normalized densitometric values (protein/actin loading control) are shown. B., C. Representative western blot with densitometric values after co-immunoprecipitation show exposure to SAHA (1 μM) or LB-205 (2 μM) resulting in increased HIF-ubiquitin association. Normalized densitometric values (protein/HIF loading control) are shown.
SAHA reduces tumor burden and hypoxia signaling in tumor xenografts
We used an HRE-Luc expressing Hep3B mouse xenograft model to validate SAHA's tumor-suppressive abilities against Hep3B, while also observing differences in real-time hypoxia signaling activity. Cells were validated in-vitro to report HIF-1/2α signal in pseudohypoxic (CoCl2) conditions (Figure 6A). Five million Hep3B HRE-Luc cells were subcutaneously injected in the right flank of SCID mice forming solid tumor masses within 10 days of implantation. Every other day (Q.O.D) administration of SAHA into the mouse peritoneum resulted in significant reductions of tumor volume and excision weight compared to non-treated controls (Figure 6B and 6C). We analyzed in-vivo hypoxia signaling activity at study endpoint, and found that mice receiving SAHA had diminished Hep3B HRE-Luc BLI signals (Figure 6D). Analysis of excised tumors confirmed that BLI signals originated from the tumors, with a significantly decreased average BLI vs control (p < 0.05) (Figure 6E). Intratumoral expression of the downstream hypoxia signaling mediators EPO and VEGFA were significantly decreased in Hep3B HRE-Luc xenografts (p < 0.01, p < 0.05, respectively) (Figure 6F and 6G). SAHA's tumor-suppressive effect was replicated in a pseudohypoxic 786-O renal cell carcinoma mouse xenograft model characterized by constitutive HIF signaling (even in the absence of hypoxia) due to absent pVHL protein, which is necessary for ubiquitin-dependent degradation of HIF [68, 69]. We found a significant reduction in 786-O tumor volume with SAHA therapy starting at day 8 post-injection, becoming more pronounced until the study endpoint (p < 0.05, p < 0.001, respectively) (Supplementary Figure S2).
Figure 6. SAHA suppressed hypoxia signaling in Hep3B cell xenografts in vivo.
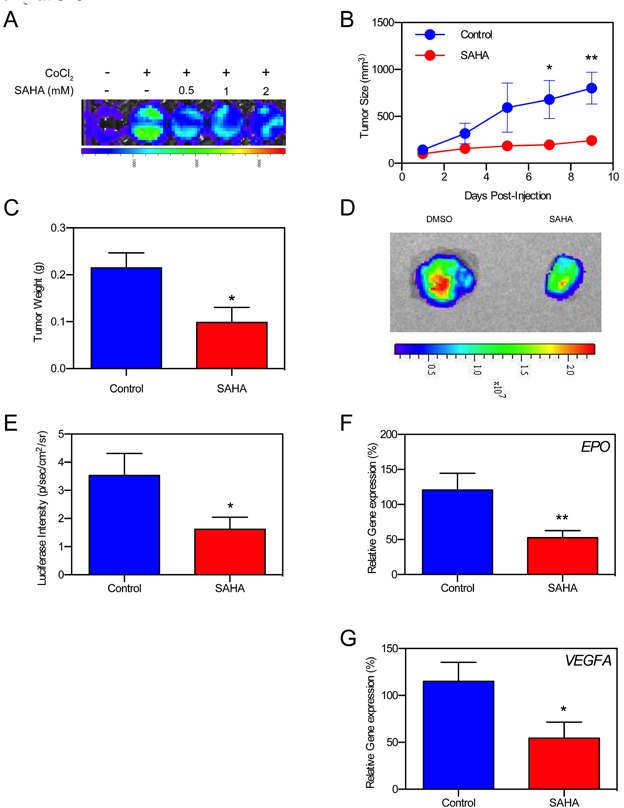
A. In-vitro luciferase assay shows decreased HRE activity in Hep3B HRE-Luc cells in the presence of SAHA. B. Tumor growth curves of Hep3B HRE-Luc cell xenografts treated with SAHA show significantly decreased tumor size at 7- and 9-days post-implantation (p < 0.05, p < 0.01, respectively). C. Average tumor weight of excised xenografts is significantly reduced in SAHA-treated mice compared to control mice (p < 0.05) at study endpoint. D. Example luminescence of excised Hep3B HRE-Luc cell xenografts at study endpoint. E. HRE-luc luminescence is significantly decreased in SAHA treated mice compared to control (p < 0.05) at study endpoint. F., G. VEGFA and EPO expression is significantly decreased in tumors resected from SAHA treated mice at study endpoint. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 7. HDACis interfere with hypoxia signaling by affecting Hsp90 acetylation and HIF-α nuclear translocation.
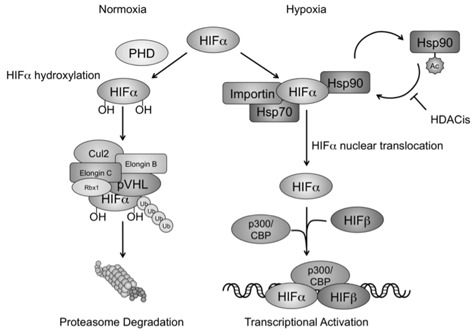
In normoxic conditions, HIF-1/2α are first modified by prolyl hydroxylase (PHD) for protein hydroxylation, and removed via the VHL associated proteasomal degradation pathway. In hypoxic conditions, HIF-α recruits Hsp70 and Hsp90 in the cytoplasm, and interacts with the karyopherin importin for nuclear translocation. Nucleated HIF-α further recruits other cofactors such as HIF-b and p300/CBP, and initiates gene transcription and hypoxia signaling. HDACis act to increase accumulation of the acetylated form of Hsp90. The reduction of Hsp90 chaperone activity and HIF-a recognition results in HIF-α nuclear translocation and transcriptional activation.
DISCUSSION
HDACis are an important class of chemotherapeutics that mediate much of their anti-neoplastic activity through suppression of the hypoxia signaling pathway. The FDA-approved class I/IIb/IV HDACi agent SAHA has shown efficacy in pre-cinical settings against a variety of cancer sub-types including pancreatic, breast, prostate, colon, and liver; as well as in clinical settings for the treatment of cutaneous T cell lymphoma [70]. However, its mechanism of HIF signaling suppression has yielded multiple hypotheses without a clear indication of relative influences. Much of the complexity results from different cell lines used across studies, and from results obtained from alternate-class HDACis grouped together with SAHA. We sought to clarify the mechanism of SAHA-mediated hypoxia suppression using a variety of different cell lines, as well as an in-vivo model capable of demonstrating real-time hypoxia signaling. While our findings support many of the conclusions obtained from several other studies, they also support a novel mechanism of SAHA-mediated hypoxia suppression. Our study demonstrates that SAHA disrupts Hsp90 function through direct Hsp90 acetylation, and contributes to decreased HIF-Hsp90 affinity, diminished HIF-Importin interaction, and decreased intranuclear HIF levels. These findings suggest a mechanism of SAHA-mediated hypoxia suppression involving interference with HIF's ability to translocate the nuclear membrane.
Studies have shown that elevated HIF-1 activity stimulates malignant transformation by promoting angiogenesis, epithelial-mesenchymal transition, invasion, metastasis, and resistance to traditional chemotherapy agents [1–3]. SAHA is implicated in interfering with HIF-1α signaling at the post-translational level through class I and class IIb HDAC inhibition [32, 42]. We found that SAHA administration at low- and moderate-doses is associated with decreased expression of HIF-1α and HIF-2α without a corresponding decrease in HIF-1α or HIF-2α mRNA across a variety of cell types (Figure 1A-1C). These findings support prior studies that demonstrated HIF-1α downregulation after administration of SAHA [17, 35, 38]. Kong attributed HIF-1α downregulation after SAHA administration to disruption of the Hsp70/90 chaperone axis [54]. We compared HIF levels after administration of SAHA, with administration of the Hsp90 inhibitor GA, and the Class I/IIb/IV HDACi LB-205. We observed a similar decrease in HIF-1α and HIF-2α expression in each treatment group, suggesting that SAHA-mediated downregulation of HIF is largely attributable to dysfunction of Hsp90. GA and LB-205 similarly decreased HRE-activity, and downstream hypoxia transcript expression (Figure 3).
Previous studies have shown that the class IIb HDAC6 directly acetylates the middle domain lysine residue of Hsp90, rendering it largely non-functional [58–60]. Our findings support moderate-dose SAHA leading to Hsp90 acetylation, Hsp90 dysfunction, and increased HIF degradation. We observed with exposure to SAHA a larger precipitant composed of Hsp90 and anti-acetylated lysine compared to hypoxia-only controls (Figure 4C), a smaller precipitant composed of HIF-1/2α and Hsp90 than hypoxia-only controls (Figure 4A-4B), and decreased HIF-1α and HIF-2α levels in cells with recombinant acetyl-Hsp90 mimetics relative to Hsp90-only controls (Figure 4D). WT Hsp90 Hep3B HRE-luc cells additionally demonstrated significantly increased HRE activity during hypoxic conditions than Hep3B HRE-luc cells with acetyl-Hsp90 recombinants in the presence of hypoxia (p < 0.0001) (Supplementary Figure S1).
Reports are mixed regarding the manner in which HIF-1α undergoes degradation in response to SAHA. Whereas Kong described SAHA-mediated HIF-1α degradation occuring in a ubiquitin-independent proteosomal manner [54], other groups proposed HIF degradation due to a ubiquitin-dependent proteosomal process resulting from direct HIF acetylation [38, 49, 50]. Our findings support SAHA-mediated HIF degradation occurring through increased HIF ubiquitination. We observed Hep3B cells exposed to hypoxic conditions+SAHA+MG132 to have elevated levels of HIF compared to hypoxia+SAHA only (Figure 5A). We also found an increased HIF-ubiquitin aggregate in Hep3B cells exposed SAHA+hypoxia relative to hypoxia-alone (Figure 5B-5C). SAHA-associated HIF degradation might be generalizable to class I/IIb/IV HDACis, as we found a similar degree of HIF-ubiquitin association with LB-205 as with SAHA.
Hsp90 was investigated in this study since it is a known target of SAHA that is heavily involved in HIF-1/2a signaling. SAHA decreases tumor neovascularization, causes abnormal tumor vascular morphology, and interferes with endothelial cell migration, proliferation, and survival at least partly due to decreased hypoxia signaling [71–74]. Pharmacologic inhibition of Hsp90 with Geldanamycin demonstrates similar in-vitro and in-vivo findings also through downregulation of the hypoxia signaling cascade [75–77]. Conditional knockout models of Hsp90 could provide another degree of validation regarding SAHA-mediated hypoxia signaling interference occurring through Hsp90 impairment. Conventional Hsp90 knockout models are generally non-viable in eukaryotic cells. The role of Hsp70 or the other Heat Shock proteins on HIF signaling after SAHA exposure could also be investigated. Hsp70 in particular has been implicated in altering HIF signaling after Hsp90 inactivation [78–80]. However, it not a direct target of SAHA, and likely plays an indirect role, if at all, during SAHA exposure [81]. Our study found absent HIF-Importin association after SAHA exposure (Figure 4E-4F), which was out of proportion to the degree of HIF downregulation seen after exposure to either hypoxia+acetyl-Hsp90 mimetics (Figure 4D) or hypoxia+SAHA-alone (Figure 2A-2B). If other HSP proteins are involved in permitting HIF signaling during SAHA-mediated Hsp90 inactivation, at least some HIF-Importin association would likely have been seen.
Future studies could perform reciprocal immunoprecipitation of Hsp90/HIF/Importin to determine whether they form a tertiary complex near the nuclear membrane. Currently, the molecular arrangement of these molecules is unknown. Our in-vivo HCC xenograft demonstrated significantly decreased HRE-luciferase intensity (p < 0.05) (Figure 6D-6E), and significantly less intratumoral EPO and VEGFA expression (p < 0.01 and p < 0.05, respectively) at the study endpoint (Figure 6E). Other studies could focus on this aspect and perform short-interval intratumoral RT-PCR of hypoxia signaling transcripts and/or imaging of HRE-luciferase activity to assess hypoxia signaling differences as the tumors grow in size. Real-time tumor oxygenation/perfusion could also be investigated using PtO2 polarographic electrodes, or via non-invasive imaging methods such as MRI-perfusion. MRI with contrast could possibly be used to identify whether changes in intratumoral vessel permeability occur over time.
In summary, we report that the HDAC inhibitor SAHA regulates hypoxia signaling by directly acetylating Hsp90 causing an increase in ubiquitin-mediated HIF degradation, and an attenuation of HIF-Importin association. In-vitro findings were recapitulated in an in-vivo model that demonstrated diminished hypoxia signaling in animals exposed to SAHA monotherapy. SAHA additionally exhibited potent in-vivo tumor suppressive activity against Hep3B and 786-O xenografts. We believe these findings highlight the role of Importin in SAHA-mediated hypoxia signaling suppression, and provide preclinical support for testing SAHA in clinical trials for HCC.
MATERIALS AND METHODS
Cell culture
Hep3B, HepG2, U2OS, MG63 and U87 MG cells were purchased from American Type Culture Collection (ATCC, Manassas, Virginia, USA) and cultured according to manufacturer's instructions. Hypoxia (1% O2, 5% CO2) conditions were established using an oxygen station (In VIVO2; Baker Ruskinn Tech, Stanford, Maine, USA). The Hep3B HRE-Luc cell line was established by infecting Hep3B cells with Cignal Lenti HIF Reporter (Luc) lentivirus (QIAGEN, Venlo, Limburg). Cells were selected in the presence of 5 mg/mL puromycin (Sigma, St. Louis, Missouri, USA). The luciferase signal was determined spectrophotometrically (Molecular Devices; Sunnyvale, CA).
Quantitative PCR
Total RNA was extracted from cell pellets using the RNeasy Mini Kit (QIAGEN). Genomic DNA was removed through in-column DNaseI digestion (QIAGEN). cDNA was reverse-transcribed using Super Script III First-Strand Synthesis SuperMix (Invitrogen, Carlsbad, California, USA). The cDNA products were analyzed on an Eco Real-Time PCR System (Illumina, San Diego, California, USA). The primer sets used in the present study were: EDN1 (Origene HP205717), EPO (Origene HP200740), GLUT1 (Origene HP209446), VEGFA (QIAGEN QT01682072), and ACTB (Promega G5740). Data analysis was performed using Microsoft Excel and GraphPad Prism (version 6.0d, San Diego, California, USA).
Western blot analysis
Cell pellets were harvested and lysed in RIPA lysis buffer supplemented with Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and 0.5% SDS. Protein was quantified using the Bio-Rad Protein Assay Kit (Bio-Rad, Berkeley, California). Samples were separated by NuPAGE Bis-Tris 4–12% gel (Invitrogen, Carlsbad California) and transferred onto PVDF membranes (Millipore, Billerica, Massachusetts, USA). The membranes were probed with primary antibody and detected through an HRP-conjugated species-specific secondary antibody and an ECL kit (Pierce, Waltham, Massachusetts, USA). The primary antibodies used in this study include: antibodies against HIF-1α [82–85], HIF-2α (Novus, Littleton, Colorado, USA), ubiquitin (Abcam, Cambridge, UK), Hsp90 (Cell Signaling Technology, Danvers, Massachusetts, USA), acetyl-lysine (Millipore, Billerica, Massachusetts, USA) and β-Actin (Santa Cruz Biotechnology, Santa Cru.z, California, USA). Antibodies were chosen based on existence of thorough external validation and widespread use in other well-respected peer-reviewed journals. Three independent experiments were performed for each representative immunoblot. Normalized densitometric values (protein/loading control) were calculated and displayed.
DNA cloning and site-directed mutagenesis
Human HSP90AB1 gene was incorporated into the pCMV6-entry vector (Origene) as previously described [61, 62]. Mutagenesis of the K286 site was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). The nucleotide sequence of the HSP90AB1 gene was verified by analyzing the entire coding regions through Sanger sequencing. The HSP90AB1 dominant negative vector (Hsp90β-D88N) was a gift from William Sessa (Department of Pharmacology and Vascular Biology and Therapeutics Program, Boyer Center for Molecular Medicine, Yale University School of Medicine, New Haven, CT).
Immunoprecipitation
Immunoprecipitation was performed per manufacturer's instructions. Cell pellets were lysed in RIPA buffer with Protease Inhibitor Cocktail and 0.5% SDS. Total cell lysate was precipitated using the DynaBeads Protein G Immunoprecipitation Kit (Invitrogen) and antibodies against FLAG-tag (Origene, Rockville, Maryland, USA) or HA (Sigma-Aldrich, St. Louis, Missouri, USA). Precipitated protein was eluted and resolved by Western blot analysis. Three independent experiments were performed for each representative immunoblot provided in the figures. Normalized densitometric values (protein/loading control) were obtained and displayed.
In vivo studies
Animal experiments were approved for use and care of animals under the guidelines of NIH animal protocol (1349-13). Six to eight-week-old female nude athymic mice (nu/nu) were obtained, with each mouse weighing approximately 20 grams at the onset of the experiment. Mice were injected subcutaneously in right flank with 5×106 Hep3B HRE-Luc cells. After the xenografts reached 0.5 cm (day 1), animals were randomized to 2 groups of 3 animals each. Animals were treated intraperitoneally with vehicle alone or with SAHA at a dose of 5mg/kg on days 1, 3, 5, 7 and 9. Tumors were measured every other day using calipers. Tumor volumes were calculated from 2-dimensional measurements using the formula (tumor volume=length×width2). The animals were imaged at day 9 post-xenograft by bioluminescence imaging (BLI). Mice were anesthetized with isoflurane for imaging, and euthanized using CO2 inhalation or cervical dislocation. Tumors from the mice were imaged after excision, and expression profiles were assessed for HIFs downstream gene expression by qPCR using the above-described RT-PCR protocol.
SUPPLEMENTARY MATERIALS FIGURES
Acknowledgments
This work was funded by the Intramural Research Program at the National Institute of Neurological Disorders and Stroke and the National Institute for Child Health and Human Development at the National Institutes of Health (NIH) and the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer, Inc., The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities, Inc. and Mr. and Mrs. Joel S. Marcus and the Howard Hughes Medical Institute, as well as other private donors. For a complete list, please visit the Foundation at: http://fnih.org/work/education-training-0/medical-research-scholarsprogram.
Footnotes
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
- 1.Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102:789–95. doi: 10.1038/sj.bjc.6605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fer N, Melillo G. The HIF-1alpha-c-Myc pathway and tumorigenesis: evading the apoptotic gate-keeper. Cell Cycle. 2011;10:3228. doi: 10.4161/cc.10.19.17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warfel NA, El-Deiry WS. HIF-1 signaling in drug resistance to chemotherapy. Curr Med Chem. 2014;21:3021–8. doi: 10.2174/0929867321666140414101056. [DOI] [PubMed] [Google Scholar]
- 5.Doublier S, Belisario DC, Polimeni M, Annaratone L, Riganti C, Allia E, Ghigo D, Bosia A, Sapino A. HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: a potential model of the chemo-resistance of invasive micropapillary carcinoma of the breast. BMC Cancer. 2012;12:4. doi: 10.1186/1471-2407-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–9. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–76. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 8.Zhao CX, Luo CL, Wu XH. Hypoxia promotes 786-O cells invasiveness and resistance to sorafenib via HIF-2alpha/COX-2. Med Oncol. 2015;32:419. doi: 10.1007/s12032-014-0419-4. [DOI] [PubMed] [Google Scholar]
- 9.Qing G, Simon MC. Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev. 2009;19:60–6. doi: 10.1016/j.gde.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin R, Smyrk TC, Reed NR, Schmidt RL, Schnelldorfer T, Chari ST, Petersen GM, Tang AH. Combining clinicopathological predictors and molecular biomarkers in the oncogenic K-RAS/Ki67/HIF-1alpha pathway to predict survival in resectable pancreatic cancer. Br J Cancer. 2015;112:514–22. doi: 10.1038/bjc.2014.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13:2780–6. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology (Williston Park) 2005;19:7–16. [PubMed] [Google Scholar]
- 14.Saltz LB, Lenz HJ, Kindler HL, Hochster HS, Wadler S, Hoff PM, Kemeny NE, Hollywood EM, Gonen M, Quinones M, Morse M, Chen HX. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25:4557–61. doi: 10.1200/JCO.2007.12.0949. [DOI] [PubMed] [Google Scholar]
- 15.Shojaei F, Ferrara N. Antiangiogenic therapy for cancer: an update. Cancer J. 2007;13:345–8. doi: 10.1097/PPO.0b013e31815a7b69. [DOI] [PubMed] [Google Scholar]
- 16.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–88. [PubMed] [Google Scholar]
- 18.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 19.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 20.Mwakwari SC, Patil V, Guerrant W, Oyelere AK. Macrocyclic histone deacetylase inhibitors. Curr Top Med Chem. 2010;10:1423–40. doi: 10.2174/156802610792232079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–91. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 22.Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 23.Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Bates SE. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10:997–1008. doi: 10.1586/era.10.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turtoi A, Peixoto P, Castronovo V, Bellahcene A. Histone deacetylases and cancer-associated angiogenesis: current understanding of the biology and clinical perspectives. Crit Rev Oncog. 2015;20:119–37. doi: 10.1615/critrevoncog.2014012423. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidanes GM, Bonilla CY, Toczyski DP. Complicated tails: histone modifications and the DNA damage response. Cell. 2005;121:973–6. doi: 10.1016/j.cell.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Polo SE, Almouzni G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 2005;220:1–9. doi: 10.1016/j.canlet.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 29.Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–81. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 31.Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X, Chen X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013;32:599–609. doi: 10.1038/onc.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 33.Castronovo V, Peixoto P, Bellahcene A, Turtoi A. Histone deacetylases and cancer-associated angiogenesis: current understanding of the biology and clinical perspectives. Crit Rev Oncog. 2015;20:119–37. doi: 10.1615/critrevoncog.2014012423. 1dc3cf1839473109,021d61097174634a. [DOI] [PubMed] [Google Scholar]
- 34.Hutt DM, Roth DM, Vignaud H, Cullin C, Bouchecareilh M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS One. 2014;9:e106224. doi: 10.1371/journal.pone.0106224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang C, Huntoon K, Ksendzovsky A, Zhuang Z, Lonser RR. Proteostasis modulators prolong missense VHL protein activity and halt tumor progression. Cell Rep. 2013;3:52–9. doi: 10.1016/j.celrep.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV, Castronovo V. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21:427–36. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 37.Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U, Zeiher AM, Dimmeler S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–44. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 38.Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 39.Michaelis M, Michaelis UR, Fleming I, Suhan T, Cinatl J, Blaheta RA, Hoffmann K, Kotchetkov R, Busse R, Nau H, Cinatl J., Jr Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol. 2004;65:520–7. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 40.Patenaude A, Fuller M, Chang L, Wong F, Paliouras G, Shaw R, Kyle AH, Umlandt P, Baker JH, Diaz E, Tong J, Minchinton AI, Karsan A. Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res. 2014;74:2402–11. doi: 10.1158/0008-5472.CAN-12-4038. [DOI] [PubMed] [Google Scholar]
- 41.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–34. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 42.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 43.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–92. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–58. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–63. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MH, Zimmermann B, Brandes RP, Mione M, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–8. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lian ZR, Xu YF, Wang XB, Gong JP, Liu ZJ. Suppression of histone deacetylase 11 promotes expression of IL-10 in Kupffer cells and induces tolerance following orthotopic liver transplantation in rats. J Surg Res. 2012;174:359–68. doi: 10.1016/j.jss.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 48.Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, Younes A. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood. 2011;117:2910–7. doi: 10.1182/blood-2010-08-303701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–20. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 50.Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem. 2011;286:38095–102. doi: 10.1074/jbc.M111.257055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 2005;579:6428–32. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray-Rust TA, Oldham NJ, Hewitson KS, Schofield CJ. Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS Lett. 2006;580:1911–8. doi: 10.1016/j.febslet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–93. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brady RO, Yang C, Zhuang Z. An innovative approach to the treatment of Gaucher disease and possibly other metabolic disorders of the brain. J Inherit Metab Dis. 2013;36:451–4. doi: 10.1007/s10545-012-9515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu J, Yang C, Chen M, Ye DY, Lonser RR, Brady RO, Zhuang Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci U S A. 2011;108:21200–5. doi: 10.1073/pnas.1119181109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett. 1999;460:251–6. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 58.Yang Y, Rao R, Shen J, Tang Y, Fiskus W, Nechtman J, Atadja P, Bhalla K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008;68:4833–42. doi: 10.1158/0008-5472.CAN-08-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aoyagi S, Archer TK. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005;15:565–7. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 60.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 61.Yang C, Rahimpour S, Lu J, Pacak K, Ikejiri B, Brady RO, Zhuang Z. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci U S A. 2013;110:966–71. doi: 10.1073/pnas.1221046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miao RQ, Fontana J, Fulton D, Lin MI, Harrison KD, Sessa WC. Dominant-negative Hsp90 reduces VEGF-stimulated nitric oxide release and migration in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:105–11. doi: 10.1161/ATVBAHA.107.155499. [DOI] [PubMed] [Google Scholar]
- 63.Groulx I, Lee S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 2002;22:5319–36. doi: 10.1128/MCB.22.15.5319-5336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD, Pause A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci U S A. 1999;96:12436–41. doi: 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonicalzi ME, Groulx I, de Paulsen N, Lee S. Role of exon 2-encoded beta -domain of the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2001;276:1407–16. doi: 10.1074/jbc.M008295200. [DOI] [PubMed] [Google Scholar]
- 66.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 67.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–7. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 68.Corn PG, McDonald ER, 3rd, Herman JG, El-Deiry WS. Tat-binding protein-1, a component of the 26S proteasome, contributes to the E3 ubiquitin ligase function of the von Hippel-Lindau protein. Nat Genet. 2003;35:229–37. doi: 10.1038/ng1254. [DOI] [PubMed] [Google Scholar]
- 69.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93:10595–9. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4:505–24. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ugur HC, Ramakrishna N, Bello L, Menon LG, Kim SK, Black PM, Carroll RS. Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J Neurooncol. 2007;83:267–75. doi: 10.1007/s11060-007-9337-z. [DOI] [PubMed] [Google Scholar]
- 72.Yang B, Yu D, Liu J, Yang K, Wu G, Liu H. Antitumor activity of SAHA, a novel histone deacetylase inhibitor, against murine B cell lymphoma A20 cells in vitro and in vivo. Tumour Biol. 2015;36:5051–61. doi: 10.1007/s13277-015-3156-1. [DOI] [PubMed] [Google Scholar]
- 73.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou H, Jiang S, Chen J, Su SB. Suberoylanilide hydroxamic acid suppresses inflammation-induced neovascularization. Can J Physiol Pharmacol. 2014;92:879–85. doi: 10.1139/cjpp-2014-0117. [DOI] [PubMed] [Google Scholar]
- 75.de Candia P, Solit DB, Giri D, Brogi E, Siegel PM, Olshen AB, Muller WJ, Rosen N, Benezra R. Angiogenesis impairment in Id-deficient mice cooperates with an Hsp90 inhibitor to completely suppress HER2/neu-dependent breast tumors. Proc Natl Acad Sci U S A. 2003;100:12337–42. doi: 10.1073/pnas.2031337100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lang SA, Klein D, Moser C, Gaumann A, Glockzin G, Dahlke MH, Dietmaier W, Bolder U, Schlitt HJ, Geissler EK, Stoeltzing O. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther. 2007;6:1123–32. doi: 10.1158/1535-7163.MCT-06-0628. [DOI] [PubMed] [Google Scholar]
- 77.Kaur G, Belotti D, Burger AM, Fisher-Nielson K, Borsotti P, Riccardi E, Thillainathan J, Hollingshead M, Sausville EA, Giavazzi R. Antiangiogenic properties of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin: an orally bioavailable heat shock protein 90 modulator. Clin Cancer Res. 2004;10:4813–21. doi: 10.1158/1078-0432.CCR-03-0795. [DOI] [PubMed] [Google Scholar]
- 78.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25:207–17. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem. 2010;285:3651–63. doi: 10.1074/jbc.M109.068577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ehrlich ES, Wang T, Luo K, Xiao Z, Niewiadomska AM, Martinez T, Xu W, Neckers L, Yu XF. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2009;106:20330–5. doi: 10.1073/pnas.0810571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Emanuele S, Lauricella M, Carlisi D, Vassallo B, D’Anneo A, Di Fazio P, Vento R, Tesoriere G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor Bortezomib. Apoptosis. 2007;12:1327–38. doi: 10.1007/s10495-007-0063-y. [DOI] [PubMed] [Google Scholar]
- 82.Ridiandries A, Bursill C, Tan J. Broad-Spectrum Inhibition of the CC-Chemokine Class Improves Wound Healing and Wound Angiogenesis. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18010155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sobhan PK, Zhai Q, Green LC, Hansford LM, Funa K. ASK1 regulates the survival of neuroblastoma cells by interacting with TLX and stabilizing HIF-1alpha. Cell Signal. 2017;30:104–17. doi: 10.1016/j.cellsig.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 84.Yoon C, Chang KK, Lee JH, Tap WD, Hart CP, Simon MC, Yoon SS. Multimodal targeting of tumor vasculature and cancer stem-like cells in sarcomas with VEGF-A inhibition, HIF-1alpha inhibition, and hypoxia-activated chemotherapy. Oncotarget. 2016;7:42844–58. doi: 10.18632/oncotarget.10212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Mori H, Yao Y, Learman BS, Kurozumi K, Ishida J, Ramakrishnan SK, Overmyer KA, Xue X, Cawthorn WP, Reid MA, Taylor M, Ning X, Shah YM, et al. Induction of WNT11 by hypoxia and hypoxia-inducible factor-1alpha regulates cell proliferation, migration and invasion. Sci Rep. 2016;6:21520. doi: 10.1038/srep21520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.