

ABSTRACT
Patients with primary solid malignancies frequently exhibit signs of systemic inflammation. Notably, elevated levels of neutrophils and their associated soluble mediators are regularly observed in cancer patients, and correlate with reduced survival and increased metastasis formation. Recently, we demonstrated a mechanistic link between mammary tumor-induced IL17-producing γδ T cells, systemic expansion of immunosuppressive neutrophils and metastasis formation in a genetically engineered mouse model for invasive breast cancer. How tumors orchestrate this systemic inflammatory cascade to facilitate dissemination remains unclear. Here we show that activation of this cascade relies on CCL2-mediated induction of IL1β in tumor-associated macrophages. In line with these findings, expression of CCL2 positively correlates with IL1Β and macrophage markers in human breast tumors. We demonstrate that blockade of CCL2 in mammary tumor-bearing mice results in reduced IL17 production by γδ T cells, decreased neutrophil expansion and enhanced CD8+ T cell activity. These results highlight a new role for CCL2 in facilitating the breast cancer-induced pro-metastatic systemic inflammatory γδ T cell – IL17 – neutrophil axis.
KEYWORDS: Breast cancer, CCL2, immunosuppression, neutrophils, tumor-induced inflammation, tumor-associated macrophages, γδ T cells
Introduction
Over 90% of breast cancer deaths are due to complications as a consequence of metastasis.1 There is an urge for the identification of new therapeutic targets through a better understanding of the molecular mechanisms underlying breast cancer metastasis formation. Emerging evidence indicates that metastasis is regulated to a great extent by reciprocal interactions between cancer cells and immune cells in the tumor microenvironment.2,3 In addition to a local inflammatory microenvironment, tumors frequently induce a systemic inflammatory state in distant organs through the release of various mediators that mobilize and activate immune cells to support metastasis.2,4 As such, systemic inflammation represents an interesting target to prevent and/or to treat metastatic disease.
Previously, were ported that neutrophils exert pro-metastatic functions by suppressing anti-tumor CD8+ T cells in the K14cre;Cdh1F/F;Trp53F/F (KEP) conditional mouse model of invasive breast cancer.5 The systemic expansion and polarization of these immunosuppressive neutrophils is elicited by tumor-associated macrophage (TAM)-derived interleukin (IL)1β that activates IL17-producing γδ T cells leading to increased systemic levels of G-CSF, a cytokine known for its role in granulopoiesis.6 However, the mediators that initiate this systemic inflammatory cascade from the primary tumor are unknown.
In the current study, we identify the pro-inflammatory chemokine (C-C motif) ligand 2 (CCL2) as an important mammary tumor-derived factor that stimulates the γδ T cell – IL17 – neutrophil axis. CCL2 is a cytokine largely known for its involvement in the recruitment of CCR2+ monocytes from the bone marrow to other sites in the body where they differentiate into macrophages.7 In breast cancer patients, high CCL2 expression is linked to macrophage infiltration and poor prognosis.8,9 Here, we show that CCL2 initiates the γδ T cell – IL17 – neutrophil axis by promoting the expression of TAM-derived IL1β. In human breast cancers, CCL2 expression is positively correlated with IL1Β and macrophage marker CD68 across all breast cancer subtypes, supporting our findings that these two cytokines are co-dependent. In vivo blockade of CCL2 in mammary tumor-bearing KEP mice results in reduced IL1β expression in tumor-associated macrophages and reduced IL17-producing γδ T cells, impaired expansion of immunosuppressive neutrophils and activation of CD8+ T cells. These data identify CCL2 as a key regulator of the mammary tumor-induced immunosuppressive systemic inflammatory γδ T cell – IL17 – neutrophil axis that drives metastasis.
Results
Mammary tumor-bearing K14cre;Cdh1F/F;Trp53F/F (KEP) mice show elevated intratumoral and systemic CCL2 levels
Previously, we analyzed the expression profile of a panel of cytokines and chemokines in KEP mammary tumors and mammary glands from wild-type mice.5 Among these molecules, CCL2 was the most upregulated cytokine in KEP tumor tissue (Fig. 1A). We also found increased CCL2 serum levels in mammary tumor-bearing KEP mice compared to wild-type littermates (Fig. 1B). RNA in situ hybridization analysis showed that Ccl2 mRNA in KEP mammary tumors is expressed in both stromal cells and tumor cells (Fig. 1C). Ccl2 expression in wild-type mammary glands was almost undetectable (Fig. 1C). Gene expression analysis on sorted cell populations from KEP tumors revealed that many cell types express Ccl2 (Fig. 1D), but due to their high abundance in KEP tumors, macrophages and tumor cells comprise the main cellular source (Fig. 1E).
Figure 1.
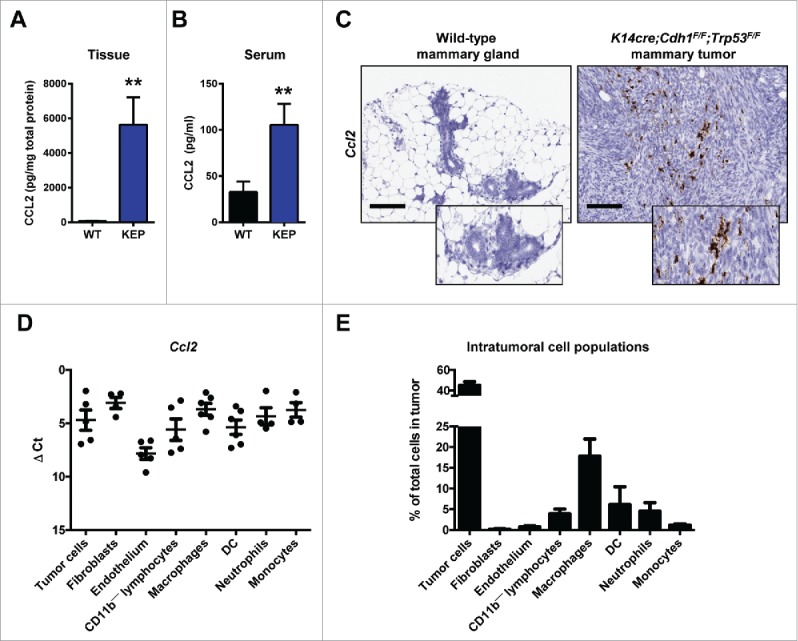
CCL2 expression in mammary tumor-bearing K14cre;Cdh1F/F;Trp53F/(F)(KEP) mice. (A) Protein expression of CCL2 in KEP mammary tumors compared to wild-type mammary glands was determined using a Luminex multiplex cytokine array (n = 5 per group; Mann-Whitney U test). (B) Serum levels of CCL2 in wild-type mice and mammary tumor-bearing KEP mice (n = 6 per group; Mann-Whitney U test). (A-B) are determined by a Luminex-based cytokine array. (C) RNA in situ hybridization of Ccl2 mRNA in wild-type mammary gland (left) and KEP mammary tumors (right). Representative images are shown. Scale bar 100 μm. (D, E) Tumor cells (CD31—CD45—CD11b—), lymphocytes (CD45+CD11b—), fibroblasts (PDGFRβ+CD31—CD45—CD11b—), endothelial cells (CD31+CD45—CD11b—), macrophages (CD11b+F4/80+), dendritic cells (DC) (CD11b+F4/80—CD11c+), neutrophils (CD11b+F4/80—Ly6G+Ly6Clo) and monocytes (CD11b+F4/80—Ly6G—Ly6Chi) were isolated from KEP mammary tumors using FACS (n = 6 per group). D) Ccl2 gene expression was determined by quantitative RT-PCR and corrected for β-actin. E) Quantification of intratumoral cell populations by flow cytometry. (** p < 0.01). All data are mean ± s.e.m.
CCL2 influences breast cancer metastasis
Several studies report a pro-metastatic role for CCL2 in breast cancer by recruiting monocytes and macrophages to primary tumors and metastatic sites.10-12 To determine the functional significance of CCL2 during metastasis in the KEP model, we used our previously described KEP-based model of spontaneous breast cancer metastasis.5,13 Mice bearing orthotopically transplanted KEP mammary tumors were treated with anti-CCL2 in a neo-adjuvant and adjuvant setting (Fig. 2A). Neo-adjuvant CCL2 blockade did not affect primary tumor growth (Fig. 2B), but increased the metastatic burden in the lungs (Fig. 2C) without affecting metastasis-related survival (Fig. 2D). These data corroborate previous findings that cessation of CCL2 blockade can enhance metastasis due to a cytokine rebound effect.14 To circumvent this undesirable effect, adjuvant treatment was initiated after surgical removal of the primary tumor and continued until animals were sacrificed due to clinical signs of metastatic disease. Conversely to our previous findings, adjuvant CCL2 blockade reduced metastatic burden in the lung (Fig. 2E), albeit not statistically significant, and resulted in a modest survival benefit (Fig. 2F). Likewise, CCL2 blockade in KEP mice bearing spontaneously arising mammary tumors also resulted in decreased pulmonary metastases (Fig. 2H), without affecting primary tumor growth (Fig. 2G). Together, the data generated in both the transplantation-based metastasis model and the genetically engineered KEP model, suggest that CCL2 functions as a pro-metastatic cytokine. However, these data also emphasize the complexity of targeting CCL2 in breast cancer metastasis, as described by others.14,15
Figure 2.

Effect of neo-adjuvant and adjuvant CCL2 blockade on spontaneous breast cancer metastasis. (A) Schematic representation of the KEP-based mouse model for spontaneous breast cancer metastasis treated with neo-adjuvant or adjuvant anti-CCL2. (B) Primary tumor growth kinetics upon neo-adjuvant CCL2 blockade (n = 15 per group). (C) Quantification of lung metastatic nodules in mice treated with neo-adjuvant anti-CCL2 (n = 8) versus controls (n = 11) that succumb due to respiratory distress. (*p < 0.05, Mann-Whitney U test). (D) Metastasis-related survival of mice treated with neo-adjuvant anti-CCL2 versus controls (n = 15 per group). Animals that succumb due to local relapse of the primary tumor are censored. (p = 0.7362) Statistical analysis was conducted using Log-rank test. (E) Representative images of cytokeratin-8-stained lung sections, and quantification of lung metastatic nodules in mice treated with adjuvant anti-CCL2 (n = 8) or controls (n = 10) that succumb due to respiratory distress. Scale bar 5 mm. (p = 0.1649) Statistical analysis was conducted using Mann-Whitney U test. (F) Metastasis-related survival of mice treated with adjuvant anti-CCL2 (n = 14) compared to controls (n = 13). (p = 0.0606) Statistical analysis was conducted using the Log-rank test. (G) Primary tumor growth kinetics of transgenic KEP mice bearing spontaneous mammary tumors treated with anti-CCL2 (n = 6) compared to controls (n = 15). (H) Proportion of mammary-tumor bearing transgenic KEP mice bearing spontaneous pulmonary metastasis after CCL2 blockade (n = 21) or controls (n = 52). (p = 0.2437) Statistical analysis was conducted using Chi-square test. All data are mean ± s.e.m.
CCL2 drives the systemic inflammatory γδ T cell – IL17 – neutrophil axis
To understand the mechanisms of CCL2 action during breast cancer progression and metastasis, we analyzed the expression of the CCL2 receptor, CCR2, on immune cell populations in the circulation of mammary tumor-bearing KEP mice. Flow cytometric analysis revealed that monocytes and γδ T cells express high levels of CCR2 (Fig. 3A and Fig. S1A). Unlike other reports,10,11 we found no effect of antibody-mediated CCL2 neutralization on the proportion of circulating monocytes in KEP mice (Fig. S1B).
Figure 3.
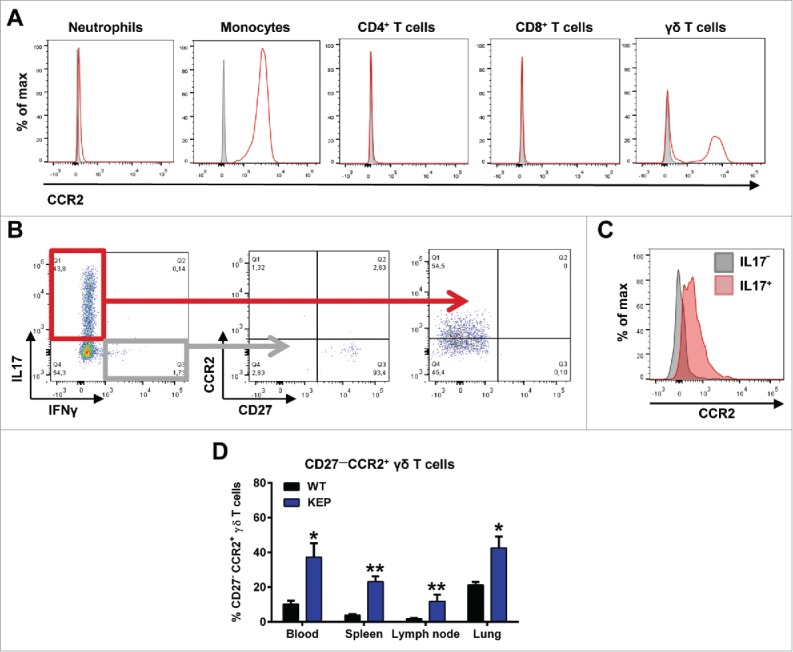
CCR2 is expressed on monocytes and IL17-producing CD27— γδ T cells. (A) Representative flow cytometry histograms showing CCR2 expression (red) compared to fluorescence minus one (FMO) controls (gray) on circulating immune cell populations in mammary tumor-bearing KEP mice. Gating strategy is described in Fig. S1A. (B) Representative dot plots of CCR2 and CD27 expression on IL17- and IFNγ-producing γδ T cells in lungs of genetically engineered KEP mice (tumor ∼225 mm2) measured by flow cytometry. (C) Representative histogram of CCR2 expression on IL17+ (red) and IL17— γδ T cells (gray). (D) Quantification of the proportion of CD27—CCR2+ cells gated on total γδ T cells in different organs of wild-type (n = 5) versus KEP mice (tumor ∼225 mm2) (n = 7). (*p < 0.05, **p < 0.01, Mann-Whitney U test). All data are mean ± s.e.m.
Because spontaneous metastasis in the KEP model is driven by IL17-producing γδ T cells and subsequent expansion of immunosuppressive neutrophils,5 we assessed whether CCL2 affects the activation of γδ T cells. Based on the expression of co-stimulatory factor CD27, γδ T cells can be phenotypically subdivided into IFNγ-producing CD27+ γδ T cells and IL17-producing CD27— γδ T cells.16 In mammary tumor-bearing KEP mice we could find these distinct subpopulations of γδ T cells and we observed that CCR2 expression is restricted to the IL17-producing CD27— γδ T cell population (Fig. 3B, C). The proportion of CD27—CCR2+ γδ T cells within the total γδ T cell population was significantly increased throughout all organs analyzed in mammary tumor-bearing KEP animals compared to wild-type littermates (Fig. 3D).
Blockade of CCL2 with neutralizing antibodies in KEP mice bearing spontaneously arising mammary tumors did not affect total γδ T cell proportions (Fig. S1C), but resulted in a significant reduction of the percentage of IL17-producing γδ T cells in blood, lymph nodes and lungs (Fig. 4A). Consistently, a 2-fold reduction in IL17 serum levels was also observed in mammary tumor-bearing KEP mice treated with anti-CCL2 (Fig. 4B).
Figure 4.
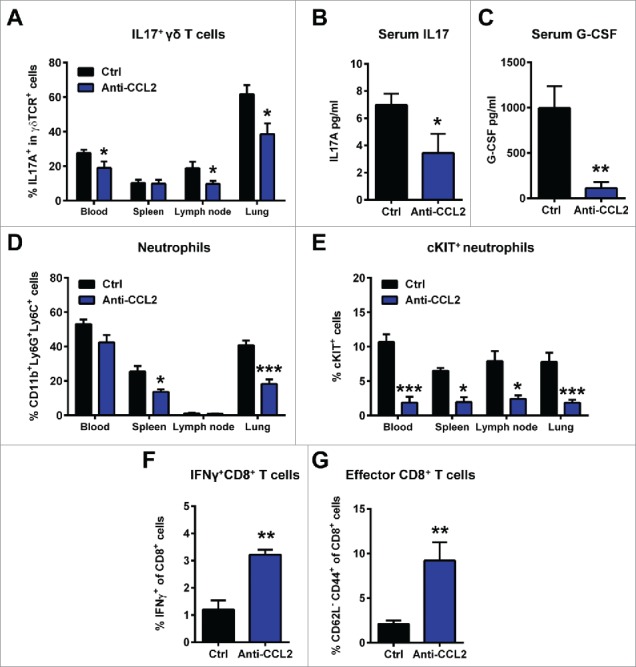
Mammary tumor-derived CCL2 promotes systemic inflammation characterized by IL17-producing γδ T cells, neutrophil expansion and suppression of T cells. KEP mice were treated with anti-CCL2 or PBS (Ctrl) during primary tumor growth starting at 25 mm2. Animals were sacrificed when tumors reached 225 mm2 and organs were collected for flow cytometric analysis. Proportions of IL17+ cells gated on total γδ T cells in blood, spleen, lymph nodes and lungs of KEP mice treated with anti-CCL2 (n = 8) and controls (n = 10) (A). Serum levels of IL17A (B) and G-CSF (C) in KEP mice determined by cytometric bead array (n = 6 per group). Flow cytometric analysis of the proportions of total CD11b+Ly6G+Ly6C+ neutrophils (gated on total CD45+ cells) (D) and cKIT+ neutrophils (gated on total neutrophils) (E) in blood, spleen, lymph nodes and lungs of KEP mice treated with anti-CCL2 (n = 6) and controls (n = 10). Flow cytometric analysis of intracellular IFNγ staining in CD8+ T cells (F) and the proportion of CD62L—CD44+ effector CD8+ T cells of total CD8+ T cells (G) in lungs of mice bearing orthotopically transplanted KEP tumors (∼100 mm2) treated with anti-CCL2 (n = 6) or controls (n = 5). Gating strategy is described in Fig. S1E, F. (*p < 0.05, **p < 0.01, ***p < 0.001, Mann-Whitney U test). All data are mean ± s.e.m.
We previously showed that in the KEP model, IL17-producing γδ T cells increase systemic G-CSF levels, resulting in systemic accumulation of immunosuppressive neutrophils.5 Consistent with the reduced IL17 serum levels, CCL2 blockade in KEP mice also decreased G-CSF serum levels (Fig. 4C). In addition, flow cytometric analysis revealed that the total proportion of CD11b+Ly6G+Ly6C+ neutrophils was reduced in various organs after CCL2 blockade (Fig. 4D). As observed in metastatic breast cancer patients,17 one hallmark of KEP tumor-induced neutrophils is the expression of haematopoietic stem cell marker cKIT on a proportion of these cells.5 In anti-CCL2 treated KEP mice the proportion of cKIT+ neutrophils was significantly reduced in all organs (Fig. 4E), indicating that CCL2 blockade reverts the tumor-induced emergence of circulating immature neutrophils. Of note, we excluded that these changes in immune parameters are a consequence of Fc receptor-mediated activation of immune cells by the Fc part of the anti-CCL2 antibody, by showing that the isotype-matched chimeric rat/mouse C1322 control antibody did not influence any of the immune parameters (Fig. S2).
Tumor-educated neutrophils in mammary tumor-bearing KEP mice exert their pro-metastatic function through suppression of CD8+ T cells.5 Since CCL2 blockade reduced mammary tumor-induced expansion of neutrophils in KEP mice, we hypothesized that CCL2 blockade would alleviate CD8+ T cell suppression in metastatic lungs. To answer this question, we turned to the spontaneous metastasis model based on orthotopic transplantation of KEP tumors in mice, since the penetrance of lung metastases in this model is high. Indeed, the proportion of interferon (IFN)-γ producing CD8+ T cells (Fig. 4F and Fig. S1E) and the proportion of CD44+CD62L— CD8+ T cells (Fig. 4G and Fig. S1F) in the lungs was significantly increased in anti-CCL2 treated animals compared to controls, while total CD8+ T cell proportions remained unaffected (Fig. S1D). Together these data demonstrate that CCL2 contributes to mammary tumor-induced immunosuppression at distant metastatic sites, which is associated with activation of IL17-expressing γδ T cells and G-CSF-dependent expansion of immunosuppressive neutrophils.
CCL2 is not sufficient to induce IL17 expression from γδ T cells
To understand how CCL2 activates the γδ T cell – IL17 – neutrophil axis, we assessed whether CCL2 is sufficient to induce IL17 expression from γδ T cells in vivo, since these cells express the CCR2 receptor (Fig. 3A and Fig. S1A). We treated wild-type tumor-free mice with recombinant murine CCL2 (rCCL2) and analyzed the proportion of IL17+ γδ T cells in the circulation. Administration of rCCL2 did not induce IL17 expression from γδ T cells (Fig. S3A) and did not expand the neutrophil population (Fig. S3B). rCCL2 did increase circulating CD11b+Ly6Chi monocytes confirming that rCCL2 was functional in vivo (Fig. S3C). Similar results were obtained in γδ T cell-deficient Tcrd—/— mice where rCCL2 induced an increase in blood monocytes but did not elicit neutrophil expansion (Fig. S3D), demonstrating that CCL2 is not sufficient to induce neutrophil expansion in the absence of γδ T cells.
We took another approach by sorting CD27— and CD27+ γδ T cells from mammary tumor-bearing KEP mice. These cells were cultured ex vivo in the presence or absence of rCCL2. While the positive control rIL23 18 induced IL17 expression from CD27— γδ T cells, rCCL2 did not (Fig. S3E). As expected, CD27+ γδ T cells did not produce IL17 (Fig. S3E). Together, these results indicate that CCL2 is not sufficient to induce IL17 expression from γδ T cells or to induce the expansion of neutrophils, and thus might require a cancer-associated intermediate cell type or mediator.
CCL2 induces IL1β expression from CCR2+ TAMs to drive the γδ T cell – IL17 – neutrophil axis
Hypothesizing that CCL2 exerts its effect via an intratumoral component, we next examined the presence of potential CCL2-responsive cells at the primary tumor site. Flow cytometric analysis of mammary tumors in the conditional KEP mice revealed that KEP cancer cells do not express CCR2 (Fig. S4B). As expected, CCR2 is abundantly expressed on CD11b+F4/80+CD206+ TAMs and CD11b+F4/80—Ly6G—Ly6Chi monocytes, and to a lesser extent on CD11b+F4/80—Ly6G+Ly6Clo neutrophils (Fig. 5A, B and Fig. S4A, B). Nevertheless, antibody-mediated neutralization of CCL2 did not alter the intratumoral accumulation of these myeloid cells (Fig. 5C).
Figure 5.

CCL2-induced IL1β expression by CCR2+ tumor-associated macrophages activates the γδ T cell – IL17 – neutrophil axis. (A) Representative histograms of CCR2 expression (red) compared to FMO (gray) on intratumoral CD11b+F4/80+CD206+ TAMs, CD11b+F4/80—Ly6G—Ly6Chi monocytes and CD11b+F4/80—Ly6G+Ly6Clo neutrophils. (B) Quantification of delta median fluorescence intensity (MFI) (MFI stained sample – MFI of FMO) of CCR2 on different populations of tumor-infiltrating myeloid cells. (C) Quantification of tumor-infiltrating immune populations in tumors (∼225 mm2) of genetically engineered KEP mice treated with anti-CCL2 (n = 7) or controls (n = 3). (D, E) TAMs were sorted from orthotopically transplanted KEP mammary tumors (∼225 mm2) treated with anti-CCL2 (n = 5) or controls (n = 4). Transcripts of Arg1, Cd206, decoy Il1r2 and Nos2 (D) and Il1β, Tgfβ, Il6 and Il23p19 (E) were determined by quantitative RT-PCR and normalized to β-actin. (F) Experimental set up of rescue of anti-CCL2 induced phenotypes with recombinant IL1β. Genetically engineered KEP mice were treated with anti-CCL2 or PBS and for 3 consecutive days with recombinant IL1β (rIL1β). 24 hours after the last injection with rIL1β and anti-CCL2 animals were sacrificed and lungs were collected for flow cytometric analysis. The proportion of IL17+ cells gated on total γδ T cells (G) and CD11b+Ly6G+Ly6C+ neutrophils gated on CD45+ cells (H) in lungs of KEP mice treated with control (n = 9), anti-CCL2 (n = 8), anti-CCL2 + rIL1β (n = 7) and rIL1β (n = 6). (*p < 0.05, **p < 0.01, ***p < 0.001, Mann-Whitney U test). All data are mean ± s.e.m.
Previously, we showed that IL1β induces IL17 expression in γδ T cells in mammary tumor-bearing KEP mice.5 Antibody-mediated neutralization of IL1β in tumor-bearing KEP mice inhibited IL17 production by γδ T cells and normalized systemic neutrophil levels.5 We identified neutrophils and TAMs as the main producers of Il1β, but due to their abundance at the primary tumor site (Fig. 1E), TAMs can be appointed as the main source of IL1β in the tumor microenvironment.5 We therefore hypothesized that CCL2 might influence the phenotype and polarization state of macrophages, including their IL1β production. Therefore, we sorted TAMs from anti-CCL2-treated and control KEP tumors and examined the expression of several genes that have been associated with the polarization of TAMs.19 We found no significant changes in gene expression of Arg1, Cd206, Decoy Il1r2 and Nos2 in TAMs upon CCL2 blockade compared to controls (Fig. 5D). Interestingly, we observed a significant decrease in TAM-derived Il1β mRNA, while expression of other known inducers of IL17, like Tgfβ, Il6 and Il23p19 (18) in TAMs was unaffected upon CCL2 blockade in vivo (Fig. 5E). Moreover, Il1β expression in tumor cells, lymphocytes and neutrophils sorted from primary KEP tumors, was unaffected by CCL2 blockade (Fig. S5A).
Consistent with these in vivo findings, expression of Il1β in bone marrow-derived macrophages cultured in vitro in the presence of KEP tumor cell-conditioned medium (KEPCM) was significantly reduced upon CCL2 blockade (Fig. S5B, C), indicating that CCL2 induces IL1β expression in TAMs. Moreover, IL1β blockade in KEP mice did not affect systemic CCL2 levels, ruling out that IL1β acts upstream of CCL2 (Fig. S5D).
To further confirm that CCL2 activates the γδ T cell – IL17 – neutrophil axis via TAM-derived IL1β, we performed in vivo rescue experiments in which mammary tumor-bearing KEP animals treated with anti-CCL2 or controls were reconstituted with recombinant murine IL1β (rIL1β) (Fig. 5F). Intracellular flow cytometry analyses revealed that reconstitution with rIL1β reversed the anti-CCL2-induced reduction of IL17-producing γδ T cells (Fig. 5G) and restored neutrophil accumulation in KEP lungs (Fig. 5H). Together these and our previous results demonstrate that tumor-derived CCL2 locally induces IL1β expression by TAMs, which can activate a systemic cascade of inflammatory events that was previously found to facilitate breast cancer metastasis (Fig. 6).5
Figure 6.
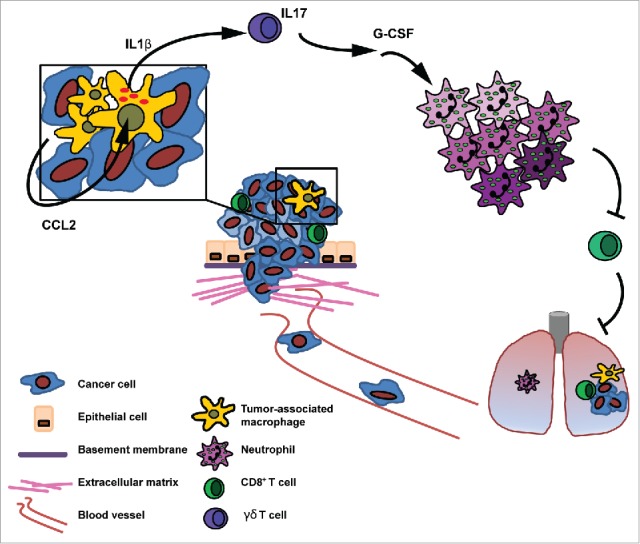
Mammary tumor-derived CCL2 promotes systemic inflammation via TAM-derived IL1β. Mammary tumors elicit a systemic inflammatory cascade via the expression of CCL2. This cascade is initiated at the primary tumor where CCL2 induces the expression of IL1β in TAMs leading to the systemic induction of IL17 production by γδ T cells, G-CSF-dependent expansion and polarization of neutrophils (indicated by color shades) and suppression of CD8+ T cell activity. By inducing this cascade of events tumors elicit an immunosuppressive state in distant organs which was described previously to facilitate the formation of metastatic disease.5
Correlation between CCL2 and IL1Β gene expression levels in human breast cancer
To determine whether there is support in human breast cancer patients for the causal link between CCL2 and IL1β as observed in the conditional KEP mouse model, we took advantage of gene expression data from tumors obtained from treatment naïve breast cancer patients. CCL2 and IL1B expression are highly enriched in basal-like tumors when compared to other subtypes of human breast cancer (Fig. 7A, B). Gene expression analysis of 2 independent data sets (METABRIC20 and 295 NKI21) confirmed these results (Fig. S6A-F). Consistent with our data obtained in the KEP model, the expression of CCL2 and IL1Β transcripts in treatment naïve human breast cancers is positively correlated across all breast cancer subtypes (Fig. 7C).
Figure 7.
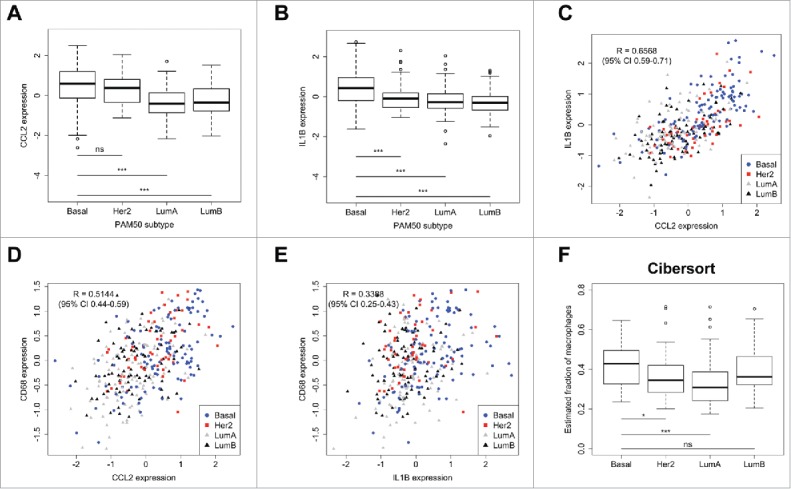
Gene expression of CCL2 and IL1B is positively correlated in human breast cancer. (A-B) Gene expression of CCL2 (A) and IL1B (B) in different subtypes of treatment naïve human breast cancer (Basal n = 106; Her2 n = 52; LumA n = 107; LumB n = 86 patients). Statistical significance was determined by Mann-Whitney U test. (C) CCL2 and IL1B gene expression are highly correlated across all subtypes of human breast cancer. CCL2 (D) and IL1B (E) gene expression in human breast cancer correlates with macrophage marker CD68. (F) Estimated fraction of macrophages in different breast cancer subtypes as determined by Cibersort. 22,23 Also see Fig. S5G. (*p < 0.05, **p < 0.01, ***p < 0.001, Mann-Whitney U test). All data are mean ± s.e.m.
Based on gene expression of CD45, we found that basal-like tumors, together with Her2+ tumors, show the highest leukocyte influx across breast cancer subtypes (Fig. S6G). Interestingly, CCL2 expression, and IL1B to a lesser extent, correlated with macrophage marker CD68 (Fig. 7D, E), suggesting that macrophage-rich tumors express higher levels of CCL2 and IL1β. To assess whether CCL2 and IL1β expression were correlated with increased proportions of macrophages in human breast cancer, we performed Cibersort analysis – a computational analysis of the intratumoral immune composition based on gene expression data.22,23 In line with the previous results, this analysis revealed that across all subtypes, basal-like breast tumors contain the highest proportions of macrophages (Fig. 7F and Fig. S6H). Together these results support the link between CCL2 and IL1β in human breast tumors.
Discussion
The role of CCL2 in breast cancer metastasis is controversial. Several studies report a pro-metastatic role through the recruitment and/or polarization of inflammatory monocytes and macrophages.10-12,24,25 However, other studies demonstrate anti-metastatic activity of CCL2, by activating neutrophils to kill disseminated tumor cells in an H2O2-mediated manner.26 Our data obtained in the genetically engineered KEP model and transplantation-based spontaneous breast cancer metastasis model show that CCL2 blockade can have both pro- and anti-metastatic effects, depending on the timing of therapeutic intervention. As reported by others,14 we show that neo-adjuvant CCL2 inhibition followed by cessation of the therapy increases the formation of pulmonary metastasis, whereas continued CCL2 blockade in an adjuvant treatment setting inhibits metastasis formation, indicating the challenge of targeting CCL2 during metastasis.15,27,28 Since targeting the CCL2/CCR2 signaling pathway is of clinical interest, it is crucial to better understand the molecular mechanisms of CCL2 action in different mouse models of human cancer.
We here report a novel mechanism by which CCL2 functions during breast cancer progression, namely via the induction of a systemic neutrophilic inflammatory cascade which we have demonstrated before to facilitate metastasis.5 We identified CCL2 as a driver of this systemic inflammatory cascade by inducing IL1β expression in TAMs. Upon CCL2 blockade, the systemic proportions of IL17+ γδ T cells and neutrophils were reduced, resulting in increased activity of CD8+ T cells. In line with our findings, therapeutic targeting of macrophages by interfering with CCL2/CCR2 signaling in experimental models has resulted in increased anti-tumor T cell responses.29-31 While some studies report direct T cell suppression by macrophages,30,31 the role of neutrophils in CCL2-mediated immunosuppression remains elusive. Here, we report that CCL2 contributes to tumor-associated immunosuppression by promoting mammary tumor-induced systemic neutrophil expansion and polarization. In line with our finding, it was reported that CCL2 promotes the accumulation and immunosuppressive properties of polymorphonuclear-myeloid-derived suppressor cells (PMN-MDSCs), which share many features with neutrophils, in a mouse model for colorectal carcinogenesis.32 Together, our findings and previous studies provide evidence that targeting CCL2/CCR2 signaling could relieve systemic immunosuppression and unleash anti-tumor immune responses.
Our experiments shed light on the multi-step mechanism underlying the interaction between CCL2 and immunosuppressive neutrophils, by showing that CCL2 promotes IL1β expression in TAMs, which triggers a cascade of downstream systemic events involving IL17 expression by γδ T cells leading to G-CSF-induced expansion of pro-metastatic neutrophils. Interestingly, the connection between CCL2/CCR2 signaling and IL1β is also important in non-tumor settings. In a model for microbiota-induced intestinal inflammation, CCR2 signaling mediates NLRP3 inflammasome-dependent release of IL1β from monocytes triggering inflammation upon epithelial injury.33 Whether CCL2-induced IL1β production in TAMs in the KEP model requires the NLRP3 inflammasome remains to be investigated.
Also, CCL2 has been described to activate and mobilize γδ T cells in various inflammatory conditions including allergy and sepsis.34,35 Furthermore, inflammation-induced CCL2 expression has been shown to recruit IL17-producing CCR2+ γδ T cells that are activated by IL1β and IL23 in a mouse model for rheumatoid arthritis.36 These striking similarities between non-tumor and tumor-induced inflammation hint toward a more general causal link between CCL2, IL1β and γδ T cell signaling in various inflammatory conditions. Intriguingly, anti-tumor γδ T cells have been shown to infiltrate tumors in a CCL2-mediated manner in the B16 melanoma inoculation model.37 In this model, CCL2 directly affected the migration and recruitment of γδ T cells, and the role of IL1β was not assessed. Whether and how these opposite functions of CCL2 on tumor biology are dictated by the genetic make-up of tumors, tumor type, tumor stage and/or other cancer cell-intrinsic or -extrinsic properties remains to be established.
Several independent clinical studies show that expression of CCL2, IL17, the intratumoral presence of macrophages, γδ T cells, and systemic neutrophil accumulation each correlate with poor prognosis in breast cancer patients.8,38-41 Moreover, expression of IL1β is elevated in human invasive breast cancers compared to healthy tissue.42 In line with these reports, we show in human treatment naïve breast cancers that CCL2 and IL1B gene expression are highly correlated and are most pronounced in macrophage-rich tumors. The findings in our previous 5 and current studies suggest that these inflammatory cells and mediators are causally linked, and that interruption of this systemic inflammatory cascade can be a potential therapeutic target to relieve tumor-induced systemic immunosuppression. A recent phase 1b clinical trial in patients with pancreatic cancer revealed that therapeutic targeting of CCL2/CCR2 signaling in combination with a chemotherapy regimen has clinical activity and resulted in reduced immunosuppression and an increase in the number of tumor-infiltrating lymphocytes.43 Together these results advocate for the exploration of CCL2/CCR2 targeting drugs for the treatment of metastasized breast cancer.
Materials and methods
Patient material and Cibersort
Biopsies of primary breast tumors were collected before treatment from women who received neo-adjuvant chemotherapy at the Netherlands Cancer Institute between 2000 and 2013 as part of ongoing clinical trials, or were treated off protocol according to the standard arms of one of these studies (NCT00448266, NCT01057069). The studies have been approved by the ethical committee and informed consent was obtained from all patients. Biopsies were taken using a core needle and were snap-frozen in liquid nitrogen. RNA was isolated from samples with a tumor percentage > 50% and analyzed on a microarray or using RNAseq (details are available in Supplementary Materials and methods). The microarray data were generated and analyzed as described previously,44 and made available through the GEO database, accession GSE34138. To determine the relative abundance of immune cells in our samples, we analyzed the microarray data using CIBERSORT (22 and https://cibersort.stanford.edu/).
Animal studies
The generation of K14cre;Cdh1F/F;Trp53F/F (KEP) mice has been described in detail.45 KEP mice were backcrossed to the FVB/N background. Mammary tumor formation was monitored twice weekly by palpation and caliper measurements. For transplantation studies female FBV/N mice (10-12 weeks) were purchased from Charles River Laboratories. Orthotopic transplantation of KEP tumors was performed as described earlier.13 Female Tcrd—/— mice on the FVB/N background were kindly provided by A. Hayday.46 Animals were kept in open cages and food and water were provided ad libitum. Animal experimental procedures were approved by the Animal Ethics Committee of the Netherlands Cancer Institute and performed in accordance with national and institutional guidelines for Animal Care and Use.
In vivo CCL2 and IL1β neutralization
Mammary tumor-bearing KEP animals were injected intraperitoneally with chimeric rat/mouse anti-mouse CCL2 (Janssen Pharmaceuticals, C1142) twice weekly dosed at 10 mg/kg or 50 mg anti-ILβ (BioXCell, BE0246) twice weekly starting from a tumor size of 25 mm2 and continued until animals were sacrificed once their primary tumor reached 225 mm2. Control mice received equal amounts of isotype-matched control antibodies (Janssen Pharmaceuticals, C1322) or PBS. For metastasis studies, FVB/N animals bearing orthotopically transplanted KEP tumors were treated in the neo-adjuvant setting with anti-CCL2 starting from a tumor size of 6 mm2 until the primary tumor was surgically removed (∼225 mm2). For adjuvant CCL2 blockade, treatment with anti-CCL2 was initiated 3 d after surgical removal of the primary tumor and continued until animals had to be sacrificed due to clinical signs of metastatic disease. Animals were randomized before initiating treatment.
Surface and intracellular staining for flow cytometry
Tissues were collected in ice-cold PBS. Blood samples were collected in tubes containing heparin (Leo Pharma) and treated with NH4 lysis buffer. Tumors and lungs were mechanically chopped using a McIlwain Tissue Chopper (Mickle Laboratory Engineering) and digested for 1 hour at 37°C in a digestion mix of 3 mg/ml collagenase type A (Roche, 11088793001) and 25 μg/ml DNAse (Invitrogen, 18068–015) or 30 min at 37°C in 100 μg/ml Liberase (Roche, 5401127001) respectively, in serum-free DMEM (Invitrogen). Reactions were terminated by addition of DMEM containing 8% FCS. Cell suspensions were dispersed through a 70 μm cell strainer (BD Falcon, 352350). All single cell suspensions were treated with NH4 lysis buffer to remove red blood cells.
For ex vivo cytokine stimulation, single cells were collected at 1500 rpm for 5 min in a round bottom 96-wells tissue culture plate (Thermo Scientific) in IMDM containing 8% FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin (Invitrogen) and 0.5% β-mercaptoethanol. Cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/ml) and ionomycin (1 μM) in the presence of Golgi-Plug™ (BD Biosciences, 555029) for 3 h at 37°C.
For flow cytometric staining, either stimulated or unstimulated single cells were collected at 1500 rpm for 5 min and resuspended in PBS containing 1% BSA (Sigma-Aldrich). Single cell suspensions were plated in round bottom 96-wells plates (Thermo Scientific) and incubated for 30 min in the dark at 4°C with different combinations of fluorescently labeled monoclonal antibodies. For intracellular staining cells were washed twice with PBS containing 1% BSA and fixed and permeabilized using the Cytofix/Cytoperm™ kit (BD Biosciences, 554714) according to manufacturer's instructions. Cells were subsequently incubated for 30 min in the dark at 4°C with antibodies against IFNγ and IL17A. Fixable Viability Dye APC eFluor® 780 (eBioscience, 65–0865) or 7AAD viability staining solution (eBioscience, 00–6993) was added to exclude dead cells. Flow cytometric analysis was performed on a BD LSRII using Diva Software (BD Biosciences). Data analyses were performed using FlowJo Software version 10.0 (Tree Star Inc.).
The following antibody panels were used:
Myeloid panel – CD45-eFluor605NC (1:50; clone 30-F11), CD11b-eFluor650NC (1:400; clone M1/70), Ly6G-AlexaFluor700 (1:400; clone 1A8; BD Pharmingen), Ly6C-eFluor450 (1:400; clone HK1.4), F4/80-APC-eFluor780 (1:200; clone BM8), VEGFR1-APC (1:50; clone 141522; R&D Systems), cKIT-PE-Cy7 (1:400; clone 2B8), CCR2-PE (1:50; clone 475301; R&D Systems), CXCR4-PerCP-eFluor 710 (1:400; clone 2B11), CD49d-FITC (1:400; clone R1–2) or Gr1-FITC (1:400; clone RB6–8C5), 7AAD.
Lymphoid panel I – CD45-eFluor605NC (1:50; clone 30-F11), CD11b-eFluor650NC (1:400; clone M1/70), CD3-PE-Cy7 (1:200; clone 145–2C11), CD4-APC-eFluor780 (1:200; clone GK1.5), CD8-PerCP-eFluor710 (1:400; clone 53–6.7), γδTCR-FITC (1:400; clone GL3; BD Biosciences), CD49b-APC (1:400; clone DX5), IL17A-PE (1:200; clone eBio17B7), IFNγ-eFluor450 (1:200; clone XMG1.2), 7AAD.
Lymphoid panel II – CD45-eFluor605NC (1:50; clone 30-F11), CD11b-APC-eFluor780 (1:200; clone M1/70), CD3-PE-Cy7 (1:200; clone 145–2C11), CD4-APC-eFluor780 (1:200; clone GK1.5), CD8-PerCP-eFluor710 (1:400; clone 53–6.7), γδTCR-PE (1:400; clone GL3), CD49b-APC (1:400; clone DX5), CD62L-AlexaFluor700 (1:400; clone MEL-14), CD44-FITC (1:400; clone IM7; BD Pharmingen), IFNγ-eFluor450 (1:200; clone XMG1.2), CD19-APC-eFluor780 (1:200; clone eBio1D3), Fixable Viability Dye eFluor® 780.
Phenotyping γδ T cells panel I – CD27-PE-Cy7 (1:200; clone LG.7F9), γδTCR-FITC (1:400; clone GL3; BD Biosciences), CD45-eFluor605NC (1:50; clone 30-F11), CD3-eFluor450 (1:200; clone 145–2C11), CCR2-PE (1:50; clone 475301; R&D Systems), CD8-PerCP-eFluor710 (1:400; clone 53–6.7), CD4-APC-eFluor780 (1:200; clone GK1.5), CD19-APC-eFluor780 (1:200; clone eBio1D3), CD11b-APC-eFluor780 (1:200; clone M1/70), Fixable Viability Dye eFluor® 780.
Phenotyping γδ T cells panel II – CD27-PE-Cy7 (1:200; clone LG.7F9), γδTCR-FITC (1:400; clone GL3; BD Biosciences), CD45-eFluor605NC (1:50; clone 30-F11), CD3-eFluor450 (1:200; clone 145–2C11), CCR2-PE (1:50; clone 475301; R&D Systems), IL17A-APC (1:50, clone TC11–18H10; BD Pharmingen), IFNγ-eFluor450 (1:200; clone XMG1.2). Fixable Viability Dye eFluor® 780.
All antibodies were obtained from eBiosciences, unless indicated otherwise.
In vivo rescue with recombinant proteins
For CCL2 rescue experiments, female wild-type or Tcrd—/— mice (10-12 weeks of age) were injected intravenously (i.v.) with 1 μg/day recombinant murine CCL2 (Peprotech, 250-10) in 100 μl sterile PBS or vehicle for 5 consecutive days. On the last day animals were sacrificed 1 hr after rCCL2 or vehicle administration and blood and lungs were collected and processed for flow cytometric analysis. For the IL17+ γδ T cell read out, lung and blood cells were pooled to gain sufficient amounts of cells. For neutrophil and monocytes read-out, only blood was used.
For IL1β rescue experiments, mammary tumor-bearing KEP animals were treated twice weekly with anti-CCL2 (C1142 Janssen Pharmaceuticals) by intraperitoneal injection dosed at 10 mg/kg starting from a tumor size of 25 mm2 until animals were sacrificed. When tumors reached a size of ∼130 mm2 animals were injected intraperitoneally (i.p.) with 0,5 μg/day recombinant murine IL1β (Peprotech, 211-11B) for 3 consecutive days. Animals were sacrificed 24 hrs after the last injection and organs were collected and processed for flow cytometry.
Cytokine analysis
Multiplex quantification of inflammatory cytokines and chemokines was performed using the premixed 32-plex Mouse Immunology Multiplex assay (Milliplex-Map Millipore, MCYTMAG-70K-PX32). Assays and tissue preparations were performed according to manufacturer's recommendations. 100 μg of total protein from lysed tissues was used for measurements. Fluorescence was measured on a Luminex FlexMap3D System using xPonent 4.0 software (Luminex Corporation). IL-17A and G-CSF levels in serum or culture supernatant were measured by BD Cytometric Bead Array (CBA) Flex Set (BD Biosciences, mouse IL-17A, 560283; mouse G-CSF, 560152). Assays were performed according to manufacturer's recommendations. Flow cytometric analysis was performed on a Cyan flow cytometer using Summit Software (Beckman Coulter Inc.). Data analyses were performed using FlowJo Software version 10.0 (Tree Star Inc.). CCL2 serum levels were measured by ELISA (R&D Systems, DY479) according to manufacturer's recommendations.
RNA in situ hybridization
Tissues were fixed in 10% neutral buffered formalin for 24 hrs, embedded in paraffin (FFPE), and sectioned at 5 µm. Localization of Ccl2 mRNA in KEP mammary tumors was examined by performing RNA in situ hybridization on fresh FFPE slides using RNAscope 2.0 FFPE assay (Advanced Cell Diagnostics). As controls, probes against DapB (negative control) and PPIB (positive control) were used. Assay was performed as described in (47). Stained slides were digitally processed using the Aperio ScanScope (Aperio, Vista) and captured using ImageScope software version 11.0.2 (Aperio, Vista).
Fluorescence activated cell sorting
Single cell suspensions from KEP mammary tumors were prepared as described above. CD11b-APC (clone M1/70; eBioscience) myeloid cells were isolated by anti-APC beads over a magnetic column (Milteny). The CD11b+ fraction was stained with F4/80-PE (clone BM8; eBioscience), Ly6C-eFluor450 (clone HK1.4; eBioscience), CD11c-PE-Cy7 (clone HL3; BD Bioscience) and Ly6G-FITC (clone 1A8; BD Pharmingen). The CD11b— fraction was stained with CD45-PerCp-Cy5.5 (clone 30-F11; eBioscience), CD31-FITC (clone 390; eBioscience), PDGFRβ-PE (clone APB5; eBioscience) and sorted using a BD FACS Aria II, and collected in Trizol for further analysis.
The following populations were identified based on the expression of the following surface markers: tumor cells (CD31—CD45—CD11b—), lymphocytes (CD45+CD11b—), fibroblasts (PDGFRβ+CD31—CD45—CD11b—), endothelial cells (CD31+CD45—CD11b—), macrophages (CD11b+F4/80+), dendritic cells (DC) (CD11b+F4/80—CD11c+), neutrophils (CD11b+F4/80—Ly6G+Ly6Clo), and monocytes (CD11b+F4/80—Ly6G—Ly6Chi). All cells were collected in Trizol for further analysis.
For γδ T cell sorts, single cells from KEP spleen and lymph nodes were pooled, collected at 1500 rpm for 5 min and stained for 30 min in the dark at 4°C with CD3-FITC (eBioscience; clone 145-2C11) in PBS containing 1% BSA. After staining, cells were collected at 1500 rpm for 5 min and suspended in IMDM containing 2% FCS, 100 IU/mL penicillin, 100 μg/mL streptomycin (Invitrogen) and 0.5% β-mercaptoethanol. Subsequently, cells were pre-sorted for CD3+ T cells using a BD FACS Aria II and collected in 100% FCS. Next, cells were collected at 1500 rpm for 5 min and stained for 30 min in the dark at 4°C with γδ TCR-PE (clone GL3; eBioscience) and CD27-PE-Cy7 (clone LG.7F9, eBioscience) in PBS containing 1% BSA. After staining, cells were sorted for CD27+ and CD27— γδ T cells and collected in 100% FCS for further use.
Ex vivo culture of γδ T cells
Sorted γδ T cells were cultured 1:1 with irradiated splenocytes (40 Gy) in flat bottom 96-wells tissue culture plate (Thermo Scientific) in IMDM containing 8% FCS, 100 IU/mL penicillin, 100 μg/mL streptomycin (Invitrogen) and 0.5% β-mercaptoethanol. T cells were activated by addition of Dynabeads Mouse T-activation CD3/CD28 beads (Thermo Scientific, 11456D). Culture medium was supplemented with recombinant murine IL-23 (10 ng/mL; purified by the NKI protein facility) or 50 ng/mL recombinant murine CCL2 (Peprotech, 250-10). After 48 hours of culture, supernatant was collected and stored in -20°C until further use.
In vitro culture of bone marrow-derived macrophages (BMDM)
Bone marrow was obtained from femurs and tibia of female wild-type mice and cultured for 6–8 d in RPMI containing 8% FCS, 100 IU/mL penicillin, 100 μg/mL streptomycin (Invitrogen) supplemented with 20 ng/ml murine CSF-1 (Peprotech, 315-02). For experiments, BMDM were primed overnight in medium containing 100 ng/ml LPS. The next day, BMDM were washed and cultured in control RPMI or RPMI 1:1 supplemented with conditioned medium from KEP tumor cell lines (KEPCM) with or without anti-CCL2 (5 μg/ml). KEP tumor cell line-conditioned media was generated by culturing KEP tumor cell lines in serum-free RPMI for 48 hours. Supernatant was centrifuged to exclude cellular debris before use in BMDM experiments. After 24 hours, BMDM were collected and RNA was extracted using RNeasy columns (Qiagen, 74104). Il1β expression levels were determined by RT-PCR. Fold change was calculated using the formula 2−(ΔCt — X[ΔCtWT]).
Real-time polymerase chain reaction (RT-PCR)
RNA was extracted from FACS-sorted immune cell populations using Trizol-chloroform method. RNA was cleaned with DNase (Invitrogen) and the yield was measured by using Nanodrop. cDNA first-strand synthesis was performed using Cloned AMV First-Strand cDNA Synthesis Kit (Invitrogen, 12328) using Oligo(dT) primers. qRT-PCR analysis was performed using LightCycler 480 SYBR Green I Master (Roche Applied Sciences) according to the manufacturer's instructions. Briefly, 20 ng cDNA was dissolved in 1x LightCycler 480 SYBR Green Master mix containing 500 nM of forward and reverse primers (see Table S1). For quantification the delta Ct method was used: ΔCt sample − ΔCt reference gene. All transcripts were normalized to β-actin.
Immunohistochemistry
Formalin-fixed tissues were processed by routine procedures. Lung metastases were detected as described previously.5,13 Briefly, one lung section of each animal was used for detection of metastatic nodules using anti-cytokeratin 8 (clone Troma1; Developmental Studies HybridomaBank, University of Iowa) with citrate antigen retrieval. Only mice that were sacrificed due to respiratory distress were included in this analysis. The number of cytokeratin 8+ metastatic nodules in the lung was blindly scored by at least 2 researchers. Stained slides were digitally processed using the Aperio ScanScope (Aperio) and captured using ImageScope software version 11.0.2 (Aperio). Brightness and contrast for representative images were adjusted equally among groups.
Statistical analysis
Data analyses were performed using GraphPad Prism version 6.01 (GraphPad Software Inc.). Applied analyses are indicated in the corresponding legends. No statistical methods were used to determine sample sizes. Sample sizes were based on previous experience with the models.5,13,48 Differences with a p<0.05 were considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
P.D. is an employee of Janssen Research and Development, USA. The authors declare no additional competing financial interests.
Acknowledgments
This work was supported by a European Research Council Consolidator award (INFLAMET 615300) and grants from the Dutch Cancer Society (2011-5004); Worldwide Cancer Research (AICR 11–0677); the Netherlands Organization for Scientific Research NWO VIDI (917.96.307); the European Union (FP7 MCA-ITN 317445 TIMCC) to K.E.dV., and a Dutch Cancer Society grant (2013-6007) to L.F.A.W. We thank L. Mulder for the preparation of the patient samples, and J. Borst, T. Schumacher and J. Jonkers for discussions. We thank the core facilities at the Netherlands Cancer Institute. We thank A. Hayday for Tcrd—/— mice.
Author contributions
K.K., S.B.C., and K.E.dV. conceived the ideas and designed the experiments. K.K., S.B.C., N.J.M.V., K.V., M.C., C.W.D., C.H., M.D.W., and C.S. performed the experiments. K.K., S.B.C., N.J.M.V., K.V., M.C., C.W.D., C.H., M.D.W. and K.E.dV. analyzed the data. Computational analysis and collection of patient data was performed by M.H., E.H.L. and L.F.A.W. P.D. provided the CCL2 neutralizing antibody and control antibody. K.K., S.B.C. and K.E.dV. wrote the paper.
References
- 1.Weigelt B, Peterse JL, van't Veer LJ. Breast cancer metastasis: Markers and models. Nat Rev Cancer 2005; 5:591-602; PMID:16056258; https://doi.org/ 10.1038/nrc1670 [DOI] [PubMed] [Google Scholar]
- 2.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014; 16:717-27; PMID:25082194; https://doi.org/ 10.1038/ncb3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol 2015; 15:73-86; PMID:25614318; https://doi.org/ 10.1038/nri3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: Neutral no more. Nature Reviews Cancer 2016; 16:431-46; PMID:27282249; https://doi.org/ 10.1038/nrc.2016.52 [DOI] [PubMed] [Google Scholar]
- 5.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015; 522:345-8; PMID:25822788; https://doi.org/ 10.1038/nature14282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 2013; 34:81-9; PMID:23000011; https://doi.org/ 10.1016/j.it.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 7.Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol 1995; 155:5769-76; PMID:7499865 [PubMed] [Google Scholar]
- 8.Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, Koike M, Inadera H, Matsushima K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res 2000; 6:3282-9; PMID:10955814 [PubMed] [Google Scholar]
- 9.Saji H, Koike M, Yamori T, Saji S, Seiki M, Matsushima K, Toi M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001; 92:1085-91; PMID:11571719; https://doi.org/ [DOI] [PubMed] [Google Scholar]
- 10.Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475:222-5; PMID:21654748; https://doi.org/ 10.1038/nature10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshimura T, Howard OMZ, Ito T, Kuwabara M, Matsukawa A, Chen K, Liu Y, Liu M, Oppenheim JJ, Wang JM. Monocyte chemoattractant protein-1/CCL2 produced by stromal cells promotes lung metastasis of 4T1 murine breast cancer cells. PLoS ONE 2013; 8:e58791; PMID:23527025; https://doi.org/ 10.1371/journal.pone.0058791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura T, Qian B-Z, Soong D, Cassetta L, Noy R, Sugano G, Kato Y, Li J, Pollard JW. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med 2015; 212:1043-59; PMID:26056232; https://doi.org/ 10.1084/jem.20141836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau C-S, Hollmann MW, Jonkers J, de Visser KE. A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res 2013; 73:353-63; PMID:23151903; https://doi.org/ 10.1158/0008-5472.CAN-11-4208 [DOI] [PubMed] [Google Scholar]
- 14.Bonapace L, Coissieux M-M, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014; 515:130-3; PMID:25337873; https://doi.org/ 10.1038/nature13862 [DOI] [PubMed] [Google Scholar]
- 15.Li M, Knight DA, A Snyder L, Smyth MJ, Stewart TJ. A role for CCL2 in both tumor progression and immunosurveillance. Oncoimmunology 2013; 2:e25474; PMID:24073384; https://doi.org/ 10.4161/onci.25474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol 2009; 10:427-36; PMID:19270712; https://doi.org/ 10.1038/ni.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cattin S, Fellay B, Pradervand S, Trojan A, Ruhstaller T, Rüegg C, Fürstenberger G. Bevacizumab specifically decreases elevated levels of circulating KIT+CD11b+ cells and IL-10 in metastatic breast cancer patients. Oncotarget 2016; 7:11137-50; PMID:26840567; https://doi.org/ 10.18632/oncotarget.7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: Mechanisms and therapeutic potential. Clinical Science 2012; 122:487-511; PMID:22324470; https://doi.org/ 10.1042/CS20110496 [DOI] [PubMed] [Google Scholar]
- 19.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004; 25:677-86; PMID:15530839; https://doi.org/ 10.1016/j.it.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 20.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486:346-52; PMID:22522925; https://doi.org/ 10.1038/nature10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347:1999-2009; PMID:12490681; https://doi.org/ 10.1056/NEJMoa021967 [DOI] [PubMed] [Google Scholar]
- 22.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015; 12:453-7; PMID:25822800; https://doi.org/ 10.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21:938-45; PMID:26193342; https://doi.org/ 10.1038/nm.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu X, Kang Y. Chemokine (C-C Motif) Ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 2009; 284:29087-96; PMID:19720836; https://doi.org/ 10.1074/jbc.M109.035899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang L-CS, Singhal S, Snyder LA, Albelda SM. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am J Respir Cell Mol Biol 2011; 44:230-7; PMID:20395632; https://doi.org/ 10.1165/rcmb.2010-0080OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011; 20:300-14; PMID:21907922; https://doi.org/ 10.1016/j.ccr.2011.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchem JB, DeNardo DG. Battle over CCL2 for control of the metastatic niche: Neutrophils versus monocytes. Breast Cancer Res 2012; 14:315; PMID:22809105; https://doi.org/ 10.1186/bcr3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavender N, Yang J, Chen S-C, Sai J, Johnson CA, Owens P, Ayers GD, Richmond A. The Yin/Yan of CCL2: A minor role in neutrophil anti-tumor activity in vitro but a major role on the outgrowth of metastatic breast cancer lesions in the lung in vivo. BMC Cancer 2017; 17:88; PMID:28143493; https://doi.org/ 10.1186/s12885-017-3074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kudo-Saito C, Shirako H, Ohike M, Tsukamoto N, Kawakami Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis 2013; 30:393-405; PMID:23143679; https://doi.org/ 10.1007/s10585-012-9545-6 [DOI] [PubMed] [Google Scholar]
- 30.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013; 73:1128-41; PMID:23221383; https://doi.org/ 10.1158/0008-5472.CAN-12-2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, Song H, Xie D, Jiang X, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017; 66:157-67; PMID:26452628; https://doi.org/ 10.1136/gutjnl-2015-310514 [DOI] [PubMed] [Google Scholar]
- 32.Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Reports 2015; 12:244-57; PMID:26146082; https://doi.org/ 10.1016/j.celrep.2015.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo S-U, Kamada N, Muñoz-Planillo R, Kim Y-G, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 2015; 42:744-55; PMID:25862092; https://doi.org/ 10.1016/j.immuni.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penido C, Costa MFS, Souza MC, Costa KA, Candéa ALP, Benjamim CF, Henriques MDGMO. Involvement of CC chemokines in gammadelta T lymphocyte trafficking during allergic inflammation: The role of CCL2/CCR2 pathway. Int Immunol 2008; 20:129-39; PMID:18056919; https://doi.org/ 10.1093/intimm/dxm128 [DOI] [PubMed] [Google Scholar]
- 35.Costa MF, De S, de Negreiros CBT, Bornstein VU, Valente RH, Mengel J, Henriques MDG, Benjamim CF, Penido C. Murine IL-17+ Vγ4 T lymphocytes accumulate in the lungs and play a protective role during severe sepsis. BMC Immunol 2015; 16:36; PMID:26037291; https://doi.org/ 10.1186/s12865-015-0098-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akitsu A, Ishigame H, Kakuta S, Chung S-H, Ikeda S, Shimizu K, Kubo S, Liu Y, Umemura M, Matsuzaki G, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vγ6(+)γδ T cells. Nat Commun 2015; 6:7464; PMID:26108163; https://doi.org/ 10.1038/ncomms8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lança T, Costa MF, Gonçalves-Sousa N, Rei M, Grosso AR, Penido C, Silva-Santos B. Protective role of the inflammatory CCR2/CCL2 chemokine pathway through recruitment of type 1 cytotoxic γδ T lymphocytes to tumor beds. J Immunol 2013; 190:6673-80; PMID:23686489; https://doi.org/ 10.4049/jimmunol.1300434 [DOI] [PubMed] [Google Scholar]
- 38.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res 1996; 56:4625-9; PMID:8840975 [PubMed] [Google Scholar]
- 39.Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Wevers E, Schwartz T, Hunborg P, Varvares MA, Hoft DF, et al. Tumor-infiltrating γδ T lymphocytes predict clinical outcome in human breast cancer. J Immunol 2012; 189:5029-36; PMID:23034170; https://doi.org/ 10.4049/jimmunol.1201892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noh H, Eomm M, Han A. Usefulness of pretreatment neutrophil to lymphocyte ratio in predicting disease-specific survival in breast cancer patients. J Breast Cancer 2013; 16:55-9; PMID:23593082; https://doi.org/ 10.4048/jbc.2013.16.1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen WC, Lai YH, Chen HY, Guo HR, Su IJ, Chen HHW. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology 2013; 63:225-33; PMID:23738752; https://doi.org/ 10.1111/his.12156 [DOI] [PubMed] [Google Scholar]
- 42.Jin L, Yuan RQ, Fuchs A, Yao Y, Joseph A, Schwall R, Schnitt SJ, Guida A, Hastings HM, Andres J, et al. Expression of interleukin-1beta in human breast carcinoma. Cancer 1997; 80:421-34; PMID:9241076; https://doi.org/ [DOI] [PubMed] [Google Scholar]
- 43.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 2016; 17:651-62; PMID:27055731; https://doi.org/ 10.1016/S1470-2045(16)00078-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Ronde JJ, Lips EH, Mulder L, Vincent AD, Wesseling J, Nieuwland M, Kerkhoven R, Vrancken Peeters M-JTFD, Sonke GS, Rodenhuis S, et al. SERPINA6, BEX1, AGTR1, SLC26A3, and LAPTM4B are markers of resistance to neoadjuvant chemotherapy in HER2-negative breast cancer. Breast Cancer Res Treat 2013; 137:213-23; PMID:23203637; https://doi.org/ 10.1007/s10549-012-2340-x [DOI] [PubMed] [Google Scholar]
- 45.Derksen PWB, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006; 10:437-49; PMID:17097565; https://doi.org/ 10.1016/j.ccr.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 46.Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE, Hayday AC. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001; 294:605-9; PMID:11567106; https://doi.org/ 10.1126/science.1063916 [DOI] [PubMed] [Google Scholar]
- 47.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 2012; 14:22-9; PMID:22166544; https://doi.org/ 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med 2012; 18:344-6; PMID:22395693; https://doi.org/ 10.1038/nm.2652 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.