

ABSTRACT
LTX 315 is an oncolytic peptide with potent immunological properties. In the present study, we demonstrate that intratumoral treatment with LTX-315 resulted in a complete regression and systemic immune response in a rat fibrosarcoma model. The treatment was T-cell dependent, and also resulted in an abscopal effect as demonstrated by the regression of distal non-treated lesions. Significant infiltration of CD8+ T cells was observed in both treated and non-treated lesions, as shown by immunohistochemical and flow cytometric analysis. LTX-315 rapidly killed the cells in vitro with a lytic mode of action followed by the subsequent release of Danger-Associated Molecular Pattern (DAMP) molecules such as HMGB1, ATP and Cytochrome c. Together, our data demonstrate that LTX-315 represents a new approach to cancer immunotherapy, which has the potential as a novel immunotherapeutic agent.
KEYWORDS: Sarcoma, DAMP, Immunotherapy, oncolytic peptide, LTX-315
Introduction
Host defense peptides, commonly also denoted as cationic antimicrobial peptides (CAPs), are found in virtually all species of life and are a part of the innate immune system.1 CAPs vary in amino acid sequence and chemical structure, but in general they are short (< 40aa), are abundant in cationic residues and acquire amphipathic properties. Their amphipathic nature and cationic charge are both important modalities for their ability to interact with, and destabilize anionic lipid membranes in cancer cells.2 Although their most studied function is the defense against microorganism such as bacteria, CAPs also have been shown to exert activities against cancer cells.3,4
We have described previously the development of a 9-mer oncolytic peptide LTX-315.5,6 The peptide is highly effective against both drug-resistant and drug-sensitive cancer cells, and displays a lower activity toward normal non-malignant cells.7 The mechanism of cell death includes a membranolytic effect on the cellular plasma membrane, as well as cytosolic organelles such as mitochondria, Golgi and lysosomes.8,9 The membranolytic mode of action leads to a subsequent release of danger-associated molecular pattern molecules (DAMPs) and tumor antigens that recruit and activate dendritic cells with a subsequent T-cell activation and specific immune response.9 Intratumoral (i.t.) treatment with LTX-315 results in growth inhibition, complete regression and long-lasting tumor-specific immune responses in a variety of different experimental animal models.10,11
In the present study, we investigated the oncolytic effect of LTX-315 in a rat fibrosarcoma model, based on a spontaneously transformed rat mesenchymal stem cell line (rTMSC).12 The transformed cell line maintained features of mesenchymal stem cells, and at the same time possessed properties and characteristics of cancer stem cells (CSCs). Side population (SP) isolation and tumorsphere cultivation of the transformed cells confirmed the presence of putative CSCs among the rTMSCs. In vivo studies demonstrated functional heterogeneity among the transformed rMSCs, and indicated the SP subset as responsible for tumor development. Tumor-initiating cells/cancer stem cells are associated with an increased resistance to conventional chemotherapy and radiation,13 making the tumor cell subset a suitable target for LTX-315 treatment.
Here, we report that i.t. injections with LTX-315 resulted in a complete regression of established fibrosarcomas. The treatment resulted in a T-cell-dependent and tumor-specific immune response associated with the infiltration of CD8+ T cells into the tumor bed. Furthermore, T-cell infiltration and regression was also observed in non-treated distal lesions, hence indicating a strong abscopal effect of the treatment with LTX-315.
Results
LTX-315 has a rapid mode of action compared with conventional chemotherapeutics
The cytotoxic effect of LTX-315 was compared with commonly used chemotherapeutic drugs (dacarbazine, temozolomide and cisplatin), using the MTT assay. LTX-315 exhibited a more rapid cytotoxic effect against the murine rTMSC cell line compared with the chemotherapeutic drugs, as shown by an IC50 value of 7 μM 2 h post-treatment, compared with >100 μM for all the chemotherapeutics. A significant anticancer effect for the chemotherapeutic drugs was first observed after 24 h (Fig. 1).
Figure 1.
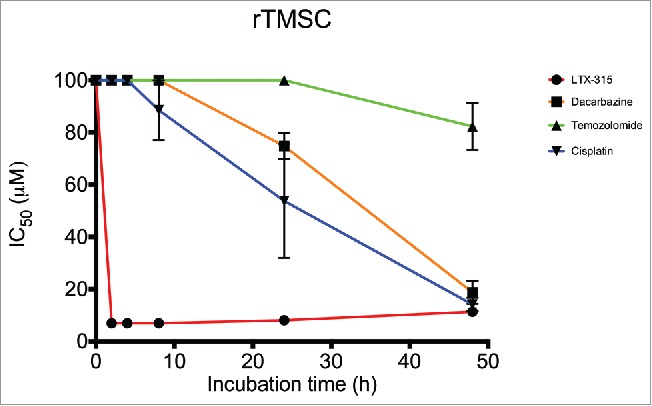
In vitro cytotoxicity of LTX-315 compared with conventional chemotherapeutics. Cytotoxicity data demonstrating IC50 values of LTX-315 (red line) and 3 different chemotherapeutic drugs, dacarbazine (orange line), temozolomide (green line) and cisplatin (blue line) after designated time points (2, 4, 8, 24 and 48 h). For each time point, the mean of 3 experiments are presented.
Rat TMSC cells treated in vitro with LTX-315 release DAMPs
To study whether LTX-315 was able to induce the release of DAMPs, which is one of the requirements for immunogenic cell death, the release of ATP, cytochrome c and HMGB1 from LTX-315-treated rTMSC cells was measured. rTMSC cells treated with 17 μM LTX-315 exhibited a gradual release of all DAMPs, with an increasing concentration of ATP, cytochrome c and HMGB1 as time progressed. Untreated control cells showed little or no release of ATP, HMGB1 and Cytochrome c (Fig. 2A-C).
Figure 2.

LTX-315 treatment leads to extracellular release of DAMPs in vitro. Rat TMSCs were treated in vitro with 17 μM of LTX-315 (IC50) for selected time points (5, 10, 30 and 60 min), and analyzed for the release of ATP, Cytochrome c and HMGB1. (A) LTX-315-treated cells showed a rapid release of ATP into the supernatant with a peak around 10 min before a subsequent descent after 30 min. (B) Similarly, LTX-315-treated cells displayed an increase in cytochrome c release related to incubation time with a significant amount in the supernatant after 30 and 60 min. (C) Rat TMSCs treated with LTX-315 showed a translocation of the HMGB1 protein from the lysate (L) to the supernatant (S) using Western blot. Control cells showed no translocation after 60 min. All experiments were performed in triplicate, and quantitative data are presented as a mean ± SD.
Intratumoral injections with LTX-315 induce a complete regression and systemic immune responses against rTMSCs
To investigate the effect of LTX-315 in the rTMSC model, we injected LTX-315 into established subcutaneous lesions in PVG rats (Fig. 3A). The efficacy of LTX-315 treatment was monitored by bioluminescence imaging (BLI) of luciferase-transfected rTMSCs, including the measurement of tumor size. In 6 out of 6 rats receiving intratumoral treatment with LTX-315, tumor tissue necrosis was observed and complete regression (CR) was obtained 20 d after tumor inoculation. In control rats receiving saline injections, luciferase activities reflecting tumor growth gradually increased with time until termination at day 9 (Fig. 3B and C).
Figure 3.

Intratumoral administration of LTX-315 inhibits rTMSC growth (A). The time schedule of LTX-315 injections and tumor rechallenge experiments. Tumors were established by an s.c. inoculation of 5·105 rTMSCs in the right flank of syngeneic PVG rats. On days 3, 5, 6 and 7 after tumor inoculation, experimental rats (n = 6) were given an i.t. injection of 1 mg LTX-315. Age-matched control rats (n = 6) received injections of saline in parallel. Antitumor responses were assessed by bioluminescence imaging (BLI) of luciferase-expressing tumor cells, images were acquired at the indicated days post-tumor inoculation. (B) Representative whole-body images of rats during treatment with LTX-315 or saline vehicle control. Total luciferase activity is summed up and depicted in figure (C), showing tumor progression and response to LTX-315 treatment. The bold line refers to treated rats, while the dashed line represents the pattern for saline vehicle. A mean ± SD in the treatment group is compared with the controls. The 6 treated rats designated as tumor-free were rechallenged s.c. with 2·104 rTMSCs in the opposite flank in week 6, while other naïve rats were included as controls (n = 6). (D) Representative images of previously treated rats and controls after the second s.c. challenge with tumor cells and (E) graph charting the tumor growth. Values are expressed as a mean ± SD of hexaplicates in treated and control rats. The experimental animals survived the second challenge without signs of tumor growth, and were exposed to a third challenge of tumor cells in week 13 administered by an i.p. inoculation of 2·105 rTMSCs. A control group was included (n = 6). (F) Representative images of animals initially treated with LTX-315 after i.p. challenge, including control rats. (G) Graph charting intraperitoneal tumor progression in treated and control rats. The data are the mean ± SD of hexaplicates.
We then examined whether persistent protective immune responses could be achieved after LTX-315 treatment by rechallenging the rats s.c. with rTMSCs in the opposite flank 6 weeks posttreatment. In 6 out of 6 animals previously cured by LTX-315, tumor growth was inhibited and the rats were tumor-free on day 6, whereas the control rats developed tumors and were killed on day 6 (Fig. 3D and E). Similar results were obtained with rTMCS cells that were not transfected with dual reporter gene luciferase and GFP (data not shown).
To assess whether LTX-315 was able to elicit a systemic antitumor response, rats were given a third challenge i.p. 13 weeks post-initial treatment. After tumor cell implantation, spread bioluminescence activities were observed in the peritoneal cavity. The luciferase activities gradually decreased until day 8, as the tumor cells were eradicated in 6 out of 6 animals previously treated with LTX-315. In contrast, progressive tumor growth in the peritoneal cavities was observed in control rats, terminated on day 8 (Fig. 3D and E). The data demonstrate that LTX-315 treatment induced a complete regression and protective systemic immune responses against rTMSCs.
LTX-315 treatment induces long-term persistence of protective immune responses
To evaluate whether durable immunologic memory responses were developed in the treated rats, the rats were rechallenged both s.c. and i.p. with rTMSCs 60 weeks after treatment (Fig. 4A). In 8 out of 8 rats initially cured by LTX-315, rapid tumor regression occurred in the subcutaneous site (Fig. 4B and C) and in the peritoneal cavity (Fig. 4D and E), whereas the control rats developed subcutaneous and peritoneal tumors and were killed on day 18. The inhibition of tumor growth in long-term survivors suggests that intratumoral LTX-315 treatment induced long-term protective systemic immune responses.
Figure 4.
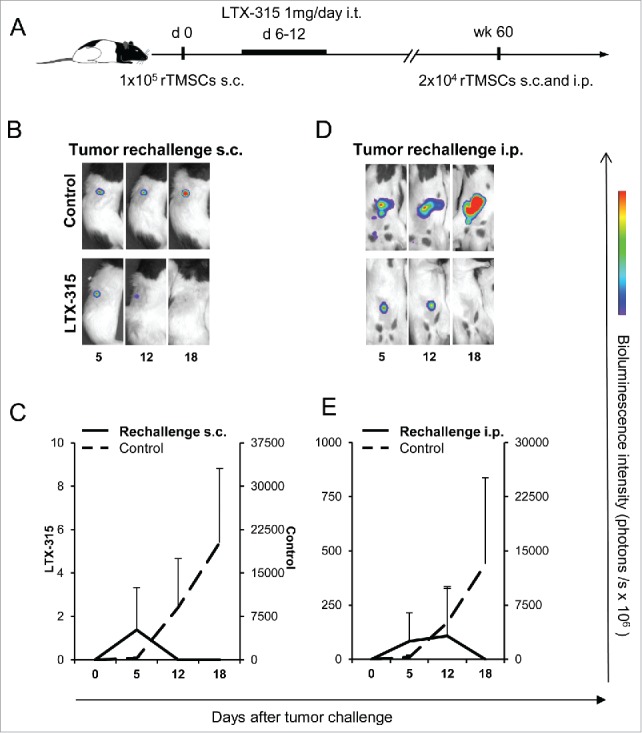
Initial LTX-315 treatment elicits long-term tumor protection. (A) Time schedule of experiment to study long-term effects of LTX-315 therapy. Rats were s.c. inoculated with 1·105 rTMSCs in the upper flank. At day 6, rats in the treatment group (n = 8) received an i.t. injection of 1 mg LTX-315 daily for 7 consecutive days. Rats in the control group (n = 6) remained untreated, and were killed after gross tumor development. The LTX-315 treated rats were considered cured and housed for 60 weeks before s.c. and i.p. rechallenge with 2 × 104 rTMSCs. Other control rats (n = 6 for each tumor site) were included. Representative luminescence images after (B) s.c. and (D) i.p. tumor rechallenge, total photon flux is summarized (mean ± SD) and depicted (C, E).
Local LTX-315 treatment causes complete regression of non-treated tumors at distant sites
Based on the results of the rTMSC rechallenge experiments and our recent findings that LTX-315 could induce a strong systemic protective immune response in the B16 melanoma model,10 we aimed to investigate whether the intratumoral administration of LTX-315 could induce effects against non-targeted lesions. For the evaluation of this hypothesis, we established tumors at 3 different sites to mimic tumor cell dissemination and the formation of metastasis (Fig. 5A). A tumor in one flank, defined as the primary site, received i.t. injections of LTX-315. The second lesion in the opposite flank and a third lesion within the peritoneal cavity were left untreated. In 12 out of 12 rats, i.t. injections of LTX-315 eradicated the primary lesion within 11 d after tumor cell inoculation (Fig. 5B and C). Complete regression of the secondary lesion and the third peritoneal tumor were observed on day 19 (Fig. 5D and E) and day 9 (Fig. 5F and G), respectively. Control rats developed tumors and were terminated on day 9 (primary lesion), day 19 (secondary lesion) and day 14 (peritoneal tumor). We observed that LTX-315 treatment eradicated both the treated lesion and the disseminated tumors, thereby indicating a strong and immediate systemic effect of LTX-315 therapy.
Figure 5.

The abscopal effect of LTX-315 in a 3-tumor model. (A) Tumor cells (2·105 rTMSCs) were inoculated on day ÷2 s.c. in the right flank (primary treated lesion), day 0 (1·104 rTMSCs) s.c. in the contralateral flank (secondary non-treated lesion) and (1·104 rTMSCs) i.p. (tertiary non-treated lesion) into PVG rats (n = 12). The primary lesion was treated with 1 mg of LTX-315 i.t. daily for 7 d. Control rats (n = 3) for each tumor site were included. Representative whole-body bioluminescence images of LTX-315 treated and untreated rats; (B) primary lesion, (D) secondary lesion and (F) peritoneal tumor are presented. The mean ± SD of tumor burden is plotted (treated; bold line, untreated; dashed line) for the primary (C), the secondary (E) and the third (G) tumor. Results are presented as a mean ± SD.
Adoptive transfer of splenocytes from donors cured by LTX-315 protects against rTMSC tumor growth
We further examined whether the protective immune response induced by LTX-315 could be transferred to naïve recipients by adoptive transfer of splenocytes from cured rats (Fig. 6A). The lymphoid compartment of recipient animals was depleted by total body irradiation (TBI) before adoptive transfer, while the transfer of splenic lymphocytes from cured rats inhibited subcutaneous tumor growth in 15 out of 15 recipients (Fig. 6B). In contrast, 5 out of 5 rats receiving splenocytes from naïve animals were not protected and were terminated due to gross tumor size. Thus, protective immune responses against rTMSCs can be adoptively transferred from previously treated rats to naïve recipients.
Figure 6.
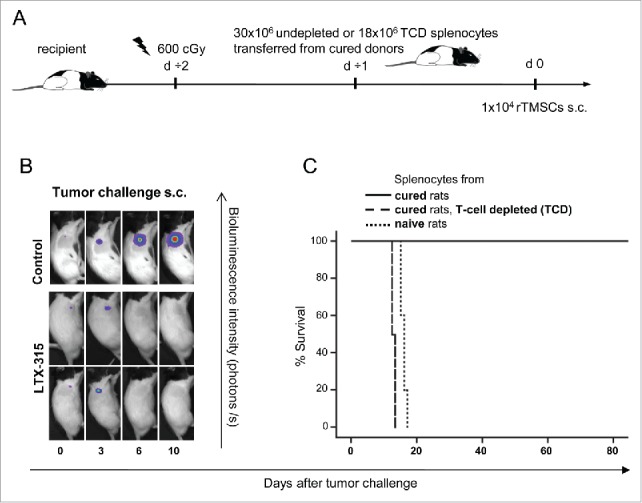
T cells originated from adoptively transferred splenocytes of donors cured by LTX-315 and inhibit the development of rTMSCs in naïve recipients. (A) Recipient rats were conditioned with total body irradiation (TBI) of 600 cGy one day before adoptive spleen cell transfer. Surviving rats were rechallenged twice after the initial LTX-315 treatment was used as a splenocyte donor. A group of rats (n = 15) received isolated spleen cells (30·106) from cured rats. Another group of rats (n = 6) were given T-cell depleted (TCD) splenocytes (18·106) from cured donors, whereas spleen cells (30·106) from naïve rats were transferred to rats in the control group (n = 5). One day after splenocyte transfer, animals were s.c. inoculated with 1·104 rTMSCs. The measurement of tumor development was tracked by bioluminescence imaging. (B) Representative images of recipient rats receiving spleen cells from cured donors (LTX-315), or from naïve donors (control) at designated days after tumor challenge. (C) Survival curve comparing tumor-free survival in animals receiving splenocytes from cured donors (solid line), TCD fraction of splenocytes (dashed line) or splenocytes from naïve donors (dotted line).
To determine whether the growth-inhibitory response in recipient rats receiving splenocytes from cured donors could be attributed to T cells, a group of rats were given T-cell-depleted (TCD) spleen cells. The transfer of TCD splenocytes did not protect against tumor development in 6 out of 6 rats, and there were only minute differences in survival between recipients receiving TCD spleen cells (12. 5 ± 0.5 days) and those transferred with splenocytes from naïve rats (15.8 ± 0.8 days) (Fig. 6C). The lack of tumor inhibition in the recipients of TCD splenocytes from cured donors suggests an essential protective role for T cells in LTX-315 mediated-antitumor immune responses.
The protective immune response induced by LTX-315 is tumor-specific
After the adoptive transfer of splenocytes from animals cured by LTX-315, the specificity of the antitumor immune responses was investigated by challenging the recipient rats with rTMSCs in one flank and a different syngeneic tumor cell line, Roser's T-cell leukemia (RL) cells, in the contralateral flank (Fig. 7A). In 3 out of 3 rats, we observed a rapid and complete regression of the rTMSC tumor, while the RL tumor in the opposite flank developed and grew large (Fig. 7B). A flow cytometric analysis of the harvested RL tumors confirmed a dominant leukemic CD3+CD4+ cell population (Fig. 7C), distinct from tumor-infiltrating lymphocytes (TILs) in rTMSC tumors (Fig. 8B). The growth inhibition of the rTMSC tumor, coincident with the tumor progression of the irrelevant RL tumor, indicates that intratumoral LTX-315 treatment leads to the induction of tumor-specific immune responses.
Figure 7.

Anti-tumor immune responses induced by LTX-315 are tumor-specific. (A) After the adoptive transfer of splenocytes from cured rats, surviving rats (n = 3) were s.c. rechallenged with 1·105 rTMSCs in the right flank and subsequently s.c. inoculated with a different syngeneic tumor cell line, Roser's T-cell leukemia (RL, 1·105 cells) in the contralateral flank. Tumor growth was monitored by measurements of tumor diameter using calipers. (B) A schematic presentation of growth curves (mean ± SD), demonstrating specificity and the inhibition of rTMSC growth. On day 30, when the left-flank RL tumors reached 100 mm2 in size, the tumors were resected and processed for flow cytometric characterization. (C) Representative dot plot showing the morphological characteristics of single-cell suspensions from RL tumors. Gating on the lymphocyte population and staining with anti-CD3 and anti-NKR-P1A revealed the frequency of T (CD3+ NKR-P1A−) cells. Phenotypic analysis of the T cell fraction is shown in the histograms, indicating the percentage of CD4+ and CD8+ T cells. A standard 4-h 51 Cr release assay was performed to quantitatively measure the cytolytic effect of tumor-specific T cells. T cells were enriched from the spleens of long-term survivors (week 60) after the adaptive transfer of tumor-reactive splenocytes. One naïve rat was included as a control. Without in vitro activation, isolated T cells were tested in cytotoxicity assays at a varying effector: target (E: T) ratios against (D) rTMSCs and (E) RL target cells. The results are reported as a mean of cpm ± SD of technical triplicates. ****P < 0.0001, with Student's t test.
Figure 8.

Infiltrating lymphocytes are recruited into the tumor after LTX-315 treatment. (A) Schedule of experiment to study cell infiltration and modification of cell composition in tumors after LTX-315 treatment. Rats were s.c. inoculated with 2·105 rTMSCs in one flank day ÷2 and 2·104 rTMSCs in the opposite flank on day 0. Rats (n = 8) with pre-established tumors were given i.t. injections of 1 mg LTX-315 daily for 3 subsequent days. Tumor-bearing rats in the control group (n = 6) received saline injections in parallel. Single-cell suspensions were prepared from fresh tumor tissue at different time points during the week after LTX-315 treatment. (B) Representative dot plots of the morphological characteristics (SSC vs. FSC) exhibited by progressing tumors and LTX-315-treated tumors. Schematic illustration of gating: Tumor-infiltrating lymphocytes determined by forward and side scatter properties, stained with relevant antibodies for flow cytometric analysis to identify NK (NKR-P1A+CD3−) cell and T (CD3+NKR-P1A−) cell populations, including CD4+ and CD8+ T cell subsets. The percentage of Tregs was derived from the percentage of FoxP3+ cells within the total CD4+ T cell population. (C) Bar graphs showing the percentage of TIL subsets in primary treated lesion (light gray bars), secondary lesion (dark gray bars) of treated rats compared with lesions from the control group (black bars). Graphs show the mean ± SD. *P < 0.5, **P < 0.1 with Student's t test. (D) Representative images of primary and secondary tumors from treated rats vs. untreated controls stained for CD3 or CD8 (brown) and counterstained with DAPI (blue).
To further evaluate the specificity of the immune response, we measured the cytotoxic activity of isolated T cells from previously LTX-315-treated rats compared with naïve controls against rTMSCs and the irrelevant RL cells using a 4 h 51Cr release assay (Fig. 7D and E). Without in vitro stimulation, T cells obtained from the spleens of cured animals were able to recognize and lyse rTMSC targets. Significant rTMSC-specific cytolysis of T cells were noted compared with T cells from naïve animals (effector: target ratio of 100:1, cured rats; 9.73 ± 1.70, naïve rats, 1.07 ± 0.85, P < 0.0001, 50:1, cured rats; 5.16 ± 1.18, naïve rats; 0.00 ± 0.0, P < 0.0001), whereas negligible cytotoxic activity against RL targets was observed. When purified T cells from naïve spleens were used as effector cells, no or negligible cytolytic activity against rTMSCs or RL cells was detected. Taken together, both the in vivo and the in vitro data confirm that T cells were responsible for the persistent tumor-specific immune responses in recipients transferred with splenocytes from LTX-315-cured donors.
LTX-315 treatment leads to increased tumor infiltration of CD8+ T cells
To investigate the cellular mechanisms underlying LTX-315-mediated regression and long-term protective immune responses, we analyzed the cellular composition of treated tumors by flow cytometry and immunohistochemistry. During the week after the last treatment, tumors were resected at different time points, and thereafter TILs were phenotyped in both LTX-315-treated and untreated animals (Fig. 8A). The gating strategy for T (CD3+NKR−P1A−) cells, CD4+ and CD8+ T cells, CD4+FoxP3+T cells and NK (NKR-P1A+CD3−) cells is shown (Fig. 8B).
Tumor-infiltrating lymphocytes in response to growing tumors were observed in untreated animals (Fig. 8C). A significantly higher proportion of NK cells were also present in progressive untreated tumors versus treated regressive tumors (untreated; 9.28% ± 6.22, treated; 2.26 ± 0.89, P < 0.5), which may reflect an NK-mediated early immune response in progressive tumors. The spontaneous infiltration of lymphocytes was insufficient to inhibit tumor growth, and tumor control was dependent on adaptive immunity and the accumulation of CD8+ T cells in the tumor microenvironment. The percentage of T cells was significantly increased after LTX-315 treatment compared with untreated tumors (treated; 28.35 ± 11.78, untreated; 13.58 ± 7.84, P < 0.5). Similarly, the percentage of T cells within secondary tumor tissue increased 3-fold compared with untreated rats (treated; 39.83 ± 17.4, untreated; 11.83 ± 10.25, P < 0.01). Within the CD3+T cell population, CD8+cells accounted for the major fraction in primary and secondary tumors of treated rats vs. untreated controls (primary tumor of treated rats; 62.77 ± 13.3, untreated rats; 37.33 ± 3.48, P < 0.01, secondary tumor of treated rats; 45.8 ± 5.4, untreated rats; 34.67 ± 3.36, P < 0.1). There was no significant difference in the relative percentage of infiltrating CD4+ T cells and CD4+Foxp3+ regulatory T cells (Tregs) between the primary tumors of treated and untreated animals. In the secondary tumors of untreated animals, we found a higher percentage of CD4+T cells compared with treated rats (treated; 38.0 ± 4.0, untreated; 54.7 ± 9.0, P < 0.01) and Tregs (treated; 2.21 ± 1.43, untreated; 5.17 ± 2.50, P < 0.5). In line with other reports, the higher percentage of Tregs in secondary lesions in untreated vs. treated rats is associated with immune suppression and poor prognosis in many types of solid tumors.14
Akin to untreated rats, a FACS analysis of LTX-315-treated rats revealed significantly elevated levels of CD3+ and CD8+ tumor-infiltrating T cells, the major immune effector cell population15 that correlated with tumor regression. Immunohistochemical analysis was consistent with the flow cytometry data demonstrating an increased infiltration of CD3+ and CD8+ cells observed in primary and secondary tumor tissues from LTX-315-treated rats (Fig. 8D).
Discussion
The identification, isolation and characterization of CSCs in various types of cancers expedite the development of targeted therapeutic strategies aiming to eradicate CSCs to prevent relapse and metastasis.16,17 The new class of local immunotherapy for cancer, oncolytic peptides, gain more selectivity against cancer cells vs. normal cells due to the higher abundance of anionic membrane components of cancer cells compared with normal cells.18 Moreover, oncolytic peptides are equally effective against cancer cells displaying dormancy and drug resistance. We have previously reported the design of an oncolytic peptide LTX-315, which induces membrane permeabilization and interaction with intracellular targets such as mitochondria, ultimately resulting in tumor cell lysis.5,6,8,9,19 Intratumoral treatment with LTX-315 induces complete tumor regression and tumor-specific immune protection in a variety of different experimental animal models.10,11 In the present study, the oncolytic effects of LTX-315 were investigated in rTMSC tumors composed of heterogeneous population of cells with various degrees of differentiation.12 Intratumoral treatment with LTX-315 caused a complete regression (CR) in all treated animals, and no adverse effects on behavior and physiology were observed. The generation of long-term and systemic immune responses was verified by tumor rejection and protection from rTMSCs after rechallenging rats up to 14 months after the initial LTX-315 treatment.
The abscopal effect, a rare event usually observed during localized radiation therapy or combination therapy,20-22 was further evaluated in a 3-tumor model. Upon treatment of a single tumor with LTX-315, rapid tumor regression was observed not only in the treated lesion, but also in the untreated, distant lesions. The abscopal response, associated with a systemic anti-tumor response to local therapy, was clearly T-cell-mediated. Whereas rTMSC growth could be impeded by adoptively transferred splenocytes from LTX-315-cured animals, TCD splenocytes did not affect tumor growth. The tumor-specificity of the splenic T cells was demonstrated by the in vivo and in vitro inhibition of rTMSCs, concurrently with the lack of tumor suppression and cytotoxicity vs. an irrelevant, syngeneic (RL) tumor cell line.
LTX-315 exerted rapid kill kinetics compared with conventional chemotherapeutic drugs against the rTMSC cell line in vitro, hence demonstrating the lytic mode of action of the peptide. The cytolytic activity and immunomodulatory properties of LTX-315 have recently been described (8, 9, 19, 23, 24), involving the perturbation of both the cell membrane and the mitochondria accompanied by the release of DAMPS such as ATP, Cytochrome c and HMGB1, thus signaling tissue damage to the surrounding tumor microenvironment. The LTX-315-mediated tumor cell lysis and subsequent release of membrane-bound and intracellular components might augment the level of tumor-associated antigens and neoantigens available for T-cell priming by dendritic cells. Of note, aberrant mitochondrial RNA transcripts may contribute to changes associated with the phenotype of a transformed cell, and may therefore serve as a source of tumor antigen recognized by T cells.25 This process of immunogenic cell death (ICD) may turn the LTX-315-treated tumors into in situ vaccines eliciting tumor-specific T cells.26,27
The impact of the host immune cells in the tumor microenvironment has emerged as a critical determinant of tumor depression or progression and the response to therapy.28,29 The conventional classification of solid tumors (TNM) summarizes data on tumor (T) nodes (N) and metastatic sites (M), and pays no attention to the effect of the immune responses following therapy.30 The Immunoscore concept considers the interactions between tumor cells and host immune cells, and uses measurement of the type, number and location of tumor-infiltrating immune cells.31 Accumulating evidence indicates that the density and ratio of tumor-infiltrating T cells appear to be prognostic factors for clinical outcomes.32
Immunohistochemistry analyses combined with flow cytometry were performed to examine the phenotype of TILs in LTX-315-treated and untreated tumor sites. Tumors injected with vehicle exhibited a viable tissue with limited necrosis and a low frequency of TILs. The intratumoral injection of LTX-315 induced extensive tumor necrosis and promoted a substantial infiltration of CD8+T cells into the tumor environment and parenchyma. The abscopal response was reflected in the increase of cytotoxic T cells in distant, untreated lesions. The presence of high densities of cytotoxic T cells correlated with tumor regression in the treated animals. Flow cytometry analysis verified the significantly increased CD8+ T-cell infiltration in both the primary and secondary tumor site, indicating that LTX-315 provokes and mediates the migration of tumor-specific T cells into distant untreated sites, thereby intervening the process of metastasis formation. In monitoring the anti-tumor effects of LTX-315, we observed increased levels of TILs and alterations in the composition of the tumor T cell subsets. A novel approach in cancer immunotherapy refers to tumors as hot or cold, depending on how densely they are infiltrated in immune cells.33 In a cold tumor, the recruitment of DCs and cytotoxic T cells is insufficient to mount an effective anti-tumor response.34
Hot tumors with high frequencies of tumor-infiltrating cells and consistent antigen exposure may escape an efficient immune response by the upregulation of checkpoint inhibitory ligands. Antibody-based checkpoint inhibitors (CPI) have emerged as a promising approach in targeting negative T -cell regulators.35 Antibodies blocking the suppression of T-cell receptor signaling through cytotoxic lymphocyte antigen 4 (CTLA-4), or interfering with T-cell exhaustion via programmed cell death 1 receptors (PD-1), have the potential to reinvigorate an antitumor immune response. Cold tumors are resistant to checkpoint inhibitors alone, leaving a large fraction of patients unresponsive to immune checkpoint therapy. The therapeutic benefit is achieved in hot tumors with pre-existing tumor-infiltrating CD8+ T cells.36
The present study in the rat fibrosarcoma model indicates that LTX-315 therapy-induced ICD can convert cold (low Immunoscore) tumors, including distant, untreated tumors, to hot (high Immunoscore) tumors. Indeed, the application of CTLA-4, in combination with LTX-315, demonstrated a synergistic effect in a murine model.11 Therefore, it is likely that LTX-315 may synergize with checkpoint inhibitors and other treatment combinations to help mount a persistent anti-tumor immune response. To the establishment of platforms for combination therapies, LTX-315 provides a treatment regimen optimized for both cytotoxic and immune activating effects. LTX-315 is currently being tested in combination with anti-CTLA-4 in anti-PD-1 refractory patients in melanoma patients, and with anti-PD1 in breast cancer patients.
Material and methods
Cell lines and reagents
Following the long-term culturing of bone marrow-derived rat mesenchymal cells, a transformed mesenchymal cell (rTMSC) line with maintained stemness properties was developed. The cell line formed soft-tissue fibrosarcomas in immunocompromised mice and syngeneic immunocompetent rats.12
Roser's leukemia is a transplantable radiation-induced acute T-cell leukemia induced in PVG rats.37
LTX-315 (K-K-W-W-K-K-W-Dip-K-NH2) was purchased from Bachem AG and prepared on request. Dip is the aromatic non-coded amino acid β-diphenylalanine. Dacarbazine (D2390), temozolomide (T2577) and cis-diammineplatinum(II) dichloride (P4394) were all purchased from Sigma-Aldrich.
In vitro cytotoxicity
A colorimetric 3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyltetrazodium bromide (MTT) viability assay38 was used to assess the in vitro cytotoxicity of LTX-315, as well as a selection of cytostatic drugs against the rTMSC cell line. Pre-cultured cells were transferred to a 96-well plate in subconfluent concentrations at a volume of 100 μl/well (culture media). Cells were left overnight in a cell incubator under conditions of 37 °C; >95% humidity and 5% CO2. Wells were plated in triplicate. LTX-315 and the different cytostatic drugs were dissolved in a serum-free RPMI 1640 and diluted to a concentration range of 7–100 μM and 10–100 μM, respectively. The cytostatic drugs were originally dissolved in dimethyl sulfoxide (DMSO) and later in a serum-free RPMI 1640, yielding a final DMSO concentration ranging from 0.5% in the 100 μM solution to 0.05% in the 10 μM solution. Cells were washed once with serum-free RPMI 1640 before incubation with the peptide solutions or cytostatic drugs for 2, 4, 8, 24 and 48 h, respectively. Cells in a serum-free media alone were used as a negative control, while cells treated with 1% Triton X-100 (Sigma-Aldrich) in a serum-free media were positive controls. A 10 μl MTT solution (5 mg MTT per ml phosphate buffered saline) was added to each well, and the incubation was continued for an additional 2 h. Seventy microliters of solution were removed from each well, and 100 μl of 0.04 M HCl in isopropanol was added before the plates were shaken in an orbital shaker for 1 h at room temperature to help facilitate formazan crystal solubilization. The absorbance was measured at 590 nm on a microtiter plate reader (Thermomax Molecular Devices). The percentage of viability was calculated as the A590 nm of peptide-treated cells relative to the negative control (100% living cells), and expressed as a 50% inhibitory concentration (IC50).
Release of ATP
The supernatant of LTX-315-treated rTMSCs was analyzed using an ENLITEN ATP luciferase assay system (Promega, FF2000). Cells were seeded (1 × 104/well) in a 96-well plate, and treated with the IC50 concentration of LTX-315 (17 μM) for varying time points (5, 10, 30 and 60 min). All experiments were performed in duplicate a total of 3 times. Negative controls were untreated rTMSCs exposed to a serum-free medium alone. Samples were analyzed using a Luminoscan RT luminometer according to the manufacturer's protocol.
Release of cytochrome c
Rat TMSCs were seeded (5 × 103/well) in a 96-well plate, and treated with 17 μM LTX-315 for different time points (5, 10, 30 and 60 min). Supernatants were collected and concentrated as with the HMGB1 release experiments. Samples were analyzed using a Quantikine ELISA Rat/Mouse Cytochrome c Elisa kit (R&D Systems, MCTC0) following the manufacturer's instructions, and samples were run in duplicate for a total of 3 experiments and expressed as ng/ml cytochrome c released.
Release of HMGB1
Rat TMSCs (2 × 105/well) were seeded in a 6-well plate, treated with 17 μM LTX-315 (IC50) and incubated at 37 °C, 5% CO2 and >95% humidity for different time points (5–60 min). Serum-free RPMI 1640 was used as a negative control. Supernatants were collected after centrifugation at 1,400 g for 5 min and up-concentrated using Amicon Ultra-0.5 Centrifugal Filter units with Ultracel-50 membrane (Millipore). Cell lysates were washed with serum-free RPMI 1640 and collected in a Mastermix containing a 2x NuPAGE LDS Sample buffer (Invitrogen), a 1x NuPAGE Sample Reducing Agent (Invitrogen) and 50% sterile H2O. Supernatants and cell lysates were boiled in a reducing NuPAGE LDS sample buffer, resolved on NuPAGE Novex 4–12% Bis–Tris Gels, and electrotransferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore). Membranes were hybridized with HMGB1 antibody (rabbit polyclonal to HMGB1-ChIP Grade; Abcam, ab18256), followed by a horseradish peroxidase (HRP)-conjugated secondary antibody (goat polyclonal anti-rabbit IgG; Abcam, ab6721) and developed by Western blot luminol reagent (Santa Cruz Biotechnology) according to the manufacturer's instructions.
Animal experiments
Rats of the inbred Piebald Virol Glaxo (PVG.RT7a, abbreviated PVG) strain were used interchangeably with the PVG.RT7b strain (in this study abbreviated as PVG). The strains are identical except for one irrelevant epitope of the leukocyte common antigen (LCA/CD45) family. PVG rats were purchased from Harlan (the Netherlands) and PVG, and7B rats were obtained from in-house breeding at the Institute of Basic Medical Sciences (IMB, University of Oslo, Norway). During the experiments, male rats, weighing 240–270 g each, were kept in groups of 2 to 3 animals per cage under climate-controlled conditions, with 12 h light/dark cycles and an ambient temperature. The rats were housed in an enriched individually ventilated cage (IVC) system with free access to standard rodent chow and water ad libitum. The animals were anesthetized during the experimental procedures with either 2.5% Isoflurane gas (Baxter Medical AB) or received s.c. injections (0.4 ml/kg) of fentanyl/fluanisone (Hypnorm; VetaPharma Ltd.), which provided a sufficient degree of sedation and analgesia. The animals were monitored daily and large-tumor-bearing rats were killed with CO2. All procedures performed were conducted under FOTS numbers 1957 and 5917, and approved by the Experimental Animal Board under the Ministry of Agriculture of Norway and in compliance with The European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes. The laboratory animal facilities are subjected to a routine health-monitoring program, and were screened for common pathogens according to a modification of the Federation of European Laboratory Animal Science Association's recommendation.
Tumor treatment
Pre-cultured rTMSCs were harvested in a serum-free RPMI-1640 and s.c. inoculated into the right flank of PVG rats. Established tumors (25 mm2 mean tumor size) were i.t. injected with LTX-315 (dissolved in sterile H2O with 0.9% NaCl) or with vehicle (sterile H2O with 0.9% NaCl). Treatment doses of LTX-315, 50μl at 20 mg/ml, were given using a 0.5 ml syringe (Myjector U-100; Terumo) needle 0.5 × 16 mm (Fine-Ject; Henke-Sass, Wolf GmbH). Tumor size was measured 3 times a week using a caliper, and expressed as the area of an ellipse [(maximum dimension/2) x (minimum dimension/2)]. Animals were terminated when the tumor exceeded 400 mm2.
Whole imaging technique
The rTMSCs were engineered with a retroviral vector (kindly provided by Dr. Rainer Löw, EUFETS AG, Germany) to express firefly luciferase and EGFP. After transfection with rTMSCs, the GFP-positive cells were collected by FACS sorting, and were used for all in vivo experiments. Tumor growth was monitored by bioluminescent imaging using the IVIS 200 Spectrum CT System (Caliper Life Sciences). Anesthetized rats were i.p. injected with a 150 mg kg−1 body weight of D-luciferin (Biosynth). After 10 min, the luminescence emitted from the luciferase rTMSCs was scanned by designing an auto program, whereas data was analyzed with the Living Image v4.4 software (Caliper Life Science).
Preparation of single cell suspensions from solid tumors
Tumors were gently minced with a razor blade, and cut into small pieces (4 mm2). Tumor tissue was incubated with Liberase TM (Thermolysin Medium; Roche Diagnostics) at a concentration of 0.18 Wünsch units/ml in 10 ml MEM media (Sigma-Aldrich) at 37°C for 60 min with gentle agitation. The enzymatic digestion was terminated by the addition of 2 ml 4°C FCS (Invitrogen). The cell suspension was filtered through a 70 μM mesh (Cell Strainer; BD), washed in PBS, and cells were then directly used for flow cytometric staining.
Antibodies and FACS analysis
The mouse monoclonal antibodies against CD4 (OX38), CD5 (OX19), CD6 (OX52) and CD8 (OX38) were isolated from culture supernatants from hybridomas, and were kind gifts from the MRC Cellular Immunology Unit, Oxford, UK. They were conjugated at IMB according to standard protocols. Fluorochrome-conjugated mAbs against CD3 (G4.18, 559975) and NKR-P1A (10/78, 555009) were obtained from BD Biosciences, including PerCP Streptavidin (554064). For the Treg analysis, cells were surface stained for CD3 and CD4, then permeabilized and stained for FoxP3 using Anti-mouse/rat Foxp3 Staining Set (eBioscience, 72–5775–40). A 4-color panel consisting of reagents in FITC/ Al488, PE, PerCP Streptavidin and Al647 was applied and analyzed on a FACS Calibur (BD) equipped with the CellQuest software (BD). Dot plot and histogram gates were set using isotype control antibodies.
Immunohistochemistry
Formalin-fixed and paraffin-embedded tissue sections were deparaffinized in xylene and graded alcohols, hydrated and washed in PBS. After antigen retrieval in a sodium citrate buffer (pH 6) in a microwave oven, the endogenous peroxidase was blocked by 0.3% H2O2 for 15 min. Sections were incubated overnight at 4 °C with primary antibody, rabbit polyclonal anti-CD3 (clone A0452, Dako) or mouse monoclonal anti-CD8 (ab33786, Abcam). As a secondary antibody, the anti-rabbit-horseradish peroxidase (HRP) SuperPicTure Polymer detection kit (Invitrogen) or Envision system HRP-anti-mouse (Dako) was used. A matched isotype control was used as a control for nonspecific background staining.
Splenocyte isolation and adoptive transfer
The animals were killed, and spleens from treated and naïve rats were harvested through abdominal incision. Splenocytes were isolated by density gradient centrifugation with Lymphoprep according to the manufacturer's instruction (Axis-Shield PoC). A fraction of splenocytes was T-cell depleted (TCD) using the MACS cell separation system (Miltenyi Biotec). The splenocytes were incubated with saturating concentrations of biotinylated mAbs, anti-CD5 (OX19) and anti-CD6 (OX52). After antibody-staining, cells were selected using appropriated diluted anti-biotin microbeads. Labeled cells were separated using the magnetic cell separator QuadroMACS. Isolated T cells were collected from the bound fraction, while TCD splenocytes were obtained from the unbound fraction of the LS column.
One day before the adoptive transfer of splenocytes, the rats were conditioned with a total body irradiation (TBI) of 600 cGy from a 137Cs source (Gammacell 3000 Elan, MDS Nordion). Isolated splenocytes were resuspended in PBS and adjusted to a concentration of 30·106 cells/ml or 18·106 cells/ml (TCD). The day after, rats were s.c. inoculated with tumor cells.
In vitro T cell cytotoxicity assay
Freshly isolated splenic CD3+ T cells from long-term survivors without further in vitro activation were assessed for cytolytic activity against rTMSCs and RL cells in a 4-h 51Cr-release assay. Target cells (10·106 cells ml-1) were incubated with 3.7 MBq of Na251CrO4 ml-1 for 1 h at 37°C. 51Cr-labeled target cells (1·105 cells ml-1) and serial dilutions of effector cells at the indicated E:T ratios were plated in 100µl of a complete RPMI 1640 in U-bottomed 96-well plates.51 Cr-release was measured after incubation for 4 h at 37°C. Supernatants were harvested with a Titertek harvesting system (Skatron), and counted in a gamma counter (Beckmann). The percentages lysis was determined using the formula (experimental cpm - spontaneous cpm) x 100/ (maximum cpm - spontaneous cpm). Spontaneous cpm was measured by incubating targets in a medium alone, and was <15% of total cpm.
Statistics
Data are expressed as a mean ± SD. Survival was estimated with the Kaplan–Meier and log-rank tests for survival distribution. Statistical differences between the 2 groups were analyzed by a 2-tailed Student's t test, and a P < 0.05 was considered to be statistically significant. Statistical analyses were performed using GraphPad Prism software (version 6, GraphPad).
Disclosure of potential conflicts of interest
The authors declare the following competing financial interest(s): B.S., K.A.C, B.E.H. and Ø.R. are shareholders in Lytix Biopharma. B.S. and Ø.R. are employed by Lytix Biopharma.
References
- 1.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002; 415(6870):389-95; PMID:11807545; https://doi.org/ 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- 2.Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 1999; 1462(1–2):55-70; https://doi.org/ 10.1016/S0005-2736(99)00200-X [DOI] [PubMed] [Google Scholar]
- 3.Gaspar D, Veiga AS, Castanho MARB. From antimicrobial to anticancer peptides. A review. Frontiers in Microbiology 2013; 4:294; https://doi.org/ 10.3389/fmicb.2013.00294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur J Pharmacol 2009; 625(1–3):190-4; PMID:19835863; https://doi.org/ 10.1016/j.ejphar.2009.08.043 [DOI] [PubMed] [Google Scholar]
- 5.Haug BE, Camilio KA, Eliassen LT, Stensen W, Svendsen JS, Berg K, Mortensen B, Serin G, Mirjolet JF, Bichat F, et al.. Discovery of a 9-mer Cationic Peptide (LTX-315) as a Potential First in Class Oncolytic Peptide. J Med Chem 2016; 59(7):2918-27; PMID:26982623; https://doi.org/ 10.1021/acs.jmedchem.5b02025 [DOI] [PubMed] [Google Scholar]
- 6.Camilio KA, Rekdal Ø, Sveinbjörnsson B. LTX-315 (Oncopore™): A short synthetic anticancer peptide and novel immunotherapeutic agent. Oncoimmunology 2014; 3:e29181; PMID:25083333; https://doi.org/ 10.4161/onci.29181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mader JS, Salsman J, Conrad DM, Hoskin DW. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol Cancer Ther 2005; 4(4):612-24; PMID:15827335; https://doi.org/ 10.1158/1535-7163.MCT-04-0077 [DOI] [PubMed] [Google Scholar]
- 8.Eike L-M, Yang N, Rekdal Ø, Sveinbjørnsson B. The oncolytic peptide LTX-315 induces cell death and DAMP release by mitochondria distortion in human melanoma cells. Oncotarget 2015; 6(33):34910-23; PMID:26472184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H, Forveille S, Sauvat A, Yamazaki T, Senovilla L, Ma Y, Liu P, Yang H, Bezu L, Müller K, et al.. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis 2016; 7(3):e2134; https://doi.org/ 10.1038/cddis.2016.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camilio KA, Berge G, Ravuri CS, Rekdal Ø, Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunology, Immunotherapy 2014; 63(6):601-13; https://doi.org/ 10.1007/s00262-014-1540-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki T, Pitt JM, Vetizou M, Marabelle A, Flores C, Rekdal O, Kroemer G, Zitvogel L. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ 2016; 23(6):1004-15; PMID:27082453; https://doi.org/ 10.1038/cdd.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang M-Y, Nestvold J, Rekdal Ø, Kvalheim G, Fodstad Ø. A novel rat fibrosarcoma cell line from transformed bone marrow-derived mesenchymal stem cells with maintained in vitro and in vivo stemness properties. Exp Cell Res 2017; 352(2):218-24; PMID:28189639; https://doi.org/ 10.1016/j.yexcr.2017.02.005 [DOI] [PubMed] [Google Scholar]
- 13.Kim Y, Joo KM, Jin J, Nam D-H. Cancer Stem Cells and Their Mechanism of Chemo-Radiation Resistance. Int J Stem Cells 2009; 2(2):109-14; PMID:24855529; https://doi.org/ 10.15283/ijsc.2009.2.2.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shang B, Liu Y, Jiang S-j, Liu Y. Prognostic value of tumor-infiltrating FoxP3(+) regulatory T cells in cancers: A systematic review and meta-analysis. Sci Rep 2015; 5:15179; PMID:26462617; https://doi.org/ 10.1038/srep15179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gooden MJM, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br J Cancer 2011; 105(1):93-103; PMID:21629244; https://doi.org/ 10.1038/bjc.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan Q, Li Q, Liu S, Ning N, Zhang X, Xu Y, Chang AE, Wicha MS. Targeting cancer stem cells using immunologic approaches. Stem cells (Dayton, Ohio) 2015; 33(7):2085-92; PMID:25873269; https://doi.org/ 10.1002/stem.2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K, Huang Y-h, Chen J-l. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol Sin 2013; 34(6):732-40; PMID:23685952; https://doi.org/ 10.1038/aps.2013.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris F, Dennison SR, Singh J, Phoenix DA. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med Res Rev 2013; 33(1):190-234; PMID:21922503; https://doi.org/ 10.1002/med.20252 [DOI] [PubMed] [Google Scholar]
- 19.Zhou H, Forveille S, Sauvat A, Sica V, Izzo V, Durand S, Müller K, Liu P, Zitvogel L, Rekdal Ø, et al.. The oncolytic peptide LTX-315 kills cancer cells through Bax/Bak-regulated mitochondrial membrane permeabilization. Oncotarget 2015; 6(29):26599-614; PMID:26378049; https://doi.org/ 10.18632/oncotarget.5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mole RH. Whole Body Irradiation—Radiobiology or Medicine? Br J Radiol 1953; 26(305):234-41; PMID:13042090; https://doi.org/ 10.1259/0007-1285-26-305-234 [DOI] [PubMed] [Google Scholar]
- 21.Levy A, Chargari C, Marabelle A, Perfettini J-L, Magné N, Deutsch E. Can immunostimulatory agents enhance the abscopal effect of radiotherapy? Eur J Cancer 2016; 62:36-45; https://doi.org/ 10.1016/j.ejca.2016.03.067 [DOI] [PubMed] [Google Scholar]
- 22.Demaria S, Formenti SC. Can abscopal effects of local radiotherapy be predicted by modeling T cell trafficking? J Immunother Cancer 2016; 4:29; https://doi.org/ 10.1186/s40425-016-0133-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forveille S, Zhou H, Sauvat A, Bezu L, Müller K, Liu P, Zitvogel L, Pierron G, Rekdal Ø, Kepp O, et al.. The oncolytic peptide LTX-315 triggers necrotic cell death. Cell Cycle 2015; 14(21):3506-12; PMID:26566869; https://doi.org/ 10.1080/15384101.2015.1093710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sistigu A, Manic G, Vitale I. LTX-315, CAPtivating immunity with necrosis. Cell Cycle 2016; 15(9):1176-7; PMID:26986689; https://doi.org/ 10.1080/15384101.2016.1160609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voo KS, Zeng G, Mu J-B, Zhou J, Su X-Z, Wang R-F. CD4+ T-Cell Response to Mitochondrial Cytochrome in Human Melanoma. Cancer Res 2006; 66(11):5919-26; PMID:16740732; https://doi.org/ 10.1158/0008-5472.CAN-05-4574 [DOI] [PubMed] [Google Scholar]
- 26.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al.. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3(9):e955691; PMID:25941621; https://doi.org/ 10.4161/21624011.2014.955691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017; 17(2):97-111; PMID:27748397; https://doi.org/ 10.1038/nri.2016.107 [DOI] [PubMed] [Google Scholar]
- 28.Emens LA, Silverstein SC, Khleif S, Marincola FM, Galon J. Toward integrative cancer immunotherapy: Targeting the tumor microenvironment. J Transl Med 2012; 10(1):70; PMID:22490302; https://doi.org/ 10.1186/1479-5876-10-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedl P, Alexander S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell. 2011 Nov 23; 147(5):992-1009; doi: 10.1016/j.cell.2011.11.016 [DOI] [PubMed] [Google Scholar]
- 30.Broussard EK, Disis ML. TNM Staging in Colorectal Cancer: T Is for T Cell and M Is for Memory. J Clin Oncology 2011; 29(6):601-3; https://doi.org/ 10.1200/JCO.2010.32.9078 [DOI] [PubMed] [Google Scholar]
- 31.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, Lugli A, Zlobec I, Hartmann A, Bifulco C, et al.. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathology 2014; 232(2):199-209; PMID: 24122236; https://doi.org/ 10.1002/path.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer 2012; 12(4):298-306; PMID:22419253; https://doi.org/ 10.1038/nrc3245 [DOI] [PubMed] [Google Scholar]
- 33.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al.. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006; 313(5795):1960-4; PMID:17008531; https://doi.org/ 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 34.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515(7528):568-71; PMID:25428505; https://doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12(4):252-64; PMID:22437870; https://doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14(10):1014-22; PMID:24048123; https://doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nestvold J, Stokland A, Naper C, Rolstad B. Phenotype and Natural Killer Cell Sensitivity of a Radiation-Induced Acute T-Cell Leukaemia (Roser Leukaemia) in PVG Rats. Scand J Immunol 2004; 60(1–2):153-8; PMID:15238084; https://doi.org/ 10.1111/j.0300-9475.2004.01436.x [DOI] [PubMed] [Google Scholar]
- 38.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65(1):55-63; PMID:6606682; https://doi.org/ 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]