

ABSTRACT
The identification of universal tumor-specific antigens shared between multiple patients and/or multiple tumors is of great importance to overcome the practical limitations of personalized cancer immunotherapy. Recent studies support the involvement of DEPDC1 in many aspects of cancer traits, such as cell proliferation, resistance to induction of apoptosis and cell invasion, suggesting that it may play key roles in the oncogenic process. In this study, we report that DEPDC1 expression is upregulated in most types of human tumors, and closely linked to a poorer prognosis; therefore, it might be regarded as a novel universal oncoantigen potentially suitable for targeting many different cancers. In this regard, we report the identification of a HLA-A*0201 allele-restricted immunogenic DEPDC1-derived epitope, which is able to induce cytotoxic T lymphocytes (CTL) exerting a strong and specific functional response in vitro toward not only peptide-loaded cells but also triple negative breast cancer (TNBC) cells endogenously expressing the DEPDC1 protein. Such CTL are also therapeutically active against human TNBC xenografts in vivo upon adoptive transfer in immunodeficient mice. Overall, these data provide evidence that this DEPDC1-derived antigenic epitope can be exploited as a new tool for developing immunotherapeutic strategies for HLA-A*0201 patients with TNBC, and potentially many other cancers.
KEYWORDS: Cancer immunotherapy, cytotoxic T lymphocytes, DEPDC1, tumor antigen, triple negative breast cancer
Abbreviations
- aa
amino acid
- aAPC
artificial antigen-presenting cells
- BLI
bioluminescence
- CAR
chimeric-antigen receptor
- CTL
cytotoxic T lymphocytes
- DC
dendritic cells
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HLA
human leucocyte antigen
- HS
human serum
- ICI
immune checkpoint inhibitors
- IFN-gamma
interferonγ
- LCL
lymphoblastoid cell line
- luc
luciferase
- MFI
mean fluorescence intensity
- MHC
major histocompatibility complex
- NK cells
natural killer cells
- NSG
NOD/SCID gamma
- PBMC
peripheral blood mononuclear cells
- PMA
phorbol 12-myristate 13-acetate
- rhIL-2
recombinant human interleukin 2
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- TCR
T-cell receptor
- TNBC
triple negative breast cancer
- TSA
tumor-specific antigens
Introduction
In recent years, the field of cancer immunotherapy has considerably expanded with the introduction of several new treatment options, including cancer vaccines, adoptive cell transfer, chimeric-antigen receptor (CAR) T-cell therapy, monoclonal antibodies and immune checkpoint inhibitors (ICI).1 Although some patients with advanced cancer may respond to single-agent immunotherapy, for the majority, however, monotherapy is relatively ineffective.2 Hence, multiple combined therapeutic approaches are likely required to achieve complete remission and cure.
As tumors grow, they accumulate mutations leading to potential generation of neoantigens that influence the response of patients to ICI. Indeed, T cells recognizing clonal neoantigens were detected in non-small cell lung cancer and melanoma patients with durable clinical benefits after therapy with anti-PD-1 and anti-CTLA-4 blockades, suggesting that therapy with ICI enhances neoantigen-specific T-cell reactivity.3-5 Accordingly, the combination therapy of ICI with cancer vaccines or the adoptive transfer of enriched populations of neoantigen-reactive T cells may function synergistically to induce more effective antitumor immune responses.6 For example, in patients not responding to ICI because of the lack of tumor-infiltrating lymphocytes, cancer vaccines may induce effector T-cell infiltration into the tumors.7 These findings support therapeutic developments targeting clonal neoantigens to design personalized immunotherapies to treat patients with advanced cancer.
Despite this evidence, the identification of expressed nonsynonymous somatic mutations by whole-exome sequencing of tumor and normal DNA, which allows the selection of candidate neoepitopes to be exploited for developing personalized immunotherapies, is typically unwieldy, time-consuming and expensive.8 To overcome the practical limitations of personalized cancer immunotherapy, the identification of universal tumor-specific antigens (TSA) shared between multiple patients and/or multiple tumors has been gaining renewed interest and great importance. Furthermore, the selection of TSAs playing a key role in tumor cell proliferation and survival is considered crucial to overcome immune escape, as their downregulation or loss is expected to impair tumor progression.9 In this regard, encouraging results have been achieved in clinical trials, such as a recent pilot study in leukemia patients exploring the use of allogenic CD8+ T cells with activity against the Wilms tumor antigen 1 (WT1).10 Similar approaches have been applied in ovarian cancer11 and melanoma.12
Among the potential TSAs, the poorly characterized DEPDC1 protein is a very promising one. First identified by Kanehira et al.,13 DEPDC1 (Dishevelled EGL-10 and Pleckstrin domain containing 1) has been shown to be significantly overexpressed in a great majority of bladder cancer cells, but not present in normal human tissues except the testis. While the biologic functions of DEPDC1 are still far from being completely understood, a few published reports have focused on the role played in inhibiting tumor cell growth, activating anti-apoptotic pathways14-17 and regulating mitotic progression.18 Interestingly, DEPDC1 has been reported to be a direct downstream target of the mutant p53 pathway, and it is a member of a group of genes relevant in the regulation of the migration and invasion of breast cancer cells, and therefore in the enhancement of tumor aggressiveness.19 Collectively, these studies support the involvement of DEPDC1 in many aspects of cancer biology, such as cell proliferation, resistance to induction of apoptosis and cell invasion, suggesting that it may play key roles in the oncogenic process.
Based on these observations, the clinical exploitation of DEPDC1 as an immunological target for vaccination in HLA-A*2402 subjects has started both in bladder cancer patients,,2021 and patients with gastrointestinal, lung or cervical tumors,22 with promising results.
Here, we report that DEPDC1 expression is upregulated in most types of human tumors, and particularly well represented in triple negative breast cancer (TNBC), an aggressive form of neoplasia that occurs in approximately 15% of all breast cancer patients23 and still lacks proper therapeutic solutions. Together with a leading role in tumorigenesis, the broad and cancer-specific expression of DEPDC1 suggests that it might be regarded as a novel universal oncoantigen potentially suitable for targeting many different cancers. In this regard, we also report the identification of an immunogenic DEPDC1-derived epitope restricted to the HLA-A*0201 molecule, which can foster a strong and specific CTL response in vitro. Such CTLs recognize the endogenously processed and presented DEPDC1-derived peptide on tumor cells, and are therapeutically active against human TNBC xenografts in vivo upon adoptive transfer. Thus, this DEPDC1-derived antigenic epitope can represent a new tool for the development of immunotherapeutic strategies for HLA-A*0201 patients with TNBC, and potentially many other cancers.
Results
DEPDC1 is widely expressed in tumors but not in normal tissues
The Oncomine database was interrogated for DEPDC1 gene expression.24,25 Fourteen independent data sets referred to different tumor histotypes showed an upregulation of DEPDC1 mRNA levels in primary cancers, as compared with normal counterpart tissues (p = 2.15E−7), (Fig. 1A).26-38 Moreover, DEPDC1 expression turned out to be associated with pathologic and prognostic parameters independently from tumor type. Indeed, advanced stage (Table S1) and high-grade tumors (Table S2) overexpressed DEPDC1 mRNA as compared with early stage or low-grade tumors, respectively. Additionally, higher DEPDC1 mRNA levels were detected in primary tumor tissues from patients with metastatic events than in patients with no recurrence, and were related to a worst overall survival (Table S3). Noteworthy, 13 independent data sets of human TNBC showed overexpression of DEPDC1 compared with other breast cancer histotypes (Table S4).
Figure 1.

DEPDC1 is upregulated in different human cancers. (A) DEPDC1 mRNA expression in normal (white) and tumor tissues (gray) as reported from microarray studies in the Oncomine database. Breast: (t-test = 10.953; p-value = 1.181E−14);26 bladder (t-test = 6.217; p-value = 8.82E−9);27 brain (t-test = 9.929; p-value = 7.56E−12);28 cervix (t-test = 7.688; p-value = 9.13E−9);29 colon (t-test = 3.978; p-value = 9.98E−5);30 esophagus (t-test = 10.994; p-value = 8.17E−19);31 stomach (t-test = 7.378; p-value = 4.54E−10);32 nasopharynx (t-test = 5.832; p-value = 1.71E−6);33 leukemia (t-test = 4.400; p-value = 8.57E−4);34 liver (t-test = 6.180; p-value = 2.18E−7);35 lung (t-test = 16.310; p-value = 7.19E−18);36 skin (t-test = 5.874; p-value = 3.54E−5);37 ovary (t-test = 14.643; p-value = 2.12E−7) (TCGA Ovarian, No Associated Paper, 2013); pancreas (t-test = 4.794; p-value = 8.57E−6).38 Numbers above each box plot refer to the number of samples reported. (B) Endogenous expression of DEPDC1 protein in several human tumor cell lines (left panels) and in different normal human tissues (right panels), as assessed by western blot analysis. MDA-MB-231shDEPDC1 refers to cells with DEPDC1 silenced by shRNA, and served as an internal negative control.
Consistent with these findings, DEPDC1 protein was found to be expressed in a large set of human tumor cell lines of different histotypes, such as breast, bladder, brain, cervix, colon, stomach, leukemia, liver, lung and melanoma cell lines (Fig. 1B), thus supporting the concept that DEPDC1 can be regarded as a potential universal tumor-associated antigen, whose expression is strictly linked to the neoplastic status. Conversely, DEPDC1 was not found to be expressed in a set of normal different human tissues (bladder, breast, cervix, kidney, ovary, placenta, prostate and uterus; Fig. 1B), consistent with data reported by Kanehira et al.13 We failed, however, to visualize DEPDC1 protein in testis, even though its specific mRNA had been previously detected by Northern blot, albeit at low levels.13
In silico prediction of HLA-A*0201-restricted DEPDC1-derived peptides and assessment of their MHC stabilizing properties
DEPDC1 amino acid (aa) sequence was analyzed for potential HLA-A*0201-restricted peptides using three epitope prediction algorithms available online, namely BIMAS, NetMHC and NetCTL. Different nine-aa peptides were classified according to their predicted ability to stabilize the MHC allele. The 10 peptides with the highest prediction score were chosen and synthesized for further studies (Table 1). To evaluate their ability to stabilize the HLA-A*0201 allele, the DEPDC1-derived peptides were incubated with antigen-processing deficient T2 cells. Then, HLA-A*0201 expression levels were measured and compared with those induced by incubation of T2 cells with an irrelevant peptide (Table 1 and Fig. S1). DEPDC1-derived peptides induced different levels of HLA-A*0201 expression, being DEPDC1#1 and DEPDC1#5 those with the highest ability to stabilize the MHC molecule.
Table 1.
Candidate peptides derived from DEPDC1 sequence, their predicted binding affinities to HLA-A*0201 and stabilization ratios.
| Peptide | Start position | Peptide sequence | BIMAS score | NetMHC affinity (nm) | NetCTL score | Stabilization ratio on T2 cells |
|---|---|---|---|---|---|---|
| DEPDC1#1 | 673 | FLMDHHQEI | 728.022 | 6.05 | 1.4641 | 3.7 |
| DEPDC1#2 | 580 | SMLTGTQSL | 57.085 | 20.15 | 1.2956 | 2.6 |
| DEPDC1#3 | 588 | LLQPHLERV | 133.255 | 47.31 | 1.2127 | 2.5 |
| DEPDC1#4 | 574 | SLLPASSML | 79.041 | 37.72 | 1.1822 | 2.5 |
| DEPDC1#5 | 282 | FLDLPEPLL | 39.307 | 21.60 | 1.1503 | 2.9 |
| DEPDC1#6 | 289 | LLTFEYYEL | 54.474 | 41.23 | 1.1172 | 2.3 |
| DEPDC1#7 | 524 | YINTPVAEI | 15.177 | 165.46 | 1.0234 | 2.0 |
| DEPDC1#8 | 786 | ALFGDKPTI | 38.601 | 83.19 | 1.0145 | 1.3 |
| DEPDC1#9 | 618 | LLMRMISRM | 71.872 | 58.86 | 1.0108 | 2.1 |
| DEPDC1#10 | 562 | RLCKSTIEL | 21.362 | 127.36 | 0.9906 | 1.2 |
DEPDC1#5 peptide induces interferon-gamma (IFNγ) production in peptide-stimulated T-cell cultures
Monocytes were obtained from peripheral blood mononuclear cells (PBMC) of healthy donors, and were induced to differentiate into dendritic cells (DC; data not shown) to act as antigen-presenting cells for T-cell stimulation. The lymphocytes isolated from the PBMC of the same healthy donors were immunomagnetically enriched for CD8+ T cells (80 ± 1.5% of total lymphocytes), being the remaining contaminant CD4+ T cell subset only fractional (7.6 ± 5.9%). To induce the expansion of antigen-reactive T-cell populations, CD8+ T cells were stimulated weekly by autologous DC pulsed with DEPDC1-derived peptides. At the third round of stimulation, T cell cultures were screened for IFNγ production in response to HLA-A*0201-positive lymphoblastoid cells (LCL) pre-loaded with different DEPDC1-derived peptides. The most responsive CD8+ population was observed after stimulation with the DEPDC1#5 peptide. LCL cells loaded with an irrelevant peptide (HLA-A*0201-restricted Gag-1777–85, SLYNTVATL) or left unpulsed did not induce IFNγ production (Figs. 2 and S2). Taken together, these results led us to focus on and further characterize DEPDC1#5 peptide-stimulated T-cell cultures. After three stimulation rounds the bulk population was essentially composed of CD8+ T cells (79.4 ± 1.5%; n = 24 donors, Fig. 3A), mainly characterized by an effector memory phenotype (78.48 ± 7% of CCR7−/CD45RA−; n = 11 donors; Fig. 3B). Natural killer (NK) cells were essentially undetectable in these cultures. DEPDC1#5 peptide-specific CD8+ T cells were quantified by tetramer staining throughout the culture period. The amount of CD8+ T cells expressing a DEPDC1#5/HLA-A*0201-specific TCR reached a peak (1 ± 0.5%) after three in vitro stimulations, and then slightly declined after the fourth stimulation (Figs. 3C and S3).
Figure 2.
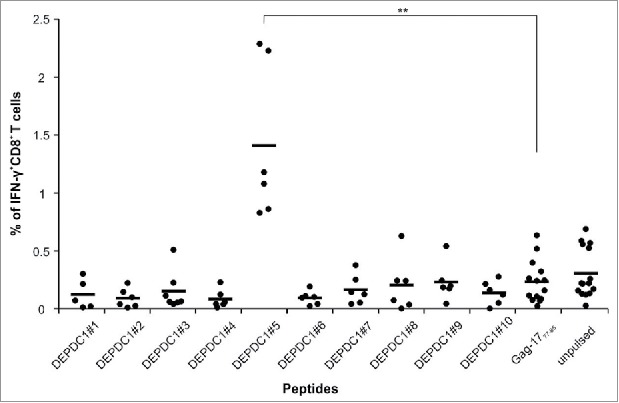
Identification of DEPDC1-derived immunogenic epitopes. The IFNγ production from CD8+ T cells stimulated for three times with autologous DCs pulsed with each DEPDC1-derived peptide was measured in response to unpulsed or pulsed LCL cells, by cytokine intracellular staining. LCL cells loaded with Gag-1777–85 peptide served as a negative control. Data are presented as the percentage of CD8+ T cells positive for IFNγ staining (**p < 0.01; not statistically significant (p > 0.05) if not indicated).
Figure 3.
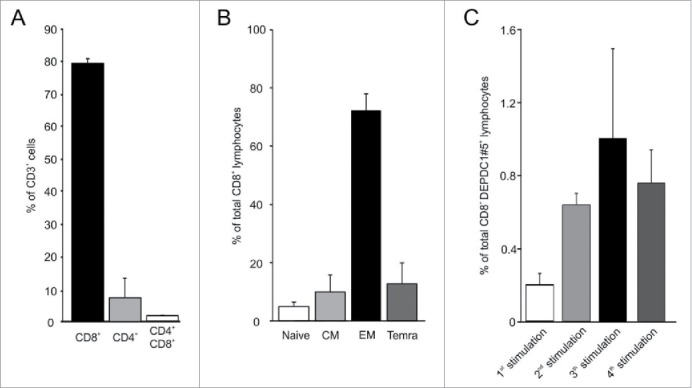
Characterization of DEPDC1#5 peptide-stimulated T cell cultures. (A) Flow cytometry analysis of T cells subsets in cell cultures after three stimulations with autologous DC pulsed with DEPDC1#5 peptide. The mean percentage and standard deviation are shown (n = 24 healthy donors). (B) Percentages of CD8+ T cells within subsets defined by the expression of CD45RA and CCR7 differentiation markers (CD45RA+CCR7+ naive, CD45RA−CCR7+ central memory (CM), CD45RA−CCR7− effector memory (EM) and CD45RA+CCR7− terminally differentiated effector memory (Temra) cells; n = 11 healthy donors)). (C) Tetramer staining of CD8+ T cells after sequential rounds of stimulation. The mean percentage ± SD of CD8+/tetramer+ lymphocytes in cultures is shown for each stimulation.
DEPDC1#5 peptide-stimulated CTL are HLA-A*0201-restricted, strictly antigen-specific and recognize an endogenously processed epitope in tumor cells
Antigen specificity of CTLs stimulated with DEPDC1#5 peptide was assessed against the MDA-MB-231 TNBC cell line, which is HLA-A*0201-positive and endogenously expresses DEPDC1 (Fig. 4A). Upon challenge with these cells, a consistent fraction of DEPDC1#5 peptide-induced CTLs was stimulated to produce IFNγ (Fig. 4B). On the other hand, the percentage of cytokine-producing CD8+ CTLs sharply dropped in response to MDA-MB-231 cells either pretreated with the anti-MHC class I W6/32 blocking antibody, or silenced for DEPDC1 expression by a specific short hairpin RNA (shDEPDC1). A control scrambled siRNA (shCTRL) produced no relevant effects on recognition. Additionally, a very limited IFNγ response was observed against the human embryonic kidney 293 cell line, which endogenously express DEPDC1 (Fig. 4A) but is devoid of the HLA-A*0201 molecule. Conversely, stable transfection of 293 cells with the HLA-A*0201 allele (Fig. S4) led to a prompt and sustained recognition of target cells, revealed by the increased percentage of IFNγ-producing CD8+ T cells (Fig. 4B).
Figure 4.
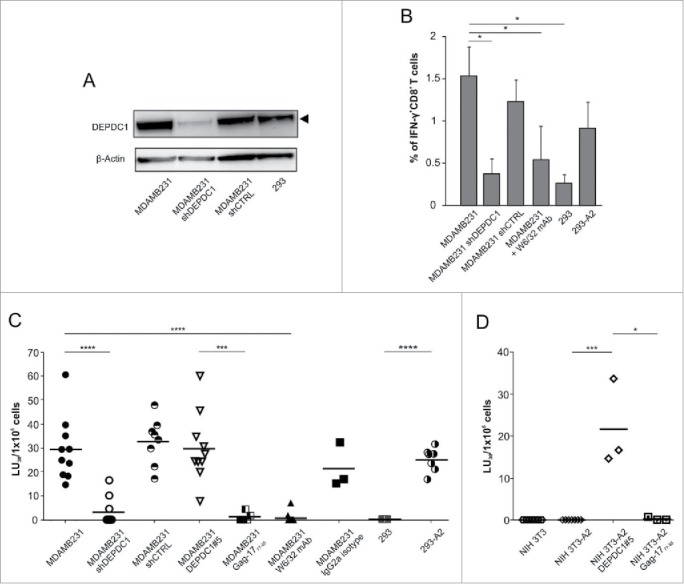
Functional characterization of DEPDC1#5 peptide-stimulated CTLs. (A) Western blot analysis of DEPDC1 protein expression in MDA-MB-231 and 293 cell lines. MDA-MB-231shDEPDC1 are silenced for DEPDC1, while shCTRL refer to cells silenced with a scrambled control siRNA. (B) Intracellular IFNγ staining of CD8+ T cells stimulated for three times with autologous DEPDC1#5 peptide-pulsed DC. Data are presented as the mean percentage ± SD of CD8+ T cells positive for intracellular IFNγ staining (*p< 0.05; n = 3 healthy donors). (C, D) Lytic activity and specificity of DEPDC1#5 peptide-stimulated CTL. Cytotoxicity of DEPDC1#5 peptide-stimulated CD8+ T cells generated as in (B) was analyzed by a 6-h chromium release assays against the reported targets. Results are expressed as LU30 × 106 effectors (*p < 0.05; ***p < 0.001; ****p < 0.0001; not statistically significant (p > 0.05) if not indicated).
In support of these data, the cytolytic activity of DEPDC1#5 peptide-stimulated CTLs was assessed in a classical 51Cr-release assay (Figs. 4C and S5A). In particular, CTLs exerted a high cytotoxicity against MDA-MB-231 cell line (Fig. 4C); such lytic effect was significantly reduced upon DEPDC1 silencing, while silencing with a shCTRL did not affect CTL-dependent cytotoxicity, thus confirming antigen specificity. To further demonstrate that CTL activity was specific for the DEPDC1#5 peptide, MDA-MB-231 cells were pulsed with DEPDC1#5 or Gag-1777–85 peptides, and tested for susceptibility to lysis. In this condition, exogenous peptides bind to HLA-A*0201 and replace the endogenously presented peptides. CTLs exhibited a significantly higher lytic activity against DEPDC1#5-pulsed MDA-MB-231 cells as compared with the same cells pulsed with the irrelevant HLA-A*0201-restricted peptide. As a confirmation of the HLA-A*0201 restriction, incubation of MDA-MB-231 cells with the W6/32 blocking antibody led to a significant reduction of cytotoxicity, an effect that was not observed with the addition of an isotype control antibody. Furthermore, DEPDC1#5 peptide-stimulated CTLs were significantly more cytotoxic against HLA-A*0201 transfected 293 cells, which acquire the ability to present DEPDC1-derived peptides in the context of the HLA-A*0201 molecule, compared with wild type 293 cells that lack the expression of this allele.
The critical aspects of peptide specificity and HLA recognition were further confirmed by assessing the lytic activity of DEPDC1#5 peptide-stimulated CTLs against the NIH 3T3 murine fibroblast cell line, in which neither human DEPDC1 nor HLA-A*0201 are endogenously expressed (data not shown and Fig. S4). CTLs did not disclose any relevant activity against wild type NIH 3T3 cells, but cytotoxicity significantly increased when target cells were simultaneously transfected with HLA-A*0201 (Fig. S4) and pulsed with DEPDC1#5 peptide; HLA-A*0201-transfected NIH 3T3 cells unloaded or pulsed with an irrelevant peptide were essentially not recognized (Fig. 4D).
On the other hand, the capacity of DEPDC1#5-stimulated CTLs to recognize the naturally processed and presented epitope was further validated using as targets additional tumor cell lines of different histotypes, namely A375 (melanoma), HepG2 (hepatocellular carcinoma) and U87MG (glioblastoma). Such cells turned out to be sensitive to killing (Fig. S5A), as expected from both DEPDC1 (Fig. 1B) and HLA-A*0201 (Fig. S5B) expression, with cell lysis apparently correlating with the expression levels of the HLA-A*0201 molecule.
DEPDC1#5 peptide-stimulated CTLs restrain tumor growth in vitro
To investigate the ability to induce long-term effects in vitro, DEPDC1#5 peptide-stimulated CTLs were co-cultured with MDA-MB-231 or NIH 3T3 in an outgrowth assay (Fig. 5). After 4 weeks, both MDA-MB-231 and NIH 3T3 reached the full confluency in the absence of lymphocytes. Conversely, the addition of DEPDC1#5 peptide-stimulated CTLs completely inhibited the growth of MDA-MB-231 cells, even at very low effector/target (E/T) ratios. Control CTLs stimulated with an irrelevant peptide were not able to repress MDA-MB-231 growth, and served as control for antigen specificity (Ctrl-CTL). No effects were observed on NIH 3T3 cell growth.
Figure 5.
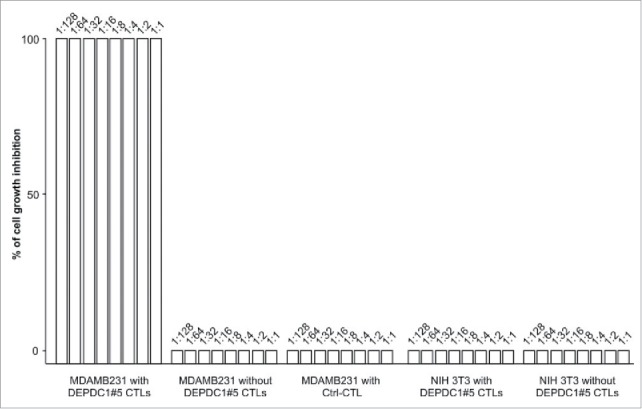
In vitro tumor growth restraining activity by DEPDC1#5 peptide-stimulated CTLs. The figure reports the inhibition of MDA-MB-231 outgrowth by DEPDC1#5 peptide-stimulated T cells after 4 weeks of culture. MDA-MB-231 cultured alone or in presence of control CTL (Ctrl-CTL), and NIH 3T3 cells alone or co-cultured with DEPDC1#5 peptide-stimulated CTL were used as negative controls. Results are expressed as percentage of target cell growth inhibition and the numbers above each bar refer to the target:CTL ratio (n = 3 healthy donors).
Adoptively transferred DEPDC1#5 peptide-stimulated CTLs inhibit breast cancer growth and restrain the metastasis process
Before proceeding with experiments aimed at assessing the in vivo therapeutic efficacy of anti-DEPDC1 T cells upon adoptive transfer, unrelated control effectors were generated. Thus, CTL populations directed against the HLA-A2-restricted Melan-A26–35*A27L peptide, which was derived from the melanocyte lineage-specific Melan-A protein expressed in most of primary and metastatic melanomas,39 were obtained following the same protocol used to generate DEPDC1#5 peptide-stimulated CTLs. The amount of CD8+ T cells expressing a Melan-A26–35*A27L/HLA-A*0201-specific TCR was quantified using tetramer staining throughout the culture period (25.7 ± 19.44% at the third round of stimulation, n = 3 healthy donors) (Fig. S6A and B). The cytotoxic activity of Melan-A26–35*A27L peptide-stimulated CTLs was assessed in a classical 51Cr-release assay to confirm that they did not recognize MDA-MB-231 cells alone or pre-loaded with the DEPDC1#5 peptide (Fig. S6C). Conversely, when breast cancer cells were pulsed with the Melan-A26–35*A27L peptide, the cytotoxicity was significantly increased.
Thereafter, the in vivo therapeutic efficacy of DEPDC1#5 peptide-stimulated and control CTLs was evaluated against MDA-MB-231 cells stably transduced with the firefly luciferase (luc) reporter gene, and injected into the mammary fat pad of NOD/SCID common γ chain knockout (NSG) female mice. Three days after inoculation, the tumor was detected by bioluminescence (BLI), and a group of mice (n = 9) received intra-tumor injections of DEPDC1#5 peptide-stimulated CTLs for a total of three doses, whereas another group (n = 8) received CTL cultures stimulated against the irrelevant Melan-A26–35*A27L peptide; the untreated mice (n = 18) were injected with PBS (Fig. 6A). While a progressive increase in tumor growth was observed in the two groups of control mice, DEPDC1#5 peptide-stimulated CTL treatment was able to delay the growth of primary tumor (Fig. 6B). When MDA-MB-231 cells are injected orthotopically, metastases are frequently observed 3–4 weeks later in the axillary lymph nodes and lungs.40-42 In this regard, mice treated with DEPDC1#5 peptide-stimulated CTLs showed a significant reduction in peripheral metastatic colonization as compared with untreated mice and to mice treated with Melan-A-specific CTLs, when examined at the same primary tumor size (Fig. 6C), thus clearly indicating that DEPDC1#5 peptide-stimulated CTLs were also effective in inhibiting metastases.
Figure 6.
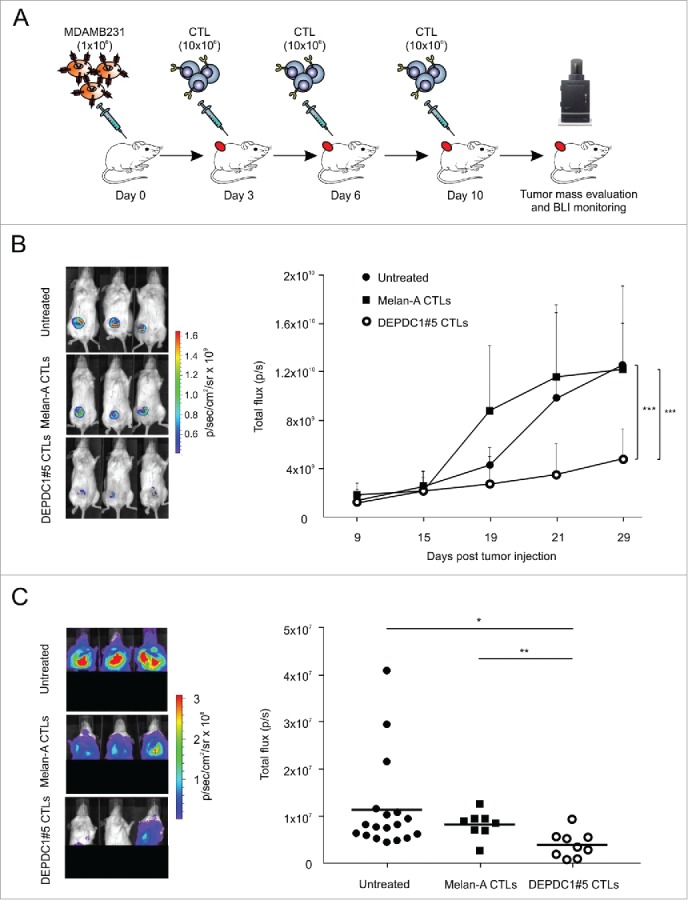
Assessment of therapeutic efficacy in vivo of DEPDC1#5 peptide-stimulated CTL. (A) On day 0, NSG mice were injected into the mammary fat pad with 1 × 106 luc-transduced MDA-MB-231 cells, and were treated intra-tumorally at days 3, 6 and 10 with 1 × 107 DEPDC1#5 peptide-stimulated CTLs (n = 9) or Melan-A peptide-stimulated CTL (n = 8). The untreated group of mice received three injections of PBS1X (n = 18). (B) Tumor growth was monitored by BLI measurement as photon flux. Left panels show the primary tumor BLI of three representative mice for each group after 29 d from tumor injection, while right panel reports tumor growth of all animals at different time points (mean of photons/second ± SD; ***p < 0.001; the ANOVA was used for statistical analysis). (C) Distant metastatic colonization was evaluated by BLI when a primary tumor size of about 700 mm3 was reached in each group. The left panels show the BLI of distant metastases in three representative mice for each group, while right panel reports the photons/second detected in each individual mouse belonging to the different groups (*p < 0.05; **p < 0.01).
Discussion
Tumor-specific mutations can be envisaged as ideal targets for cancer immunotherapy since they are not present in healthy tissues and can potentially be recognized as neoantigens by the T-cell repertoire. However, mutated protein-based personalized vaccines are hampered by the fact that in each patient the tumor possesses a unique set of mutations, which must be first identified with enormous time-consuming and expensive molecular screenings. Therefore, the identification of TSA shared by multiple patients and different types of cancers is likely destined to become again crucial for overcoming these problems and making immunotherapy universally applicable.
The involvement of DEPDC1 in many cancer traits suggests its key role in tumor cell growth and survival,13,43 hence supporting a low possibility of antigen downregulation. Therefore, we investigated DEPDC1 gene expression among human cancer tissues using the Oncomine database, and found that it is overexpressed almost ubiquitously in human cancers as compared with healthy counterpart tissues. This aspect, together with its involvement in the oncogenic process, strongly indicates that DEPDC1 can be regarded as a novel universal oncoantigen, potentially suitable for targeting many different tumors. Additionally, the pathologic and prognostic value of DEPDC1 upregulation is consistently supported by its association with the most common clinic-pathological variables associated to cancer aggressiveness, such as the presence of regional nodal metastasis, invasion of distant organs and presence of poorly differentiated or undifferentiated abnormal-looking cells, and a significant correlation was demonstrated with both overall and disease-free survival. According to the predominant DEPDC1 overexpression in most aggressive tumors, its upregulation was found to be particularly relevant in TNBCs, which are characterized by a poorer prognosis respect other forms of breast cancer, and associated with an increased risk of recurrence within 3 y from diagnosis and an increased 5-y mortality rate.23 In this regard, our study shows that the identified HLA-A*0201-restricted DEPDC1-derived peptide could be exploited for immunotherapy against TNBCs, which unfortunately still suffer from the lack of therapeutic options already available for other breast cancer subtypes.44
We decided to focus on the HLA-A*0201 allele as it is the most common HLA class I subtype in the Caucasian population and the second most common in the Japanese population.45,46 Therefore, 10 HLA-A*0201-restricted DEPDC1-derived peptides were identified by integrating the results of three epitope prediction programs based on the prediction of peptide binding affinity to MHC, on the probability of peptide proteasomal cleavage and on the efficiency of peptide transport trough the endoplasmic reticulum. One of the peptides tested had the ability to induce a specific and relevant functional CTL response against target cells; indeed, DEPDC1#5-stimulated CTLs produced IFNγ in presence of both target cells artificially loaded with the DEPDC1#5 peptide, and tumor cells endogenously expressing the DEPDC1 protein. Furthermore, despite the small percentage of tetramer-positive T cells, the DEPDC1#5-stimulated CTL populations exerted a relevant and specific cytotoxic activity against TNBC cells, indicating that they naturally process the DEPDC1 antigen and present the epitope in association with the HLA-A*0201 molecules, thus becoming susceptible to the CTL attack. These findings suggest that even a small fraction of antigen-specific T cells can exert a significant lytic function, likely due to the recycling potentialities of the effectors, ultimately leading to a marked enhancement of the overall activity of DEPDC1#5-specific lymphocytes.
Apparently, the functional response mediated by DEPDC1#5-stimulated CTLs was not a short-term effect, as completely inhibited in vitro breast cancer cell growth even at very low E/T ratios. Moreover, in a TNBC mouse model DEPDC1-stimulated CTL delayed breast tumor growth and reduced peripheral colonization despite the apparently limited number of DEPDC1#5-specific CD8+ T cells injected, thus demonstrating the effectiveness of the CTL therapy also in restraining the metastasis process. Conversely, CTL populations directed against the well-known immunogenic Melan-A protein,39 which is not expressed in MDA-MB-231 cells,47 had no therapeutic effects despite the higher amount of Melan-A26–35*A27L specific CD8+ T cells infused, thus excluding a possible therapeutic effect of non-specific lymphocytes.
The limited number of DEPDC1#5-specific T cells obtained after the in vitro stimulations could represent a potential limitation for ACT. Nonetheless, it must be considered that our DEPDC1#5-specific CTL cultures were generated from PBMC of healthy donors, where the precursor frequency is almost undetectable, while priming of DEPDC1-specific T cells could occur in tumor patients. In this regard, a preliminary survey on a very limited number of TNBC patients provided evidence of a potential response in one patient, thus suggesting the need of further studies. Alternatively, a higher number of DEPDC1#5-specific T cells could be obtained by stimulating cultures with a Rapid Expansion Protocol,48 or with cellular or acellular artificial Antigen Presenting Cells (aAPC),49 even though these protocols generate a large number of non-specific responders, the interactions between T cells and aAPC are weaker than those between the lymphocytes and the natural autologous APC.
Taking together, the results of this study emphasize the idea that DEPDC1 can be considered as a universal tumor antigen, and might represent a potential and safe target for cancer immunotherapy, as its overexpression is confined only in tumor cells and not in normal tissues. In support of this concept, results of three vaccination clinical studies based on a HLA-A24-restricted DEPDC1-derived peptide have shown a good tolerability and provision of clinical benefit to patients with both bladder cancer20,21 and other different solid tumors.22
Materials and methods
cDNA microarrays analysis
The Oncomine database, a repository for published cancer-profiling cDNA microarray data25 (http://www.oncomine.org), was explored for DEPDC1 mRNA expression in various human tumor tissues, and in the corresponding non-neoplastic tissues. Statistical analysis of the differences in DEPDC1 expression was accomplished through the use of the ONCOMINE algorithms, which allow multiple comparisons among different studies.24,50 The fold change and gene rank were defined as “all,” whereas the data type was restricted to mRNA. Only studies with results achieving a p <0.05 were considered.
Western blot
Total cell extracts from tumor cells were obtained with a buffer containing 50-mM Tris–HCl pH 7.5, 2-mM EDTA, 150-mM NaCl, 1% NP-40 and 1% protease inhibitor cocktail (Roche Biochemical Reagents). Protein concentration was determined by BCA Protein Assay Micro Kit (Serva Electrophoresis). Lysates were resolved by SDS-sample buffer (4% sodium dodecyl sulfate (SDS), 200-mM Tris–HCl pH 6.8, 3% β-Mercaptoethanol, 6% glycerol and 0.6% Bromophenol Blue, all from Sigma-Aldrich), boiled for 5 min before loading in NuPage Novex 4–12% Bis-Tris Midi Protein gels (Invitrogen), and then transferred to the nitrocellulose membrane (PerkinElmer). DEPDC1 expression was assessed in non-neoplastic samples using a commercially available tissue microarray (ProSci) containing nine normal human tissues (bladder, breast, cervix, kidney, ovary, placenta, prostate, testis and uterus). Western blot analysis was performed according to standard procedures using a rabbit anti-DEPDC1 polyclonal antibody (1:500, Abcam) and HRP-conjugated goat anti-rabbit IgG (diluted 1:3,000, Abcam) secondary antibody. The membrane was developed with ECL detection reagents (ThermoFisher Scientific) and visualized using chemiluminescence. Signal intensity was measured by a Bio-Rad XRS chemiluminescence detection system (Lyfe Science Group).
Cell lines
The following cell lines were used in this study: the human TNBC cell line MDA-MB-231 (HLA-A*0201+), and the related derivatives MDA-MB-231 shDEPDC1 and MDA-MB-231 shCTRL that have been stably transduced with a short hairpin RNA (siRNA) for DEPDC1 silencing or with a control siRNA, respectively; the human TNBC cell line MDA-MB-468; MCF-7, a human breast cancer cell line; the human bladder cancer cell line SW-780; U87MG, a human glioblastoma cell line; HeLa, a human cervix adenocarcinoma cell line; HCT-15, a human colorectal adenocarcinoma cell line; MKN-45, a human gastric cancer cell line; K562, a human chronic myelogenous leukemia cell line; HepG2, a human hepatocellular carcinoma cell line; A549, a human lung carcinoma cell line; A375, a human melanoma cell line; the human embryonic kidney cell line 293 (HLA-A*0201−) and 293-A2 [stably expressing the HLA-A*0201 molecule upon pcDNA3/neo plasmid-mediated transfection using Lipofectamine2000 (Invitrogen)]; the T2 cells (HLA-A*0201+), a TAP-deficient human hybrid B/T lymphoblastic cell line; the mouse fibroblast cell line NIH 3T3 and NIH 3T3-A2 (stably expressing HLA-A*0201 upon retroviral transduction). Cells were maintained in DMEM (MDA-MB-231, MDA-MB-468, MCF-7, SW-780, U87MG, HeLa, HepG2, A549, A375, 293 and NIH 3T3) or RPMI 1640 (HCT-15, MKN-45, K562, T2) medium (EuroClone) supplemented with 10% Fetal Bovine Serum (FBS, Gibco), 2 mM L-Glutamine, 10 mM HEPES, 100-U/mL Penicillin and 100 U/mL Streptomycin (all from Lonza), at 37 °C in a 5% CO2 atmosphere.
Peptide selection and synthesis
Several 9-mer HLA-A*0201 binding motifs from DEPDC1 protein sequence were selected based on a integration of their score using the HLA peptide binding prediction algorithms available at BIMAS (http://www-bimas.cit.nih.gov/molbio/hla_bind/),51 NetMHC (http://www.cbs.dtu.dk/services/NetMHC/)52 and NetCTL (www.cbs.dtu.dk/services/NetCTL/)53 websites. A total of 10 HLA-A*0201-restricted peptides were chosen. The DEPDC1-derived HLA-A*2402-restricted peptide EYYELFVNI served as negative control in the T2 stabilization assays. All peptides were synthesized by CRIBI (Padova, Italy) with a purity of 98% verified by mass spectrometry analysis. The peptide SLYNTVATL (HIV-1 p17 Gag, aa 77–85; named Gag-1777–85) was used as negative control in functional assays, as it was described previously as an optimal HLA-A2-restricted CTL epitope.54-56 The HLA-A2-restricted peptide ELAGIGILTV (Melan-A/MART-1 analog, aa 26–35*A27L; named Melan-A26–35*A27L) was used for the selection and expansion of Melan-A-specific T cells for in vivo experiments.39
T2 stabilization assay
Binding and stabilization of HLA-A*0201 molecule by DEPDC1-derived peptides was evaluated using T2 cells.57 T2 cells were stripped in 0.131 M citric acid, 0.066 M Na2HPO4 (pH 3.3) for 45 s, washed and resuspended in serum-free culture media. A total of 2 × 105 cells were incubated with 3 μg/mL β2-microglobulin (Sigma-Aldrich) and 100 μg/mL peptide in a final volume of 500 μL for 4 h at 37 °C. Cells were then washed and stained with the FITC-conjugated HLA-A*0201 monoclonal antibody BB7.2 (BioLegend) before cytometry evaluation (FACSCalibur, BD Biosciences). Stabilization was calculated by dividing the Mean Fluorescence Intensity (MFI) of peptide-pulsed T2 cells with the MFI of T2 cells loaded with a negative control peptide (EYYELFVNI) with no predicted binding affinity to HLA-A*0201.58
Generation of peptide-specific T cells from healthy donors
CD8+ T cells and monocyte-derived DC were obtained from peripheral blood of HLA-A*0201 positive healthy donors. PBMC were isolated by Ficoll-Plaque PLUS (GE Healthcare) density gradient centrifugation, and then separated by adherence to plastic culture flasks (Falcon) for 1.5 h at 37 °C in 5% CO2 to enrich the monocyte fraction. Non-adherent cells were collected and purified for CD8+ T cells using MACS CD8+ microbeads (Miltenyi Biotec) according to manufacturer's instructions. Isolated CD8+ T cells were cryopreserved until further use. The adherent fraction was differentiated into mature DC by culture in RPMI 1640 supplemented with 8% Human AB serum (HS; Aurogene), 2 mM L-Glutamine, 10 mM HEPES, 100 U/mL Penicillin, 100 U/mL Streptomycin and 50 μM 2β-mercaptoethanol for 7 d with the addition of 1,000 U/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 500 U/mL IL-4 (PeproTech). After 5 d, 1 μg/mL of LPS was added, and the DCs' mature phenotype at day 7 was confirmed by flow cytometry analysis of the expression of CD14 (clone TUK4; Miltenyi), CD83 (clone HB15; Miltenyi), CD86 (clone FM95; Miltenyi), CD80 (clone L307.4; BD Biosciences), CCR7 (clone G043H7; BioLegend), HLA-ABC (clone W6/32; BioLegend) and HLA-DR (clone L243; BioLegend) markers. Mature DCs were pulsed with 20 μg/mL of synthesized peptides in presence of 3 μg/mL β2-microglobulin (Sigma) in RPMI 1640 medium supplemented with 3% HS at 37 °C for 4 h. These peptide-pulsed DC were then irradiated (55 Gy) and mixed at 1:10 ratio with autologous CD8+ T cells in a complete medium supplemented with 8% HS, 10 ng/mL IL-7 (PeproTech) and 20 U/mL rhIL-2 (Proleukine, Novartis). After 2 d, half of medium was replaced with fresh medium supplemented with 100 U/mL rhIL-2. On days 7, 14 and 21, T cells were restimulated with the autologous peptide-pulsed DC as described above.
Intracellular cytokine staining for IFNγ detection
IFNγ production in response to peptide epitopes was measured by intracellular cytokine staining after blocking the cellular secretion using BD Cytofix/Cyotperm kit, according to the manufacturer's instructions. HLA-A*0201-positive LCL or tumor target cell lines were used as stimulators. LCL cells were pulsed overnight with each peptide (10 μg/mL), washed and incubated at 1:1 ratio with peptide-specific CTLs stimulated three times with autologous peptide-pulsed DC. The Gag-1777–85 peptide served as negative control, while phorbol 12-myristate 13-acetate (PMA; 40 ng/mL)/ionomycin (4 μg/mL) (Sigma Aldrich) as positive control. After 1 h, cellular cytokine secretion was blocked by the addiction of GolgiStop (2 μM) and the incubation was allowed to continue for 5 h at 37 °C. Cells were then stained with APC-conjugated anti-CD8a antibody (clone RPA-T8; BioLegend), washed and fixed. After permeabilization, cells were stained with a PE-conjugated anti-IFNγ antibody (clone B27; BioLegend) for 30 min at 4 °C, and flow cytometry analysis was performed (FACSCalibur, BD Biosciences).
MHC-biomonomer and MHC-tetramer preparation
The synthesis of MHC-peptide tetrameric complexes has been reported previously.59 Briefly, the MHC HLA-A*0201 heavy chain, β2m and epitope peptide (DEPDC1#5 or Melan-A26–35*A27L peptides) were subjected to a refolding in vitro in Tris–HCl pH 8, L-Arginine-HCl, NaEDTA, oxidized glutathione and reduced glutathione (Sigma-Aldrich). The complex was isolated through dialysis, concentrated and purified by HPLC to separate monomers from unconjugated components. Monomers were then enzymatically biotinylated by the enzyme BirA. The biomonomers obtained were purified and then quantified with a spectrophotometer. The tetramers were finally assembled with PE-conjugated extravidin (Sigma-Aldrich) for subsequent use in flow cytometry analysis.
Peptide-stimulated T-cell characterization
T cells selected and expanded in vitro were characterized using the following fluorochrome-conjugated monoclonal antibodies: APC-conjugated CD3 (clone UCHT1; BioLegend) and CD8+ (clone RPA-T8; BioLegend); FITC-conjugated CD16 (clone 3G8; BioLegend) and CCR7 (clone G043H7; BioLegend); PerCP-Cy5.5-conjugated CD56 (clone B159; BD Biosciences) and CD45RA (clone HI100; BioLegend); BV421-conjugated CD8+ (clone RPA-T8; BD Biosciences); APC-H7-conjugated CD4+ (clone RPA-T4; BD Biosciences); PE-conjugated CD4+ (clone RPA-T4; BioLegend). For the tetramer staining, CTLs were incubated with the PE-conjugated HLA-A*0201-DEPDC1#5 or PE-conjugated HLA-A*0201-Melan-A26–35*A27L tetramers. Samples were analyzed by an LSRII flow cytometer (BD Biosciences) and evaluated with FlowJo software (TreeStar, Inc.).
51Cr-release assay
DEPDC1#5 peptide-specific CTL cytotoxic activity was assessed using a 6 h 51Cr-release assay. Briefly, a total of 1 × 106 target cells were labeled with 100 μCi of Na251CrO4 for 1 h at 37 °C and washed twice with culture medium. Depending on the experiment, target cells were incubated with 10 μM of DEPDC1#5, Gag-1777–85 or Melan-A26–35*A27L peptides for 40 min at 37 °C and then washed twice. For blocking experiments, target cells were incubated for 30 min on ice with 10 μg/mL anti-MHC-class I blocking antibody (W6/32 clone; BioLegend) or the relative isotype control (mouse IgG2a, κ isotype ctrl; BioLegend). Target and effector cells were then plated in a 96-well U-bottom plate at the indicated E/T ratio in a total volume of 200 μL. After 6 h of incubation at 37 °C, 30 µL supernatant was transferred on a scintillation plate (PerkinElmer), and measured using a Top Count gamma counter (PerkinElmer). The percentage of lysis was calculated as follows: % Specific lysis = (experimental release − spontaneous release)/(maximal release − spontaneous release) × 100. Spontaneous and maximal releases were obtained by incubating target cells in medium alone or in RPMI 2% SDS, respectively. Cytotoxicity was expressed as lytic units 30 (LU30) or lytic units 20 (LU20), where 1 LU30 or 1 LU20 were defined as the number of effector cells capable of killing 30% or 20% of target cells, respectively.60 Results were expressed in number of LU30 or LU20 per 106 responder cells.
Outgrowth assays
Target MDA-MB-231 or NIH 3T3 cells were serially double diluted into replicate flat-bottom 96-wells plate starting from 3,000 cells/well, and T cells were added where indicated at 3,000 cells/well. Plates were incubated at 37 °C in 5% CO2 with weekly refeeding, and target cells outgrowth was scored after 4 weeks.
In vivo experiments of adoptive immunotherapy
On day 0, 6–8 weeks old female NSG (Charles River) mice were anesthetized (1–3% isoflurane, Merial Italia), and injected into the mammary fat pad with 1 × 106 MDA-MB-231 cells transduced with a lentiviral vector coding for the Firefly Luciferase reporter gene.61 At day 3, mice were injected intra-tumorally with 10 × 106 DEPDC1-specific or Melan-A-specific CTL stimulated three times with autologous peptide-pulsed DC. Another group of mice received only PBS1X as negative controls. The same treatment was repeated for two additional times at 3–4 d interval.
Tumor growth was monitored over time by bioluminescence analysis. In particular, anesthetized animals were given the substrate D-Luciferin (PerkinElmer) by intraperitoneal injection at 150 mg/kg in PBS (Sigma). The light emitted from the bioluminescent tumors or metastasis was detected using a cooled charge-coupled device camera mounted on a light-tight specimen box (IVIS Lumina II Imaging System; PerkinElmer). Imaging times ranged from 15 s to 8 min. Regions of interest from displayed images were identified around the tumor sites or at peripheral metastasis region, and were quantified as total photon counts or photon/s using Living Image® software (PerkinElmer). For the peripheral metastatic colonization detection, the lower portion of each animal was shielded before reimaging to minimize the bioluminescence from primary tumor, thus allowing the signals from metastatic regions to be observed in vivo.
Statistical analysis
Results were analyzed for statistical significance by using paired or unpaired Student's t-test and ANOVA, as appropriate (****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05). Histograms represent mean values ± SD. In scatter-plot graphs, symbols indicate different samples or assays, and horizontal bars represent means ± SD. Statistical analysis was performed using Sigmaplot and GraphPad Prism 4.0 software.
Study approval
Anonymized human buffy coats were obtained from the Blood Bank of Padova Hospital, and donors provided their written informed consent to participate in this study. Procedures involving animals and their care were in conformity with institutional guidelines that comply with national and international laws and policies (D.L. 26/2014 and subsequent implementing circulars), and the experimental protocol (Authorization no. 1143/2015-PR) was approved by the Italian Ministry of Health.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank A. Testa for reading the manuscript and for discussions.
Funding
This work was partly supported by grants from the Italian Association for Cancer Research (AIRC, IG-17035) to A. R., AIRC Special Program Molecular Clinical Oncology “5 per mille” (ID 10016) to A. R. and G. D. S., and Progetto di Ricerca di Ateneo 2015, University of Padova, to A. R. We acknowledge the support by the Italian Ministry of Health (RF-2011-02346976), the Italian Ministry of University and Research (PRIN-2015-8KZKE3), Cariplo Foundation (Grant no. 2014-0812), and Beneficentia-Stiftung, to G. D. S.
References
- 1.Neves H, Kwok HF. Recent advances in the field of anti-cancer immunotherapy. BBA Clin 2015; 3:280-8; PMID:26673349; https://doi.org/ 10.1016/j.bbacli.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015; 14(8):561-84; PMID:26228759; https://doi.org/ 10.1038/nrd4591 [DOI] [PubMed] [Google Scholar]
- 3.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA et al.. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014; 20(19):5064-74; PMID:24714771; https://doi.org/ 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al.. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348(6230):124-8; PMID:25765070; https://doi.org/ 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al.. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351(6280):1463-9; PMID:26940869; https://doi.org/ 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melero I, Berman DM, Aznar MA, Korman AJ, Pérez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015; 15(8):457-72; PMID:26205340; https://doi.org/ 10.1038/nrc3973 [DOI] [PubMed] [Google Scholar]
- 7.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res 2014; 74(11):2974-85; PMID:24728077; https://doi.org/ 10.1158/0008-5472.CAN-13-2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geynisman DM, Chien C-R, Smieliauskas F, Shen C, Shih Y-CT. Economic evaluation of therapeutic cancer vaccines and immunotherapy: a systematic review. Hum Vaccin Immunother 2014; 10(11):3415-24; PMID:25483656; https://doi.org/ 10.4161/hv.29407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14(2):135-46; PMID:24457417; https://doi.org/ 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 10.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY et al.. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 2013; 5(174):174ra27; PMID:23447018; https://doi.org/ 10.1126/scitranslmed.3004916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright SE, Rewers-Felkins KA, Quinlin IS, Phillips CA, Townsend M, Philip R, Dobrzanski MJ, Lockwood-Cooke PR, Robinson W. Cytotoxic T-lymphocyte immunotherapy for ovarian cancer: a pilot study. J Immunother 2012; 35(2):196-204; PMID:22306908; https://doi.org/ 10.1097/CJI.0b013e318243f213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA 2002; 99(25):16168-73; PMID:12427970; https://doi.org/ 10.1073/pnas.242600099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehira M, Harada Y, Takata R, Shuin T, Miki T, Fujioka T, Nakamura Y, Katagiri T. Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene 2007; 26(44):6448-55; PMID:17452976; https://doi.org/ 10.1038/sj.onc.1210466 [DOI] [PubMed] [Google Scholar]
- 14.Harada Y, Kanehira M, Fujisawa Y, Takata R, Shuin T, Miki T, Fujioka T, Nakamura Y, Katagiri T. Cell-permeable peptide DEPDC1-ZNF224 interferes with transcriptional repression and oncogenicity in bladder cancer cells. Cancer Res 2010; 70(14):5829-39; PMID:20587513; https://doi.org/ 10.1158/0008-5472.CAN-10-0255 [DOI] [PubMed] [Google Scholar]
- 15.Chung S, Kijima K, Kudo A, Fujisawa Y, Harada Y, Taira A, Takamatsu N, Miyamoto T, Matsuo Y, Nakamura Y. Preclinical evaluation of biomarkers associated with antitumor activity of MELK inhibitor. Oncotarget 2016; 7(14):18171-82; PMID: 26918358; https://doi.org/ 10.18632/oncotarget.7685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Jiang Y, Jiang M, Zhang J, Yang B, She Y, Wang W, Deng Y, Ye Y. Protocadherin 10 inhibits cell proliferation and induces apoptosis via regulation of DEP domain containing 1 in endometrial endometrioid carcinoma. Exp Mol Pathol 2016; 100(2):344-52; PMID:26970279; https://doi.org/ 10.1016/j.yexmp.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 17.Sendoel A, Maida S, Zheng X, Teo Y, Stergiou L, Rossi CA, Subasic D, Pinto SM, Kinchen JM, Shi M et al.. DEPDC1/LET-99 participates in an evolutionarily conserved pathway for anti-tubulin drug-induced apoptosis. Nat Cell Biol 2014; 16(8):812-20; PMID:25064737; https://doi.org/ 10.1038/ncb3010 [DOI] [PubMed] [Google Scholar]
- 18.Mi Y, Zhang C, Bu Y, Zhang Y, He L, Li H, Zhu H, Li Y, Lei Y, Zhu J. DEPDC1 is a novel cell cycle related gene that regulates mitotic progression. BMB Rep 2015; 48(7):413-18; PMID:25902835; https://doi.org/ 10.5483/BMBRep.2015.48.7.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A et al.. A Pin1/Mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011; 20(1):79-91; PMID:21741598; https://doi.org/ 10.1016/j.ccr.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 20.Obara W, Ohsawa R, Kanehira M, Takata R, Tsunoda T, Yoshida K, Takeda K, Katagiri T, Nakamura Y, Fujioka T. Cancer peptide vaccine therapy developed from oncoantigens identified through genome-wide expression profile analysis for bladder cancer. Jpn J Clin Oncol 2012; 42(7):591-600; PMID:22636067; https://doi.org/ 10.1093/jjco/hys069 [DOI] [PubMed] [Google Scholar]
- 21.Obara W, Eto M, Mimata H, Kohri K, Mitsuhata N, Miura I, Shuin T, Miki T, Koie T, Fujimoto H et al.. A phase I/II study of cancer peptide vaccine S-288310 in patients with advanced urothelial carcinoma of the bladder. Ann Oncol 2017; 28(4):798-803; PMID:27998971; https://doi.org/ 10.1093/annonc/mdw675 [DOI] [PubMed] [Google Scholar]
- 22.Murahashi M, Hijikata Y, Yamada K, Tanaka Y, Kishimoto J, Inoue H, Marumoto T, Takahashi A, Okazaki T, Takeda K et al.. Phase I clinical trial of a five-peptide cancer vaccine combined with cyclophosphamide in advanced solid tumors. Clin Immunol 2016; 166–167:48-58; PMID:27072896; https://doi.org/ 10.1016/j.clim.2016.03.015 [DOI] [PubMed] [Google Scholar]
- 23.Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol 2012; 23(Suppl_6):vi7-12; PMID:23012306; https://doi.org/ 10.1093/annonc/mds187 [DOI] [PubMed] [Google Scholar]
- 24.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA 2004; 101(25):9309-14; PMID:15184677; https://doi.org/ 10.1073/pnas.0401994101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6(1):1-6; PMID:15068665; https://doi.org/ 10.1016/S1476-5586(04)80047-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006; 9(2):121-32; PMID:16473279; https://doi.org/ 10.1016/j.ccr.2006.01.013 [DOI] [PubMed] [Google Scholar]
- 27.Lee J-S, Leem S-H, Lee S-Y, Kim SC, Park ES, Kim SB, Kim SK, Kim YJ, Kim WJ, Chu IS. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol 2010; 28(16):2660-7; PMID:20421545; https://doi.org/ 10.1200/JCO.2009.25.0977 [DOI] [PubMed] [Google Scholar]
- 28.Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC et al.. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008; 26(18):3015-24; PMID:18565887; https://doi.org/ 10.1200/JCO.2007.15.7164 [DOI] [PubMed] [Google Scholar]
- 29.Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res 2007; 67(21):10163-72; PMID:17974957; https://doi.org/ 10.1158/0008-5472.CAN-07-2056 [DOI] [PubMed] [Google Scholar]
- 30.Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One 2010; 5(10):e13091; PMID:20957034; https://doi.org/ 10.1371/journal.pone.0013091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su H, Hu N, Yang HH, Wang C, Takikita M, Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ et al.. Global gene expression profiling and validation in esophageal squamous cell carcinoma and its association with clinical phenotypes. Clin Cancer Res 2011; 17(9):2955-66; PMID:21385931; https://doi.org/ 10.1158/1078-0432.CCR-10-2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, Calcagnile A, Sera F, Saieva C, Ottini L, Palli D et al.. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer 2009; 45(3):461-9; PMID:19081245; https://doi.org/ 10.1016/j.ejca.2008.10.032 [DOI] [PubMed] [Google Scholar]
- 33.Sengupta S, den Boon JA, Chen I-H, Newton MA, Dahl DB, Chen M, Cheng YJ, Westra WH, Chen CJ, Hildesheim A et al.. Genome-wide expression profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal carcinoma. Cancer Res 2006; 66(16):7999-8006; PMID:16912175; https://doi.org/ 10.1158/0008-5472.CAN-05-4399 [DOI] [PubMed] [Google Scholar]
- 34.Choi YL, Tsukasaki K, O'Neill MC, Yamada Y, Onimaru Y, Matsumoto K, Ohashi J, Yamashita Y, Tsutsumi S, Kaneda R et al.. A genomic analysis of adult T-cell leukemia. Oncogene 2007; 26(8):1245-55; PMID:16909099; https://doi.org/ 10.1038/sj.onc.1209898 [DOI] [PubMed] [Google Scholar]
- 35.Wurmbach E, Chen Y, Khitrov G, Zhang W, Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J et al.. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007; 45(4):938-47; PMID:17393520; https://doi.org/ 10.1002/hep.21622 [DOI] [PubMed] [Google Scholar]
- 36.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC et al.. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One 2010; 5(4):e10312; PMID:20421987; https://doi.org/ 10.1371/journal.pone.0010312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS et al.. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics 2008; 1:13; PMID:18442402; https://doi.org/ 10.1186/1755-8794-1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L et al.. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009; 16(3):259-66; PMID:19732725; https://doi.org/ 10.1016/j.ccr.2009.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen O, Lupu A, Schmidt S, Condomines M, Belle S, Maier A, Hose D, Neuber B, Moos M, Kleist C et al.. Melan-A/MART1 analog peptide triggers anti-myeloma T-cells through crossreactivity with HM1.24. J Immunother 2009; 32(6):613-21; PMID:19483648; https://doi.org/ 10.1097/CJI.0b013e3181a95198 [DOI] [PubMed] [Google Scholar]
- 40.Iorns E, Drews-Elger K, Ward TM, Dean S, Clarke J, Berry D, El Ashry D, Lippman M. A new mouse model for the study of human breast cancer metastasis. PLoS One 2012; 7(10):e47995; PMID:23118918; https://doi.org/ 10.1371/journal.pone.0047995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R et al.. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol Cell 2014; 56(5):617-29; PMID:25454946; https://doi.org/ 10.1016/j.molcel.2014.10.013 [DOI] [PubMed] [Google Scholar]
- 42.Rustighi A, Zannini A, Tiberi L, Sommaggio R, Piazza S, Sorrentino G, Nuzzo S, Tuscano A, Eterno V, Benvenuti F et al.. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol Med 2014; 6(1):99-119; PMID:24357640; https://doi.org/ 10.1002/emmm.201302909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kassambara A, Schoenhals M, Moreaux J, Veyrune JL, Rème T, Goldschmidt H, Hose D, Klein B. Inhibition of DEPDC1A, a bad prognostic marker in multiple myeloma, delays growth and induces mature plasma cell markers in malignant plasma cells. PLoS One 2013; 8(4):e62752; PMID:23646139; https://doi.org/ 10.1371/journal.pone.0062752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363(20):1938-48; PMID:21067385; https://doi.org/ 10.1056/NEJMra1001389 [DOI] [PubMed] [Google Scholar]
- 45.Cao K, Hollenbach J, Shi X, Shi W, Chopek M, Fernández-Viña MA. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum Immunol 2001; 62(9):1009-30; PMID:11543903; https://doi.org/ 10.1016/S0198-8859(01)00298-1 [DOI] [PubMed] [Google Scholar]
- 46.Itoh Y, Mizuki N, Shimada T, Azuma F, Itakura M, Kashiwase K, Kikkawa E, Kulski JK, Satake M, Inoko H. High-throughput DNA typing of HLA-A, -B, -C, and -DRB1 loci by a PCR-SSOP-Luminex method in the Japanese population. Immunogenetics 2005; 57(10):717-29; PMID:16215732; https://doi.org/ 10.1007/s00251-005-0048-3 [DOI] [PubMed] [Google Scholar]
- 47.Nerlich AG, Bachmeier BE. Density-dependent lineage instability of MDA-MB-435 breast cancer cells. Oncol Lett 2013; 5(4):1370-74; PMID:23599796; https://doi.org/ 10.3892/ol.2013.1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin J, Sabatino M, Somerville R, Wilson JR, Dudley ME, Stroncek DF, Rosenberg SA. Simplified method of the growth of human tumor infiltrating lymphocytes in gas-permeable flasks to numbers needed for patient treatment. J Immunother 2012; 35(3):283-92; PMID:22421946; https://doi.org/ 10.1097/CJI.0b013e31824e801f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turtle CJ, Riddell SR. Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer J 2010; 16(4):374-81; PMID:20693850; https://doi.org/ 10.1097/PPO.0b013e3181eb33a6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P et al.. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007; 9(2):166-80; PMID:17356713; https://doi.org/ 10.1593/neo.07112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 1994; 152(1):163-75; PMID:8254189 [PubMed] [Google Scholar]
- 52.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2015; 32(4):511-7; PMID:26515819; https://doi.org/ 10.1093/bioinformatics/btv639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larsen M V, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform 2007; 8:424; PMID:17973982; https://doi.org/ 10.1186/1471-2105-8-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsomides TJ, Walker BD, Eisen HN. An optimal viral peptide recognized by CD8+ T cells binds very tightly to the restricting class I major histocompatibility complex protein on intact cells but not to the purified class I protein. Proc Natl Acad Sci USA 1991; 88(24):11276-80; PMID:1722325; https://doi.org/ 10.1073/pnas.88.24.11276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsomides TJ, Aldovini A, Johnson RP, Walker BD, Young RA, Eisen HN. Naturally processed viral peptides recognized by cytotoxic T lymphocytes on cells chronically infected by human immunodeficiency virus type 1. J Exp Med 1994; 180(4):1283-93; PMID:7523570; https://doi.org/ 10.1084/jem.180.4.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrer E, Harrer T, Barbosa P, Feinberg M, Johnson RP, Buchbinder S, Walker BD. Recognition of the highly conserved YMDD region in the human immunodeficiency virus type 1 reverse transcriptase by HLA-A2-restricted cytotoxic T lymphocytes from an asymptomatic long-term nonprogressor. J Infect Dis 1996; 173(2):476-9; PMID:8568316; https://doi.org/ 10.1093/infdis/173.2.476 [DOI] [PubMed] [Google Scholar]
- 57.Nijman HW, Houbiers JG, Vierboom MP, van der Burg SH, Drijfhout JW, D'Amaro J, Kenemans P, Melief CJ, Kast WM. Identification of peptide sequences that potentially trigger HLA-A2.1-restricted cytotoxic T lymphocytes. Eur J Immunol. 1993; 23(6):1215-9; PMID:7684681; https://doi.org/ 10.1002/eji.1830230603 [DOI] [PubMed] [Google Scholar]
- 58.Vikman S, Giandomenico V, Sommaggio R, Oberg K, Essand M, Tötterman TH. CD8+ T cells against multiple tumor-associated antigens in peripheral blood of midgut carcinoid patients. Cancer Immunol Immunother 2008; 57(3):399-409; PMID:17717663; https://doi.org/ 10.1007/s00262-007-0382-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mandruzzato S, Rossi E, Bernardi F, Tosello V, Macino B, Basso G, Chiarion-Sileni V, Rossi CR, Montesco C, Zanovello P. Large and dissimilar repertoire of Melan-A/MART-1-specific CTL in metastatic lesions and blood of a melanoma patient. J Immunol 2002; 169(7):4017-4024; PMID:12244204; https://doi.org/ 10.4049/jimmunol.169.7.4017 [DOI] [PubMed] [Google Scholar]
- 60.Bryant J, Day R, Whiteside TL, Herberman RB. Calculation of lytic units for the expression of cell-mediated cytotoxicity. J Immunol Methods 1992; 146(1):91-103; PMID:1735786; https://doi.org/ 10.1016/0022-1759(92)90052-U [DOI] [PubMed] [Google Scholar]
- 61.Keyaerts M, Verschueren J, Bos TJ, Tchouate-Gainkam LO, Peleman C, Breckpot K, Vanhove C, Caveliers V, Bossuyt A, Lahoutte T. Dynamic bioluminescence imaging for quantitative tumour burden assessment using IV or IP administration of D: -luciferin: effect on intensity, time kinetics and repeatability of photon emission. Eur J Nucl Med Mol Imaging 2008; 35(5):999-1007; PMID:18180921; https://doi.org/ 10.1007/s00259-007-0664-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.