

Abstract
Background
Differences in the gut microbiota and breath methane production have been observed in chronic constipation, but the relationship between colonic microbiota, transit, and breath tests remains unclear.
Methods
In 25 healthy and 25 constipated females we evaluated the sigmoid colonic mucosal and fecal microbiota using 16S rRNA gene sequencing, abundance of hydrogenogenic FeFe (FeFe-hydA) and hydrogenotrophic (methyl coenzyme M reductase A [mrcA] and dissimilatory sulfite reductase A [dsrA]) genes with real-time qPCR assays, breath hydrogen and methane levels after oral lactulose, and colonic transit with scintigraphy.
Key Results
Breath hydrogen and methane were not correlated with constipation, slow colon transit, or with abundance of corresponding genes. After adjusting for colonic transit, the abundance of FeFehydA, dsrA, and mcrA were greater (P<.005) in colonic mucosa, but not stool, of constipated patients. The abundance of the selected functional gene targets also correlated with that of selected taxa. The colonic mucosal abundance of FeFe-hydA, but not mcrA, correlated positively (P<.05) with breath methane production, slow colonic transit, and overall microbiome composition. In the colonic mucosa and feces, the abundance of hydrogenogenic and hydrogenotrophic genes were positively correlated (P<.05). Breath methane production was not associated with constipation or colonic transit.
Conclusions & Inferences
Corroborating our earlier findings with 16S rRNA genes, colonic mucosal but not fecal hydrogenogenic and hydrogenotrophic genes were more abundant in constipated versus healthy subjects independent of colonic transit. Breath gases do not directly reflect the abundance of target genes contributing to their production.
Keywords: breath hydrogen, breath methane, constipation, genes, hydrogen, lactulose, methane, microbiota, microbiome, transit
Graphical Abstract
Abbreviated abstract: The relationship between colonic microbiota, transit, and breath hydrogen and methane production in chronic constipation is unclear. Corroborating our earlier findings with 16S rRNA genes, colonic mucosal but not fecal hydrogenogenic and hydrogenotrophic genes were more abundant in constipated than healthy subjects independent of colonic transit. Breath gas excretion after lactulose was not correlated with the abundance of target genes contributing to their production.
Considerable evidence exists which highlights associations between the colonic microbiota and chronic constipation (1). Colonic mucosal and fecal microbiota differ between constipated patients and healthy people (2–4). Breath methane, which is derived from intestinal methanogenesis, is increased in constipated patients (5), associated with slow colon transit, and with altered colonic microbiota (2, 4).
An earlier study suggested that breath hydrogen and methane excretion after lactulose ingestion were proportional to the production of these gases measured with whole-body calorimetry (7). However, more recently, breath methane excretion was not an accurate marker of colonic methane production in healthy people and irritable bowel syndrome (IBS) patients, nor was it associated with the clinical features or colonic transit in IBS (8). Indeed, several aspects of microbial carbohydrate fermentation limit the utility of using breath gases to assess the abundance or activity of hydrogen-producing (hydrogenogenic) microbes or intestinal gas production. First, preformed gas in hard stools may be released in response to treatment with a laxative {Di Stefano, 2014 #3753}, thereby affecting the accuracy of breath tests. Second, the proportion of H2 excreted in breath depends on total production. At a low production rate (<200 mL/day), 65% of H2 is exhaled (7), but at a high rate (>500 mL/day), most was expelled as flatus and only 25% was exhaled (7). Third, the magnitude of fermentation varies among carbohydrates (7). Fourth, hydrogen from hydrogenogenic microbes is utilized by hydrogenotrophic microbes (6), which reduces the accumulation of hydrogen, thereby decreasing feedback inhibition of fermentation.
Microbial hydrogenogenesis and hydrogenotrophy involves the reversible oxidation of H2 by hydrogenase harboring organisms. Categorized by their metal functional cores, hydrogenases are phylogenetically widespread and functionally diverse and include both hydrogen evolving and hydrogen uptake [NiFe] hydrogenases, hydrogen evolving [FeFe] hydrogenases, and hydrogen uptake [Fe]-only hydrogenases (10). Hydrogenotrophic microbes utilize hydrogen to convert carbon dioxide to acetate (i.e., acs, fhs genes) (11), methane (mcrA gene) (12), and hydrogen sulfide (dsrA gene) (13). Conceivably, low methane production may reflect low hydrogenotrophic methanogenic abundance or low substrate availability (i.e., hydrogen), perhaps due to fewer hydrogenogenic microbes. Measuring the colonic abundance of hydrogenogenic and hydrogenotrophic microbes may distinguish amongst these possibilities. Previous studies have characterized hydrogenogens (14) or methanogens (12) with functional genomics, but none have done so concurrently.
This study aimed to (i) compare the abundance of hydrogenogenic and hydrogenotrophic genes in colonic mucosa and feces between healthy people and constipated patients, and to assess the relationship between gene abundance and (ii) colonic transit, (iii) breath hydrogen and methane production, and (iv) overall composition and specific microbiota.
METHODS
Participants
As detailed previously, 25 women who had Rome III symptom criteria for IBS-C or functional constipation (FC) and 25 healthy people were recruited between February 2013 and April 2014 at Mayo Clinic (4). All participants were nonsmokers aged 18–80 years, who had no clinical evidence of significant systemic disease, inflammatory bowel disease, gastrointestinal cancer, gastric, intestinal, or colonic resection, or antibiotic use within 30 days. This study was approved by the Mayo Clinic Institutional Review Board, and written informed consent was obtained from all participants.
Assessment of Dietary Intake
Briefly, a registered dietitian advised participants to maintain a stable diet for 1 week before and throughout the study; to follow a low fiber diet and avoid interfering foodstuffs including FODMAPs for 24 hours prior to lactulose breath test; and to complete a food record for 3 days before stool collection (4). After the breath test, they resumed their regular diet. Food records were analyzed with ESHA Food Processor software (Version 10.14, ESHA Research, Salem, OR).
Lactulose breath test
After an overnight fast, an antiseptic mouthwash (30 mL) was provided immediately before the procedure to preclude lactulose fermentation by oropharyngeal bacteria. Participants ingested lactulose syrup (10 g). Breath samples were collected every 15 minutes for 60 minutes, and then every 30 minutes for the next 120 minutes. Breath hydrogen and methane concentrations were measured with model SC Quintron gas chromatograph (Quintron Instrument Company, Milwaukee, WI), and summarized as area under the curve (AUC). A positive breath test was defined by an increase in breath hydrogen and methane exceeding 20 ppm over baseline (15).
GI and colonic transit by scintigraphy
Colonic transit was measured by asking participants to ingest a methacrylate-coated, delayed-release capsule containing indium-111 (111In) adsorbed on activated charcoal particles (4). The delayed-release coating dissolves in the alkaline pH of the terminal ileum, releasing 111In in the cecum. Gastric and small bowel transit was evaluated with two scrambled eggs labelled with technetium-99m (99mTc) sulfur-colloid that were eaten with one slice of whole wheat bread and one glass of whole milk (300 kcal total). Established approaches were used to summarize the gastric emptying (GE) half time (t1/2), small intestinal transit (i.e., proportion of 99mTc in the colon at 6 hours) and colonic transit, expressed as the geometric center (GC) at specified times. The GC is the weighted average of counts in the different colonic regions (ascending, transverse, descending, rectosigmoid colon) and stool, respectively, 1 to 5. A higher GC reflects a faster colonic transit.
Stool collection
Using stool kits and standardized instructions, patients collected stool samples according to the Manual of Procedures for Human Microbiome Project (Version Number 11.0, 29 March 2010). Stool samples were frozen and stored in −20°C freezer. One patient required an enema, and one patient required a laxative before providing a stool sample.
Colonic biopsies
After 1–2 Fleet (C.B. Fleet Company Inc, Lynchburg, VA) enemas, a flexible sigmoidoscopy were performed. Using 2 mm forceps, five mucosal biopsies were obtained from normal colorectal mucosa in the sigmoid colon. These biopsies were snap frozen and stored in a −70°C freezer.
Sequencing and Analytical Methods
DNA was extracted from stool with a commercial kit (MoBio DNA extraction kit, Carlsbad, CA) following standard Human Microbiome Project guidelines (16). After extraction, total DNA was quantified using a Qubit assay kit (Life Technologies Corporation, NY, USA), with an average yield of 4.1 (range 0–22.5) ng/μL. 16S rRNA-based sequencing was performed with an Illumina MiSeq sequencer (Illumina Inc., San Diego, CA). Phylotype profiles of the microbiota from healthy and constipated populations were generated using deep rDNA hypervariable tag sequencing of the hypervariable V3–V5 region of the small subunit (SSU) rRNA gene, which has been validated for use with human microbiota and is the preferred technique in the Human Microbiome Project. With the longer reads from the MiSeq (300×300 paired end reads), sequencing included the V3–V5 regions, thereby optimizing the phylogenetic analysis (17); the 300 base pair reads ensured optimal phylogenetic identification. Barcoding of samples prior to sequencing yielded a range of 3,116 – 425,701 reads per sample, ensuring detection of both dominant (core microbiota) and poorly represented taxa (variable microbiota). Paired end reads were stitched, aligned, and classified using a custom pipeline (TORNADO v2.0) (18). Briefly, low base quality reads were either trimmed or discarded (19), and these reads were not classified as a bacteria kingdom (20) or matched to the bacteria 16S rRNA secondary structure (21). To evaluate the microbial diversity and abundance, UPARSE was used for Operational Taxonomical Units (OTU) clustering (22), and FastTree was used for phylogeny (23). The 16S data were clustered into OTUs at 97% sequence similarity, and the taxonomy was assigned using the Ribosomal Database Project classifier.
PCR amplification of functional gene targets
Stool and mucosa DNA were extracted with a MoBio DNA extraction kit and QIAmp Stool DNA mini kit, respectively. Stool and sigmoid colonic mucosal biopsy microbial abundance were quantified using Real-Time quantitative PCR assays performed with a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA), and expressed as gene copies per nanogram of DNA. Because a recent metagenomic survey observed that [FeFe] hydrogenases made up the vast majority of hydrogenases in the human microbiome (14), this was the focus of hydrogenogenic genes. The functional hydrogenogenic FeFe (FeFe-hydA) gene (24) and hydrogenotrophic methyl coenzyme M reductase A (mcrA) (25) and dissimilatory sulfite reductase A (dsrA) genes (26) were targeted (27). Standard curves were constructed using cloned 16S rRNA and functional genes or amplified PCR products.
Statistical analysis
Demographic features, diet, transit, and gene abundance in healthy people and constipated patients were compared using the Student’s t test or Wilcoxon test as appropriate. Results with P values <.05 were considered significant. Univariate regression with permutation based on F-statistics assessed differences in breath tests, and gene abundance between health and constipation. Associations between (i) breath tests and diet and separately with colonic transit, and (ii) gene abundance and demographic factors (i.e., age and body mass index [BMI]), diet, colonic transit, and breath methane production were evaluated with multiple variable regression with permutation based on the F-statistic, all adjusted for constipation status. Associations between breath tests, gene abundance, demographic factors (i.e., age and BMI), diet, and colonic transit were assessed by multivariable models after adjusting for constipation status. Variables were log transformed as appropriate. Associations between hydrogenogenic genes and breath hydrogen production, and separately, hydrogenotrophic genes and breath methane production, were assessed after adjusting for methane production and hydrogen production, respectively. To assess the differences between microbiota profiles, i.e., β diversity, we used both unweighted and weighted Unifrac distances (‘GUniFrac’ function in the R package ‘GUniFrac’) and the Bray-Curtis dissimilarity. These metrics capture different parameters, with unweighted UniFrac reflecting differences in community membership (i.e., presence or absence of OTU), while the weighted UniFrac and Bray-Curtis dissimilarity better reflect differences in OTU abundance.(28) To prevent artifacts in the analysis that can arise from unequal sequencing depth, rarefaction was performed on the OTU table before calculating the unweighted UniFrac distance. In order to identify associations between microbial community profiles and variables of interest (e.g., constipation status, colonic transit), we used the Microbiome Regression-Based Kernel Association Test (MiRKAT) that adjusts for potential confounding covariates (29).
In addition to performing an association analysis with the OTU-based microbiome profiles, we also tested for associations between specific taxa and variables of interest. This was done by comparing relative taxa abundance and variables of interest using a linear model with adjustments for covariates where appropriate. Because the taxa data is not normally distributed, assessment of statistical significance considered 1,000 permutations with the F-statistic as the test statistic. Associations were evaluated at the phylum, family, and genus level. The false discovery rate (FDR) control was performed based on the Benjamini-Hochberg procedure to correct for multiple testing, i.e., ‘p.adjust’ in R. Analysis was confined to taxa with a prevalence greater than 10% and a maximum proportion (relative abundance) greater than 0.002. An FDR-adjusted P value (or Q-value) less than 5% was considered to be significant. Associations between gene abundance and microbiota, and separately with breath tests, were assessed in multivariable models, after adjusting for both constipation and colonic transit. Spearman correlation coefficients assessed associations between abundance of genes in stool and colonic mucosa. All statistical analyses were performed in R-3.0.2 (R Development Core Teams, Vienna, Austria).
RESULTS
Participant characteristics
The results of symptoms, diet, colonic transit, breath hydrogen and methane excretion, and phylogenetic assessment of the colonic and mucosal microbiome with 16S rRNA gene techniques in these participants have been published (4). This paper focuses on the results of functional genes and their relationship with other characteristics in this cohort.
Of the 25 patients, 13 had symptoms of FC, 6 had IBS-C, and 6 had mixed IBS, with predominant constipation. Compared to patients, healthy participants also consumed more calories (P=.005), carbohydrate (P=.054), protein (P=.002), fat (P=.03), and fiber (P=.01) when expressed as an absolute amount, but not as a proportion of the total calorie intake (Table 1). Gastric emptying at 2 hours was lower (P=.005) in constipated patients than in healthy participants. Fourteen patients (9 with FC, 3 with IBS-C, and 2 with mixed IBS) but only 2 controls (P=.0001) had delayed colonic transit. Colonic transit (GC24) was directly correlated with total calorie intake (P=.03) and total fiber intake (P=.01), and inversely correlated with age (P=.03).
Table 1.
Summary of Patient Characteristics *
| Variable | Healthy (n=25) | Constipated (n=25) | P value |
|---|---|---|---|
| Age, y | 39±10 | 48±15 | .02 |
| BMI, kg/m2 | 26±4 | 25±4 | .13 |
| Total caloric intake, kcal | 1597±402 | 1265±350 | .005 |
| Carbohydrate, g | 188±54 | 155±62 | .054 |
| Protein, g | 75±19 | 60±26 | .002 |
| Fat, g | 60±26 | 46±15 | .03 |
| Fiber, g | 17±13 | 12±4 | .01 |
| Carbohydrate (% of total calories) | 47±9 | 49±12 | .24 |
| Protein (% of total calories) | 20±6 | 20±7 | .88 |
| Fat (% of total calories) | 33±6 | 32±6 | .85 |
| Breath methane (AUC, ppm* min) | 1488±2895 | 4100±6656 | .20 |
| Breath hydrogen (AUC, ppm* min) | 7154±4716 | 6483±5824 | .44 |
| Gastric emptying, % | |||
| 2 hours | 59±18 | 47±11 | .005 |
| 4 hours | 93±10 | 90±12 | .11 |
| Small intestinal transit (Colonic filling [%] at 6 hours) | 45±27 | 48±26 | .74 |
| Colonic transit, GC24 | 2.6±1.1 | 1.6±0.8 | .0006 |
| Colonic transit, GC48 | 3.9±0.9 | 2.8±1.0 | .001 |
Abbreviations: AUC, area under the curve; BMI, body mass index; GC24, geometric center of colonic transit at 24 hours; GC48, geometric center of colonic transit at 48 hours
All data presented as mean (SD)
Relationship between breath tests and other parameters
Breath methane excretion was numerically greater in constipated subjects than in controls at all timepoints (Figure 1). However, breath hydrogen and methane excretion (AUC) were not different (P=.20) between patients and controls. After adjusting for constipation status, a positive hydrogen breath test was associated with higher dietary protein intake (P=.05) and slow colonic transit (P<.05). Breath methane excretion (AUC) was positively associated with dietary fiber intake (P<.05).
Figure 1.

Breath methane concentration after lactulose intake.
Relationship between abundance of functional genes in the colonic mucosa and other parameters
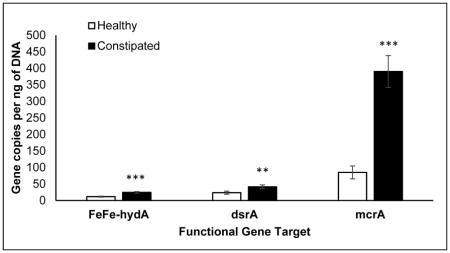
The colonic mucosal abundance of hydrogenogenic and hydrogenotrophic genes was not associated with age, sex, or body mass index (BMI) (data not shown). However, these genes (FeFe-hydA, mcrA, and dsrA) were more abundant in constipated patients than healthy people (Figure 2), even after adjusting for colonic transit. The abundance of FeFe-hydA was inversely correlated with methane production (AUC) (r=−0.37, P=.02) and directly correlated with slow colonic transit (r=0.26, P=.047). No other associations between these genes and diet, colonic transit, and breath methane or hydrogen excretion were significant.
Figure 2. Differences in mucosal gene abundance in health and constipation.

P values are adjusted for colonic transit. **P<0.005, ***P<0.001; FeFehydA, FeFe hydrogenase; dsrA, dissimilatory sulfite reductase A; mcrA, methyl coenzyme M reductase A
Relationships between abundance of functional gene targets in stool and other parameters
The abundance of hydrogenogenic and hydrogenotrophic genes in feces was not associated with age, sex, or BMI (data not shown). None of the genes in stool were associated with constipation or colonic transit. The abundance of fecal mcrA was positively associated with a positive hydrogen breath test (r=0.31, P=.04). The abundance of fecal FeFe-hydA was positively associated with dietary carbohydrate intake (r=0.37, P=.01). Associations between the abundance of fecal FeFe-hydA and breath hydrogen production remained nonsignificant even after adjusting for methane production and separately, for abundance of individual hydrogenotrophic genes. Conversely, associations between the abundance of individual hydrogenotrophic genes and breath methane production remained nonsignificant even after adjusting for hydrogen production and separately, abundance of fecal FeFe-hydA. No other associations between the fecal abundance of these genes and diet, colonic transit, and breath methane or hydrogen excretion were statistically significant.
Associations between abundance of functional genes and the colonic mucosal microbiota
The abundance of FeFe-hydA and dsrA in the colonic mucosa were separately associated with the overall microbiome composition assessed with both unweighted and weighted Unifrac distance (Table 2).
Table 2.
Association Between Mucosal Gene Abundance and Overall Mucosal Microbiota Composition *
| Gene | Unweighted Unifrac | Weighted Unifrac | Bray-Curtis dissimilarity |
|---|---|---|---|
| Mucosal | |||
| FeFe-hydA | .002 | .02 | .07 |
| dsrA | .002 | .004 | .08 |
| mcrA | .03 | .19 | .007 |
| Fecal | |||
| FeFe-hydA | .21 | .54 | .30 |
| dsrA | .37 | .16 | .28 |
| mcrA | .79 | .66 | .49 |
All values are P values from the Microbiome Regression-Based Kernel Association Test
The abundance of mcrA in the colonic mucosa was associated with the overall microbiome composition assessed with unweighted but not weighted Unifrac distance. The abundance of several taxa in the mucosa correlated with the abundance of FeFe-hydA, mcrA, and dsrA in tissue (Table 3).
Table 3.
Association Between Mucosal Gene Abundance and Mucosal Microbiota Abundance
| Gene | Taxon | Spearman’s correlation coefficient | P value | FDR-adjusted P value |
|---|---|---|---|---|
| FeFe-hydA | Phyla | |||
| Actinobacteria | −0.22 | .006 | .02 | |
| Bacteroidetes * | 0.23 | .05 | .08 | |
| Proteobacteria | −0.58 | .002 | .01 | |
| Family | ||||
| Actinobacteria;Propionibacteriaceae | −0.35 | .006 | .09 | |
| Bacteroidetes;Odoribacteraceae † | −0.06 | .03 | .24 | |
| Bacteroidetes;Sphingobacteriaceae | 0.46 | .02 | .19 | |
| Proteobacteria;Comamonadaceae † | −0.52 | .001 | .03 | |
| Genus | ||||
| Actinobacteria;Propionibacterium | −0.35 | .003 | .06 | |
| Bacteroidetes;Odoribacter † | −0.19 | .02 | .19 | |
| Proteobacteria;Comamonas † | −0.25 | .02 | .19 | |
| Proteobacteria;Delftia † | −0.45 | .002 | .06 | |
|
| ||||
| dsrA | Phyla | |||
| Bacteroidetes * | 0.33 | .04 | .09 | |
| Proteobacteria | −0.50 | .02 | .08 | |
| Family | ||||
| Bacteroidetes;Barnesiellaceae | 0.04 | .03 | .28 | |
| Proteobacteria;Bradyrhizobiaceae | −0.28 | .04 | .28 | |
| Proteobacteria;Comamonadaceae † | −0.43 | .02 | .26 | |
| Proteobacteria;Rhizobiaceae | −0.48 | .02 | .26 | |
| Genus | ||||
| Bacteroidetes;Odoribacter † | −0.30 | .02 | .14 | |
| Bacteroidetes;Sphingobacterium | 0.28 | .008 | .14 | |
| Firmicutes;Ruminococcus | −0.22 | .03 | .23 | |
| Proteobacteria;Agrobacterium † | −0.48 | .01 | .14 | |
| Proteobacteria;Bradyrhizobium | −0.28 | .05 | .28 | |
| Proteobacteria;Delftia † | −0.48 | .009 | .14 | |
|
| ||||
| mcrA | Phylum | |||
| Actinobacteria | −0.25 | .04 | .11 | |
| Proteobacteria | −0.36 | .02 | .08 | |
| Family | ||||
| Bacteroidetes;Odoribacteraceae † | −0.14 | .01 | .17 | |
| Bacteroidetes;Paraprevotellaceae | −0.39 | .02 | .18 | |
| Proteobacteria;Comamonadaceae † | −0.44 | .03 | .23 | |
| Proteobacteria;Pseudomonadaceae | 0.30 | .04 | .25 | |
| Proteobacteria;Rhizobiaceae | −0.48 | .004 | .12 | |
| Genus | ||||
| Actinobacteria;Propionibacterium | −0.36 | .04 | .27 | |
| Bacteroidetes;Odoribacter † | −0.28 | .003 | .06 | |
| Firmicutes;Butyrivibrio | −0.05 | .05 | .30 | |
| Proteobacteria;Agrobacterium † | −0.48 | .003 | .06 | |
| Proteobacteria;Delftia † | −0.43 | .008 | .10 | |
| Proteobacteria;Pseudomonas * | 0.30 | .04 | .27 | |
Abbreviations: FDR, false discovery rate
More and † less abundant in colonic mucosa of constipated patients with 16sRNA
The genera Sphingobacterium and Pseudomonas correlated positively with dsrA and mcrA respectively, before, but not after, adjusting for false discovery rate. As previously reported, 16sRNA analyses disclosed that some of these microbes were also differentially (i.e., more or less) abundant in the colonic mucosa of constipated patients (4). In contrast, the abundance of these genes in stool was not associated with the composition of stool microbiome (data not shown).
Correlations between abundance of functional gene targets in stool and colonic mucosa
In the colonic mucosa, the abundance of FeFe-hydA correlated positively with the abundance of hydrogenotrophic genes. The abundance of hydrogenotrophic genes also correlated positively with each other (Figure 3).
Figure 3. Correlations among abundance of genes in colonic mucosa.

Values represent Spearman correlation coefficients. **P<.005, †P<.001; FeFehydA, FeFe hydrogenase; dsrA, dissimilatory sulfite reductase A; mcrA, methyl coenzyme M reductase A
In stool samples, the abundance of FeFe-hydA was positively correlated with mcrA (ρ=0.21, P<.05). The abundance of fecal FeFe-hydA tended to correlate with its abundance in mucosa (ρ=0.30, P=.06).
DISCUSSION
We observed previously that phylogenetic assessment of mucosal microbiota distinguished constipated and healthy subjects with 94% accuracy independent of colonic transit (4). Extending those findings, this study found that hydrogenogenic and hydrogenotrophic gene targets were more abundant in the colonic mucosa of constipated patients, and provides several insights into the relationship between gene abundance, colonic transit, and gases (30).
Gaseous byproducts of colonic fermentation have been implicated in IBS in general, and IBS-C in particular (5). Methane delivered at very high concentrations increased ileal muscle contractility in an ex vivo model of the guinea pig ileum and delayed small bowel transit in dogs (31). In this study, breath methane excretion was numerically greater in patients than in controls at individual time points. However, the overall breath methane excretion profile was not associated with constipation or slow colonic transit. One explanation is that only a subset of healthy individuals, approximately 30% in our population, produce methane at quantities detectable in breath (32), which is lower than the corresponding proportion (53%) in a study from Spain (33). The abundance of individual hydrogenogenic genes was not significantly associated with breath hydrogen production, even after adjusting for its utilization, i.e., to produce methane. Similarly, the abundance of individual hydrogenotrophic genes was not associated with breath methane production, even after adjusting for the availability of hydrogen, its substrate. Taken together, these observations suggest that breath methane and hydrogen excretion is not a useful measure of the abundance of hydrogenogenic and hydrogenotrophic genes in humans.
The abundance of hydrogenotrophic and hydrogenogenic genes was significantly correlated in both colonic mucosa and feces. This is beneficial because the hydrogenotrophic process prevents accumulation of hydrogen, which would thermodynamically restrict further fermentation (6). Thus, hydrogen removal ensures fermentation of substrates to more oxidized end products, hence increasing the energy yield of fermentative microbes. The abundance of the methanogenic gene mcrA and the hydrogenogenic gene FeFe-hydA, were respectively not correlated with breath methane and hydrogen production, perhaps because breath methane and likely also hydrogen, are not accurate markers of colonic methane and hydrogen production (8). Further, the abundance of the methanogenic gene mcrA or the hydrogenogenic gene FeFe-hydA, may not reflect their activity (36).
The colonic mucosal abundance of hydrogenogenic and hydrogenotrophic genes were associated with the overall composition of the colonic mucosal microbiome evaluated with unweighted and weighted analyses. Indeed, several taxa that correlated with the abundance of genes in this study were also differentially abundant in the mucosal microbiota of healthy and constipated persons (4). Moreover, taxa that were more abundant (i.e., Bacteroidetes, Proteobacteria; Pseudomonas) in constipated patients were positively correlated with FeFe-hydA, dsrA, and mcrA. Conversely, taxa that were less abundant (i.e., Proteobacteria; Comamonas, Proteobacteria; Delftia, Proteobacteria; Agrobacterium) in constipated patients inversely correlated with FeFe-hydA, dsrA, and mcrA. Hence, it is conceivable that differences in these taxa at least partly explain why hydrogenogenic and hydrogenotrophic genes were more abundant in constipation.
Relationships between taxa abundance and functional gene targets may provide insights into the contribution of individual organisms to the overall microbial hydrogen economy. For example, first, some Bacteroidetes express the gene FeFe-hydA, which may explain the correlation between this phylum and FeFe-hydA (24, 37). Second, taxa belonging to phyla Proteobacteria and Actinobacteria correlated inversely with FeFe-hydA. To speculate, the colonic abundance of these phyla may be inversely correlated with the abundance of the phylum Firmicutes, which are strong hydrogen producers (6). Third, Bacteroidetes encode sulfatases (38), which liberate sulfate from sulfated mucins. Sulfate-reducing bacteria (SRB) contain dsrA, which utilizes sulfate and hydrogen to produce hydrogen sulfide. Sulfate promotes the growth of SRB (39). Perhaps this explains the correlation between Bacteroidetes and dsrA-harboring SRB, which generate sulfate and synthesize hydrogen sulfide, respectively. Subject to the caveat that the phylogenetic assessment relied on 16S rRNA gene sequences while the functional gene abundance was evaluated with qPCR techniques, taxa that are known hydrogen producers (e.g., Roseburia spp. and Ruminococcus spp) were not significantly different between healthy and constipated subjects. Refining our understanding of the relationships between specific taxa and functional genes will require metagenomic approaches.
In contrast to the colonic mucosa, few correlations were observed between hydrogenogenic and hydrogenotrophic gene abundance in stool and constipation or the stool microbiome. Correlations between the abundance of mcrA and a positive hydrogen breath test and between the abundance of the hydrogenogenic gene FeFe-hydA and the hydrogenotrophic gene mcrA were significant, likely due to the syntrophic nature of organisms which create these respective metabolites. Similar to the phylogenetic assessment of the microbiome (4), the abundance of gene targets in stool and colonic mucosa were not correlated, which reinforces the importance of obtaining both fecal and colonic mucosal samples. Dietary carbohydrate intake was associated with the abundance of FeFe-hydA in feces, presumably because fermentable carbohydrates are a major substrate for microbial hydrogen production (40).
Limitations of this study include target gene activity depends on factors other than their abundance, such as substrate availability, environment, and the evolution of the organism (41). While microbial communities are ubiquitous throughout the colon, there may be regional differences (6). The current sample size provided 80% power to detect a relatively large effect size, that is 0.8SD (within-group standard deviation) between groups. Hence, a type 2 error may explain why some differences were not statistically significant and may have also limited our ability to identify significant relationships, for instance, between the abundance of genes and the composition of specific microbiota as also between the abundance of FeFe hydA and breath hydrogen excretion. The relationship between the abundance of mrcA and 16S rRNA genes of methane producing microbes (i.e., Archaea) was not evaluated.
In conclusion, similar to our prior phylogenetic assessment of microbiota, the functional genes associated with H2 production and utilization in the colonic mucosa were more abundant in constipated patients than healthy people, independent of colonic transit. While the abundance of hydrogenogenic and hydrogenotrophic genes was correlated, the abundance of these genes was not associated with the breath levels of their respective gases after lactulose ingestion. Breath methane was not related with constipation or colonic transit. A larger study integrating phylogenetic, transcriptomic, functional genomic, and metagenomics techniques with physiologic parameters is necessary to more fully resolve how microbes generating and disposing of H2 impact colonic function.
KEY POINTS.
Mucosal but not fecal abundance of hydrogenogenic and hydrogenotrophic genes are greater in constipated than healthy people even after adjusting for slow colonic transit.
These differences can be partly explained by differences in the abundance of specific taxa between healthy and constipated people.
Breath hydrogen and methane were not observed to be (i) significantly different between health and constipation, (ii) associated with slow colonic transit, or (iii) correlated with colonic mucosal or fecal abundance of functional genes encoding enzymes contributing to their production.
Acknowledgments
Financial Support: Dr. Bharucha was supported in part by grant R01 DK78924 from the National Institutes of Health, US Department of Health and Human Services. This project was supported by the Center for Individualized Medicine at Mayo Clinic, Mayo Clinic-University of Illinois Strategic Alliance for Technology-Based Healthcare, and in part by grant number UL1 TR000135 from the National Center for Advancing Translational Sciences. Patricia Wolf was supported by a predoctoral fellowship from Mayo Clinic-University of Illinois Alliance for Technology-Based Healthcare.
Disclosures: None of the authors have any conflicts of interest that are relevant to the manuscript.
Glossary
- BMI
body mass index
- FC
functional constipation
- GC
geometric center
- GE
gastric emptying
- IBS
irritable bowel syndrome
- MiRKAT
Microbiome Regression-Based Kernel Association Test
- 99mTc
technetium-99m
Footnotes
Author contributions:
Study concept and design: Bharucha and Gaskins
Acquisition of data: Bharucha, O’Connor, Wolf.
Analysis and interpretation of data: Bharucha, Chen, Chia, Gaskins, O’Connor, Parthasarathy, Wolf.
Drafting of the manuscript: Bharucha, Gaskins, Parthasarathy, Wolf.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical expertise: Chen
Obtained funding and study supervision: Bharucha and Gaskins.
References
- 1.Ohman L, Tornblom H, Simren M. Crosstalk at the mucosal border: importance of the gut microenvironment in IBS. Nat Rev Gastroenterol Hepatol. 2015;12:36–49. doi: 10.1038/nrgastro.2014.200. [DOI] [PubMed] [Google Scholar]
- 2.Attaluri A, Jackson M, Valestin J, Rao SSC. Methanogenic flora is associated with altered colonic transit but not stool characteristics in constipation without IBS. Am J Gastroenterol. 2010;105:1407–1411. doi: 10.1038/ajg.2009.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeffery IB, O’Toole PW, Ohman L, et al. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut. 2012;61:997–1006. doi: 10.1136/gutjnl-2011-301501. [DOI] [PubMed] [Google Scholar]
- 4.Parthasarathy G, Chen J, Chen X, et al. Relationship Between Microbiota of the Colonic Mucosa vs Feces and Symptoms, Colonic Transit, and Methane Production in Female Patients With Chronic Constipation. Gastroenterology. 2016;150:367–379. e361. doi: 10.1053/j.gastro.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunkel D, Basseri RJ, Makhani MD, Chong K, Chang C, Pimentel M. Methane on breath testing is associated with constipation: a systematic review and meta-analysis. Dig Dis Sci. 2011;56:1612–1618. doi: 10.1007/s10620-011-1590-5. [DOI] [PubMed] [Google Scholar]
- 6.Carbonero F, Benefiel AC, Gaskins HR. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat Rev Gastroenterol Hepatol. 2012;9:504–518. doi: 10.1038/nrgastro.2012.85. [DOI] [PubMed] [Google Scholar]
- 7.Christl SU, Murgatroyd PR, Gibson GR, Cummings JH. Production, metabolism, and excretion of hydrogen in the large intestine. Gastroenterology. 1992;102:1269–1277. [PubMed] [Google Scholar]
- 8.Di Stefano M, Mengoli C, Bergonzi M, et al. Breath Methane Excretion Is not An Accurate Marker of Colonic Methane Production in Irritable Bowel Syndrome. Am J Gastroenterol. 2015;110:891–898. doi: 10.1038/ajg.2015.47. [DOI] [PubMed] [Google Scholar]
- 9.Sellin JH. A Breath of Fresh Air. Clinical Gastroenterology & Hepatology. 2016;14:209–211. doi: 10.1016/j.cgh.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Wolf PG, Biswas A, Morales SE, Greening C, Gaskins HR. H metabolism is widespread and diverse among human colonic microbes. Gut Microbes. 2016;7:235–245. doi: 10.1080/19490976.2016.1182288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovell CR, Leaphart AB. Community-level analysis: key genes of CO2-reductive acetogenesis. Methods in enzymology. 2005;397:454–469. doi: 10.1016/S0076-6879(05)97028-6. [DOI] [PubMed] [Google Scholar]
- 12.Scanlan PD, Shanahan F, Marchesi JR. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC microbiology. 2008;8:79. doi: 10.1186/1471-2180-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson GR, Macfarlane S, Macfarlane GT. Metabolic Interactions Involving Sulfate-Reducing and Methanogenic Bacteria in the Human Large-Intestine. Fems Microbiology Ecology. 1993;12:117–125. [Google Scholar]
- 14.Greening C, Biswas A, Carere CR, et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H is a widely utilised energy source for microbial growth and survival. Isme J. 2016;10:761–777. doi: 10.1038/ismej.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saad RJ, Chey WD. Breath testing for small intestinal bacterial overgrowth: maximizing test accuracy. Clinical Gastroenterology & Hepatology. 2014;12:1964–1972. doi: 10.1016/j.cgh.2013.09.055. quiz e1119–1920. [DOI] [PubMed] [Google Scholar]
- 16.Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome research. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeraldo P, Chia N, Goldenfeld N. On the suitability of short reads of 16S rRNA for phylogeny-based analyses in environmental surveys. Environ Microbiol. 2011;13:3000–3009. doi: 10.1111/j.1462-2920.2011.02577.x. [DOI] [PubMed] [Google Scholar]
- 18.Jeraldo P, Kalari K, Chen X, et al. IM-TORNADO: a tool for comparison of 16S reads from paired-end libraries. PLoS One. 2014;9:e114804. doi: 10.1371/journal.pone.0114804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1. 0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 23.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd ES, Spear JR, Peters JW. [FeFe] hydrogenase genetic diversity provides insight into molecular adaptation in a saline microbial mat community. Appl Environ Microbiol. 2009;75:4620–4623. doi: 10.1128/AEM.00582-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallam SJ, Girguis PR, Preston CM, Richardson PM, DeLong EF. Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol. 2003;69:5483–5491. doi: 10.1128/AEM.69.9.5483-5491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein M, Friedrich M, Roger AJ, et al. Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. Journal of bacteriology. 2001;183:6028–6035. doi: 10.1128/JB.183.20.6028-6035.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereyra LP, Hiibel SR, Prieto Riquelme MV, Reardon KF, Pruden A. Detection and quantification of functional genes of cellulose- degrading, fermentative, and sulfate-reducing bacteria and methanogenic archaea. Appl Environ Microbiol. 2010;76:2192–2202. doi: 10.1128/AEM.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao N, Chen J, Carroll IM, et al. Testing in Microbiome-Profiling Studies with MiRKAT, the Microbiome Regression-Based Kernel Association Test. Am J Hum Genet. 2015;96:797–807. doi: 10.1016/j.ajhg.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quigley EM, Spiller RC. Constipation and the Microbiome: Lumen Versus Mucosa! Gastroenterology. 2016;150:300–303. doi: 10.1053/j.gastro.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 31.Pimentel M, Lin HC, Enayati P, et al. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1089–1095. doi: 10.1152/ajpgi.00574.2004. [DOI] [PubMed] [Google Scholar]
- 32.Levitt MD, Furne JK, Kuskowski M, Ruddy J. Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements. Clin Gastroenterol Hepatol. 2006;4:123–129. doi: 10.1016/j.cgh.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Vega AB, Perello A, Martos L, et al. Breath methane in functional constipation: response to treatment with Ispaghula husk. Neurogastroenterology & Motility. 2015;27:945–953. doi: 10.1111/nmo.12568. [DOI] [PubMed] [Google Scholar]
- 34.Miller TL, Wolin MJ. Methanosphaera stadtmaniae gen. nov., sp. nov: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol. 1985;141:116–122. doi: 10.1007/BF00423270. [DOI] [PubMed] [Google Scholar]
- 35.Fricke WF, Seedorf H, Henne A, et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. Journal of bacteriology. 2006;188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xing D, Ren N, Rittmann BE. Genetic diversity of hydrogen-producing bacteria in an acidophilic ethanol-H2-coproducing system, analyzed using the [Fe]-hydrogenase gene. Appl Environ Microbiol. 2008;74:1232–1239. doi: 10.1128/AEM.01946-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benjdia A, Martens EC, Gordon JI, Berteau O. Sulfatases and a radical S-adenosyl-L-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J Biol Chem. 2011;286:25973–25982. doi: 10.1074/jbc.M111.228841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci U S A. 2013;110:13582–13587. doi: 10.1073/pnas.1312524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levitt MD, Hirsh P, Fetzer CA, Sheahan M, Levine AS. H2 excretion after ingestion of complex carbohydrates. Gastroenterology. 1987;92:383–389. doi: 10.1016/0016-5085(87)90132-6. [DOI] [PubMed] [Google Scholar]
- 41.Preidis GA, Hotez PJ. The newest “omics”--metagenomics and metabolomics--enter the battle against the neglected tropical diseases. PLoS Negl Trop Dis. 2015;9:e0003382. doi: 10.1371/journal.pntd.0003382. [DOI] [PMC free article] [PubMed] [Google Scholar]