

Abstract
Systemically administered chemotherapeutic drugs are often ineffective in the treatment of invasive brain tumors due to poor therapeutic index. Within gliomas, despite the presence of heterogeneously leaky microvessels, dense extracellular matrix and high interstitial pressure generate a “blood-tumor barrier” (BTB), which inhibits drug delivery and distribution. Meanwhile, beyond the contrast MRI-enhancing edge of the tumor, invasive cancer cells are protected by the intact blood-brain barrier (BBB). Here, we tested whether brain-penetrating nanoparticles (BPN) that possess dense surface coatings of polyethylene glycol (PEG) and are loaded with cisplatin (CDDP) could be delivered across both the blood-tumor and blood-brain barriers with MR image-guided focused ultrasound (MRgFUS), and whether this treatment could control glioma growth and invasiveness. To this end, we first established that MRgFUS is capable of significantly enhancing the delivery of ~60 nm fluorescent tracer BPN across the blood-tumor barrier in both the 9L (6-fold improvement) gliosarcoma and invasive F98 (28-fold improvement) glioma models. Importantly, BPN delivery across the intact BBB, just beyond the tumor edge, was also markedly increased in both tumor models. We then showed that a CDDP loaded BPN formulation (CDDP-BPN), composed of a blend of polyaspartic acid (PAA) and heavily PEGylated polyaspartic acid (PAA-PEG), was highly stable, provided extended drug release, and was effective against F98 cells in vitro. These CDDP-BPN were delivered from the systemic circulation into orthotopic F98 gliomas using MRgFUS, where they elicited a significant reduction in tumor invasiveness and growth, as well as improved animal survival. We conclude that this therapy may offer a powerful new approach for the treatment invasive gliomas, particularly for preventing and controlling recurrence.
Keywords: focused ultrasound, blood-brain barrier, blood-tumor barrier, glioma, nanoparticle, cisplatin
Graphical abstract
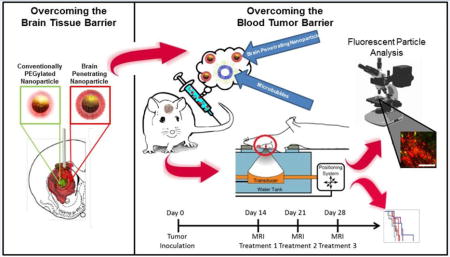
Introduction
Glioblastoma multiforme (GBM) is an aggressive brain tumor that accounts for 67% of all primary brain tumors [1]. Due to the highly invasive nature of the disease, cancer cells often lie beyond the visible tumor boundary, which makes full surgical resection difficult [2]. A significant percentage (90%) of patients develop tumor recurrence at or near the surgical site and many tumors develop drug resistance [3]. Despite advances in drug development, the five year survival rate for GBM is 12% and has remained virtually unchanged over the past decade [1].
Cis-diamminedichloroplatinum (Cisplatin, CDDP) is a powerful chemotherapeutic used as a first-line therapy for several types of cancer, including testicular, ovarian, bladder and lung cancers [1]. It has also been used as an adjuvant in the treatment of pediatric brain tumors [4]. However, adult GBM patients treated with CDDP suffer severe kidney and neurotoxicity, even at sub-therapeutic drug concentrations [5–7]. Methods that reduce off-target toxicity and/or increase local delivery to permit a decrease in systemic dose would greatly increase CDDP’s utility in the treatment of brain tumors [5].
While direct injection of CDDP can improve therapeutic outcomes, this requires an invasive procedure and still produces significant neurotoxicity [8]. Biodegradable nanoparticle formulations can shield healthy tissues from the toxic effects of CDDP, especially during systemic administration, while enhancing therapeutic efficacy [1,9]. When combined with a strategy to increase NP concentration at the target site, this drug delivery approach provides the ability to achieve sustained therapeutic CDDP levels in the tumor while minimizing both systemic and local off-target effects.
However, drug delivery to the brain presents unique difficulties [10]. Invasive cancer cells exist within otherwise healthy brain tissue, where they are protected from systemically administered drugs by the blood-brain barrier (BBB). The BBB regulates the transport of most molecules to and from the brain and prevents the vast majority of CNS therapeutics from reaching their target [11,12]. The BBB within the tumor may be impaired, but this impairment is often heterogeneous [2,13] and produces an unfavorable pressure gradient for drug penetration [14]. Additionally, brain tumors have a higher cell density and collagen content than normal brain tissue, further limiting drug penetration [15,16]. Together, these factors generate what is now commonly referred to as the “blood-tumor barrier” (BTB). Transcranial MR-guided focused ultrasound (MRgFUS) is currently the only treatment modality capable of achieving safe, non-invasive, reversible BBB and BTB disruption in a targeted manner [17–22].
Nonetheless, once therapeutics are delivered across the BBB and/or BTB using MRgFUS, they must still penetrate brain tissue through the complex brain extracellular space (ECS) in order to provide more uniform drug delivery to the tumor, including invasive cells. To address this challenge, we recently developed “brain penetrating nanoparticles” (BPNs), which are drug-or DNA plasmid-loaded nanoparticles that possess non-adhesive surfaces (a result of exceptionally dense coatings with poly(ethylene glycol (PEG)) which enable particles up to 114 nm [23] and 70 nm [24] in size to rapidly spread within normal brain parenchyma and brain tumors, respectively. Importantly, when considering the systemic administration of BPN, the dense PEG coat also offers long circulation times because rapid clearance by the reticuloendothelial system (RES) is minimized [25].
In this study, we tested the hypothesis that MRgFUS will increase the delivery of drug-loaded BPN across the BBB/BTB in an intracranial rat model of glioma, yielding reduced tumor invasiveness and improved tumor growth control and animal survival. We used both fluorescently labelled PEGylated polystyrene (PS-PEG) BPN and biodegradable cisplatin-loaded BPNs (CDDP-BPN) composed of PAA/PAA-PEG blends to evaluate the efficacy of the therapy. We show that MRgFUS significantly improves BPN delivery and distribution in the tumor and that MRgFUS in combination with CDDP-BPN improves tumor growth control and animal survival. This is the first MRgFUS study demonstrating efficacy with a systemically administered drug-loaded biodegradable polymeric nanoparticle in the treatment of GBM.
Material and Methods
PEGylation of Polyaspartic Acid (PAA) Polypeptide
A co-polymer of PAA-PEG was synthesized with 27 kDa PAA (200 apsartic acid units, Alamanda Polymers, Huntsville, AL) of which C- and N-terminus are amide and amine, respectively, and 5 kDa methoxy-PEG-amine (mPEG-NH2, Creative PEGworks, Winston Salem, NC). Briefly, PAA was reacted with mPEG-NH2 at a 1:10 molar ratio with an addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Invitrogen, Carlsbad, CA; equimolar with PEG). The reaction was carried out in 200 mM borate buffer (pH 8.5) for 24 hours at room temperature followed by dialysis against deionized water using a 50 kDa MWCO dialysis device (Spectrum Lab, Rancho Dominguez, CA) for 120 hours. The solution was lyophilized to obtain a powder of purified PEG-conjugated PAA (PAA-PEG) which was then stored at −20°C until use. The PAA:PEG ratio was confirmed using nuclear magnetic resonance (NMR) to be ~1:10:1 H NMR (500 MHz, D2O): δ 2.70–2.80 (br, -CHCH2COOH) 3.55–3.75 (br, -CH2CH2O-), 4.40–4.55 (br, NHCHCH2-) and 3.3–3.4 (s, -OCH2CH2CH3). A representative NMR spectrum confirming PEGylation of PAA is provided in Fig S1. Immediately prior to nanoparticle (NP) formulation, the lyophilized polymers were dissolved in ultrapure distilled water.
Fluorescent Labeling of PAA Polymer
For fluorescent labeling of polymer, AlexaFluor 647-cadaverine (AF647; Thermo Fisher Scientific) or AlexaFluor 555-cadaverine (AF555; Thermo Fisher Scientific) was conjugated to PAA in 200 mM borate buffer (pH 8.5) for 72 hours at room temperature. The solution was dialyzed against deionized water using a 20kDa MWCO G2 Dialysis device (Spectrum Lab) for 120 hours, followed by lyophilization. The AF647- and AF555-labeled PAA (AF647-PAA and AF555-PAA, respectively) were stored at −20°C until further use.
Preparation of CDDP Loaded Nanoparticles
Un-PEGylated CDDP-loaded NP (CDDP-UPN) were formulated using a previously reported method [26] with a slight modification. Briefly, 5 mM cisplatin and 37.03 μM PAA (equivalent to 5 mM aspartic acid residues) were dissolved in RNase-free water and reacted for 72 hours. CDDP-BPN were formulated using previously reported method with appropriate modifications to improve CDDP loading [26]. As a first step, CDDP (16.7 mmol) and silver nitrate (31.73 mmol) were reacted together in 5 ml of nuclease-free water at 55°C for 3 h and at room temperature for additional 21 h. The reaction mixture was centrifuged at 15,000 rpm for 10 min to separate silver chloride precipitate formed during the reaction. The supernatant was filtered through a 0.45 μm filter to obtain aquated cisplatin. The concentration of aquated cisplatin was measured using flameless atomic absorption spectrophotometer (AAS; Perkin Elmer, Waltham, MA) and adjusted to 5 mM. PAA-PEG and PAA were dissolved in RNase-free water at a 17:1 mass ratio to obtain a total aspartic acid residue concentration of 5 mM. The pH of the mixture was then adjusted to 6.5–6.8 using 0.1 M NaOH, and reacted with 5 mM aquated cisplatin at room temperature for 72 hours. Subsequently, CDDP-UPN or CDDP-BPN were transferred to a centrifugal filtration unit (Amicon Ultra, 100 kDa MWCO; Millipore, Billerica, MA), and centrifuged at 1,000 xg for 10 min. Both CDDP-UPN and CDDP-BPN were stored at 4° C until further use. Fluorescent CDDP-UPN and CDDP-BPNB were formulated with AF555-PAA and a mixture of PAA-PEG and AF647-PAA, respectively.
Physicochemical Characterization of NP
Physicochemical characteristics of NP were determined using a Zetasizer NanoZS (Malvern Instruments, Southborough, MA). All particles were diluted in 10 mM NaCl (diluted from phosphate buffered saline) and dynamic light scattering (DLS) was employed to determine the hydrodynamic diameter and polydispersity index (PDI) at a backscattering angle of 173°. The ζ-potential, a measure of particle surface charge, was determined using laser Doppler anemometry. Quantification of drug content within the NP was conducted using AAS and the loading density was calculated as the % mass of drug in the total particle mass. The size and morphology of NP was determined using a Hitachi H7600 transmission electron microscope (TEM, Hitachi, Japan).
NP Stability Analysis
NP stability was measured in artificial cerebrospinal fluid (ACSF) and 10% fetal bovine serum (FBS). Either CDDP-UPN or CDDP-BPN were dissolved in ACSF and/or 10% FBS at 37°C and hydrodynamic diameters were determined by DLS at different time points after the initiation of incubation.
CDDP Nanoparticle Release Kinetics
To determine the CDDP release rate over time, CDDP-BPN were dispersed in 2 mL PBS (pH: 7.4) (Harvard Apparatus, Holliston, MA) within a 100 kDa MWCO dialysis tube (Spectrum Lab). The tube was then placed in a 20 mL PBS sink (simulated infinite sink) and shaken at 37°C. At specific time points, total sink volume of PBS was replaced with fresh PBS and the CDDP content in the sink was determined by AAS to generate a release kinetics curve.
Cell Culture
F98 and 9L rat glioma cells were obtained from the American Type Culture Collection (ATCC). Cells were cultured in high glucose Dulbecco modified Eagle medium (DMEM, Gibco) with 1 mM sodium pyruvate (Gibco) and 10% fetal bovine serum (Gibco) added. For each experiment, cells were brought back from cryopreservation to eliminate variations due to passage number. Cells were maintained at 37°C and 5% CO2.
In-Vitro Cell Viability Assay
F98 glioma cells were seeded at a concentration of 5,000 cells per well in 100 μL of media and allowed to attach overnight in a 96-well plate. The following day, the media was replaced with 100 μL of fresh media with 10 μL of either carrier-free CDDP solution (prepared at the concentration of 1 mg/ml by heating at 40°C and subsequent sonication) or CDDP-BPN (25 μM to 0.0625 μM in 10-fold dilutions). Cells were incubated for 3 days at 37°C and 5% CO2. To quantify the number of live cells, media was replaced with 100 μL of fresh medium and 10 μL of Dojindo Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Rockville, MD) solution was subsequently added. Cells were incubated for 2 hours at 37°C, followed by the measurement of absorbance at 450 nm using a Synergy Mx Multi-Mode Microplate Reader (Biotek, Instruments Inc. Winooski, VT). The % cell viability was normalized to the untreated cell control.
Neocortical Slice Preparation and Multiple Particle Tracking
All animal experiments were carried in accord with local Institutional Animal Care and Use Committee regulations. Healthy rat brain tissue slices were prepared according to a slightly modified protocol of a previous publication [27]. 1.5 mm thick brain tissue slices were prepared using a Zivic Mouse Brain slicer (Zivic instruments, Pittsburgh, PA) and placed in custom-made microscopy chambers. 0.5 μL of fluorescently labeled CDDP-UPN or CDDP-BPN was injected at a depth of 1 mm into the cerebral cortex using a 10 μL Hamilton Neuros Syringe (Hamilton, Reno, NV). The chambers were sealed using a coverslip to minimize convective bulk flow so that particle movement could be fully attributed to Brownian diffusion. The transport rates of particles were calculated by analyzing the particle trajectories in brain tissue slices (N = 3 for each particle type). The particle trajectories were recorded as 20 second movies at an exposure of 66 ms, using an EMCCD camera (Evolve 512; Photometrics, Tuscon, AZ) mounted on an inverted epifluorescence microscope (Axio Observer D1, Carl Zeiss, Hertfordshire, UK) equipped with a 100x oil-immersion objective (NA 1.3). MSD of particles were calculated and compared at a timescale of τ = 1 s.
Fluorescence Imaging of CDDP NP Distribution After Direct Intracranial Injection
Fluorescently labeled CDDP-UPN and CDDP-BPN (equivalent to 12 μg of CDDP) were suspended in ACSF solution at a ratio of 1:1 (total volume of 20 μL) and were loaded in a 50 μL Hamilton Neuros Syringe (Hamilton). A mixture of AF555-labeled CDDP-UPN and AF647-labeled CDDP-BPN were co-administered at the rate of 0.33 μl/min and over a period of 1 hour into the striatum of female Fisher 344 rats (200–220 g) via convection enhanced delivery (CED). A burr hole was drilled 3 mm lateral and 1 mm posterior to the bregma. The catheter was vertically mounted on a Chemyx Nanojet Injector Module (Chemyx, Stafford, TX), which was held on a small animal stereotactic frame (Stoelting, Wood Dale, IL). The catheter tip was lowered to a depth of 3.5 mm and NP were infused at a rate of 0.33 μL/min, followed by catheter withdrawal at a rate of 1 mm/min. Animals were sacrificed 1 hour post-administration and the brains were removed and immediately frozen on dry ice. Tissues were cryosectioned (Leica CM 3050S, Leica Biosystems, Buffalo Grove, II) into successive 100 μm coronal slices and imaged using a Zeiss confocal 710 laser scanning microscope through Cy5 (AF647; CDDP-BPN) and Cy3 (AF555; CDDP-UPN) channels. Confocal images of brain slices were quantified for fluorescent distribution of NP within the striatum using a MATLAB script developed in our lab. Fluorescent distribution of NP in the ventricles or white matter tracts (WMT) was not included in the quantification. The area of distribution calculated from each slice was multiplied by the slice thickness of 100 μm and summated across all images to obtain a total volume of distribution.
Tumor Inoculation
Two different cell lines were used in the in-vivo tumor studies: 9L and F98. 9L is a well-characterized glioma model that is both highly immunogenic and poorly infiltrative [28–30]. In contrast F98 gliomas are poorly immunogenic and highly infiltrative, more closely replicating the behavior of gliomas in patients [28–30]. F98 is also more sensitive to CDDP [31]. 9L cells were implanted in Sprague-Dawley rats and F98 cells were implanted in Fischer 344 rats. F98 or 9L cells were removed from the culture flask with 0.25% Trypsin-EDTA (Gibco), centrifuged at 1000 rpm for 5 minutes at 4°C and resuspended in serum-free high glucose DMEM at a concentration of 100,000 cells/mL. Cells were kept on ice for no longer than 1 hour. Animals were anesthetized with 2% isoflurane (Piramal). Their heads were shaved and aseptically prepared. Animals were then placed in a stereotaxic frame (Harvard Apparatus) and a midline incision was made along the scalp. A burr hole was made ~0.2 mm posterior from Bregma, 2.0 mm lateral from the sagittal suture. A 26 gauge needle was inserted to 4 mm below the dura and 10 uL of the cell suspension was injected into the brain over a 10 minute period using a micropump (UltraMicroPump, World Precision Instruments). The needle was left in place for an additional 3 minutes following completion of the injection, and then withdrawn at 1 mm/min. The burr hole was sealed with bone wax (Surgical Specialties) and the incision was closed with wound glue (Vetbond, 3M). Animals were given buprenorphine (DOSE, Buprenex) for the first 24 hours and antibiotic water (10 mg/kg, Baytril, Bayer) for 2 weeks following the inoculation.
Fluorescent BPN Delivery with MRgFUS
All animals were 160g–180g females from the same vendor (Envigo). Fluorescently labelled polystyrene-PEG-BPN (PS-PEG-BPN) were intravenously administered at a dose of 15 μg/g body weight immediately prior to sonication. Tumors were sonicated with a 9 spot grid at 0.6 MPa as described in detail in a forthcoming section. Animals were euthanized 24 hours after FUS treatment with pentobarbital (0.5 μl/g body weight).
CDDP-BPN Delivery with MRgFUS
To test the hypothesis that ultrasound can enhance the efficacy of drug-loaded BPN in the treatment of glioblastoma, female 160–180g Fischer 344 rats (Envigo) were inoculated with F98 glioma cells and randomly assigned to one of four groups: 0.8 MPa FUS (FUShi) + CDDP-BPN, 0.6 MPa FUS (FUSlo) + CDDP-BPN, CDDP-BPN, and control. Animals were kept on a 12/12 hr light/dark light cycle in a temperature and humidity controlled environment and were provided with food and water ad libitum. Beginning 2 weeks after F98 tumor cell inoculation, rats were imaged using contrast-enhanced T1 weighted MRI to assess tumor volume and target the delivery of CDDP-BPN with MRgFUS. Tumors were sonicated with MRgFUS in a 9 spot grid as described in detail in a forthcoming section. This contrast MR imaging and CDDP-BPN delivery protocol was repeated both 1 and 2 weeks later, corresponding to 3 and 4 weeks post-inoculation. Animals were imaged weekly following the final CDDP-BPN treatment at 4 weeks post-inoculation. CDDP-BPN were administered at a dose of 2.5 mg/kg CDDP. An additional set of animals was used to evaluate the delivery of fluorescently labelled CDDP-BPN. These animals were treated once and euthanized 24 hours after treatment. All animals were weighed and numerically scored (0–3, 0 being normal) weekly or as needed (up to twice a day) in the following categories: body weight, physical condition (lack of grooming, etc.), appearance of eyes and nose (porphyrin staining, etc.), activity, and posture. When animals reached approved humane endpoint criteria as defined by a total score of 8 or a maximum score in at least two categories, they were euthanized with pentobarbital (0.5 μl/g body weight).
Targeted Nanoparticle Delivery to Brain Tumors with MRgFUS
On the day of treatment, animals were anesthetized with an intraperitoneal injection of Ketamine (40 mg/kg, Fort Dodge) and Dexdomitor (0.2 mg/kg, Pfizer) in sterilized water. A tail vein catheter was inserted to allow intravenous (i.v.) delivery of albumin-shelled microbubbles, the nanoparticle being delivered, and MRI contrast agent. Albumin-shelled microbubbles were similar to the Optison® formulation and prepared using previously described methods [32]. Animal skulls were depilated and positioned in a degassed water bath coupled to the FUS system. Nanoparticles and MBs (1E5 MBs/g body weight) were administered immediately prior to sonication. All sonications were performed using a 1.14 MHz single element focused transducer (FUS Instruments, Toronto, CA) operating at a 0.5% duty cycle for 2 minutes. Non-derated peak negative pressure was either 0.6 MPa or 0.8 MPa. Prior to placing the animal on the MRI table, the FUS system was passively aligned with a 3T MRI system (Trio, Siemens) using an anechoic Zerdine phantom (CIRS, Inc, Norfolk, VA). After the animal was coupled to the FUS system, high resolution contrast enhanced (0.5 μl/g body weight, Magnevist, Bayer), T1-weighted scans were performed and utilized to define the tumor boundaries. A 9-spot square grid, approximately 3mm by 3mm laterally and 5 mm deep, was centered over the tumor. 5 locations were sonicated within one 2 min sonication period using interleaved sonication mode, and the remaining 4 targets were treated with an additional 2 min sonication. Sonications were spaced 6 minutes apart to allow clearance of the first dose of MBs. An additional dose of MBs was injected immediately prior to the second sonication. Immediately following the final sonication, MRI contrast agent (0.25 μl/g body weight) was delivered intravenously and the high resolution contrast enhanced T1-weigted scan was repeated to verify BTB/BBB disruption. Animals were then removed from the MRI table, given Antisedan (2 mg/kg, Zoetis) in 5 mL warm saline subcutaneously, and placed on a heating pad at 37°C for 30 minutes.
MR Image Analysis
Contrast enhanced T1-weighted images were taken weekly prior to each treatment. Enhancing tumor diameter was calculated from thresholded images taken sagittally through the tumor at its maximum diameter. Enhancing tumor volume was calculated using the formula . Percent tumor growth inhibition was calculated as where Vc and Vt are the median tumor volumes in the treated groups and control group, respectively, on day 28 and Vo is the median tumor volume at Day 14. Significance was determined using a two-way ANOVA.
Tumor invasiveness was assessed by examining the edges of enhancing tumor regions in Day 28 contrast MR images with ImageJ software. For each tumor, 8 plots of grayscale intensity as a function of radial position were generated. These plots were derived from line segments drawn orthogonal to and across tumor edges at 0°, 45°, 90°, 135°, 180°, 225°, 270°, and 315° from horizontal. These plots exhibited sigmoidal shapes. Linear region slopes (Δ grayscale value/mm) were averaged for each of the 8 sigmoidal intensity curves per tumor and designated as a representation of “tumor edge sharpness”. Linear region width, defined as the distance between the inner and outer inflection points of each sigmoidal curve, was taken as a representation of “tumor edge thickness” (mm). The radial intensity profile function in ImageJ was also used to graphically illustrate invasiveness of selected tumors from each group.
Processing and Microscopic Imaging of Sonicated Brain Tumors and Tissue
Following euthanasia, rat brains were excised for histology. In animals that received fluorescently labelled CDDP-BPN or PS-PEG-BPN, the left and right carotid arteries were cannulated and each artery was perfused with 22 mL 2% heparinized saline prior to brain excision. Brains were dessicated in a 30% sucrose solution at 4°C for 24 hours or until brains sunk. Dessicated brains were placed in O.C.T. compound (Sakura Finetek, Torrance, CA) for one hour at 21°C before freezing at −80°C. Transverse 8 μm cryosections were mounted, stained for H&E or endothelial cells (1:200 Lectin 488 for 1.5 hours at 21°C, Invitrogen), and sealed with Prolong Gold (Invitrogen, Grand Island, NY) for imaging. H&E sections were imaged on a Nikon TE300 confocal microscope equipped with a 20x oil objective, while nanoparticle delivery analysis on Lectin-stained sections was performed on a Nikon TE300 confocal microscope equipped with a 20x oil objective. An Optronics QuantiFIREXI camera was used to capture the images for analysis.
Confocal images were analyzed in MATLAB using a code developed in our lab. The nanoparticle (red channel) and endothelial (green channel) signals were separated for processing. Each channel was thresholded and converted to black and white using Otsu’s method [33]. Total nanoparticle coverage and vascular density were calculated as the total number of pixels above threshold in the red and green channels, respectively, normalized by the total number of pixels and expressed as a percentage.
Results
Physicochemical Characterization of CDDP Nanoparticles
We encapsulated CDDP in conventional un-PEGylated nanoparticles (CDDP-UPN) and in highly PEGylated nanoparticles (CDDP-BPN), each with high CDDP loading efficiency and diameters smaller than the ECM pore size within the brain [23] (Table 1). CDDP-BPN possessed a more uniform size distribution compared to the CDDP-UPN (polydispersity index of 0.15 ± 0.01 and 0.2 ± 0.03, respectively). The PEG coating on the CDDP-BPN shielded the anionic carboxyl side groups of the PAA, thereby reducing the surface charge from −35.2 ± 0.45 mV for CDDP-UPN to near neutral (−3.27 ± 0.48 mV) for CDDP-BPN. The BPN formulations encapsulated nearly twice the amount (40 ± 5 %w/w) of CDDP as the UPN formulation (20 ± 2 %w/w). Both NP formulations were spherical in shape (Fig 1A). The hydrodynamic diameter of a CDDP-BPN suspension was stable for up to 24 hours in both serum and ACSF, but CDDP-UPN showed a rapid increase in hydrodynamic diameter after 6 hours in ACSF, indicating particle aggregation (Fig 1B). Approximately ~30% of total CDDP was release from CDDP-BPN over a 7 day period (Fig 1C).
Table 1.
Physiochemical properties of CDDP-loaded nanoparticles.
| Nanoparticle type | Hydrodynamic Diameter ± SEM (nm)† | ζ-potential ± SEM (nm)‡ | Polydispersity index (PDI) ± SEM† | Cisplatin loading density ± SEM (% w/w) |
|---|---|---|---|---|
| CDDP-UPN | 65.0 ± 5.1 | −35.2 ± 0.45 | 0.2 ± 0.03 | 20 ± 2 |
| CDDP-BPN | 45.3 ± 2.5 | −3.27 ± 0.48 | 0.15 ± 0.01 | 40 ± 5 |
Hydrodynamic diameters and PDI were measured in 10 mM NaCl at pH 7.0. Data represent the mean ± SEM (n ≥ 3 measurements).
ζ-potentials were measured by laser Doppler anemometry in 10 mM NaCl at pH 7.0. Data represent the mean ± SEM (n ≥ 3 measurements).
Figure 1. Physiochemical properties and diffusion behaviors of CDDP -loaded nanoparticles.
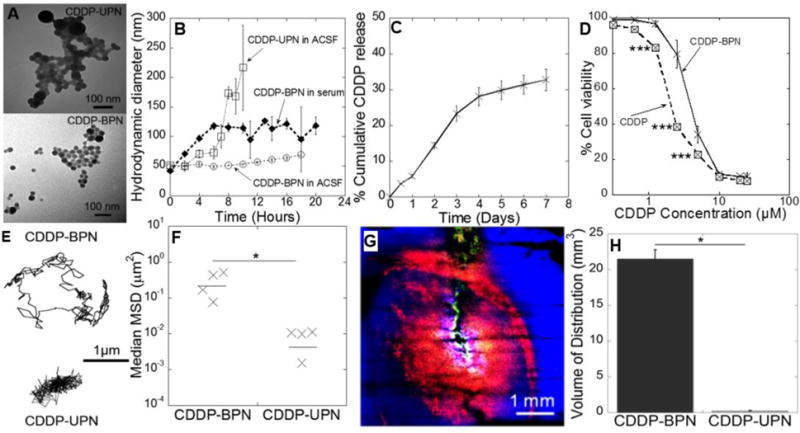
(A) Transmission electron micrograph of CDDP -BPN (upper panel) and CDDP-UPN (lower panel). Scale bars = 100 nm. (B) Stability of CDDP -BPN and CDDP-UPN in ACSF and media with 10% FBS over time. (C) In vitro drug release kinetics of CDDP-BPN in PBS (pH 7.0) at 37°C (n=3). (D) In vitro cytotoxicity of carrier-free CDDP and CDDP-BPN in F98 rat GBM cells (n=3); *** p < 0.005. (E) Representative trajectories of CDDP-BPN and CDDP-UPN diffusing in rat brain tissue ex vivo. (F) Median MSD of CDDP-BPN and CDDP-UPN at a timescale of t = 1 s in at least n = 3 rat brain tissues (> 100 particles tracked in individual samples). *P< 0.05. (G) In vivo distribution of CDDP-BPN (red) and CDDP-UPN (green) in rat brain striatum following administration via CED. Cell nuclei are stained by DAPI (blue). Scale bar = 1 mm. (H) Volume of in vivo distribution of CDDP-BPN and CDDP-UPN in rat hemispheres (n > 3 rats).
In Vitro Cytotoxicity of CDDP-BPN Against Rodent Glioma Cells
We assessed in vitro cytotoxicity of CDDP-BPN against F98 glioma cells to evaluate the effect of incorporation of CDDP into the nanoparticles on its chemotherapeutic activity. Our studies showed that F98-IC50 value of CDDP-BPN (3.7 ± 0.22 μM) was significantly higher compared to CDDP solution (1.04 ± 0.04 μM). The lower in vitro cytotoxicity (i.e. higher IC50 value) observed in case of CDDP-BPN was indicative of gradual release of CDDP from CDDP-BPN and also corroborated sustained release of CDDP observed during CDDP-BPN release kinetics. The slow release of CDDP from CDDP-BPN would be useful for reduction in the off-target toxicity into the brain microenvironment.
Dense PEGylation Enhances CDDP Nanoparticle Spreading in Healthy Brain Tissue
Mean squared particle displacements (MSD) of both CDDP-UPN and CDDP-BPN were calculated using multiple particle tracking (MPT) following NP administration in fresh rat brain slices ex vivo. Representative CDDP-BPN particle trajectories over 20 s demonstrated unhindered diffusion compared to the CDDP-UPN, which were largely immobilized (Fig 1E). At a timescale of τ = 1 s, the mean MSD of the CDDP-BPN was 160-fold greater than their un-PEGylated controls, indicating that the dense PEG coat reduced particle interactions with the ECM and permitted particle spreading over time (Fig 1F).
Dense PEGylation Enhances In-Vivo Distribution of CDDP Nanoparticles Following Intracranial Administration
To determine whether the increased NP diffusion rate ex vivo would translate into greater volume of distribution following local NP administration in vivo, we co-administered fluorescently labeled CDDP-BPN and CDDP-UPN (equivalent to 12 μg CDDP) into the rat striatum via CED and evaluated the volume of distribution (Vd) for each particle formulation (Fig 1G). CDDP-BPN distribution was homogeneous, producing a Vd of 21.5 ± 1.25 mm3, compared to only 0.25±0.025 mm3 for the CDDP-UPN formulation, a 100-fold difference (Fig 1H). Note that CDDP-UPN were used as a control for CDDP-BPN for nanoparticle characterization, but they were not tested in combination with MRgFUS targeted delivery due to their inability to widely distribute throughout the rat brain tissue following local administration.
Activation of Microbubbles with MRgFUS Opens Both the Blood-Tumor and Blood-Brain Barriers
Tumors appeared as small (1–3 mm in diameter) areas of focal enhancement prior to FUS treatment (Fig 2, Pre-FUS). Before treatment, a 9-spot (3 mm × 3 mm × 5 mm per spot) FUS sonication grid, centered over the enhancing tumor region and overlapping the surrounding brain tissue, was planned. After i.v. MB injection, FUS was applied according to the treatment plan. FUS application consistently yielded an increase in both the intensity and area of MR contrast enhancement, indicating opening of both the blood-tumor and blood-brain barriers (Fig 2, Post-FUS).
Figure 2. Activation of microbubbles with MRgFUS opens the BTB in F98 gliomas, as well as the BBB in surrounding brain tissue.
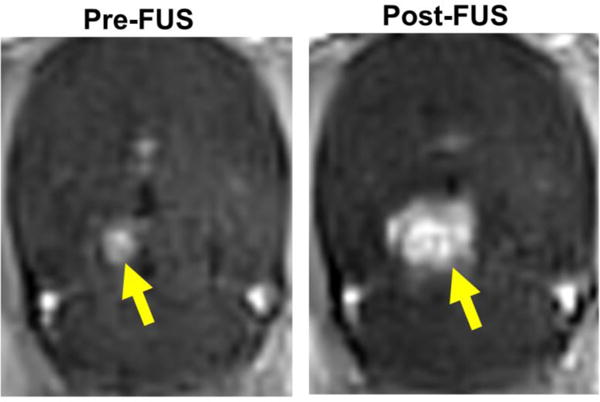
Representative contrast enhanced T1-weighted MR images of F98 glioma before (Pre-FUS) and after (Post-FUS) MRgFUS activation of microbubbles using a 9 spot grid pattern. BTB/BBB opening is indicated by increases in both the intensity and area of contrast enhancement.
MRgFUS Activation of Microbubbles Markedly Enhances PS-PEG-BPN Delivery to Gliomas and Surrounding Brain Tissue
Confocal microscopy images were used to evaluate fluorescent PS-PEG-BPN delivery in sonicated and unsonicated 9L and F98 tumors 24 hours after treatment. In 9L gliomas, 60 nm PS-PEG-BPN delivery within MRgFUS treated (FUS+) tumors was more abundant and homogeneous than in FUS− tumors (Fig 3A). In untreated (FUS−) tumors, PS-PEG-BPN deposition occurred primarily within large vessel structures (Fig 3A). PS-PEG-BPN coverage per field of view was 6.4-fold higher in FUS+ 9L tumors when compared to FUS− 9L tumors (Fig 3B). In the brain tissue just beyond the edge of the tumor border, MRgFUS treatment produced a 61.1-fold increase in PS-PEG-BPN coverage per field of view over FUS− tumors (Fig 3B). In FUS+ 9L tumors, PS-PEG-BPN coverage was significantly greater within the tumor than in the surrounding edge region.
Figure 3. MRgFUS markedly enhances the delivery of 60 nm PS-PEG-BPN across the BTB and BBB in 9L and F98 tumors.
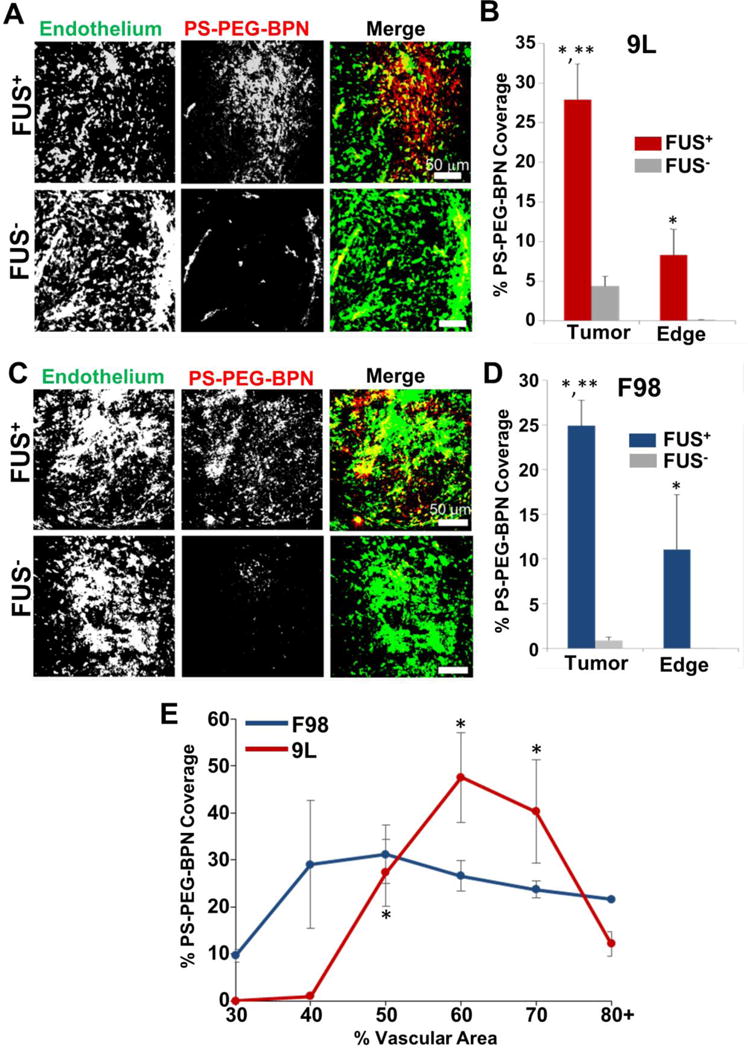
(A) Representative confocal microscopic images of 9L tumor cross-sections from FUS treated (FUS+) and untreated (FUS−) rats. PS-PEG-BPN (red) are shown in relation to tumor endothelium (green). (B) Bar graph of PS-PEG-BPN delivery to 9L tumors and tumor edge regions. N=6 per group. *P<0.05 vs. FUS− in same region. **P<0.05 vs. FUS+ in Edge region. (C) Representative confocal microscopic images of F98 tumor cross-sections from FUS treated (FUS+) and untreated (FUS−) rats. PS-PEG-BPN (red) are shown in relation to tumor endothelium (green). (D) Bar graph of PS-PEG-BPN delivery to F98 tumors and tumor edge regions. N=4 per group. *P<0.05 vs. FUS− in same region. **P<0.05 vs. FUS+ in Edge region. (E) Line graph of PS-PEG-BPN delivery to MRgFUS treated (FUS+) 9L and F98 tumors as a function of % Vascular Area. *P<0.05 vs. all other points within the 9L group.
In general, PS-PEG-BPN delivery followed a similar pattern in F98 gliomas. MRgFUS treatment markedly improved the delivery and distribution of PS-PEG-BPN (Fig 3C) within F98 tumors when compared to FUS− control tumors (Fig 3C). In FUS− tumors, PS-PEG-BPN were largely concentrated in and around vessels (Fig 3C). PS-PEG-BPN coverage per field of view was 26-fold higher within FUS+ tumors when compared to FUS− tumors (Fig 3D). In the edge region just beyond the tumor border, PS-PEG-BPN were not observed in FUS− specimens, while FUS+ F98 tumors averaged 11% PS-PEG-BPN coverage. In FUS+ animals, there was a significant increase in BPN delivery in the tumor when compared to edge tissue. Further experiments in the F98 model using fluorescently labelled CDDP-BPN demonstrated similar results, with MRgFUS producing a 30-fold increase (p<0.001) in fluorescent CDDP-BPN coverage in tumor tissue and a 75-fold increase (p<0.005) in the edge region (data not shown).
PS-PEG-BPN Delivery with MRgFUS Varies with Local Vascular Density
We also examined the relationship between percent vascular area (%VA) and PS-PEG-BPN delivery within treated and untreated tumors. MRgFUS-mediated PS-PEG-BPN delivery across the BTB varied significantly with %VA in the 9L model (Fig 3E). Maximum PS-PEG-BPN coverage occurred in tumor regions with 50–60% VA and 60–70% VA in the F98 and 9L models, respectively. Furthermore, PS-PEG-BPN coverage was at least 3 fold higher than in less vascularized regions (<40%VA) and at least 1.5 fold higher than in highly vascularized regions (>80%VA). In untreated animals, PS-PEG-BPN delivery was not affected by %VA and was similar to relatively avascular (<30%VA) regions in FUS+ tumors (data not shown).
MRgFUS Targeted CDDP-BPN Delivery Inhibits F98 Tumor Growth and Improves Survival
Contrast enhanced T1 weighted MR images were used to determine the enhancing tumor volume in each animal. Measurements were made weekly, beginning when the tumor was first visible on MRI (Day 14). Enhancing tumor volume was identical across all groups at the start of treatment (Day 14, Fig 4A). Control tumors, as well as those in rats receiving intravenous CDDP-BPN without FUS, grew rapidly between days 21 and 28. F98 tumors treated with both FUS and CDDP-BPN demonstrated delayed tumor growth between days 21 and 28 (Fig 4A). 0.8 MPa FUS + CDDP-BPN (FUShi + CDDP-BPN) produced the greatest tumor growth inhibition (61% compared to untreated control) at day 28 (Fig 4B), corresponding to a 15% improvement over 0.6 MPa FUS + CDDP-BPN (FUSlo + CDDP-BPN) and a 64% improvement compared to CDDP-BPN alone. This improvement in tumor growth control in the FUShi + CDDP-BPN group translated to a significant extension of animal survival (Fig 4C, Table S1) compared to untreated controls (15% increase in median survival time, hazard ratio 5.03). Maximum survival time was identical (32 days) for animals receiving 0.6 MPa FUS in combination with CDDP-BPN (FUSlo + CDDP-BPN) or BPN alone, but significance compared to untreated controls was not achieved. Survival was analyzed using the Kaplan-Meier method. Significance between groups was calculated using the log-rank test.
Figure 4. MR image-guided delivery of CDDP-BPN to F98 gliomas with 0.8 MPa FUS inhibits tumor growth and improves survival.
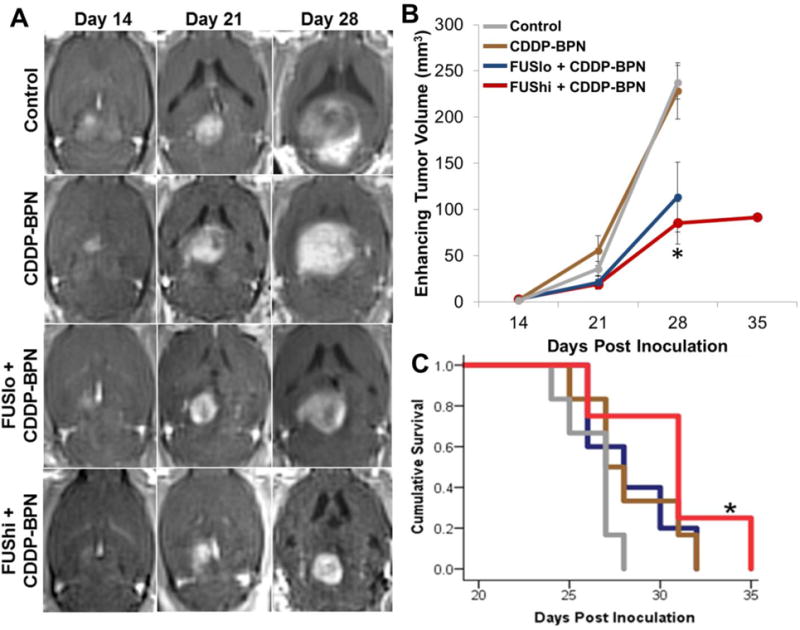
(A) Representative contrast-enhanced T1 weighted MR images taken before FUS application on Days 14, 21, and 28. Images show enhancing tumor volumes in untreated Control, CDDP-BPN, FUSlo + CDDP-BPN, and FUShi + CDDP-BPN treated rats. (B) Line graph of enhancing tumor volumes, taken from T1 weighted contrast MR images, for the 4 groups denoted in panel A. *P<0.05 vs. Control and CDDP-BPN groups. (C) Kaplan-Meier survival curves. *P<0.05 vs. Control. N≥4 for all groups.
MRgFUS Targeted CDDP-BPN Delivery Inhibits F98 Glioma Invasion
To determine whether CDDP-BPN delivery with MRgFUS affects F98 glioma invasion, we examined tumor borders using both contrast enhanced T1 weighted MRI and H&E stained cross-sections. Contrast MR images revealed that untreated F98 animals and animals receiving CDDP-BPN without FUS had tumors with diffuse borders, typical of an invasive GBM tumor [2], while animals treated with 0.8 MPa FUS in combination with BPN had smaller tumors with more defined borders (Fig 5A). These morphological differences were captured by representative normalized radial intensity plots of the contrast-enhanced tumor volume at day 28 (Fig 5B). Note that, in Fig 5B, radial position “0” corresponds to the upper inflection point of the sigmoidal intensity curve at the inner edge of the tumor. Tumor edge thickness (Fig 5C) and sharpness (Fig 5D), derived from intensity profiles of line segments drawn orthogonal to tumor boundaries, were used as representations of tumor invasiveness. Here, tumors in the Control, CDDP-BPN, and FUSlo + CDDP-BPN groups exhibited similar levels of invasiveness. However, the FUShi + CDDP-BPN group exhibited a significant reduction in edge thickness (Fig 5C) and a significant increase in edge sharpness (Fig 5D) when compared to all other groups, indicating reduced invasiveness. Moreover, examination of H&E stained cross-sections from Control and FUShi + CDDP-BPN treated F98 tumors revealed that FUShi + CDDP-BPN tumors had significantly more defined tumor edges (Fig 5E) and a 3.5-fold reduction in the number of infiltrating tumor nodules present beyond the tumor edge (Fig 5F).
Figure 5. MR image-guided delivery of CDDP-BPN with FUS reduces the invasiveness of F98 gliomas.
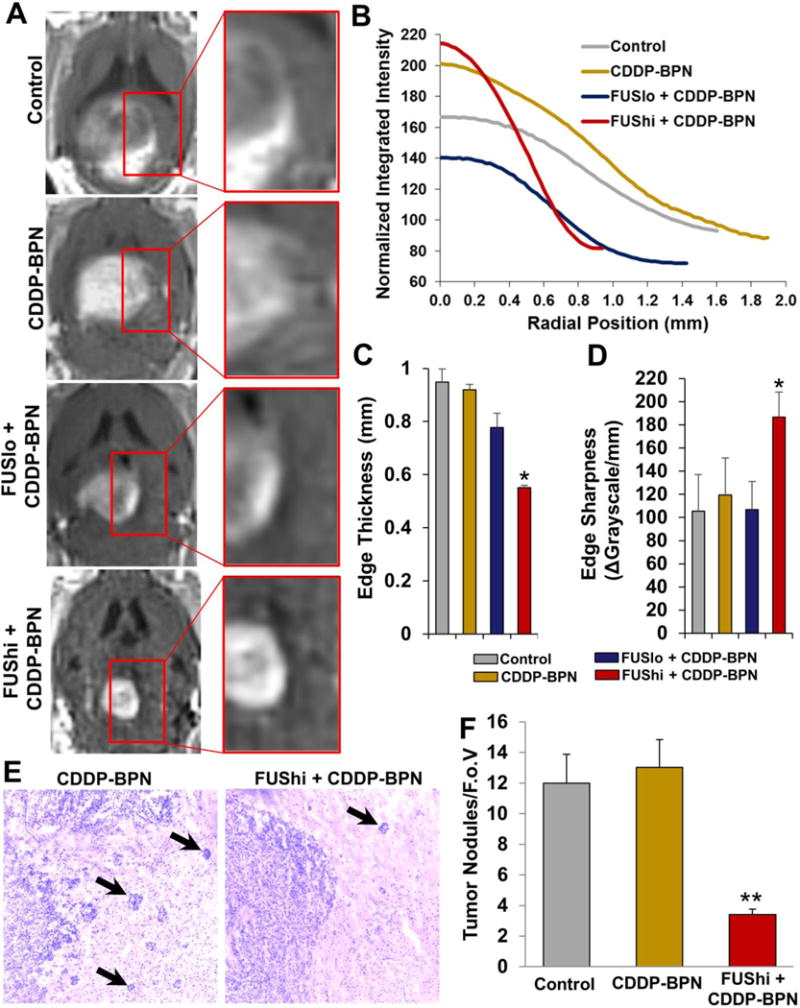
(A) Representative contrast enhanced T1 weighted MR images taken from untreated Control, CDDP-BPN, FUSlo + CDDP-BPN, and FUShi + CDDP-BPN groups on Day 28. Insets show that the edge of the contrast enhancing region is more defined in the FUShi +CDDP-BPN treated F98 tumor when compared to the other 3 groups. (B) Radial intensity profiles acquired from the contrast MR images shown in the left column of panel A. Note that the linear region of the sigmoidal radial intensity profile from the FUShi + CDDP-BPN treated F98 glioma is considerably steeper than for the other 3 groups. Meanwhile, the width of the radial intensity profile is also reduced, indicating less infiltration into surrounding brain tissue. (C) Bar graph of tumor edge thickness for all 4 groups. *P<0.05 vs. all other groups. (D) Bar graph of tumor edge sharpness for all 4 groups. *P<0.05 vs. all other groups. (E) Representative transmitted light microscopic images of H&E stained cross-sections through F98 tumors from the CDDP-BPN and FUShi + CDDP-BPN groups. Note that the tumor border in the FUShi + CDDP-BPN image is well-defined compared to the CDDP-BPN image. Infiltrating tumor nodules are denoted with black arrows. (F) Bar graph of infiltrating tumor nodules per field of view (F.o.V.). **P<0.01 vs. all other groups. N≥4 for all groups.
Discussion
Current treatment strategies for glioma are unable to effectively control tumor recurrence, the leading cause of death for patients [1]. Invasive tumor cells that lie beyond the enhancing tumor border in areas protected by the intact BBB limit the efficacy of systemically administered drugs [12,13,34,35], and locally administered therapeutics do not diffuse well beyond the resection cavity [36,37]. Standard polymeric nanoparticles can provide extended drug release; however, they are immobilized in the complex tumor parenchyma [38,39] and do not spread well within tumors. Here, we evaluated the potential of a novel combination of MRgFUS and CDDP-loaded brain penetrating nanoparticles in the treatment of GBM. We designed a polymeric nanoparticle formulation for enhanced delivery and distribution within the brain [23,24,37] following delivery across the BBB/BTB with MRgFUS [32]. These densely PEGylated BPN were shown to deliver higher concentrations of CDDP in a sustained manner to a larger population of tumor cells. Importantly, MRgFUS-mediated BBB/BTB disruption enhanced both the delivery and distribution of CDDP-BPN, improving efficacy against both the tumor bulk and infiltrating tumor cells. We conclude that this approach may provide improved therapeutic outcomes and potentially decreased rates of glioma recurrence.
CDDDP-BPN Formulation, Characterization, and Therapeutic Benefits
As a platinum-based chemotherapeutic, CDDP is a highly potent and non-specific inhibitor of DNA synthesis and repair [40]. The clinical use of CDDP in gliomas has been limited by significant neurotoxicities [6,7] which mandated the use of low CDDP doses and invasive local delivery methods [9,41]. These difficulties lead to the development of less toxic but less effective compounds [42,43]. Sustained, local release of low concentrations of CDDP can both increase the effectiveness of therapy and decrease off-target toxicity [27,41,44–47]. The particles developed here show sustained CDDP release over 7 days. In vitro cytotoxicity studies using F98 glioma cells showed that CDDP-BPN exhibited lower potency compared to CDDP solution which is most likely due to slow release of CDDP from CDDP-BPN as observed in case of in vitro release kinetics of CDDP-BPN. Several other types of polymer- and lipid-based nanoparticles containing CDDP have also shown lower in vitro potency against F98 glioma and other cancer cells compared to carrier-free CDDP solution[48–51]. We anticipate that combined with weekly MRgFUS, these CDDP-BPN could provide stable local drug concentrations with reduced systemic and local toxicities.
As we have previously reported [23,24,27,32], a dense coating of PEG reduces the particle polydispersity and the net charge at the particles’ surface, producing a monodisperse, near-neutrally charged BPN that is smaller than the average pore size within the ECS of the brain parenchyma [23]. The combination of small particle size and non-adhesive surfaces significantly increased the rate of spreading of the particles in healthy brain tissue, as measured by MSD and Vd, in agreement with previous studies with other BPN formulations [23,24,27]. The use of activated, aquated cisplatin during BPN formulation doubled drug loading efficiency and significantly extended drug release compared to our previous formulation using un-activated CDDP [27]. Additionally, these CDDP-BPN remain stable in both blood and ACSF for up to 24 hours, making them ideal candidates for MRgFUS-mediated delivery, as they will continue to circulate in the blood, accumulate, and spread within brain or tumor tissue following BBB/BTB disruption.
Improved Anti-Tumor Efficacy Achieved by MR Image-Guided Delivery of CDDP-BPN with FUS
Clinical utilization of the Gliadel® wafer demonstrated the potential of local drug administration methods, but poor drug diffusion into the surrounding tissue limits the therapeutic efficacy of such approaches [52–54]. Furthermore, local administration techniques are highly invasive and associated with significant risks [1,55], particularly for therapeutics that require multiple treatments. MRgFUS is the only treatment modality capable of producing non-invasive, localized, reversible BBB disruption [19,21,22,32,56,57] and has been shown to increase the delivery of therapeutics across the BTB as well [17,58–60]. In agreement with this body of work, as well as our previous work [22,32], we show here that MRgFUS significantly increases both the total delivery and distribution of BPN within the tumor parenchyma and surrounding normal tissue in both the 9L and F98 glioma models. The enhanced permeation and retention (EPR) effect produced very little BPN delivery in either tumor, emphasizing the need for a mechanism for improved delivery. The low efficiency of EPR-mediated delivery is further demonstrated by the performance of the CDDP-BPN group in the in-vivo MRgFUS delivery experiments, wherein tumors were of similar size and morphology to untreated F98 control tumors. FUS-mediated BTB opening has been shown to increase pinocytotic vesicle formation and upregulate caveoli-associated proteins [60]. Additionally, tumor permeability, as measured by gadolinium enhancement in both this study and studies by other investigators [58], is enhanced by MRgFUS opening of the BTB. These mechanisms yield increased BPN delivery and uptake within the tumor.
Here, we demonstrate that three weekly treatments with 0.8 MPa MRgFUS + CDDP-BPN significantly improved tumor growth control and animal survival. We previously determined [22,32] that 0.6 MPa was sufficient to produce significant enhancements in BPN delivery across the BBB in normal tissue, and we demonstrated here that 0.6 MPa significantly increased BPN delivery across the BTB in both the 9L and F98 glioma models. However, presumably abnormal tumor vasculature [30] and parenchyma [61] combined with the aggressive nature of the F98 tumor model [28,29,61] required the use of slightly higher pressure to achieve a treatment effect. In addition to reducing tumor volume, animals treated with MRgFUS + CDDP-BPN had more defined tumor boundaries. While MRgFUS + CDDP-BPN was not curative, it did control the growth and infiltration of the tumor, indicating that this may be a viable treatment option following surgical de-bulking.
Relationship Between BPN Delivery and Tumor Vascularity
While MRgFUS-mediated delivery produced more homogeneous BPN distribution throughout the tumor, BPN delivery varied with vascular density in both tumor models. This spatial variation may reflect changes in the vasculature itself, as it has been shown that FUS-induced BBB opening is dependent on vessel size, and vessels over 30 μm in diameter do not demonstrate BBB opening following FUS [62,63]. Furthermore, areas with high vascular density may have higher interstitial pressures [64] and reduced extracellular space available, thus reducing BPN uptake. Fluid diffusion is known to vary throughout gliomas [65]. Optimal BPN delivery occurred at slightly different vascular densities in the two models, and the degree of dependence of BPN delivery on vascular density was different as well. This likely reflects inherent differences in the characteristics of the tumors, as F98 gliomas are known to co-opt existing vasculature, while 9L gliomas are highly angiogenic and significantly alter vascular networks [30]. These observations may help determine optimal treatment schedules, as well as which patients may benefit most from MRgFUS.
Comparisons to Previous Studies of MRgFUS-Targeted Drug Delivery to Brain Tumors
Previous work has demonstrated that repeated treatments and higher FUS pressures produce improvements in tumor growth control and animal survival [17,66–68]. In the less infiltrative 9L model, a single high-pressure treatment (0.8 MPa) of FUS with liposomal doxorubicin produced a significant 24% increase in median survival time, but was not curative [68]. A follow-up study in the same model utilized three weekly FUS + liposomal doxorubicin treatments (again at 0.8 MPa) to achieve complete tumor regression in 3/8 animals [67]. Doxorubicin liposomes are large (~100nm), and these studies did not determine whether FUS was able to deliver intact liposomes across the BBB/BTB. Given the FUS pressures used, it was postulated that FUS disrupted the liposomes [69] in the tumor vasculature and enhanced the delivery of the released doxorubicin across the BBB/BTB. Indeed, FUS has been shown to increase delivery of free doxorubicin across the BTB in preclinical glioma models [70,71]. A similar study, also in the 9L glioma model, showed that a single 0.75 MPa FUS + temozolomide (TMZ) treatment increased survival by 3 days [72]. Our FUS+CDDP-BPN treatment yielded similar results to treatment with FUS+TMZ, but it was less effective than FUS+ liposomal doxorubicin. However, we were able to demonstrate delivery of intact 60 nm particles (much larger than either free doxorubicin or TMZ) across the BTB and our studies were performed in the more infiltrative F98 glioma model rather than the 9L model [28]. Additionally, therapy was not initiated in our study until 14 days post-inoculation, due to a reliance on MR imaging for targeting. At this time point, the F98 tumors were well established and had compromised BBB function. Utilizing an alternative imaging method to verify tumor engraftment would permit initializing treatment earlier and would likely improve treatment outcome, particularly since it has been shown that MRgFUS produces greater improvements in BTB disruption in earlier stage tumors[58]. Alternatively, slower-growing glioma models like the 9L [29] may provide a larger therapeutic window. Delivery of similar CDDP-BPN to orthotopic F98 glioma using CED achieved efficacy only when treatment began within one week of inoculation [27]. To our knowledge, the only other study to treat F98 glioma with FUS utilized transfected cells expressing HSV1-tk, which makes them highly susceptible to the small-molecule antiviral drug ganciclovir. In this model, FUS + ganciclovir produced up to 80% tumor regression, compared to 5% for ganciclovir alone [73]. However, it should be noted that these transfected cells were several fold less aggressive than wild-type F98 cells, most likely due to increased immunogenicity. Additional strategies could be employed to improve the efficacy of MRgFUS-mediated delivery of BPN, including utilizing BPNs loaded with other chemotherapeutics [74,75], immunotherapeutics [76,77], or plasmids [22,25,78–80]. Finally, to maintain treatment consistency both within and between the FUSlo+CDDP-BPN and FUShi+CDDP-BPN groups, we always used a 3×3 sonication grid for FUS-mediated BPN delivery, independent of whether this treatment volume matched the size of the F98 tumor being treated. It is highly probable that increasing FUS treatment volume in accord with tumor size, as has been done in studies by other investigators [67,68], would lead to improved treatment efficacy.
Conclusions
This is the first study demonstrating that MRgFUS can be used to enhance the delivery and efficacy of a polymeric nanoparticle in the treatment of glioma. BPN delivery with MRgFUS can improve antitumor efficacy by: (i) providing deeper brain tissue penetration of drug than is achieved with polymer implants (located only at the periphery in the surgical cavity), (ii) enabling multiple dosing regimens without the need for repeat surgery, and (iii) facilitating local delivery for tumors with high surgical risk. MR guidance adds the ability to identify and target specific regions, reducing off-target toxicity within the brain. We anticipate that MRgFUS-mediated delivery of CDDP-BPN may be a promising treatment approach to prevent tumor recurrence in patients with high grade gliomas.
Supplementary Material
Figure S1: Representative NMR spectrum confirming PEGylation of PAA. 1H NMR spectrum of PAA (upper panel), PEG (middle panel) and PAA-PEG (lower panel) in D2O.
Table S1: Survival analysis.
Acknowledgments
This work was supported by National Institutes of Health Grants R01CA164789, R01EB020147, R01CA204968 and R01CA197111, and the Focused Ultrasound Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corso CD, Bindra RS. Success and failures of combined modalities in GBM: Old Problems and New Directions. Semin Radiat Oncol. 2016 doi: 10.1016/j.semradonc.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Ideguchi M, Kajiwara K, Goto H, Sugimoto K, Nomura S, Ikeda E, Suzuki M. MRI findings and pathological features in early-stage glioblastoma. J Neurooncol. 2015;123:289–97. doi: 10.1007/s11060-015-1797-y. [DOI] [PubMed] [Google Scholar]
- 3.Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol. 2014;71:1437–44. doi: 10.1001/jamaneurol.2014.1701. [DOI] [PubMed] [Google Scholar]
- 4.Hargrave DR, Zacharoulis S. Pediatric CNS tumors: current treatment and future directions. Expert Rev Neurother. 2007;7:1029–12. doi: 10.1586/14737175.7.8.1029. [DOI] [PubMed] [Google Scholar]
- 5.Kim IH, Park CK, Heo DS, Kim CY, Rhee CH, Nam DH, Lee SH, Han JH, Lee SH, Kim TM, Kim DW, Kim JE, Paek SH, Kim DG, Kim IA, Kim YJ, Kim JH, Park BJ, Jung HW. Radiotherapy followed by adjuvant temozolomide with or without neoadjuvant ACNU-CDDP chemotherapy in newly diagnosed glioblastomas: a prospective randomized controlled multicenter phase III trial. J Neurooncol. 2011;103:595–602. doi: 10.1007/s11060-010-0427-y. [DOI] [PubMed] [Google Scholar]
- 6.Silvani A, Gaviani P, Lamperti EA, Eoli M, Falcone C, Dimeco F, Milanesi IM, Erbetta A, Boiardi A, Fariselli L, Salmaggi A. Cisplatinum and BCNU chemotherapy in primary glioblastoma patients. J Neurooncol. 2009;94:57–62. doi: 10.1007/s11060-009-9800-0. [DOI] [PubMed] [Google Scholar]
- 7.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Pérez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 8.Huo T, Barth RF, Yang W, Nakkula RJ, Koynova R, Tenchov B, Chaudhury AR, Agius L, Boulikas T, Elleaume H, Lee RJ. Preparation, biodistribution and neurotoxicity of liposomal cisplatin following convection enhanced delivery in normal and F98 glioma bearing rats. PLoS One. 2012;7:e48752. doi: 10.1371/journal.pone.0048752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheleg SV, Korotkevich EA, Zhavrid EA, Muravskaya GV, Smeyanovich AF, Shanko YG, Yurkshtovich TL, Bychkovsky PB, Belyaev SA. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J Neurooncol. 2002;60:53–9. doi: 10.1023/a:1020288015457. [DOI] [PubMed] [Google Scholar]
- 10.Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012;72:648–72. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- 11.Krol S. Challenges in drug delivery to the brain: Nature is against us. J Control Release. 2012:1–11. doi: 10.1016/j.jconrel.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 12.Drapeau A, Fortin D. Chemotherapy Delivery Strategies to the Central Nervous System: neither Optional nor Superfluous. Curr Cancer Drug Targets. 2015 doi: 10.2174/1568009615666150616123548. [DOI] [PubMed] [Google Scholar]
- 13.Hendricks BK, Cohen-Gadol AA, Miller JC. Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg Focus. 2015;38:E10. doi: 10.3171/2015.1.FOCUS14767. [DOI] [PubMed] [Google Scholar]
- 14.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 15.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 16.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185–99. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 17.Zhao YZ, Chen LJ, Lin Q, Cai J, Yu WZ, Zhao YP, Xu CY, Mao KL, Tian FR, Li WF, Wong HL, Lu CT. Using FUS induced BBB/BTB-opening technique combined with Doxorubicin liposomes to improve glioma-targeted inhibition. Oncotarget. 2015 doi: 10.18632/oncotarget.5144. [DOI] [Google Scholar]
- 18.Frenkel V. Ultrasound mediated delivery of drugs and genes to solid tumors. Adv Drug Deliv Rev. 2008;60:1193–208. doi: 10.1016/j.addr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobus T, Vykhodtseva N, Pilatou M, Zhang Y, McDannold N. Safety Validation of Repeated Blood-Brain Barrier Disruption Using Focused Ultrasound. Ultrasound Med Biol. 2015 doi: 10.1016/j.ultrasmedbio.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hynynen K, McDannold N, Sheikov NA, Jolesz FA, Vykhodtseva N. Local and reversible blood-brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage. 2005;24:12–20. doi: 10.1016/j.neuroimage.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 21.Timbie KF, Mead BP, Price RJ. Drug and gene delivery across the blood-brain barrier with focused ultrasound. J Control Release. 2015 doi: 10.1016/j.jconrel.2015.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mead BP, Mastorakos P, Suk JS, Klibanov AL, Hanes J, Price RJ. Targeted gene transfer to the brain via the delivery of brain-penetrating DNA nanoparticles with focused ultrasound. J Control Release. 2016;223:109–17. doi: 10.1016/j.jconrel.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8:10655–64. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C. Convection Enhanced Delivery of Cisplatin-Loaded Brain Penetrating Nanoparticles Cures Malignant Glioma in Rats. J Control Release. 2017 doi: 10.1016/j.jconrel.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C, Nance EA, Mastorakos P, Chisholm J, Berry S, Eberhart C, Tyler BM, Brem H, Suk JS, Hanes J. Convection Enhanced Delivery of Cisplatin-Loaded Brain Penetrating Nanoparticles Cures Malignant Glioma in Rats. J Control Release. 2016 doi: 10.1016/j.jconrel.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT–2 and CNS–1 gliomas. J Neurooncol. 2009;94:299–312. doi: 10.1007/s11060-009-9875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doblas S, He T, Saunders D, Hoyle J, Smith N, Pye Q, Lerner M, Jensen RL, Towner RA. In vivo characterization of several rodent glioma models by 1H MRS. NMR Biomed. 2012;25:685–94. doi: 10.1002/nbm.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doblas S, He T, Saunders D, Pearson J, Hoyle J, Smith N, Lerner M, Towner RA. Glioma morphology and tumor-induced vascular alterations revealed in seven rodent glioma models by in vivo magnetic resonance imaging and angiography. J Magn Reson Imaging. 2010;32:267–75. doi: 10.1002/jmri.22263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bencokova Z, Pauron L, Devic C, Joubert A, Gastaldo J, Massart C, Balosso J, Foray N. Molecular and cellular response of the most extensively used rodent glioma models to radiation and/or cisplatin. J Neurooncol. 2008;86:13–21. doi: 10.1007/s11060-007-9433-0. [DOI] [PubMed] [Google Scholar]
- 32.Nance E, Timbie K, Miller GW, Song J, Louttit C, Klibanov AL, Shih TY, Swaminathan G, Tamargo RJ, Woodworth GF, Hanes J, Price RJ. Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood-brain barrier using MRI-guided focused ultrasound. J Control Release. 2014;189:123–32. doi: 10.1016/j.jconrel.2014.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otsu N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans Syst Man Cybern. 1979;9:62–66. doi: 10.1109/TSMC.1979.4310076. [DOI] [Google Scholar]
- 34.Puhalla S, Elmquist W, Freyer D, Kleinberg L, Adkins C, Lockman P, McGregor J, Muldoon L, Nesbit G, Peereboom D, Smith Q, Walker S, Neuwelt E. Unsanctifying the sanctuary: challenges and opportunities with brain metastases. Neuro Oncol. 2015;17:639–651. doi: 10.1093/neuonc/nov023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez A, Tatter SB, Debinski W. Neurosurgical Techniques for Disruption of the Blood-Brain Barrier for Glioblastoma Treatment. Pharmaceutics. 2015;7:175–87. doi: 10.3390/pharmaceutics7030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Patel TR, Sirianni RW, Strohbehn G, Zheng M-QQ, Duong N, Schafbauer T, Huttner AJ, Huang Y, Carson RE, Zhang Y, Sullivan DJ, Piepmeier JM, Saltzman WM, Sullivan DJ, Jr, Piepmeier JM, Saltzman WM. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A. 2013;110:11751–11756. doi: 10.1073/pnas.1304504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allard E, Passirani C, Benoit JP. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30:2302–18. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, Kracht L, Coenen HH, Sturm V, Wienhard K, Heiss WD, Jacobs AH. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54:479–87. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 39.MacKay JA, Deen DF, Szoka FC. Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005;1035:139–53. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Prestayko A. Cisplatin: Current Status and New Developments. Academic Press; Atlanta: 1979. [Google Scholar]
- 41.Debinski W, Tatter SB. Convection-enhanced delivery for the treatment of brain tumors. Expert Rev Neurother. 2009;9:1519–27. doi: 10.1586/ern.09.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldwirt L, Canney M, Horodyckid C, Poupon J, Mourah S, Vignot A, Chapelon JY, Carpentier A. Enhanced brain distribution of carboplatin in a primate model after blood-brain barrier disruption using an implantable ultrasound device. Cancer Chemother Pharmacol. 2016;77:211–6. doi: 10.1007/s00280-015-2930-5. [DOI] [PubMed] [Google Scholar]
- 43.Stordal B, Pavlakis N, Davey R. Oxaliplatin for the treatment of cisplatin-resistant cancer: a systematic review. Cancer Treat Rev. 2007;33:347–57. doi: 10.1016/j.ctrv.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93:678–87. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paraskar AS, Soni S, Chin KT, Chaudhuri P, Muto KW, Berkowitz J, Handlogten MW, Alves NJ, Bilgicer B, Dinulescu DM, Mashelkar RA, Sengupta S. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc Natl Acad Sci U S A. 2010;107:12435–40. doi: 10.1073/pnas.1007026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br J Cancer. 2011;104:593–8. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yong D, Luo Y, Du F, Huang J, Lu W, Dai Z, Yu J, Liu S. CDDP supramolecular micelles fabricated from adamantine terminated mPEG and β-cyclodextrin based seven-armed poly (l-glutamic acid)/CDDP complexes. Colloids Surfaces B Biointerfaces. 2013;105:31–36. doi: 10.1016/j.colsurfb.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 49.Saisyo A, Nakamura H, Fang J, Tsukigawa K, Greish K, Furukawa H, Maeda H. pH-sensitive polymeric cisplatin-ion complex with styrene-maleic acid copolymer exhibits tumor-selective drug delivery and antitumor activity as a result of the enhanced permeability and retention effect. Colloids Surfaces B Biointerfaces. 2016;138:128–137. doi: 10.1016/j.colsurfb.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 50.Charest G, Paquette B, Fortin D, Mathieu D, Sanche L. Concomitant treatment of F98 glioma cells with new liposomal platinum compounds and ionizing radiation. J Neurooncol. 2010;97:187–93. doi: 10.1007/s11060-009-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishiyama N, Kataoka K. Preparation and characterization of size-controlled polymeric micelle containing cis-dichlorodiammineplatinum(II) in the core. J Control Release. 2001;74:83–94. doi: 10.1016/s0168-3659(01)00314-5. [DOI] [PubMed] [Google Scholar]
- 52.Attenello FJ, Mukherjee D, Datoo G, McGirt MJ, Bohan E, Weingart JD, Olivi A, Quinones-Hinojosa A, Brem H. Use of Gliadel (BCNU) wafer in the surgical treatment of malignant glioma: a 10-year institutional experience. Ann Surg Oncol. 2008;15:2887–93. doi: 10.1245/s10434-008-0048-2. [DOI] [PubMed] [Google Scholar]
- 53.Westphal M, Ram Z, Riddle V, Hilt D, Bortey E. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 2006;148:269–75. doi: 10.1007/s00701-005-0707-z. discussion 275. [DOI] [PubMed] [Google Scholar]
- 54.Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jääskeläinen J, Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shahar T, Ram Z, Kanner AA. Convection-enhanced delivery catheter placements for high-grade gliomas: Complications and pitfalls. J Neurooncol. 2012;107:373–378. doi: 10.1007/s11060-011-0751-x. [DOI] [PubMed] [Google Scholar]
- 56.Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Non-invasive opening of BBB by focused ultrasound. Acta Neurochir Suppl. 2003;86:555–558. doi: 10.1007/978-3-7091-0651-8_113. [DOI] [PubMed] [Google Scholar]
- 57.Konofagou EE. Optimization of the ultrasound-induced blood-brain barrier opening. Theranostics. 2012;2:1223–37. doi: 10.7150/thno.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aryal M, Park J, Vykhodtseva N, Zhang YZ, McDannold N. Enhancement in blood-tumor barrier permeability and delivery of liposomal doxorubicin using focused ultrasound and microbubbles: evaluation during tumor progression in a rat glioma model. Phys Med Biol. 2015;60:2511–27. doi: 10.1088/0031-9155/60/6/2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park EJ, Zhang YZ, Vykhodtseva N, McDannold N. Ultrasound-mediated blood-brain/blood-tumor barrier disruption improves outcomes with trastuzumab in a breast cancer brain metastasis model. J Control Release. 2012;163:277–84. doi: 10.1016/j.jconrel.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia C, Zhang Z, Xue Y, Wang P, Liu Y. Mechanisms of the increase in the permeability of the blood-tumor barrier obtained by combining low-frequency ultrasound irradiation with small-dose bradykinin. J Neurooncol. 2009;94:41–50. doi: 10.1007/s11060-009-9812-9. [DOI] [PubMed] [Google Scholar]
- 61.Belloli S, Brioschi A, Politi LS, Ronchetti F, Calderoni S, Raccagni I, Pagani A, Monterisi C, Zenga F, Zara G, Fazio F, Mauro A, Moresco RM. Characterization of biological features of a rat F98 GBM model: a PET-MRI study with [18F]FAZA and [18F]FDG. Nucl Med Biol. 2013;40:831–40. doi: 10.1016/j.nucmedbio.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 62.Cho EE, Drazic J, Ganguly M, Stefanovic B, Hynynen K. Two-photon fluorescence microscopy study of cerebrovascular dynamics in ultrasound-induced blood-brain barrier opening. J Cereb Blood Flow Metab. 2011;31:1852–1862. doi: 10.1038/jcbfm.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nhan T, Burgess A, Cho EE, Stefanovic B, Lilge L, Hynynen K. Drug delivery to the brain by focused ultrasound induced blood-brain barrier disruption: Quantitative evaluation of enhanced permeability of cerebral vasculature using two-photon microscopy. J Control Release. 2013;172:274–280. doi: 10.1016/j.jconrel.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stapleton S, Mirmilshteyn D, Zheng J, Allen C, Jaffray DA. Spatial Measurements of Perfusion, Interstitial Fluid Pressure and Liposomes Accumulation in Solid Tumors. J Vis Exp. 2016 doi: 10.3791/54226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mekkaoui C, Metellus P, Kostis WJ, Martuzzi R, Pereira FR, Beregi JP, Reese TG, Constable TR, Jackowski MP. Diffusion Tensor Imaging in Patients with Glioblastoma Multiforme Using the Supertoroidal Model. PLoS One. 2016;11:e0146693. doi: 10.1371/journal.pone.0146693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobus T, Zervantonakis IK, Zhang Y, McDannold NJ. Growth inhibition in a brain metastasis model by antibody delivery using focused ultrasound-mediated blood-brain barrier disruption. J Control Release. 2016 doi: 10.1016/j.jconrel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aryal M, Vykhodtseva N, Zhang Y-Z, Park J, McDannold N. Multiple treatments with liposomal doxorubicin and ultrasound-induced disruption of blood-tumor and blood-brain barriers improve outcomes in a rat glioma model. J Control Release. 2013;169:103–11. doi: 10.1016/j.jconrel.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Treat LH, McDannold N, Zhang Y, Vykhodtseva N, Hynynen K. Improved anti-tumor effect of liposomal doxorubicin after targeted blood-brain barrier disruption by MRI-guided focused ultrasound in rat glioma. Ultrasound Med Biol. 2012;38:1716–1725. doi: 10.1016/j.ultrasmedbio.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rizzitelli S, Giustetto P, Faletto D, Castelli DD, Aime S, Terreno E. The release of Doxorubicin from liposomes monitored by MRI and triggered by a combination of US stimuli led to a complete tumor regression in a breast cancer mouse model. J Control Release. 2016;230:57–63. doi: 10.1016/j.jconrel.2016.03.040. [DOI] [PubMed] [Google Scholar]
- 70.Kovacs Z, Werner B, Rassi A, Sass JO, Martin-Fiori E, Bernasconi M. Prolonged survival upon ultrasound-enhanced doxorubicin delivery in two syngenic glioblastoma mouse models. J Control Release. 2014 doi: 10.1016/j.jconrel.2014.05.033. [DOI] [PubMed] [Google Scholar]
- 71.Treat LH, McDannold N, Vykhodtseva N, Zhang Y, Tam K, Hynynen K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int J Cancer. 2007;121:901–7. doi: 10.1002/ijc.22732. [DOI] [PubMed] [Google Scholar]
- 72.Wei KC, Chu PC, Wang HYJ, Huang CY, Chen PY, Tsai HC, Lu YJ, Lee PY, Tseng IC, Feng LY, Hsu PW, Yen TC, Liu HL. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: a preclinical study. PLoS One. 2013;8:e58995. doi: 10.1371/journal.pone.0058995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang FY, Chang WY, Lin WT, Hwang JJ, Chien YC, Wang HE, Tsai ML. Focused ultrasound enhanced molecular imaging and gene therapy for multifusion reporter gene in glioma-bearing rat model. Oncotarget. 2015;6:36260–8. doi: 10.18632/oncotarget.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang M, Yu T, Wang YY, Lai SK, Zeng Q, Miao B, Tang BC, Simons BW, Ensign LM, Liu G, Chan KWY, Juang CY, Mert O, Wood J, Fu J, McMahon MT, Wu TC, Hung CF, Hanes J. Vaginal Delivery of Paclitaxel via Nanoparticles with Non-Mucoadhesive Surfaces Suppresses Cervical Tumor Growth. Adv Healthc Mater. 2014;3:1044–1052. doi: 10.1002/adhm.201300519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, Fu J, Chen B, Xia S, Liu Y, Neisser M, Nguyen C, Lee R, Park JK, Reyes J, Hartung T, Rojas C, Rais R, Tsukamoto T, Semenza GL, Hanes J, Slusher BS, Le A. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci. 2016;113:E5328–E5336. doi: 10.1073/pnas.1611406113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sampath P, Hanes J, DiMeco F, Tyler BM, Brat D, Pardoll DM, Brem H. Paracrine immunotherapy with interleukin–2 and local chemotherapy is synergistic in the treatment of experimental brain tumors. Cancer Res. 1999;59:2107–14. [PubMed] [Google Scholar]
- 77.Rhines LD, Sampath P, DiMeco F, Lawson HC, Tyler BM, Hanes J, Olivi A, Brem H. Local immunotherapy with interleukin–2 delivered from biodegradable polymer microspheres combined with interstitial chemotherapy: a novel treatment for experimental malignant glioma. Neurosurgery. 2003;52 doi: 10.1227/01.neu.0000053211.39087.d1. 872-9-80. [DOI] [PubMed] [Google Scholar]
- 78.Mastorakos P, Zhang C, Berry S, Oh Y, Lee S, Eberhart CG, Woodworth GF, Suk JS, Hanes J. Highly PEGylated DNA Nanoparticles Provide Uniform and Widespread Gene Transfer in the Brain. Adv Healthc Mater. 2015:n/a–n/a. doi: 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suk JS, Suh J, Lai SK, Hanes J. Quantifying the intracellular transport of viral and nonviral gene vectors in primary neurons. Exp Biol Med (Maywood) 2007;232:461–9. [PubMed] [Google Scholar]
- 80.Boylan NJ, Suk JS, Lai SK, Jelinek R, Boyle MP, Cooper MJ, Hanes J. Highly compacted DNA nanoparticles with low MW PEG coatings: in vitro, ex vivo and in vivo evaluation. J Control Release. 2012;157:72–9. doi: 10.1016/j.jconrel.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Representative NMR spectrum confirming PEGylation of PAA. 1H NMR spectrum of PAA (upper panel), PEG (middle panel) and PAA-PEG (lower panel) in D2O.
Table S1: Survival analysis.