

Abstract
Magnetoencephalography (MEG), a direct measure of neuronal activity, is an underexplored tool in the search for biomarkers of Alzheimer's disease (AD). In this study, we used MEG source estimates of auditory gating generators, nonlinear correlations with neuropsychological results, and multivariate analyses to examine the sensitivity and specificity of gating topology modulation to detect AD. Our results demonstrated the use of MEG localization of a medial prefrontal (mPFC) gating generator as a discrete (binary) detector of AD at the individual level and resulted in recategorizing the participant categories in: (1) controls with mPFC generator localized in response to both the standard and deviant tones; (2) a possible preclinical stage of AD participants (a lower functioning group of controls) in which mPFC activation was localized to the deviant tone only; and (3) symptomatic AD in which mPFC activation was not localized to either the deviant or standard tones. This approach showed a large effect size (0.9) and high accuracy, sensitivity, and specificity (100%) in identifying symptomatic AD patients within a limited research sample. The present results demonstrate high potential of mPFC activation as a noninvasive biomarker of AD pathology during putative preclinical and clinical stages. Hum Brain Mapp 38:5180–5194, 2017. © 2017 Wiley Periodicals, Inc.
Keywords: Alzheimer's disease, preclinical Alzheimer's disease, mild cognitive impairment, dementia, biomarker, auditory sensory gating, prefrontal cortex, Rey–Osterreith complex figure test, magnetoencephalography, neuroimaging
INTRODUCTION
Alzheimer's disease (AD), the most prevalent aging‐related neuropathology, is marked by cerebral amyloid deposition, tauopathy, neuroinflammation, highly disrupted cholinergic transmission, and extensive neuronal loss. A wealth of evidence suggests that the pathological changes associated with AD start decades before the onset of clinical symptoms. Also, there is accruing evidence that irreversible pathological processes related to the disease have occurred prior to the mild cognitive impairment (MCI) phase, considered until recently the earliest stage of AD [Farlow, 2009; Lazarczyk et al., 2012]. Consequently, the concept of AD pathogenesis is evolving toward a view of the disease as a long‐term continuum which differs only by symptom appearance; that is, a nonsymptomatic (preclinical) AD phase and an irreversible symptomatic AD phase [Morris et al., 2001; Petersen and Morris, 2005; Sperling et al., 2011]. There are two major sets of criteria for the clinical diagnosis of Alzheimer's disease that have been published, one from an International Working Group (IWG) [Dubois et al., 2007] and the other from working groups convened by the National Institute on Aging (NIA) and the Alzheimer's Association (AA) [McKhann et al., 2011] in the United States. There are important differences in terms of how AD is conceptualized, the terminology used, and the diagnostic algorithm. The Key Symposium in Stockholm revealed harmonized IWG and NIA–AA criteria for clinical and research practice [Morris et al., 2014] which defines AD as a brain disorder regardless of clinical status. They recommended that the term symptomatic AD should be used to describe the entire clinical spectrum of AD, from the earliest symptomatic stages (MCI/prodromal AD) to the most severe. Currently, the incorporation of biomarkers into the diagnostic procedure for AD is postponed until the successful minimization of within‐ and between‐center variability establishes uniform cutoff levels and standardization processes [Morris et al., 2014]. Importantly, the consensus of the Key Symposium meeting was that AD is viewed as a disease that occurs across a continuum in which pathophysiological changes occur prior to the identification of clinical symptoms, thereby providing an opportunity to identify biomarkers of AD progression prior to the clinical onset of AD.
This concept of AD emphasizes the neurobiological advantage of early intervention; that is, it is crucial to detect very early, possibly reversible, pathological changes related to AD in cognitively intact individuals, before the occurrence of the first symptoms. Proposed state‐of‐the‐art diagnostic measures of AD have limited efficacy in detecting preclinical changes associated with the disease [Dubois et al., 2007; Holtzman et al., 2011; Lazarczyk et al., 2012; McKhann et al., 2011; Morris et al., 2014; Sperling et al., 2011] and are invasive for participants since there are risks associated with lumbar puncture (cerebrospinal fluid analysis, CSF), exposure to radiation (PET/CT), or claustrophobic time‐consuming scanning (fMRI). Therefore, there is an increasing need for additional simple, noninvasive tools that can be used to differentiate preclinical and symptomatic AD from normal aging.
The first symptom of AD is insidious dissolution of the ability to learn new information accompanied with subtle and variable amnestic impairment without any clinically detectable signs of brain injury, pointing to the existence of discrete interruption of synapses that are involved in encoding new declarative memory [Selkoe, 2002]. As longer maintenance of sensory memory traces results in more successful memory encoding [Atkinson and Shiffrin, 1968], sensory gating mechanisms—conceptualized as the neural ability to modulate its responses to subsequent stimuli—have a major role in guiding our understanding of successful encoding of new information. Auditory gating‐out [Boutros and Belger, 1992; Gjini et al., 2010] has been proposed as a mechanism of habituating to redundant auditory stimuli that protects working memory overload by preventing irrelevant information from recurrent sensory processing [Venables, 1964]. Dysfunction of the auditory gating‐out mechanism likely reduces preattentive signal‐to‐noise ratio and alert augmentation during synaptic consolidation in inchoate phases of memory encoding [Freedman et al., 1996], thus contributing to the first symptoms of AD pathology.
Recent studies [Josef Golubic et al., 2014a; Weiland et al., 2008] strongly suggest that the auditory gating topology (i.e., set of neural generators) is composed of a PFC generator in addition to the anticipated generators in bilateral auditory cortices. The study of Josef Golubic and colleagues provides evidence of a modulatory role for the mPFC generator on bilateral superior temporal gyri (STG) sources activated during gating responses. This result suggests the existence of a novel, early sensory processing loop from mPFC to auditory cortices, alongside well‐affirmed limbic (dorsal) and somatic (ventral) sensory processing pathways [Hickok and Poeppel, 2004]. The cholinergic modulation of the gating response [Adler et al.,1992; Lucas‐Meunier et al., 2003] indicates that this novel cortical pathway would likely be altered in AD pathophysiology since AD is accompanied by a deterioration of cholinergic signal transmission by selective impact on the plasticity of nAChr and mAChr synaptic receptors [Levin et al., 2006; Small et al., 2001]. On a macro scale, synaptic dysfunction is likely to cause subtle functional alterations years before meeting criteria for symptomatic AD [Knafo et al., 2016; Palop and Mucke, 2010; Um et al., 2012]. Effects associated with cholinergic alterations of the gating response may be detected using functional neuroimaging techniques with high temporal resolution, such as EEG and MEG [Baillet, 2017; Zamrini et al., 2011]. MEG is sensitive to weak neuromagnetic fields induced by coherent postsynaptic currents and, compared to EEG, provides better spatial resolution of the localized cortical sources underlying the measured magnetic fields. Consequently, MEG has an advantage in the search for early neurodegenerative biomarkers associated with synaptic alterations [Supek and Aine, 2014].
A range of EEG/MEG studies, measuring spontaneous and/or evoked brain activity, have reported changes in neural processing that correlate with the neuropathology of symptomatic AD [Bajo et al., 2010; Cheng et al., 2012; Golob et al., 2009; Green et al., 2015; Irimajiri et al., 2005; Jelic et al., 2000; Maestú et al., 2015; Stephen et al., 2006; Stam et al., 2006; Thomas et al., 2010]. Particularly, neurophysiological studies have found differences in early processing of auditory [Cheng et al., 2012; Green et al., 2015; Irimajiri et al., 2005; Thomas et al., 2010] and somatosensory stimuli [Stephen et al., 2006] in MCI/AD patients, affirming the possibility of impaired inhibition of redundant stimuli (gating‐out) and processing of distracting stimuli (gating‐in) in the initial phase of symptomatic AD. In addition, increased power in delta and theta bands, accompanied by a loss of resting‐state functional connectivity in lower alpha and beta bands have been detected in AD patients [Stam et al., 2006]. MEG studies also demonstrated disrupted connectivity among brain regions and loss of long‐distance synchronization as being responsible for some of the earliest cognitive changes in symptomatic AD patients [Bajo et al., 2010; Stam et al., 2006]. Although numerous EEG/MEG studies have identified associations between neural processing and AD pathology, the reported findings have received limited attention in the search for a biomarker of AD. While the analysis of extracranial EEG/MEG signals provides valuable information regarding the pathology‐related alterations in the amplitudes, latencies, frequency bands, spectral densities, and coherence of oscillatory brain dynamics, classification based on the difference between group means of sensor‐level measures generally cannot provide a clear boundary between normal and pathological response values and may result in low clinical significance [Merlo and Wagner, 2013].
Individual subject heterogeneity and variability of responses is the underlying reason for the low discriminatory accuracy of many proposed biomarkers [Poulson et al., 2012]. However, MEG spatiotemporal localization of cortical sources underlying extracranial magnetic field measurements shows more internal consistency and provides highly reliable and stable results of both cortical dynamics (amplitudes and latencies) and topology (cortical location) of the activated network [Aine et al., 2000; Josef Golubic et al., 2014a], enabling a search for neuropathology markers at the individual level. The sensitivity of MEG to depth, cortical extent, cortical geometry and noise level, and the stability, reliability and use of multi‐dipole localization methods for identifying cortical sources underlying early and late evoked responses has been examined in numerous simulation and empirical studies [Aine et al. 2012; Josef Golubic et al., 2014a, 2011; Sanfratello et al., 2014; Supek and Aine, 1993, 1997]. Moreover, estimated cortical source dynamics garnered from extracranial MEG measurements are analogous to intracranial cortical response profiles [Sutherling et al., 1998]. However, cortical source dynamics are not the focus of this study; this aspect was explored previously in Josef Golubic et al. [2014a].
In this study, we explored the sensitivity, specificity, and accuracy of MEG source localization of early auditory gating modulation to detect AD pathology at the individual level. We hypothesized that pathology in the symptomatic AD patients (MCI/AD) should manifest as absolute inactivation of the mPFC auditory gating generator. For healthy controls, the mPFC gating generator should be activated for both gating‐in and gating‐out phenomena (i.e., evoked by the deviant and the standard tones, respectively).
We utilized MEG source localization estimates from our earlier study which identified cortical generators subserving auditory gating processing in elderly controls and symptomatic AD patients [Josef Golubic et al., 2014a] along with results from neuropsychological tests, including Mini Mental State Exam (MMSE), Wechsler Adult Intelligence Scale‐Revised, and Rey‐Osterreith Complex Figure Test (ROCFT). Multivariate analyses such as principal component analyses (PCA) were applied to this dataset to limit the possibility of a chance influence on the discrimination results while clustering methods were used to reveal the internal structure of the data and to differentiate between‐subject categories. Nonlinear correlations between mPFC gating generator activation and psychometric test scores were also conducted. Localization of auditory gating generators was derived from noninvasive MEG measurements of evoked neuromagnetic fields acquired during a passive auditory oddball paradigm to obtain functional information from each patient/subject. MRI scans (structural information) were also obtained from each patient/subject [Josef Golubic et al., 2014a]. A multidipole, spatiotemporal algorithm was used for cortical source localization [Ranken et al., 2002] which provided the topology of the gating generators underlying recorded auditory brain responses. The passive paradigm, which is suitable for elderly participants, evokes both gating mechanisms, habituation of redundant information (standard stimuli), and preattentive memory‐based comparison processes (deviant stimuli).
MATERIALS AND METHODS
Participants
Twenty right‐handed elderly individuals (5 females and 15 males) ranging from 63 to 87 years of age (mean = 76 years) participated in the study. Ten of these individuals, recruited from the community (>65 years, mean = 73 years) with self‐reported normal status of cognitive ability, participated in the study as controls. Ten patients diagnosed as either single‐domain amnestic mild cognitive impairment (aMCI) or AD patients (mean = 78 years), recruited from the Memory Disorders Clinic at the New Mexico Veterans Health Care System, also participated.
All participants underwent neurological exams performed by Board Certified Neurologists with expertise in geriatric and behavioural neurology (J. Knoefel and J. Adair). The five AD patients met the criteria for a diagnosis of AD as defined by the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) and the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM‐IV) [McKhann et al., 1984; American Psychiatric Association, 2000]. The five aMCI patients were evaluated for memory complaints and demonstrated isolated memory impairment. Specifically, aMCI patients met modified Petersen criteria including normal general cognitive functions, indicated by age‐ and education‐adjusted MMSE scores above the 25th percentile, were not demented as defined in DSM‐IV, and were not impaired on routine activities of daily living. Their cognitive performance was at least 1.5 SD below age‐referenced means on the Hopkins Verbal Learning Test 3‐trial immediate recall or percent delayed recall.
All participants underwent a screening evaluation including MRI, laboratory tests, and modified Hachinski Ischemic Scale. For both participant groups, major anatomical abnormalities on MRI scans that would result in rejection of a subject's data include structural lesions indicating prior cortical infarct, evidence of prior significant traumatic injury, or vascular malformations. To exclude other etiologies of cognitive dysfunction, participants must not have had oxygen‐dependent chronic obstructive pulmonary disease, severe congestive heart failure, or neurological or psychiatric disease conditions that could confound study interpretations (e.g., major depression, delusions, hallucinations, Parkinsonism, epilepsy, etc.). In addition, the modified Hachinski Ischemic Scale scores included only participants indicating a low likelihood of vascular injury (<4). Further exclusion criteria included chronic neurological conditions (e.g., seizures, hemiparesis, sensory loss, and visual field deficits), gait disturbances, or sudden behavioral changes. Finally, participants were excluded if they met criteria for alcohol/substance use disorder or used any drugs that interfered with cognitive functioning (anticholinergic, antipsychotic, antiepileptic, or antidepressant drugs).
This research was approved by the Human Research Review Committee at the University of New Mexico, Health Sciences Center and by the Research and Development Committee at the New Mexico VA Health Care System and was conducted in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki. Written informed consent was obtained from all participants (HRRC# 98‐346).
Neuropsychological Evaluations
All enrolled subjects underwent a battery of psychometric tests including the MMSE and the Wechsler Adult Intelligence Scale‐Revised battery. Visual spatial memory was assessed with the ROCFT. Affective symptoms were evaluated with the Geriatric Depression Scale. Functional status was measured with the Functional Activities Questionnaire for aMCI and AD patients, completed by a knowledgeable informant who accompanied the patient to the clinic.
Experimental design, MEG, and MRI data acquisition protocols, data processing, and details of the multi‐dipole, spatiotemporal source localization of cortical generators underlying gating fields evoked by the auditory oddball paradigm were reported in our earlier study on the topology and dynamics of the auditory gating network [Josef Golubic et al., 2014a]. For ease of reference, these details can be found below.
Experimental Design and Paradigm
The passive auditory oddball task consisted of 400 frequent (1000 Hz, P = 0.8) and 100 deviant (1200 Hz, P = 0.2) binaurally presented tones. Tone duration was 200 ms. The interstimulus interval was 1 s with a jitter range of 200 ms. The tones of the oddball paradigm were presented to subjects using NBS Presentation software and were delivered inside a magnetically shielded room to the participant's ear canal using Etymotic Research ER‐3A sound transducers with plastic tubing. Adjustments in the intensity of the tones were made for each participant separately, based on the results of a previous hearing threshold test given within the shielded room to achieve a level of 60 dB SPL.
MEG and MRI Data Acquisition
Anatomical T1‐weighted MPRAGE (1.5 mm slices) and T2‐weighted Turbo Spin Echo (1.8 mm slices) magnetic resonance images were obtained for all subjects from a 1.5‐T Siemens Sonata MR scanner at the Mind Research Network (MRN) in Albuquerque, New Mexico. Magnetic fields were acquired with a CTF 275‐channel whole‐head system (VSM MedTech, Ltd.) inside a two‐layer magnetically shielded room (Vacuumschmelze, GmbH & Co. KG, Nanau, Germany) at the MRN. For artefact elimination, EOG signals were simultaneously recorded with the MEG signals. Electrodes were placed above and below the eye for monitoring eye blinks or eye movements. The subject's nasion and the left and right preauricular points were registered by the Polhemus head position device for establishing a 3‐D coordinate head frame. An additional 150 points along the surface of the head were marked to determine head shape for later precise co‐registration with anatomical MR images. Subjects were seated comfortably in a chair with the head centered in the measurement helmet. The coils were pulsed and localized to establish head position relative to the helmet. The data were digitized at 600 Hz with online filters at 0.1–200 Hz.
Data Processing and Filtering
All data were preprocessed using CTF software for deletion of large amplitude single‐epoch MEG responses, digital filtering, and artefact rejection before offline averaging. MEG epochs that contained amplitudes exceeding a threshold of 3 pT/cm and/or EOG signals >75 µV were rejected from offline averaging. All preprocessed raw data for each stimulus were used for later averaging. Averaged data were filtered at 0.1–50 Hz.
MEGAN (MEG ANalysis), an MEG data visualization and analysis tool developed by Elaine Best, in the Biophysics group, Los Alamos National Laboratory were used to remove baseline noise and for producing NetMEG files from the original CTF data format. Baseline correction was used to decrease the low frequency noise effects that result from the offsets of individual sensors and slow ambient field fluctuations. It was performed by estimating the DC offset of each sensor based on the interval −100 to 0 prior to stimulus presentation and subtracting this DC offset from each timepoint of the averaged evoked response.
Later processing was performed using MRIVIEW [Ranken, 2014] to (a) perform semiautomated segmentation of volumetric MRI data; (b) identify the best‐fitting sphere; (c) reconcile coordinate systems (MRI data with MEG coordinate space); and (d) obtain multi‐dipole source estimates using a Calibrated Start Spatio Temporal (CSST) tool [Ranken et al., 2002] for multistart multidipole MEG inverse calculations.
Source Localization
Spatiotemporal localization of early sensory processing was performed assuming point sources [Aine et al., 2012; Josef Golubic et al., 2011; Sušac et al., 2014, 2009, 2010]. The cortical sources of the auditory evoked fields (AEF) were modeled assuming multiple rotating current dipoles embedded in a spherical volume conductor [Huang et al., 1998]. Individual cortical surfaces were identified and labelled from the MRI data (slices) using a semiautomated segmentation tool within MRIVIEW. The 3D morphological operations were applied to identify the gray/white matter boundary. Segmented cortical surfaces were used to estimate the best‐fitting sphere for each individual head model. Spatiotemporal source analyses of the empirical data acquired from all MEG channels were performed using the multidipole CSST algorithm and were not constrained to any preselected cortical locations. Spatiotemporal source localization was conducted across a 30–100 ms poststimulus time window. Estimation of the time invariant parameters (spatial locations) was derived first using nonlinear minimization and kept constant for the selected time window, while a linear estimation of the associated time‐varying parameters (source strengths and orientation) was calculated for each time instance.
CSST uses a two‐stage, semiautomated multistart downhill simplex minimization [Aine et al., 2000; Huang et al., 1998] of the reduced chi‐square metric [Supek and Aine, 1993, 1997] for optimizing the locations, strengths, and orientations of activated cortical sources. Up to 12,000 starting points (for each dipole source model) were used for simplex searches and were randomly selected from the realistic head geometry (segmented cortex). This algorithm minimizes possible bias of the results toward anticipated areas of activity due to investigator selection of starting points.
The minimum model order (number of dipoles, n) was estimated using singular value decomposition (SVD) of the spatiotemporal data matrix of the selected time interval. The inverse calculations were conducted starting from the estimated minimum model order n, and continued subsequently increasing the model order (n + 1, n + 2, etc.). The adequate (best‐fitting) model was selected based on reduced chi‐square measure of goodness‐of‐fit for each model order assumed [Supek and Aine, 1993], proportion of variance explained (PVE), dipole clustering to assess location scatter (an indication of overmodeling) [Supek and Aine, 1993], the residual waveforms to assess whether additional signal remained (an indication of undermodeling), and the inspection of the estimated source time‐courses (near‐zero amplitude across entire time interval is an indication of overmodeling).
To provide confidence regions for the best‐fitting dipole solutions and to estimate effect of the measurement noise, 100 Monte Carlo simulations were conducted by using the source locations from the best‐fitting model as starting locations and adding noise determined by the sensor baseline noise variance (−100 to 0 ms).
Anatomical Locations
Anatomical locations of the best‐fitting dipole solutions were assessed by reconciling the MEG head‐centered coordinate system with the participants' MRI coordinate system. Unlike the commonly used methods that remove size differences by scaling the structural data (spatially normalized to the Talairach stereotaxic system), we retain the original individual head shapes to avoid distortion of brain characteristics. The coordinate system corresponded to the following convention: the positive x‐axis points out toward the nose, the positive y‐axis points toward the left ear, and the positive z‐axis points out the top of the head. We manually identified STG (right and left hemispheres) on individual MRI scans (Brodmann's areas (BA) 41/42). For the anterior limit of STG, we used the first slice showing the white matter tract (temporal stem) connecting the temporal lobe with the base of the brain. The posterior boundary of STG was defined as the slice where the fibers of the crux of the fornix last appeared. The location of a dipole in the anterior portion of the medial prefrontal lobe (including frontopolar (BA 10) and orbitofrontal region (BA 11)) was the criterion for the mPFC source relative to each individual subject's MRI.
Statistical Analyses
Cortical locations of each modeled source in the 30–100 ms time window were examined for each subject and for the two auditory conditions (standard and deviant tones). Variables of our multivariate dataset (scores on neuropsychological tests and neurophysiologic results of MEG source localization in a 30–100 ms time window across participants) were tested for the assumptions of the analysis of variance: the Kolmogorov–Smirnov test [Smirnov, 1948] was carried out to verify normality and Bartlett–Box test [Bartlett, 1937] was used to check homoscedasticity of variance. Post‐hoc comparisons of the ANOVA were conducted using Tukey least significance difference t test to isolate interaction effects. Two‐way repeated measures ANOVA was conducted on spatial locations (number of localized dipoles, hemisphere) of localized sources across categories of subjects (controls and symptomatic AD) and two conditions (standard/deviant tone) after Bartlett–Box test confirmed homogeneity of data variance in each group. The factors submitted to ANOVA consisted of Condition (response to Standard and Deviant tone) and Clinical Category (controls and symptomatic AD). The Mann–Whitney–Wilcoxon (MWW) rank‐sum test [Mann and Whitney, 1947] was subsequently used to test the differences in MMSE/delayed‐ROCFT scores between new categories that emerged after cluster analyses.
Principal Component Analysis
Principal component analyses were used to reduce dimensionality of the multivariate data matrix and limit the possibility of a chance influence on the discrimination of the results by decreasing the number of variables used in the analysis [Ahlgren, 1986]. The 20 subjects (observations) and results on the neuropsychological tests, together with MEG localization results identifying the cortical network underlying auditory gating in both oddball tone conditions (variables), were submitted to a principal component analysis. The SVD of covariance matrix was used for estimation of principal components (the eigenvectors of the covariance matrix). The percentage of variance explained (95%) by principal components was employed as a cutoff rule for estimating relevant variables.
Cluster Analysis
Blind cluster analyses were used to provide information regarding data structure (existence of subject categories) using a purely data‐driven approach, without any a priori hypotheses (i.e., clinical diagnosis). To check reliability and internal consistency of categories, different methods of clustering were conducted on the dominant variables. Classical clustering algorithms with Ward's minimum variance method [Ward, 1963] as amalgamation rule and with Euclidean distance and Manhattan metrics and correlation function as a measure of dissimilarity were applied to the data matrix. Then, the neuropsychological variables were extracted and clustering methods were reapplied to examine the influence of dichotomous neurophysiologic variables.
Blind cluster analyses were performed by an expert in multivariate analyses (L. Caklovic, Department of Mathematics, and University of Zagreb). No modifications, additions, or exclusion were made to the data set and neither the data nor any subsets of it were used to assess or refine the model being tested.
Correlation
Multivariate nonparametric rank correlation coefficients were calculated as a statistical measure of a relationship between principal neuropsychological (MMSE, dROCFT) and neurophysiologic (mPFC gating activation) variables. First, results of neuropsychological tests were two‐step ranked across subjects with MMSE test scores as a basic metric. Relevant neurophysiologic variables were presented by a set of binary results (1, localized mPFC generator; 0, nonlocalized mPFC gating generator). The nonlinear regression approach fits the neuropsychological test ranks to an unknown binary step function g (Heaviside function: g(t) = 0, t < c; g(t) = 1, t ≥ c) that represents neurophysiologic variables. The step function was expanded as a generalized Fourier series to estimate the cutoff Fourier coefficients (c) from the data. The Levenberg–Marquardt method [Levenberg, 1944; Marquardt, 1963] was applied to estimate nonlinear correlation coefficients.
RESULTS
Demographics by Diagnostic Category
The normality tests (Bartlett–Box test and Kolmogorov–Smirnov test) confirmed that none of the data distributions (results of neuropsychological exams, amplitudes, and latencies of evoked cortical activity) was significantly different from normal. There were no excluded data or outliers. There were no significant differences in general demographics between males and females (ANOVA age: F(1,10) = 1.66, P = 0.31; years of education: F(1,10) = 1.39, P = 0.26) or as an effect of clinical category (controls vs patients (aMCI/AD); ANOVA: age F(1, 18) = 2.48, P = 0.08; education F(1,18) = 0.89, P = 0.54). Significant differences were found for MMSE (F(1,18) = 35.18, P < 0.001), dROCFT (F(1,18) = 27.87, P < 0.001), performance IQ (IQperf) (F(1,18) = 12.22, P = 0.003), and verbal IQ (IQver) (F(1,18) = 5.38, P = 0.03) test scores as an effect of clinical category (Table 1). There were no significant differences in full scale IQ scores (F(1,18) = 1.8, P = 0.18) and ROCFT copy (F(1,18) = 2.1, P = 0.12) when comparing controls to patients.
Table 1.
Demographics by diagnostic category (mean ± standard deviation)
| Category | Controls | MCI/AD |
|---|---|---|
| N | 10 | 10 |
| Male | 7 | 8 |
| Female | 3 | 2 |
| Age | 74.4 ± 4.8 | 78.8 ± 5.3 |
| Education (years) | 15 ± 4 | 14 ± 3 |
| MMSE a | 29.4 ± 1.6 | 25.1 ± 3.6 |
| IQ | 121 ± 18 | 105 ± 31 |
| IQperf a | 128 ± 10 | 96 ± 19 |
| IQver a | 126 ± 13 | 101 ± 13 |
| ROCFT (copy) | 33 ± 4 | 26 ± 13 |
| ROCFT (delay) b | 22.0 ± 3.1 | 5.8 ± 5.2 |
Statistically significant difference P < 0.001.
Statistically significant difference P < 0.05.
Localization of Auditory Gating Generators
The stability and reliability of our spatiotemporal localization approach to identify auditory gating generators and the modulatory role of a mPFC gating generator on bilateral STG gating activity were reported earlier [Josef Golubic et al., 2014a]. The current work is focused on evaluating differences in the gating generators topography as a function of the different tone conditions, as a potential biomarker for Alzheimer‐type memory impairment.
The multidipole spatiotemporal localization of gating generators evoked by the auditory oddball paradigm was conducted in the 30–100 ms poststimulus time window for both tone conditions (standard and deviant). The ventral mPFC (BA 10 and 11) and the bilateral STG areas (BA 41/42) were identified as cortical regions where the gating generators were localized. The average location of mPFC generators in head‐centered coordinates was as follows: x = (5.58 ± 0.71) cm; y = (−0.15 ± 0.38) cm; z = (2.64 ± 0.68) cm. There was no significant y‐coordinate asymmetry of mPFC gating generators across subjects (ANOVA mPFC hemisphere: F(1,19) = 1.71, P = 0.22).
We demonstrated three different gating generator topologies identified across subjects and two conditions, based on the activation (the detection or absence) of an mPFC generator (Fig. 1). Sustained activation of STG was found for all subjects regardless of clinical category and tone condition, demonstrating insensitivity of STG gating topology to AD neuropathology. Control subjects showed two distinct topologies of the gating generators (panels A and B of Fig. 1) which differed according to the presence or absence of an mPFC gating generator evoked by the standard tone. The healthy gating topology consisted of three generators evoked by both tones: bilateral STG (green and blue) and mPFC (red), shown in panel A of Figure 1. In contrast, altered gating topology was found in a subgroup of controls, as shown in panel B of Figure 1. This type of gating topology consisted of three generators activated for the deviant tone, mPFC (red), and bilateral STG (blue and green), but the standard tone evoked only the two bilateral STG generators without mPFC activation. Panel C of Figure 1 shows a gating topology characteristic for symptomatic AD patients. This gating topology represented only bilateral STG activation for both deviant and standard tone conditions. The complete absence of mPFC generator activation is the main sign of symptomatic AD gating topology.
Figure 1.
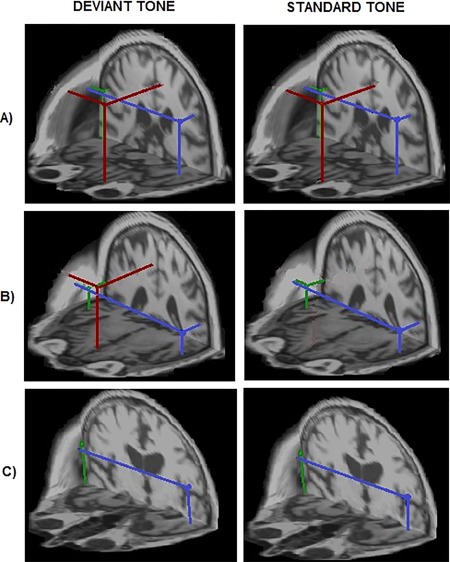
Three distinct types of auditory gating network identified in healthy controls (A), possible preclinical AD (B), and symptomatic AD patients (C). Localization of auditory gating generators estimated in the 30–100 ms time interval evoked by the tones of an oddball paradigm in three representative subjects across conditions. The best‐fitting source locations are superimposed on individual volumetric MRI head data to achieve a spatial (3D) rendering of the auditory gating topology. Panel (A) shows the healthy gating topology type where all three gating generators—mPFC (red dot) in addition to bilateral STG sources (green and blue dots; right and left STG generators, respectively)—were active in processing both tone conditions (4/10 controls). Panel (B) shows an altered gating topology type characterized by selective mPFC activation only by the deviant tone (6/10 controls and one aMCI). Panel (C) shows the third topology type, where the mPFC generator for both standard and deviant tones was missing. This type of network was found in symptomatic AD patients.
Principal Component Analysis
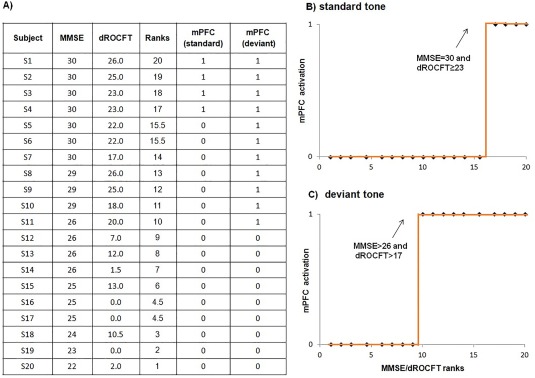
The PCA revealed four nontrivial variables whose linear combination explained 94.6% of the total variance (PVE): MMSE results, dROCFT score, mPFC activation in response to the standard tone, and mPFC activation in response to the deviant tone. Additional variables—IQperf and IQver—resulted in surpassing 95% of variance explained. Table 2 shows the individual results for the dominant variables: neuropsychological tests and MEG localization of the mPFC gating generator across the tone conditions (1, active; 0, nonactive) along with explicit coordinates of localized mPFC gating generators. As there were no significant within‐subject differences in the spatial positions of the mPFC generators evoked by the two stimulus conditions, the mPFC coordinates were averaged for individuals who showed mPFC activation for both tone conditions. Subjects were assigned numbers from 1 to 20 according to the test results achieved on the MMSE. The ROCFT drawings were scored according to Meyers and Meyers [1995], yielding a scoring range from 0 to 36 for delayed recall trial.
Table 2.
The subjects' scores on the PCA extracted variables, estimated mPFC gating generator coordinates, clinical diagnosis, and cluster category
| Subject | MMSE | dROCFT | IQper | IQver | mPFC standard | mPFC deviant | mPFC coordinate (x, y, z) | Clinical diagnosis | Cluster category |
|---|---|---|---|---|---|---|---|---|---|
| S1 | 30 | 26 | 138 | 138 | 1 | 1 | (5.98, −0.49, 2.99) | H | 1 |
| S2 | 30 | 25 | 136 | 116 | 1 | 1 | (5.16, −0.51, 2.78) | H | 1 |
| S3 | 30 | 23 | 132 | 138 | 1 | 1 | (5.91, 0.43, 2.98) | H | 1 |
| S4 | 30 | 23 | 130 | 136 | 1 | 1 | (5.93, 0.01, 1.89) | H | 1 |
| S5 | 30 | 22 | 138 | 135 | 0 | 1 | (5.15, 0.11, 3.92) | H | 2 |
| S6 | 30 | 22 | 107 | 122 | 0 | 1 | (5.19, −0.58, 2.96) | H | 2 |
| S7 | 30 | 17 | 130 | 125 | 0 | 1 | (5.21, 0.31, 2.93) | H | 2 |
| S8 | 29 | 26 | 131 | 144 | 0 | 1 | (6.95, −0.39, 2.07) | MCI | 2 |
| S9 | 29 | 25 | 116 | 110 | 0 | 1 | (5.02, −0.46, 1.56) | H | 2 |
| S10 | 29 | 18 | 124 | 129 | 0 | 1 | (4.47, −0.35, 2.04) | H | 2 |
| S11 | 26 | 20 | 126 | 103 | 0 | 1 | (6.38, 0.31, 2.89) | H | 2 |
| S12 | 26 | 12 | 99 | 93 | 0 | 0 | ‐ | MCI | 3 |
| S13 | 26 | 7 | 103 | 93 | 0 | 0 | ‐ | MCI | 3 |
| S14 | 26 | 1.5 | 103 | 100 | 0 | 0 | ‐ | MCI | 3 |
| S15 | 25 | 13 | 87 | 100 | 0 | 0 | ‐ | MCI | 3 |
| S16 | 25 | 0 | 117 | 121 | 0 | 0 | ‐ | AD | 3 |
| S17 | 25 | 0 | 117 | 121 | 0 | 0 | ‐ | AD | 3 |
| S18 | 24 | 10.5 | 92 | 110 | 0 | 0 | ‐ | AD | 3 |
| S19 | 23 | 0 | 111 | 113 | 0 | 0 | ‐ | AD | 3 |
| S20 | 22 | 2 | 55 | 81 | 0 | 0 | ‐ | AD | 3 |
Clustering
Blind cluster grouping of subjects using the principal variables disclosed three well‐defined clusters with a stable subject distribution as shown in Figure 2. Clustering results distinguished symptomatic AD patients from controls (H) but did not discriminate between aMCI and AD patients.
Figure 2.
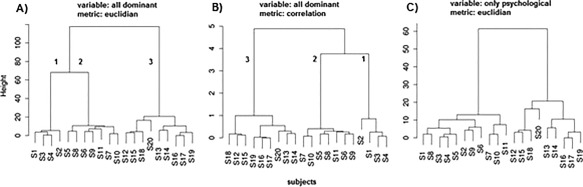
Three distinct clusters identified across subjects. Dendrograms show the results of different cluster groupings carried out on the dominant variables shown in Table 2. Variables were normalized before clustering. Subjects were assigned numbers from 1 to 20 as in Table 2 and grouped into clusters. Clustering algorithms with Euclidian distance (panel A) or correlation function (panel B) as a measure of dissimilarity yielded identical results: our sample of 20 elderly subjects had an internal structure consisting of three well‐defined and stable clusters. Panel C shows the result of clustering conducted only on dominant neuropsychological variables.
However, two distinct clusters (1 and 2) emerged across the control group. A classical clustering algorithm with Ward's minimum variance method as amalgamation rule and Euclidean distance (Fig. 2, panel A), Manhattan metric (not shown), or correlation function (Fig. 2, panel B) as a measure of dissimilarity differentiated three clusters across subjects. Subject distributions in clusters were identical for all clustering approaches. The first cluster included control subjects characterized by mPFC generator evoked by both tones: the highest MMSE scores and the highest performance on the dROCFT. The second cluster included lower functioning control subjects with selective activation of the mPFC gating generator evoked by the deviant tone and with significantly lower MMSE and dROCFT scores (MWW test; n1 = 4, n2 = 7; U1 = 0 < U(4,7) = 3; P = 0.05).
The third category coincided with the symptomatic AD patients who lacked mPFC activation regardless of the tone condition and had the lowest MMSE and dROCFT scores (MWW test, n2 = 7, n3 = 9; U2 = 0 < U(7,9) = 12, P = 0.05). Clustering applied to dominant neuropsychological variables only yielded two low‐distant clusters which did not detect an additional cluster within the control group (panel C).
Correlation Between Neuropsychological and Neurophysiological Measures
Using a multivariate, nonparametric, rank correlation along with the Levenberg–Marquardt method to estimate nonlinear correlation coefficients, we found very strong nonlinear correlations (R 2 > 0.98) between the neuropsychological MMSE/dROCFT results and neurophysiological mPFC activation for both standard and deviant tones, as shown in Figure 3. Fourier expansion of discrete function g(t) provided MMSE/dROCFT score boundaries for gating activation of the mPFC generator evoked by the oddball paradigm tones: MMSE < 26 and dROCFT < 17 indicated absolute mPFC gating inactivation (9/9); scores within 26 < MMSE ≤ 30 and 17 ≤ dROCFT < 23 indicated altered mPFC activation, that is, activation only by the deviant stimuli (7/7); and finally, the scores MMSE = 30 and dROCFT ≥ 23 correlated with mPFC activation for both tone conditions (4/4).
Figure 3.
Correlation between neuropsychological and MEG localization results. MMSE/dROCFT two‐step ranked test scores (panel A) and presence/absence of mPFC gating activation show very strong nonlinear correlation for both tone conditions, R 2 (standard) = 0.97 (panel B) and R 2 (deviant) = 0.99 (panel C); P < 0.001. Dotted line on panels B and C represent discrete step functions g(t) with marked boundary test scores which indicate mPFC activation. [Color figure can be viewed at http://wileyonlinelibrary.com]
Diagnostic Power
The absence of an mPFC gating generator evoked by a passive auditory oddball paradigm was demonstrated (Fig. 2) to have a specificity of 100% (10/10), 95% accuracy (19/20), and 90% sensitivity (9/10 symptomatic AD; 4/5 aMCI and 5/5 AD) to detect symptomatic AD patients in this sample. The contingency table yielded χ 2 = 16.36 ( = 7.88; α = 0.005) with a large effect size (r φ = 0.904). The only subject “misclassified” by the above approach (assigned as number 8 in Table 2) belonged to the symptomatic AD group with an aMCI diagnosis. The proposed biomarker detected impaired mPFC gating activity for this patient; that is, an mPFC generator was absent in this patient for the standard tone. Selective gating‐out impairment and higher test scores in comparison with other symptomatic AD patients (MMSE = 29 and dROCFT = 26) placed this individual in the lower functioning control group.
DISCUSSION
In this study, we demonstrate the use of MEG localization of a mPFC gating generator as a discrete (binary) detector of AD at the individual level. The large effect size (>0.9; binary result) enabled high statistical power despite relatively low sample size.
Our putative biomarker for identifying AD pathology is not based on the use of group means and it is not associated with statistically significant changes in a continuous variable (i.e., increase or decrease of mPFC gating activity). Its strength lies in the simplicity of using a binary value (i.e., activated or nonactivated) for the mPFC gating generator. The low sensitivity to individual heterogeneity and variability due to the binary nature of impaired mPFC activation is probably the most important property of the proposed method for AD detection.
We found three types of gating generator topologies evoked by an auditory oddball paradigm that discriminated patients (aMCI and AD) from controls, confirmed the indiscernibility between aMCI and AD patients, and differentiated two distinct gating generator topologies within the controls. To explain the presence of two different types of gating topologies within a control group (>65 years), we applied different clustering approaches to disclose potentially hidden structure in our multivariate data set (neuropsychological test scores and neurophysiological gating network topology).
Clustering based on principal variables demonstrated the existence of three stable groups across participants. Although dominant weighting in cluster forming was generated from dichotomous neurophysiologic MEG variables, subsequent statistical review of group differences on neuropsychological tests confirmed low magnitude but statistically significant differences in MMSE and dROCFT scores between participants in distinct clusters. The first group of controls, who were characterized by consistent activation of mPFC gating generator for both oddball tones, had the highest MMSE scores and the highest performance on the dROCFT was considered to be healthy elderly controls.
The second group of controls characterized by the absence of mPFC gating generator activation for the standard tone and significantly lower MMSE and dROCFT scores was considered as lower functioning controls. We speculate that these individuals may be in a possible preclinical AD phase as they show both neuropsychological and neurophysiological impairments characteristic for an AD type of dementia, although they did not yet meet clinical criteria for aMCI. Also, it has been shown that dROCFT is sensitive to early AD pathology and may be an indicator of future conversion to AD [Takayama, 2010] which additionally supports our speculation.
All participants characterized by the absence of an mPFC gating generator regardless of the tone condition and had the lowest test results overall, belonged to the patient group with aMCI or AD diagnosis (9/9). This result is in line with recent evidence showing that major pathophysiological processes associated with AD have already occurred by the time of MCI diagnosis [Lazarcyzk et al., 2012], supporting the Key Symposium group recommendation that clinically expressed disorders, from MCI to later stages, represent symptomatic AD [Morris et al., 2014].
Inactivation of the mPFC gating generator implies that we could not localize any prefrontal neuromagnetic source activity within the first 100 ms poststimulus using the multidipole CSST localization approach. A series of numerical simulations demonstrated that for a deep mPFC generator (at 3.5 cm), a dipole strength of 5 nAm represents a threshold for CSST spatiotemporal localization [Josef Golubic et al., 2014a] which is smaller than the previously reported estimated limit of the MEG detectable current moment density [Baillet et al., 2001]. Moreover, the CSST approach provides high stability and reliability of spatiotemporal localization of the auditory gating network in elderly and young participants [Josef Golubic et al., 2014a, 2014b]. It has been shown that, contrary to the widely accepted assumption that source orientation is the main limiting factor of MEG sensitivity to neural activity, the source depth and its cortical extent are the major limiting factors for detection probability [Josef Golubic et al., 2011]. Consequently, MEG sensitivity to deep and low‐signal prefrontal gating generator signals that are simultaneously active with more superficial, extended, and more tangentially oriented current dipoles originating in the STG can be increased using a high‐density sensor array. We used 275 high‐order gradiometers (CTF 275‐channel whole‐head system), which are effective at rejecting magnetic noise outside the brain, thereby augmenting detectability of low amplitude prefrontal generators. Consequently, inability to localize an mPFC gating generator by MEG CSST source localization represents a strong indication that early feedback from the mPFC to auditory sensory cortices is disrupted.
The present results agree with a recently proposed concept that introduces PFC damage, in combination with cingulate damage, as a predictive indicator for the development of AD clinical symptoms [Vogt, 2009]. It was found that PFC atrophy preceded dementia onset over a 6 year period and appeared to be a more sensitive predictive factor than hippocampal volume [Burgmans et al., 2008]. Recent findings suggest that neurofibrillary tangles and amyloid‐β accumulation are a necessary but not sufficient condition to produce clinical manifestations of AD; cognitive decline develops only with associated synaptic dysfunction [Giannakopoulos et al., 2007; Jack Jr et al., 2014; Sperling et al., 2011]. Our results suggest that synaptic activation of an mPFC source within the auditory gating network topology coincides with stages (possible preclinical and clinical) of Alzheimer's type dementia. In the symptomatic stage of disease (aMCI/AD), gating activation of the mPFC source cannot be identified for either standard or deviant tones while selective (impaired) activation of postsynaptic gating activity may signify a possible preclinical phase (i.e., mPFC activation was identified for the deviant tones only).
Our results suggest that a very early stage of alteration in auditory gating processing is associated with an absence of mPFC gating transmission that corrupts habituation to redundant inputs [Josef Golubic et al., 2014a]. The impaired mPFC processing during endogenous brain activity or during memory tasks in cognitively normal participants who were AD risk‐factor gene (APOE ɛ 4) carriers [Cuesta et al., 2015] together with evidence of reduced gating amplitude as a predictor of cerebrospinal amyloid‐β reduction in MCI patients [Green et al., 2015] strongly support our speculation that absent mPFC gating‐out activation in lower functioning controls may be associated with a possible preclinical AD phase. Also, it has been shown that PFC structures are affected by tau‐pathology in the possible preclinical stage of AD [Giannakopoulos et al., 2007]. Recent findings provide novel evidence that links p‐tau pathology in a very early phase of AD‐type memory impairment to reduced functional connectivity affecting the PFC, which is also involved in amyloid‐β‐related hypersynchronization [Canuet et al., 2015]. Synaptic loss and trans‐synaptic or transneuronal spread of pathological tau‐forms [Wu et al., 2016] through PFC regions could result in the gating deficit that we have found in a subgroup of controls which may reflect a possible preclinical phase of AD pathology.
Progressive failure in sensory gating‐out is likely to lead to an overload of working memory due to signal to noise reduction and consequently to the first symptoms of memory impairment found in AD patients [Baddeley et al., 1991]. Our results provide evidence that mPFC disengagement may take place in symtopmatic stages during both inhibition of redundant stimuli (gating‐out) and processing of distracting stimuli (gating‐in). Consistent with our results, numerous studies confirmed that symptomatic AD pathology strongly affects PFC physiology. Lower frontal–parietal correlations of glucose metabolism [Rapoport et al., 1986], frontal retention of 11C‐Pittsburgh compound [Klunk et al., 2004], and prefrontal glucose hypometabolism on 18F‐fluorodeoxyglucose PET scans [Coleman, 2005] were found in AD patients. Moreover, recent longitudinal measurements in AD patients found significant hypometabolism in PFC regions that are not strongly affected by amyloid deposition, arguing that functional impairment of mPFC may be related to longitudinal increases in amyloid deposition in remote but functionally connected brain areas such as STG cortices within the auditory gating network [Klupp et al., 2015].
As one of the major roles of mPFC is control of overall acetylcholine input from subcortical neuromodulatory systems [Van de Werd et al., 2010], absence of synchronized neuromagnetic activity in mPFC during auditory gating may reflect dysfunction of fast cholinergic signal transmission between mPFC and primary sensory areas in AD. Such mPFC gating dysfunction may result in PFC hypometabolism reported in AD patients [Coleman 2005; Klunk et al., 2004; Klupp et al., 2015; Rapoport et al., 1986]. Moreover, the subtype of alpha‐7‐nAChr bearing neurons which are highly involved in synaptic gating regulation [Freedman et al., 2003] and are especially vulnerable to β‐amyloid damage [D'Andrea and Nagele, 2006] are found in PFC regions [Poorthuis et al., 2012].
In line with our results showing a correlation of AD memory impairment and mPFC gating functionality, we also found a strong correlation (>0.98) between the presence/absence of the mPFC gating generator and performance on the dROCFT. This result enabled us to make a quantitative assessment of dROCFT scores that reflect stages of mPFC auditory gating functionality. We found scores on dROCFT ≥23 to be associated with healthy mPFC activation during both gating phenomena, while scores within 17 ≤ dROCFT < 23 indicated impaired mPFC activation to the deviant tone only, and scores of dROCFT < 17 were associated with total absence of mPFC gating generator. These results suggest the usefulness of the dROCFT scores as a measure of frontal gating functions.
Strict inclusion criteria for participation in this research study, in line with the Key symposium recommendation, suggest a greater probability that the underlying pathophysiology of patients diagnosed as aMCI and AD is of the Alzheimer's type. Additionally, broad exclusion criteria provide greater likelihood that the pathology of control participants who are possibly in a preclinical phase of dementia (still nonsymptomatic), is likely due to Alzheimer's disease (i.e., lower likelihood of having other confounding medical conditions). Nevertheless, there is always a possibility that subclinical cardiovascular or metabolic disorder may be present in these participants, such as undetected small vessel disease, high blood pressure, or hypercholesterolemia and insulin resistance, which may contribute to cognitive decline as well [Aine et al., 2014]. However, our putative marker of AD pathology specifically underlies an auditory sensory gating deficit [Cheng et al., 2012; Thomas et al., 2010]. Currently, there is no evidence for a link between corrupted auditory sensory gating and metabolic or cardiovascular pathology.
The diagnostic power of a test that uses the absence of mPFC gating activity as a biomarker of symptomatic AD showed very high effect size (0.904), specificity (100%), accuracy (95%), and sensitivity (90%) relative to clinical diagnostic category. The only subject that was misclassified by our approach was subject 8 with an aMCI diagnosis. The proposed MEG marker could have detected AD pathology in this subject in a phase characterized by the lack of mPFC gating‐out activation and preservation of mPFC gating‐in activation. Our results suggest that this type of mPFC gating dysfunction may indicate very early gating impairment and possibly mark the preclinical AD stage. Such variation supports the dynamic view of AD that considers pathology as evolving progressively over decades before the first symptoms [Dubois et al., 2007; Holtzman et al., 2011; Jack JR et al., 2014; McKhann et al., 2011; Morris et al., 2014; Petersen and Morris, 2005; Sperling et al., 2011] and suggesting a physiological continuum between absolutely healthy aging and severe AD. The dynamic model accommodates variability in the clinical expression of disease changes over time depending on individual vulnerability to the initial phases of pathology, the severity of AD degeneration on a cellular level and the efficiency of compensatory mechanisms [Morris et al., 2001]. We speculate that subject 8 might have been captured in an extended phase of transition between preclinical (nonsymptomatic) and early symptomatic stage of disease (MCI). Perhaps the compensatory mechanisms in this patient were deficient in circumventing cognitive decline; therefore, this patient may have experienced cognitive changes indicative of early features of dementia, although the underlying pathophysiology was not yet so severe. However, because we do not have access to amyloid imaging results, we acknowledge that the high sensitivity/specificity values are limited by the accuracy of the clinical diagnosis itself; that is, 95% of the time, this clinical diagnosis will reflect the underlying AD pathology as identified by MEG measures.
In conclusion, our results suggest that MEG localization of mPFC gating activation has the potential not only to detect symptomatic AD but also to become a predictor of cognitive decline thought to be related to the pathophysiological processes of AD, both at the individual level. The statistical independence of the proposed biomarker, noninvasiveness of MEG measurements, high reliability of CSST localization of auditory, and mPFC gating generators along with the nondemanding passive oddball paradigm are additional benefits for future clinical use. The potential of our proposed MEG biomarker to recognize a preclinical state could represent a major advancement in AD research as it may provide much earlier disease‐modifying intervention during presumably still reversible stages of neurodegeneration. Our MEG biomarker indicates that failure to modulate activity in the posteromedial cortices may be an early indicator of synaptic dysfunction that underlies the earliest pathological processes associated with AD.
MEG localization of a discrete mPFC gating activation is a promising AD marker at the individual level; however, this approach needs to be tested in a large independent sample and requires assessment in longitudinal clinical MEG studies. Ideally, studies would track nonsymptomatic elderly with mPFC gating impairment until the first clinical symptoms appear (associated with absence of mPFC gating activity) and finally to autopsy for confirmation of disease. It would be necessary to directly verify the correlation between MEG‐localized mPFC gating inactivation and recently proposed early AD biomarkers from National Institute on Aging‐Alzheimer's Association workgroups [McKhan et al., 2011]. Furthermore, related to the absence of a completely specific biomarker of AD, it would also be necessary to explore cortical mPFC gating dynamics in other dementias and in depression to determine the specificity to discriminate AD from other etiologies of age‐related cognitive decline.
AUTHOR CONTRIBUTIONS
SJG developed the working hypothesis, performed spatiotemporal source localization, analyzed and interpreted data, performed statistical analyses, and wrote the manuscript. CJA was responsible for the overall design and implementation of the study and was the principal investigator of the grants. JMS helped design the study and developed the task paradigm and oversaw data collection. JCA, as director of the Memory Disorders Clinic at the VA Medical Center in Albuquerque, facilitated the recruitment of symptomatic AD patients for study; together with JEK evaluated all subjects/patients (e.g., neurological exams) included in the study and both were involved in editing the manuscript. SS supervised the research and discussed the structuring of the manuscript. SS, CJA, and JMS substantially edited the manuscript. All authors read and approved the final version of the manuscript.
COMPETING INTERESTS
The authors report no biomedical financial interests or potential conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Lavoslav Caklovic (Department of Mathematics, Faculty of Science, University of Zagreb) for his assistance in statistical analyses (principal component analyses and blind clustering) and preparing the dendrogram figure.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes on Aging or the National Institutes of Health.
REFERENCES
- Adler LE, Hoffer LJ, Griffith J, Waldo MC, Freedman R (1992): Normalization by nicotine of deficient auditory sensory gating in the relatives of schizophrenics. Biol Psychiatry 32:607–616. [DOI] [PubMed] [Google Scholar]
- Ahlgren A (1986): Multivariate analysis. Science 234:530–531. [DOI] [PubMed] [Google Scholar]
- Aine CJ, Sanfratello L, Adair JC, Knoefel JE, Qualls C, Lundy SL, Caprihan A, Stone D, Stephen JM (2014): Characterization of a normal control group: Are they healthy? NeuroImage 84:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aine CJ, Huang M, Stephen J, Christner R (2000): Multistart algorithms for MEG empirical data analysis reliably characterize locations and time courses of multiple sources. NeuroImage 12:159–172. [DOI] [PubMed] [Google Scholar]
- Aine CJ, Sanfratello L, Ranken D, Best E, MacArthur JA, Wallace T, Gilliam K, Donahue CH, Montaño R, Bryant JE, Scott A, Stephen JM (2012): MEG‐SIM: A web portal for testing MEG analysis methods using realistic simulated and empirical data. Neuroinformatics 10: 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (2000): Diagnostic and Statistical Manual of Mental disorders, 4th Edition Text Revision Washington DC. [Google Scholar]
- Atkinson RC, Shiffrin RM (1968): Human memory: A proposed system and its control processes In: Spence W, Spence JT, editors. The Psychology of Learning and Motivation, Vol. 2 New York: Academic Press; pp 89–195. [Google Scholar]
- Baddeley AD, Bressi S, Della Sala S, Logie R, Spinnler H (1991): The decline of working memory in Alzheimer's disease. Brain 114:2521–2542. [DOI] [PubMed] [Google Scholar]
- Baillet S (2017): Magnetoencephalography for brain electrophysiology and imaging. Nat Neurosci 20:327–339. [DOI] [PubMed] [Google Scholar]
- Baillet S, Mosher JC, Leahy RM (2001): Electromagnetic brain mapping. IEEE Signal Proc Mag 18:14–30. [Google Scholar]
- Bajo R, Maestú F, Nevado A, Sancho M, Gutiérrez R, Campo P, Castellanos NP, Gil P, Moratti S, Pereda E, Del‐Pozo F (2010): Functional connectivity in mild cognitive impairment during a memory task: implications for the disconnection hypothesis. J Alzheimer Dis 22: 183–193. [DOI] [PubMed] [Google Scholar]
- Bartlett MS (1937): Properties of sufficiency and statistical tests. P Roy Soc Lond A Mat 160:268–282. [Google Scholar]
- Boutros NN, Belger A (1992): Midlatency evoked potentials attenuation and augmentation reflect different aspects of sensory gating. Biol Psychiatry 45: 917–922. [DOI] [PubMed] [Google Scholar]
- Burgmans S, van Boxtel MP, Smeets F, Vuurman EF, Gronenschild EH, Verhey FR, Uylings HB, Jolles J (2008): Prefrontal cortex atrophy predicts dementia over a six‐year period. Neurobiol Aging 30: 1413–1419. [DOI] [PubMed] [Google Scholar]
- Canuet L, Pusil S, López ME, Bajo R, Pineda‐Pardo JÁ, Cuesta P, Gálvez G, Gaztelu JM, Lourido D, García‐Ribas G, Maestú F (2015): Network disruption and cerebrospinal fluid amyloid‐beta and phospho‐tau levels in mild cognitive impairment. J Neurosci 35:10325–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CH, Pei‐Ning W, Wan‐Yu H, Yung‐Yang L (2012): Inadequate inhibition of redundant auditory inputs in Alzheimer's disease: An MEG study. Biol Psychol 89:365–373. [DOI] [PubMed] [Google Scholar]
- Coleman RE (2005): Positron emission tomography diagnosis of Alzheimer's disease. Neuroimag Clin N Am 15:837–846. [DOI] [PubMed] [Google Scholar]
- Cuesta P, Garcés P, Castellanos NP, López ME, Aurtenetxe S, Bajo R, Pineda‐Pardo JA, Bruña R, Marín AG, Delgado M, Barabash A, Ancín I, Cabranes JA, Fernandez A, Del Pozo F, Sancho M, Marcos A, Nakamura A, Maestú F (2015): Influence of the APOE ɛ4 allele and mild cognitive impairment diagnosis in the disruption of the MEG resting state functional connectivity in sources space. J Alzheimer Dis 44: 493–505. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG (2006): Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer's disease pyramidal neurons. Curr Pharm Des 12: 677–684. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger‐Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, Meguro K, O'brien J, Pasquier F, Robert P, Rossor M, Salloway S, Stern Y, Visser PJ, Scheltens P (2007): Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS‐ADRDA criteria. Lancet Neurol 6:734–746. [DOI] [PubMed] [Google Scholar]
- Farlow MR (2009): Treatment of mild cognitive impairment (MCI). Curr Alzheimer Res 6:362–367. [DOI] [PubMed] [Google Scholar]
- Freedman R, Adler LE, Myles‐Worsley M, Nagamoto HT, Miller C, Kisley M, McRae K, Cawthra E, Waldo M (1996): Inhibitory gating of an evoked response to repeated auditory stimuli in schizophrenic and normal subjects. Human recordings, computer simulation, and an animal model. Arch Gen Psychiatry 53:1114–1121. [DOI] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Ross RG, Waldo MC, Stevens KE, Adler LE, Leonard S (2003): The genetics of sensory gating deficits in schizophrenia. Curr Psychiatry Rep 5:155–161. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Gold G, Kövari R, von Gunten A, Imhof A, Bouras C, Hof PR (2007): Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol 113:1–12. [DOI] [PubMed] [Google Scholar]
- Gjini K, Arfken C, Boutros NN (2010): Relationships between sensory “gating out” and sensory “gating in” of auditory evoked potentials in schizophrenia: a pilot study. Schizophr Res 121:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golob EJ, Ringman JM, Irimajiri R, Bright S, Schaffer B, Medina LD, (2009): Starr A Cortical event‐related potentials in preclinical familial Alzheimer disease. Neurology 73:1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DL, Payne L, Polikar R, Moberg PJ, Wolk D, Kounios J (2015): P50: A candidate ERP biomarker of prodromal Alzheimer's disease. Brain Res 624:390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickok G, Poeppel D (2004): Dorsal and ventral streams: a framework for understanding aspects of the functional anatomy of language. Cognition 92:67–99. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011): Alzheimer's disease: the challenge of the second century. Sci Transl Med 3: 77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Aine CJ, Supek S, Best E, Ranken D, Flynn ER (1998): Multi‐start downhill simplex method for spatio‐temporal source localization in magnetoencephalography. Electroencephalogr Clin Neurophysiol 108:32–44. [DOI] [PubMed] [Google Scholar]
- Irimajiri R, Golob EJ, Starr A (2005): Auditory brain‐stem, middle‐ and long‐latency evoked potentials in mild cognitive impairment. Clin Neurophysiol 116: 1918–1929. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Preboske GM, Pankratz VS, Vemuri P, Petersen RC (2014): Age‐specific population frequencies of cerebral β‐amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: a cross‐sectional study. Lancet Neurol 13:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelic V, Johansson SE, Almkvist O, Shigeta M, Julin P, Nordberg A, Winbald B, Wahlund LO (2000): Quantitative electroencephalography in mild cognitive impairment: longitudinal changes and possible prediction of Alzheimer's disease. Neurobiol Aging 21: 533–540. [DOI] [PubMed] [Google Scholar]
- Josef Golubic S, Aine CJ, Stephen JM, Adair JC, Knoefel JE, Supek S (2014a): Modulatory role of the prefrontal generator within the auditory M50 network. NeuroImage 9:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josef Golubic S, Susac A, Grilj V, Ranken D, Huonker R, Haueisen J, Supek S (2011): Size matters: MEG empirical and simulation study on source localization of the earliest visual activity in the occipital cortex. Med Bio Eng Comput 4:545–554. [DOI] [PubMed] [Google Scholar]
- Josef Golubic S, Susac A, Huonker R, Haueisen J, Supek S (2014b): Early attentional modulation of the neural network evoked with the auditory paired‐click paradigm: An MEG study. Procedia Soc Behav Sci 126:195–196. [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B (2004): Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound‐B. Ann Neurol 55:306–319. [DOI] [PubMed] [Google Scholar]
- Klupp E, Grimmer T, Tahmasian M, Sorg C, Yakushev I, Yousefi BH, Drzezga A, Förster S (2015): Prefrontal hypometabolism in Alzheimer disease is related to longitudinal amyloid accumulation in remote brain regions. J Nucl Med 56:399–404. [DOI] [PubMed] [Google Scholar]
- Knafo S, Sánchez‐Puelles C, Palomer E, Delgado I, Draffin JE, Mingo J, Wahle T, Kaleka K, Mou L, Pereda‐Perez I, Klosi E, Faber EB, Chapman HM, Lozano‐Montes L, Ortega‐Molina A, Ordóñez‐Gutiérrez L, Wandosell F, Viña J, Dotti CG, Hall RA, Pulido R, Gerges NZ, Chan AM, Spaller MR, Serrano M, Venero C, Esteban JA (2016): PTEN recruitment controls synaptic and cognitive function in Alzheimer's models. Nat Neurosci 19:443–453. [DOI] [PubMed] [Google Scholar]
- Lazarczyk MJ, Hof PR, Bouras C, Giannakopoulos P (2012): Preclinical Alzheimer disease: identification of cases at risk among cognitively intact older individuals. BMC Med 10:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenberg K (1944): A method for the solution of certain non‐linear problems in least squares. Q Appl Math 2:164–168. [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH (2006): Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology 184:523–539. [DOI] [PubMed] [Google Scholar]
- Lucas‐Meunier E, Fossier P, Baux G, Amar M (2003): Cholinergic modulation of the cortical neuronal network. Pflugers Arch 446:17–29. [DOI] [PubMed] [Google Scholar]
- Maestú F, Peña JM, Garcés P, González S, Bajo R, Bagic A, Cuesta P, Funke M, Mäkelä JP, Menasalvas E, Nakamura A, Parkkonen L, López ME, Del Pozo F, Sudre G, Zamrini E, Pekkonen E, Henson RN, Becker JT; Magnetoencephalography International Consortium of Alzheimer's Disease (2015): A multicenter study of the early detection of synaptic dysfunction in mild cognitive impairment using magnetoencephalography‐derived functional connectivity. NeuroImage Clin 9:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann HB, Whitney DR (1947): On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 18: 50–60. [Google Scholar]
- Marquardt D (1963): An algorithm for least‐squares estimation of nonlinear parameters. SIAM J Appl Math 11: 431–441. [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984): Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34: 939–944. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011): The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer Dement 7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo J, Wagner P (2013): The tyranny of the averages and the indiscriminate use of risk factors in public health: a call for revolution. Eur J Epidemiol 28: 148. [Google Scholar]
- Meyers J, Meyers L (1995): The Meyers Scoring System for the Rey Complex Figure and the Recognition Trial: Professional Manual. Odessa, Florida: Psychological Assessment Resources. [Google Scholar]
- Morris JC, Blennow K, Froelich L, Nordberg A, Soininen H, Waldemar G, Wahlund LO, Dubois B (2014): Harmonized diagnostic criteria for Alzheimer's disease: recommendations. J Intern Med 275:204–213. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L (2001): Mild cognitive impairment represents early‐stage Alzheimer disease. Arch Neurol 58:397–405. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010): Amyloid‐β–induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Morris JC (2005): Mild cognitive impairment as a clinical entity and treatment target. Arch Neurol 62:1160–1163. [DOI] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Schak B, Wester J, de Kock CPJ, Mansvelder HD (2012): Layer‐specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex 23:148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulson RS, Gadbury L, Allison DB (2012): Treatment heterogeneity and individual qualitative interaction. Am Stat 66: 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranken DM (2014): MRIVIEW: A software package for the analysis and visualization of brain imaging data In: Supek S, Aine CJ, editors. Magnetoencephalography. Berlin, Heidelberg: Springer; pp 237–254. [Google Scholar]
- Ranken DM, Best E, Stephen JM, Schmidt DM, George JS, Wood CC, Huang M (2002): MEG/EEG forward and inverse modeling using MRIVIEW. Proceedings of Biomag 2002: 13th International Conference on Biomagnetism, 785–787.
- Rapoport SI, Horwitz B, Haxby JV, Grady CL (1986): Alzheimer's disease: Metabolic uncoupling of associative brain regions. Can J Neurol Sci 13:540–545. [DOI] [PubMed] [Google Scholar]
- Sanfratello L, Stephen J, Best E, Ranken D, Aine C (2014): MEG‐SIM web portal: A database of realistic simulated and empirical MEG data for testing algorithms In: Supek S, Aine CJ, editors. Magnetoencephalography. Berlin, Heidelberg: Springer; pp 285–307. [Google Scholar]
- Selkoe DJ (2002): Alzheimer's disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- Small DH, Mok SS, Bornstein JC (2001): Alzheimer's disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci 2:595–598. [DOI] [PubMed] [Google Scholar]
- Smirnov N (1948): Table for estimating the goodness of fit of empirical distributions. Ann Math Stat 19:279–281. [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison‐Bogorad M, Wagster MV, Phelps CH (2011): Toward definig the preclinical stages of Alzheimer's disease: Recommendation from national Institute on aging‐ Alzheimer's Asocciation workgroup on diagnostic guidelines for Alzheimer's disease. Alzheimer Dement 7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Jr , Aisen PS (2011): Testing the right target and right drug at the right stage. Sci Transl Med 3:111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam CJ, Jones BF, Manshanden I, van Cappellen van Walsum AM, Montez T (2006): Magnetoencephalographic evaluation of resting‐state functional connectivity in Alzheimer's disease. NeuroImage 32: 1335–1344. [DOI] [PubMed] [Google Scholar]
- Stephen JM, Ranken D, Best E, Adair J, Knoefel J, Kovacevic S, Padilla D, Hart B, Aine CJ (2006): Aging changes and gender differences in response to median nerve stimulation measured with MEG. Clin Neurophysiol 117:131–143. [DOI] [PubMed] [Google Scholar]
- Supek S, Aine CJ (2014): Magnetoencephalography: From Signals to Dynamic Cortical Networks. Berlin, Heidelberg: Springer‐Verlag. [Google Scholar]
- Supek S, Aine CJ (1993): Simulation studies of multiple dipole neuromagnetic source localization: model order and limits of source resolution. IEEE Trans Biomed Eng 40:529–540. [DOI] [PubMed] [Google Scholar]
- Supek S, Aine CJ (1997): Spatio‐temporal modelling of neuromagnetic data: I. Multi‐source location versus time‐course estimation accuracy. Hum Brain Mapp 5:139–153. [DOI] [PubMed] [Google Scholar]
- Sušac A, Heslenfeld D, Huonker R, Supek S (2014): Magnetic source localization of early visual mistmatch response. Brain Topogr 27: 648–651. [DOI] [PubMed] [Google Scholar]
- Sušac A, Ilmoniemi R, Pihko E, Nurminen J, Supek S (2009): Early dissociation of face and object processing: A magnetoencephalographic study. Hum Brain Mapp 30: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sušac A, Ilmoniemi R, Pihko E, Ranken D, Supek S (2010): Early cortical responses are sensitive to changes in face stimuli. Brain Res 1346:155–164. [DOI] [PubMed] [Google Scholar]
- Sutherling WW, Crandall PH, Cahan LD, Barth DS (1998): The magnetic field of epileptic spikes agrees with intracranial localizations in complex partial epilepsy. Neurology 38: 778–786. [DOI] [PubMed] [Google Scholar]
- Takayama YA (2010): Delayed recall battery as a sensitive screening for mild cognitive impairment: Follow‐up study of memory clinic patients after 10 years. J Med Dent Sci 57:177–184. [PubMed] [Google Scholar]
- Thomas C, VomBerg I, Rupp A, Seidl U, Schröder J, Roesch‐Ely D (2010): P50 gating deficit in Alzheimer dementia correlates to frontal neuropsychological function. Neurobiol Aging 31:416–424. [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012): Alzheimer amyloid‐β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Werd HJJM, Rajkowska G, Evers P, Uylings BMHBM (2010): Cytoarchitectonic and chemoarchitectonic characterization of the prefrontal cortical areas in the mouse. Brain Struct Funct 214: 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables P (1964): Input dysfunction in schizophrenia. Prog Exp Pers Psychopathol Res 1:1–47. [PubMed] [Google Scholar]
- Vogt BA (2009): Cingulate Neurobiology and Disease. New York: Oxford University Press. [Google Scholar]
- Ward JH (1963): Hierarchical grouping to optimize an objective function. J Am Stat Assoc 58: 236–244. [Google Scholar]
- Weiland BJ, Boutros NN, Moran JM, Tepley N, Bowyer SM (2008): Evidence for a frontal cortex role in both auditory and somatosensory habituation: a MEG study. NeuroImage 42:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE (2016): Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19:1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamrini E, Maestu F, Pekkonen E, Funke M, Makela J, Riley M, Bajo R, Sudre G, Fernandez A, Castellanos N, Del Pozo F, Stam CJ, van Dijk BW, Bagic A, Becker JT (2011): Magnetoencephalography as a putative biomarker for Alzheimer's disease. Int J Alzheimer Dis 280289–21547221. [DOI] [PMC free article] [PubMed] [Google Scholar]