

Abstract
Psoriasis is a chronic inflammatory skin disease affecting 2.5–6 million patients in the United States. The cause of psoriasis remains unknown. Previous human and animal studies suggest that patients with a susceptible genetic background and some stimulus, such as barrier disruption, leads to a coordinated signaling events involving cytokines between keratinocytes, endothelial cells, T cells, macrophages and dendritic cells. Ceramides are endogenous skin lipids essential for maintaining skin barrier function and loss of ceramides may underlie inflammatory and premalignant skin. Ceramides act as a double-edged sword, promoting normal skin homeostasis in the native state, but can be metabolized to sphingosine-1-phosphate (S1P), linked to inflammation and tumorigenesis. To overcome this difficulty, we synthesized solenopsin analogs which biochemically act as ceramides, but cannot be metabolized to S1P. We assess their in vivo bioactivity in a well-established mouse model of psoriasis, the KC-Tie2 mouse. Topical solenopsin derivatives normalized cutaneous hyperplasia in this model, decreased T cell infiltration, interleukin (IL)-22 transcription, and reversed the upregulation of calprotectin and Toll-like receptor (TLR) 4 in inflamed skin. Finally, they stimulated interleukin (IL)-12 production in skin dendritic cells. Thus suggesting barrier restoration has both a biochemical and physical component, and both are necessary for optimal barrier restoration.
Introduction
Psoriasis is a common autoimmune disease mediated in part by interleukin (IL) 17-biased T lymphocytes1, 2. It affects 1–2% of the US population and the mainstays of treatment are topical agents for limited disease and systemic and biologic agents for more generalized disease. Systemic agents include methotrexate, vitamin A derivatives and cyclosporine, while biologic therapies target tumor necrosis factor alpha (TNFα), IL-23 and IL-17. Biologic therapies have made a major impact on the quality of life of patients with severe psoriasis, but are expensive and predispose patients to infection and malignancy3–5. Finally, biologic agents are not indicated for the majority of patients who have mild to moderate disease. For decades, the mainstay of topical treatment for psoriasis was topical steroids. While effective in a significant number of patients, topical steroids are not a panacea for psoriasis. First, resistance (tachyphylaxis) is observed after long-term use. Second, cutaneous atrophy is a well-known side effect of long-term topical steroid use6. Finally, some lesions are highly refractory to topical steroids, especially thicker lesions. While new topical agents have been introduced, none have significantly impacted the rate of use of topical steroids. These agents include vitamin D analogs, retinoids, and calcineurin inhibitors. Barrier restoration with emollients contributes to resolution of psoriasis, but does not fully reverse the psoriatic phenotype.
Ceramides play an essential role in maintaining the barrier function of the skin and loss of barrier function increases expression of vascular endothelial growth factor (VEGF), a major mediator of angiogenesis and inflammation7. However, the failure of emollient barriers to fully reduce inflammation implies a specific biochemical role of ceramides in epidermal homeostasis. Ceramides are generated from lipid precursors and can be degraded by enzymatic hydrolysis or metabolized into sphingosine-1-phosphate (S1P), a mediator of cellular growth, carcinogenesis, and inflammation8. We have synthesized ceramide analogs that are incapable of hydrolysis or metabolism to S1P and have demonstrated that these analogs have in vivo efficacy in a well-validated murine model of psoriasis9. Gene array analysis validated by immunohistochemistry indicates that these analogs decrease production of S100A8 and S100A9 (calprotectin) and the Toll-like receptor (TLR) 4 calprotectin receptor, leading to a reduction in epidermal thickness and lymphocytic infiltration10. Calprotectin is a dimer of S100A8 and S100A9, which complexes both calcium and zinc, and may act as a pro-inflammatory factor through the sequestration of zinc. Fecal calprotectin is now used as a biomarker of colonic inflammation11–14. Gene array of treated vs. untreated inflamed skin shows downregulation of IL-22 and upregulation of AP-1 members FosB and c-Jun. Calprotectin has been implicated as the human PSORS4 locus15, 16. Given that IL-22 plays a major role in mediating both inflammation and carcinogenesis17–20, these compounds could potentially be useful in a wide variety of skin disorders. Thus, restoration of barrier function with stable ceramide analogs leads to an upregulation of IL-12, downregulation of IL-22 and stable production of IL-23, leading to an antiangiogenic phenotype. Finally, we introduce the concept of biochemical barrier restoration that explains, in part, why physical barrier restoration is, by itself, incapable of fully resolving cutaneous inflammation.
Results
Topical S12 and S14 treatment of KC-Tie2 mice decreases epidermal thickness and immune cell infiltrate
S12 and S14 are solenopsin analogs that have 12 and 14 carbon side chains respectively, on the piperidine pharmacophore. KC-Tie2 mice treated with either S12 or S14 for 28 days showed significant decreases (n = 9/grp; 28–35%; P < 0.001) in acanthosis and hyperkeratosis compared to animals treated with vehicle control (n = 8; Fig. 1). This pattern of acanthosis and hyperkeratosis is highly similar to the pathologic changes seen in human psoriasis.
Figure 1.
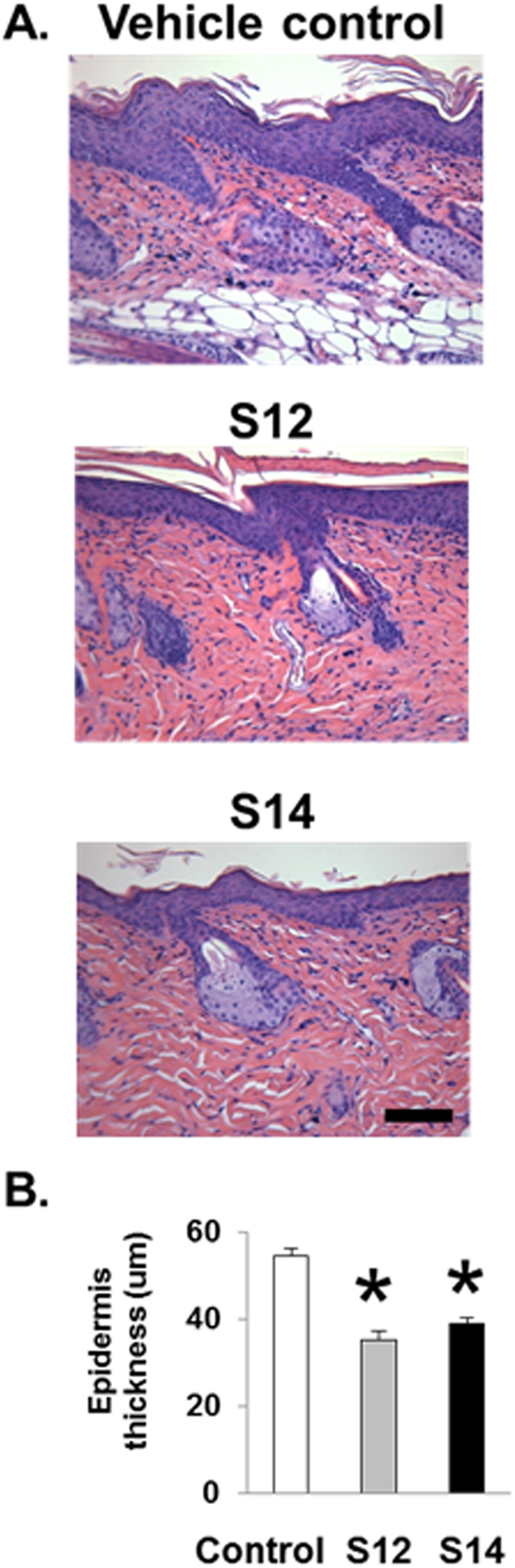
Treatment of inKC-Tie2 mice with S12 and S14 results in decreased epidermal thickness (acanthosis). (A) Representative images of H&E-stained dorsal skin sections from KC-Tie2 mice following treatment with S12, S14 or vehicle cream. (B) Quantification of epidermal thickness (μm) of H&E-stained dorsal skin sections of vehicle treated (n = 8), S12 (n = 9) and S14 (n = 9) KC-Tie2 mice. Values shown represent the mean ± SEM. Data were analyzed using a Student’s t-test. P values are as indicated. Scale bar = 50 μm.
Psoriasis is characterized by T cell infiltration. Significant decrease in CD4+ T cell infiltration (47–63%; P < 0.001) accompanied the improvement in acanthosis in both S12- and S14-treated mice (Fig. 2). In order to determine whether solenopsin analogs had differential effect on T cell and murine dendritic cells (DC), we examined the effect of treatment on KC-Tie2 mice treated with S14, but not S12. Treatment with S14 resulted in significant decreases in CD8+ T cell (47%; P < 0.01) and CD11c+ dendritic cell infiltration into dorsal skin (18%; P = 0.04) compared to vehicle controls (Fig. 3). It may be that the 14 carbon chain is an optimal length to penetrate cell membranes and inhibit the function of both CD8+ and CD11c+ dendritic cells. We have made analogs with longer side chains than 14 carbons and have found them to be not as effective9.
Figure 2.

Treatment of CD4+ T cells decrease in KC-Tie2 mice with topical S12 and S14 analogs results in decreased CD4+ infiltration. (A) Representative images of CD4+ stained dorsal skin sections from KC-Tie2 mice following treatment with S12, S14 or vehicle cream. (B) Quantification of CD4+ T cell numbers in KC-Tie2 mice treated with vehicle (n = 8), S12 (n = 9) and S14 (n = 9). Values shown represent the mean # of cells/FOV (field of view) ± SEM. Data were analyzed using a Student’s t-test. P values are as indicated. Scale bar = 50 μm.
Figure 3.

S14 causes decreased infiltration of CD8+ and CD11c+ cells compared with S12. (A) Representative images of CD8+ stained and CD11c+ stained dorsal skin sections from KC-Tie2 mice following treatment with S12, S14 or vehicle cream. (B) Quantification of CD8+ T cell and (C). Cd11c+ cell numbers in KC-Tie2 mice treated with vehicle (n = 8), S12 (n = 9) or S14 (n = 9). Values shown represent the mean # of cells/FOV (field of view) ± SEM. Data were analyzed using a Student’s t-test. P values are as indicated. Scale bar = 50 μm.
In order to gain insight into the mechanism of the efficacy of solenopsin analogs, RNA was generated from vehicle- and compound-treated skin and subjected to gene array analysis. The heat maps showed strong overlap between the genes regulated by S12 and S14 compared with controls (Figure S1) and genes commonly regulated by both analogs compared with vehicles were studied further. These include specific AP-1 subunits, including FosB and ATF3.
S12 and S14 compounds cause a decrease in IL-22, an increase in IL-12, but no changes in IL-23 production by dendritic cells
Since the solenopsin analogs were shown to decrease DC infiltration, and given DC are a source of IL-22, antiangiogenic IL-12 and proangiogenic IL-23, we wanted to investigate the effect of S12 and S14 on DC. Murine DC were pre-incubated with the compounds and then stimulated with a TLR4-ligand, LPS. The compounds significantly inhibited mRNA expression of IL-22 (Fig. 4A) and strongly induced IL-12p70 production in DC (Fig. 4B). This effect was not observed in IL-23 production (Fig. 4C).
Figure 4.
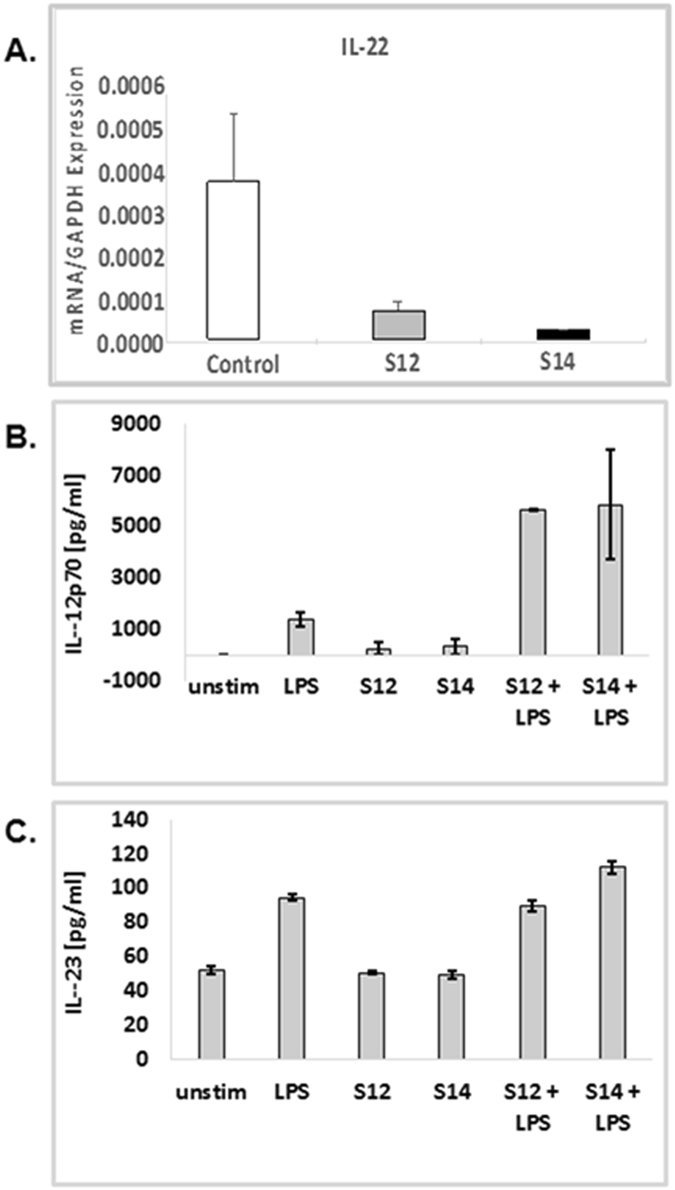
Solenopsin analogs S12 and S14 inhibit IL-22 expression. Compounds cause a significant decrease in IL-22 mRNA expression. The compounds also increase levels of IL-12 but no changes in IL-23 production by DC. Mouse DC were pre-incubated for 12 h with 10 µM compounds and then stimulated with LPS. IL-22 mRNA expression was measured by qRT-PCR. (A) Concentrations of IL-12 (B) and IL-23 (C) in the culture supernatants were determined by ELISA. Results are expressed as mean ± SD.
Solenopsin Analogs Downregulate TLR4 expression in Keratinocytes
TLR4 serves as a co-receptor for S100A8 and S100A9 and has been shown to be a prominent mediator of both inflammation and tumorigenesis10. Given that S100A8 and S100A9 are downregulated by solenopsin analogs and that S100A8/A9 expression of TLR4 could mediate a positive feedback loop, we looked at the expression of TLR4 in human HaCaT keratinocytes treated with LPS, a canonical activator of TLR4. Treatment with the solenopsin derivatives S12 and S14 led to a significant decrease in TLR4 expression (Fig. 5).
Figure 5.
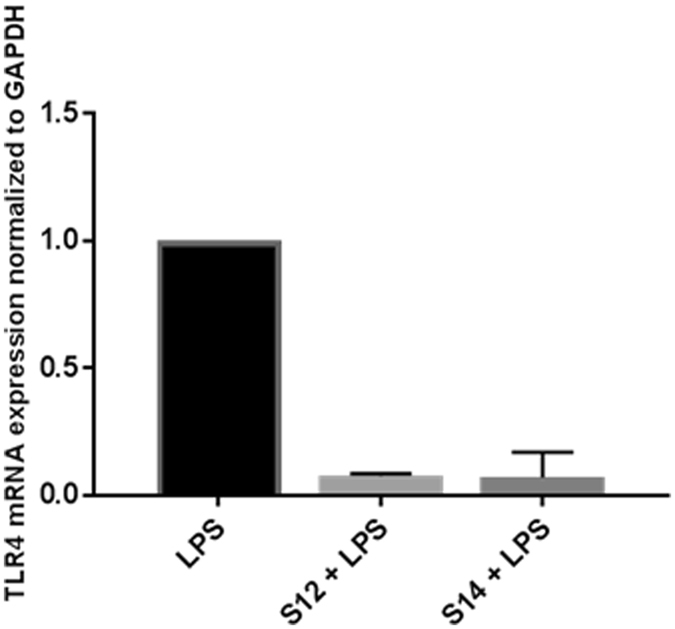
TLR4 in human HaCat keratinocytes treated with LPS, a canonical activator of TLR4. Treatment with the solenopsin derivatives S12 and S14 led to a significant decrease in TLR4 expression. Cells were treated with solenopsin analogs (in DMSO) and/or primed with LPS (10 µg/ml) for 24 h.
Discussion
Psoriasis is a common skin disorder that afflicts approximately 1% of the population3, 21. Increasing evidence of systemic involvement is suggested by epidemiologic association with other diseases, including coronary artery disease, obesity, and metabolic syndrome21. In recent years, targeting cytokines has become very popular for the treatment of severe psoriasis, with modalities targeting TNFα, IL-12, and, most recently, IL-1722–25. These modalities are reserved for the most severe cases, as they can result in systemic immunosuppression and are also costly.
Most cases of psoriasis are classified as mild or moderate and topicals are a mainstay of therapy. Topical corticosteroids remain the mainstay of therapy for the last 5 decades, with other topicals (retinoids, calcineurin inhibitors and vitamin D analogs) playing a relatively minor role in the management of psoriasis26. While effective in some cases, topical corticosteroids exhibit some negatives as well, including loss of efficacy, rebound, and skin atrophy. Therefore, alternative therapies are needed for the treatment of mild to moderate psoriasis.
Barrier disruption is a well-known clinical phenomenon in inflammatory and malignant skin disease. We have previously demonstrated that barrier disruption induces the potent angiogenic mediator VEGF, which contributes to chronic inflammation and angiogenesis7. Ceramides may also impact on the neuroendocrine environment of the skin in a manner to prevent excess inflammation and angiogenesis27, 28. The biochemical effectors of barrier disruption are not fully understood, but one candidate is a family of lipids known as ceramides, a class of fatty acid amides. Ceramides are synthesized from fatty acid precursors and degraded in part through amide hydrolysis. Ceramides have been demonstrated to have anti-inflammatory and antitumor properties. The beneficial effects of ceramides have been explained in part by the normalization of lipid rafts, which subsequently activates the tumor suppressor PTEN, as well as stimulating mitophagy29. Disordered ceramide synthesis has been shown to result in an inflammatory skin disorder in mice8, 30, 31.
In order to augment ceramide signaling, we synthesized a series of non-hydrolysable ceramide analogs that exhibit ceramide-like signaling, including normalization of lipid rafts and mitophagy9, 32, 33. Since these compounds, analogs of the ant venom compound solenopsin, lack amide groups, they cannot be hydrolyzed. We analyzed the effect of a topical 1% formulation of two analogs, S12 and S14, which we have previously found to be the most biologically-active derivatives.
In order to determine the mechanism of the anti-inflammatory activity of topical solenopsin derivatives, we performed gene array analysis on active- vs. vehicle-treated skins and looked for genes that were coordinately upregulated by both S12 and S14. The top most regulated genes were S100A8 and S100A9, which are downregulated by the derivatives. These two genes form a complex called calprotectin that protects against reactive oxygen stress, chelates zinc, and has antimicrobial properties. Of interest, calprotectin is upregulated in virtually every murine model of psoriasis, including this model, imiquimod-induced inflammation, and loss of selective AP-1 subunits from the epidermis (c-Jun/JunB), and is highly expressed in human psoriasis, but not atopic dermatitis1, 11, 34. Our derivatives also downregulate TLR4, a co-receptor for calprotectin. Examination of the promoter sequences of S100A8, S100A9, and TLR4 reveal that these genes are regulated by NFκB and AP-135, 36. It should also be noted that both S100A8 and S100A9 are highly expressed in vascular atheroma of atherosclerosis that is highly associated with psoriasis37, 38.
Traditionally, psoriasis has been thought to be mediated by CD4 lymphocytes. However, recent findings have pointed towards a distinct role of CD8 lymphocytes in psoriasis. First, CD8 lymphocytes have been found to be more abundant in psoriatic epidermis than CD4 lymphocytes39. Second, subsets of CD8 lymphocytes have been shown to be of the Tc22 lineage, which is a major producer of IL-22 in the skin.
Both S12 and S14 also upregulated the AP-1 transcriptional regulators FosB and ATF3 in gene arrays. These genes have previously been demonstrated to be downregulated in non-lesional psoriatic skin compared with normal skin, thus suggesting that even non-lesional skin in psoriasis patients may have disturbances in ceramide metabolism and that this disturbance may predispose skin to psoriatic inflammation8, 40.
Finally, solenopsin derivatives modulate DC to produce IL-12 rather than IL-23. IL-12 has been shown to be anti-angiogenic and, more recently, has been shown to have anti-psoriatic activity on its own41. Thus, it is likely that solenopsin analogs, by restoring ceramide signaling, restore normal skin homeostasis potentially through an anti-inflammatory AP-1/IL-12 pathway.
Our findings have direct relevance to human psoriasis. S100A8 is upregulated by some of the current treatments for human psoriasis, namely glucocorticoids and UVA40, 42, 43. We believe this to be a compensatory upregulation of S100A8 and related molecules in response to treatment, which may be a mechanism of resistance to anti-psoriatic therapy. S100A8 may thus serve as a biomarker for the effectiveness of anti-psoriatic drugs, and combination therapies with solenopsin analogs and other modalities for psoriasis, such as glucocorticoids and UV light, may lead to long-term remission. Further studies of these analogs as anti-psoriatic drugs are warranted in humans.
Materials and Methods
Mouse Model
K5tTA and TetosTek/Tie2 mice (on a CD1 outbred background) were crossed to generate double transgenic KC-Tie2 mice as described previously44. These mice spontaneously develop an inflammatory skin phenotype that phenocopies human psoriasis, including an ~6-fold increase in epidermal thickness (acanthosis), infiltrating CD4+ and CD8+ T cells, and a dense cutaneous myeloid cell presence (CD11c+, CD11b+ and F4/80+), along with the development of munro-like microabscesses containing GR1+ neutrophils44. The skin resembles human psoriasis at the transcriptomic45 and proteomic levels46. The cutaneous phenotype is reversible following broad immunosuppression using cyclosporine A (CsA)44 and inhibition of clinically efficacious targets, including TNFα, IL-23 or antigen cell depletion20–22. Importantly, KC-Tie2 mice fail to respond to drugs to which psoriasis patients have failed to respond47, 48. Thus, the KC-Tie2 model provides a critical tool for testing efficacy of responsiveness to potential anti-psoriasis drugs.
Adult male and female KC-Tie2 mice with established skin disease were treated with the compounded S12 (n = 9) or S14 (n = 9) analogs or control vehicle cream (n = 8) applied to their dorsal skin 1x/day for 28 days. At the completion of the experiment, mice were euthanized and dorsal skin harvested and processed as described previously44 for histological, immunostaining and transcriptomic analyses.
Animal protocols were approved by the Case Western Reserve University Institutional Animal Care and Use Committee (IACUC; Cleveland, OH) and were consistent with guidelines issued by the American Association for Accreditation of Laboratory Animal Care.
Histological and immunostaining analyses
Dorsal skin was collected and processed as described previously for histology and immunohistochemistry44. Formalin-fixed, paraffin-embedded skin was sectioned and stained with haematoxylin and eosin (H&E) using standard techniques as described44. Fresh frozen skin was sectioned and stained, as previously published44, 49, using antibodies targeting the following proteins: CD4 (clone RM4-5, Cat.#550280), CD8a (clone 53-6.7, Cat.#550281), CD11c (clone HL3, Cat.#550283; all BD Pharmingen; San Jose, CA) and F4/80 (clone BM8, Cat.#14-4801, eBioscience; San Diego, CA).
Epidermal thickness (acanthosis) measurements and immune cell quantification in dorsal skin sections was completed using microscopic images collected from the stained sections using interactive image analyses approaches as described previously44.
Gene array analysis
Dorsal skin samples from KC-Tie2 mice treated with control vehicle, S12 or S14 were excised, snap-frozen and submitted to the Emory Integrated Genomics Core for RNA extraction, quality analysis and gene expression assay. Gene expression analysis was performed using an Affymetrix GeneChipTM Microarray: Mouse Gene ST 2.0 and Gene Expression Module of GenomeStudio Software (v2011.1, Illumina).
Generation of murine bone marrow-derived myeloid dendritic cells (BMDCs)
Murine dendritic cells (DC) were generated from bone marrow cells according to the method of Guenova et al.50. Tibiae and femurs from naive mice were removed under sterile conditions. Both ends of the bones were cut off and bone marrow was collected from the medullary cavities by flushing with a PBS-filled syringe. Bone marrow was disrupted by pushing the clumps through 40 μm cell strainers using the rubber tip of a syringe plunger. Cell suspensions were centrifuged and the cell pellets were resuspended in 500 μl hypotonic ACK Lysing Buffer and incubated for 3 min to lyse the erythrocytes. Cells were centrifuged, washed with medium, and counted.
Bone marrow cells were resuspended at 2 × 105 cells/ml in DC culture medium containing 200 U/ml rmGM-CSF (Cat.#415-ML, R&D) and 10 ml were plated into 100 × 15 mm plastic petri dishes (Cat# 351029, BD). On day 3, an additional 10 ml DC culture medium containing 200 U/ml rmGM-CSF (R&D) were added and, at day 6, half of the culture supernatant was removed and centrifuged. The cell pellet was resuspended in 10 ml DC culture medium containing 200 U/ml rmGM-CSF and transferred back into the plate. At day 7, BMDCs (non-adherent cells) were harvested and used for stimulation. Cells were treated with vehicle, S12 or S14 compounds (5 µM or 10 µM) overnight and, at day 8, DC were further stimulated with lipopolysaccharide (LPS; 100 ng/ml, from Salmonella minnesota R595, Alexis Biochemicals, Lausanne, Switzerland) for 24 h.
Supernatants were collected for ELISA. For FACS analysis, BMDCs were harvested and labeled with appropriate antibodies to measure the expression of activation markers and costimulatory molecules. Cytokines were detected by ELISA using commercially available kits for determination of IL-12p70 (BD Biosciences, Cat.#555256) and IL-23 (eBioscience, Cat.#88-7230).
TLR4 Assessment
Human keratinocyte (HaCaT) cell line was obtained from UAB Skin Disease Research Center and propagated in monolayer in Dulbecco’s Modified Eagle’s Medium supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/ml penicillin and 100 μg/ml streptomycin from Life Technologies (Carlsbad, CA). The cells were maintained in 5% CO2 in a humidified incubator at 37 °C. Cells were treated with different concentrations of solenopsin analogs (in DMSO) and/or primed with LPS (10 µg/ml) for 24 h.
Total RNA was extracted from HaCaT cells using TRIzol (Life Technologies, Carlsbad, CA). The concentration of total RNA was determined by measuring the absorbance at 260 nm using a Bio-Rad Smart Spec spectrophotometer (Hercules, CA). cDNA was synthesized from 1 µg RNA using Reverse Transcriptase kit (Bio-Rad, Hercules, CA) according to kit instructions. Using iQTM SYBR Green Master Mix (Bio-Rad, Hercules, CA), cDNA was amplified by qPCR using human TLR4 primers with a Bio-Rad MyiQ thermocycler and SYBR Green detection system (Bio-Rad, Hercules, CA). The standard PCR conditions were 95 °C for 10 min followed by 40 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The expression level of TLR4 was normalized to the expression level of GAPDH in each sample. Relative mRNA expression for duplicate samples was calculated using the ΔΔCT method.
Human TLR4 Forward: AAGCCGAAAGGTGATTGTTG
Human TLR4 Reverse: CTGAGCAGGGTCTTCTCCAC
IL-22 mRNA assessed by qRT-PCR
IL-22 mRNA was assessed by qRT-PCR as previously described by Fritz et al. with primers purchased from Applied Biosystems - primers chosen to span intro-exon51.
Statistics
Comparisons between groups for acanthosis and immune cell density were performed using an unpaired, two-tailed, unequal variance Student’s t-test. Probability values less than 0.05 were considered significant. Data are presented as mean ± SEM.
Statistics for GeneChipTM Analysis
Probe level intensity values were extracted and quantile normalization was performed using the Gene Expression Module of the GenomeStudio Software (v2011.1, Illumina). Principle Component Analysis (PCA) and unsupervised hierarchical clustering were used to assess the variation in the expression profiles. The Significance Analysis of Microarrays (SAM; http://statweb.stanford.edu/~tibs/SAM/) was used to derive a refined list of genes most affected by S12 and S14 compared to the vehicle control.
Data Availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Electronic supplementary material
Acknowledgements
This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. Dr. Arbiser is supported by NIH RO1 AR47901. Dr. Ward is supported by The Lozick Discovery Grant, National Psoriasis Foundation, the Murdough Family Center of Psoriasis, the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (P30 AR39750, P50 AR055508, R01 AR063437, R01 AR062546; R21 AR063852, R01 AR069071). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
J.L.A. obtained funding and designed the experiments as well as wrote the manuscript, R.N. performed experiments, K.M. performed experiments, Y.S. performed experiments, T.B. designed experiments and performed data analysis, N.Y. designed experiments and analyzed data M.Y.B. performed experiments including preparation of topical solenopsin analogs, M.J.L. performed experiments and data analysis, S.R. performed experiments and data analysis, L.C.G. performed experiments and data analysis, I.K. assisted in synthesis of solenopsin analogs, Y.F. and N.L.W. supervised animal experiments, designed experiments, obtained funding and data analysis.
Competing Interests
Emory and Mercer University have applied for patents for solenopsin derivatives.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-10580-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Guttman-Yassky E, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. Journal of immunology. 2008;181:7420–7427. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kryczek I, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. Journal of immunology. 2008;181:4733–4741. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pariser DM, et al. National Psoriasis Foundation clinical consensus on disease severity. Archives of dermatology. 2007;143:239–242. doi: 10.1001/archderm.143.2.239. [DOI] [PubMed] [Google Scholar]
- 4.Chen YJ, et al. Psoriasis independently associated with hyperleptinemia contributing to metabolic syndrome. Archives of dermatology. 2008;144:1571–1575. doi: 10.1001/archderm.144.12.1571. [DOI] [PubMed] [Google Scholar]
- 5.Love TJ, Qureshi AA, Karlson EW, Gelfand JM, Choi HK. Prevalence of the metabolic syndrome in psoriasis: results from the National Health and Nutrition Examination Survey, 2003–2006. Arch Dermatol. 2011;147:419–424. doi: 10.1001/archdermatol.2010.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lund IM, Donde R, Knudsen EA. Persistent local cutaneous atrophy following corticosteroid injection for tendinitis. Rheumatology and rehabilitation. 1979;18:91–93. doi: 10.1093/rheumatology/18.2.91. [DOI] [PubMed] [Google Scholar]
- 7.Elias PM, et al. Epidermal vascular endothelial growth factor production is required for permeability barrier homeostasis, dermal angiogenesis, and the development of epidermal hyperplasia: implications for the pathogenesis of psoriasis. The American journal of pathology. 2008;173:689–699. doi: 10.2353/ajpath.2008.080088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tawada C, et al. Interferon-gamma decreases ceramides with long-chain fatty acids: possible involvement in atopic dermatitis and psoriasis. The Journal of investigative dermatology. 2014;134:712–718. doi: 10.1038/jid.2013.364. [DOI] [PubMed] [Google Scholar]
- 9.Karlsson I, et al. Solenopsin A and analogs exhibit ceramide-like biological activity. Vascular cell. 2015;7:5. doi: 10.1186/s13221-015-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmad I, et al. Toll-like receptor-4 deficiency enhances repair of UVR-induced cutaneous DNA damage by nucleotide excision repair mechanism. The Journal of investigative dermatology. 2014;134:1710–1717. doi: 10.1038/jid.2013.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schonthaler HB, et al. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity. 2013;39:1171–1181. doi: 10.1016/j.immuni.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Roseth AG, et al. Faecal calprotectin: a novel test for the diagnosis of colorectal cancer? Scandinavian journal of gastroenterology. 1993;28:1073–1076. doi: 10.3109/00365529309098312. [DOI] [PubMed] [Google Scholar]
- 13.Nordal HH, et al. Calprotectin (S100A8/A9) has the strongest association with ultrasound-detected synovitis and predicts response to biologic treatment: results from a longitudinal study of patients with established rheumatoid arthritis. Arthritis research & therapy. 2017;19:3. doi: 10.1186/s13075-016-1201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye, L. et al. Fecal calprotectin is a strong predictive marker of relapse in Chinese patients with Crohn’s disease: a two-year prospective study. Scandinavian journal of gastroenterology, 1–7, doi:10.1080/00365521.2017.1346704 (2017). [DOI] [PubMed]
- 15.Zenz R, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 16.Park CC, Kim KJ, Woo SY, Chun JH, Lee KH. Comparison of the Expression Profile of JunB, c-Jun, and S100A8 (Calgranulin A) in Psoriasis Vulgaris and Guttate Psoriasis. Annals of dermatology. 2009;21:35–38. doi: 10.5021/ad.2009.21.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, et al. The expression of interleukin-22 and S100A7, A8, A9 mRNA in patients with psoriasis vulgaris. J Huazhong Univ Sci Technolog Med Sci. 2007;27:605–607. doi: 10.1007/s11596-007-0533-z. [DOI] [PubMed] [Google Scholar]
- 18.Shimauchi T, et al. Serum interleukin-22 and vascular endothelial growth factor serve as sensitive biomarkers but not as predictors of therapeutic response to biologics in patients with psoriasis. J Dermatol. 2013;40:805–812. doi: 10.1111/1346-8138.12248. [DOI] [PubMed] [Google Scholar]
- 19.Ward NL, Umetsu DT. A new player on the psoriasis block: IL-17A- and IL-22-producing innate lymphoid cells. The Journal of investigative dermatology. 2014;134:2305–2307. doi: 10.1038/jid.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanemaru K, Matsuyuki A, Nakamura Y, Fukami K. Obesity exacerbates imiquimod-induced psoriasis-like epidermal hyperplasia and interleukin-17 and interleukin-22 production in mice. Experimental dermatology. 2015;24:436–442. doi: 10.1111/exd.12691. [DOI] [PubMed] [Google Scholar]
- 21.Gelfand JM, et al. Risk of myocardial infarction in patients with psoriasis. JAMA: the journal of the American Medical Association. 2006;296:1735–1741. doi: 10.1001/jama.296.14.1735. [DOI] [PubMed] [Google Scholar]
- 22.Ratner M. IL-17-targeting biologics aim to become standard of care in psoriasis. Nature biotechnology. 2015;33:3–4. doi: 10.1038/nbt0115-3. [DOI] [PubMed] [Google Scholar]
- 23.Wu, J. J. Anti-interleukin-17 monoclonal antibody ixekizumab in psoriasis. N Engl J Med367, 274–275; author reply 275, doi:10.1056/NEJMc1205835#SA2 (2012). [DOI] [PubMed]
- 24.Reich, K. Anti-interleukin-17 monoclonal antibody ixekizumab in psoriasis. The New England journal of medicine367, 274; author reply 275, doi:10.1056/NEJMc1205835#SA1 (2012). [DOI] [PubMed]
- 25.Papp KA, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 26.Arbiser JL. Angiogenesis and the skin: a primer. Journal of the American Academy of Dermatology. 1996;34:486–497. doi: 10.1016/S0190-9622(96)90444-2. [DOI] [PubMed] [Google Scholar]
- 27.Ekstrand AJ, et al. Deletion of neuropeptide Y (NPY) 2 receptor in mice results in blockage of NPY-induced angiogenesis and delayed wound healing. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6033–6038. doi: 10.1073/pnas.1135965100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huggenberger R, Detmar M. The cutaneous vascular system in chronic skin inflammation. The journal of investigative dermatology. Symposium proceedings / the Society for Investigative Dermatology, Inc. [and] European Society for Dermatological Research. 2011;15:24–32. doi: 10.1038/jidsymp.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sentelle RD, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nature chemical biology. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho Y, Lew BL, Seong K, Kim NI. An inverse relationship between ceramide synthesis and clinical severity in patients with psoriasis. Journal of Korean medical science. 2004;19:859–863. doi: 10.3346/jkms.2004.19.6.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakajima K, et al. Barrier abnormality due to ceramide deficiency leads to psoriasiform inflammation in a mouse model. The Journal of investigative dermatology. 2013;133:2555–2565. doi: 10.1038/jid.2013.199. [DOI] [PubMed] [Google Scholar]
- 32.Park J, Kaufmann GF, Bowen JP, Arbiser JL, Janda KD. Solenopsin A, a venom alkaloid from the fire ant Solenopsis invicta, inhibits quorum-sensing signaling in Pseudomonas aeruginosa. The Journal of infectious diseases. 2008;198:1198–1201. doi: 10.1086/591916. [DOI] [PubMed] [Google Scholar]
- 33.Arbiser JL, et al. Solenopsin, the alkaloidal component of the fire ant (Solenopsis invicta), is a naturally occurring inhibitor of phosphatidylinositol-3-kinase signaling and angiogenesis. Blood. 2007;109:560–565. doi: 10.1182/blood-2006-06-029934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing, X. et al. IL-17 Responses Are the Dominant Inflammatory Signal Linking Inverse, Erythrodermic, and Chronic Plaque Psoriasis. The Journal of investigative dermatology, doi:10.1016/j.jid.2016.07.008 (2016). [DOI] [PMC free article] [PubMed]
- 35.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Molecular cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Long J, He T, Belshaw R, Scott J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Scientific reports. 2015;5:12393. doi: 10.1038/srep12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sekimoto R, et al. Visualized macrophage dynamics and significance of S100A8 in obese fat. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E2058–2066. doi: 10.1073/pnas.1409480112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick MM, et al. S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. The Journal of biological chemistry. 2005;280:41521–41529. doi: 10.1074/jbc.M509442200. [DOI] [PubMed] [Google Scholar]
- 39.Vissers WH, et al. Memory effector (CD45RO+) and cytotoxic (CD8+) T cells appear early in the margin zone of spreading psoriatic lesions in contrast to cells expressing natural killer receptors, which appear late. The British journal of dermatology. 2004;150:852–859. doi: 10.1111/j.1365-2133.2004.05863.x. [DOI] [PubMed] [Google Scholar]
- 40.Carceller E, et al. Overexpression of Glucocorticoid-induced Leucine Zipper (GILZ) increases susceptibility to Imiquimod-induced psoriasis and involves cutaneous activation of TGF-beta1. Scientific reports. 2016;6:38825. doi: 10.1038/srep38825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kulig P, et al. IL-12 protects from psoriasiform skin inflammation. Nature communications. 2016;7:13466. doi: 10.1038/ncomms13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu K, et al. Regulation of S100A8 by glucocorticoids. Journal of immunology. 2005;174:2318–2326. doi: 10.4049/jimmunol.174.4.2318. [DOI] [PubMed] [Google Scholar]
- 43.Grimbaldeston MA, Geczy CL, Tedla N, Finlay-Jones JJ, Hart PH. S100A8 induction in keratinocytes by ultraviolet A irradiation is dependent on reactive oxygen intermediates. The Journal of investigative dermatology. 2003;121:1168–1174. doi: 10.1046/j.1523-1747.2003.12561.x. [DOI] [PubMed] [Google Scholar]
- 44.Wolfram JA, et al. Keratinocyte but not endothelial cell-specific overexpression of Tie2 leads to the development of psoriasis. The American journal of pathology. 2009;174:1443–1458. doi: 10.2353/ajpath.2009.080858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swindell WR, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PloS one. 2011;6:e18266. doi: 10.1371/journal.pone.0018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lundberg KC, et al. Proteomics of skin proteins in psoriasis: from discovery and verification in a mouse model to confirmation in humans. Molecular & cellular proteomics: MCP. 2015;14:109–119. doi: 10.1074/mcp.M114.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. Interleukin 6 regulates psoriasiform inflammation-associated thrombosis. JCI insight. 2016;1:e89384. doi: 10.1172/jci.insight.89384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward NL, et al. Erlotinib-induced skin inflammation is IL-1 mediated in KC-Tie2 mice and human skin organ culture. The Journal of investigative dermatology. 2015;135:910–913. doi: 10.1038/jid.2014.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward NL, et al. Depletion of antigen-presenting cells by clodronate liposomes reverses the psoriatic skin phenotype in KC-Tie2 mice. The British journal of dermatology. 2011;164:750–758. doi: 10.1111/j.1365-2133.2010.10129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guenova E, et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:2163–2168. doi: 10.1073/pnas.1416922112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fritz Y, et al. Induction of Alternative Proinflammatory Cytokines Accounts for Sustained Psoriasiform Skin Inflammation in IL-17C+IL-6KO Mice. The Journal of investigative dermatology. 2017;137:696–705. doi: 10.1016/j.jid.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).