

Abstract
Catalytic CO2 conversion to energy carriers and intermediates is of utmost importance to energy and environmental goals. However, the lack of fundamental understanding of the reaction mechanism renders designing a selective catalyst inefficient. Here we show the correlation between the kinetics of product formation and those of surface species conversion during CO2 reduction over Pd/Al2O3 catalysts. The operando transmission FTIR/SSITKA (Fourier transform infrared spectroscopy/steady-state isotopic transient kinetic analysis) experiments demonstrates that the rate-determining step for CO formation is the conversion of adsorbed formate, whereas that for CH4 formation is the hydrogenation of adsorbed carbonyl. The balance of the hydrogenation kinetics between adsorbed formates and carbonyls governs the selectivities to CH4 and CO. We apply this knowledge to the catalyst design and achieve high selectivities to desired products.
Understanding the mechanism of CO2 reduction on a catalyst surface is essential for achieving the desired product selectivity. Here, the authors show an operando kinetic analysis of CO2 hydrogenation over a palladium catalyst in order to address the factors governing the selectivity of the process.
Introduction
Heterogeneous catalytic CO2 reduction has been attracting great attention, because it not only mitigates the risks associated with increasing CO2 concentration in the atmosphere but also offers a variety of pathways for producing fuels and industrial chemicals1, 2. Under atmospheric pressure, methanation and reverse water gas shift (RWGS) reaction are the two pathways for thermocatalytic CO2 reduction on Group VIII metals2. Although methanation produces synthetic natural gas, RWGS generates CO, an important component of syngas. Considerable efforts have been devoted to developing catalysts to achieve high selectivity towards either CH4 or CO3–9. However, for designing a selective catalyst, it is very important to first have a fundamental understanding of the site requirements, elementary steps and intermediates, which can ultimately lead to the construction of viable mechanisms for both methanation and RWGS. Although intricate interplay between various surface species at different catalyst components that determine product selectivity has been recognized, controversies still exist regarding the detailed mechanism and the roles of surface species10–15. Some issues may originate from the different interrogation methods used; for example, some prior conclusions were based on kinetic and spectroscopic information acquired under transient conditions12. Under transient conditions, the perturbation of the chemical environment around the adsorbates and catalyst surface causes characteristically divergent behaviors from steady state.
Steady-state isotopic transient kinetic analysis (SSITKA) has evolved as one of the most powerful techniques allowing measurements at steady state to determine the mean surface lifetimes and abundances of intermediates leading to products16, 17. However, SSITKA itself cannot identify the chemical nature of surface species, a task that can be accomplished by operando spectroscopic measurements, e.g., Fourier transform infrared spectroscopy (FTIR). Correlating the reaction kinetics with the transformations of spectroscopically observable surface species, both simultaneously measured under steady-state reaction conditions, is vital for the complete elucidation of the complex mechanistic framework18–21. Pd-based catalysts are able to catalyze both CO2 methanation and RWGS22, 23. Our previous work on Pd/Al2O3 bifunctional catalysts has proposed the plausible pathways for both CO2 methanation and RWGS24. However, the factors that govern product selectivity (to CH4/CO) under steady-state CO2 reduction conditions are still not clear.
Here we show that CH4 and CO selectivities are governed by the balance of the hydrogenation kinetics between adsorbed formates and carbonyls over Pd/Al2O3 catalysts, which was demonstrated by a combined operando transmission FTIR spectroscopy/SSITKA investigation. Using this knowledge, we show that tailor-made catalysts can be prepared to achieve high selectivity to either of the two desired products.
Results
Steady-state isotopic transient kinetic analysis
Figure 1a, b show normalized mass spectrometry (MS) responses of gases in the effluent after the feed gas was switched at 0 s from 12CO2/H2/Ar to 13CO2/H2 at 533 K. The fast disappearance of Ar gas in <4 s indicates a short gas hold-up time for the system. The disappearance of 12CO2 was almost as fast as Ar, indicating weak interaction between 12CO2 and the catalyst and the reactor walls. In the meantime, the concentration of 13CO2 signal increased accordingly, with the concentration and the conversion (2.5%) of carbon dioxide (12CO2 + 13CO2) constant during the switch. The decaying signals of 12CH4 and 12CO and the rising signals of 13CH4 and 13CO crossed at y = 0.5, indicating constant rates for methane and carbon monoxide formation regardless of isotopic substitution. These symmetrical responses indicate that the steady state of the catalyst surface was not perturbed during the switch. The concentrations of products 12CH4 and 12CO decreased more slowly than that of 12CO2 and disappeared at ~600 s. This indicates large mean residence times for C-containing surface intermediates leading to the products, and that intermediates which equilibrate with CO2 are not involved in the rate-determining steps for CO2 methanation and RWGS.
Fig. 1.
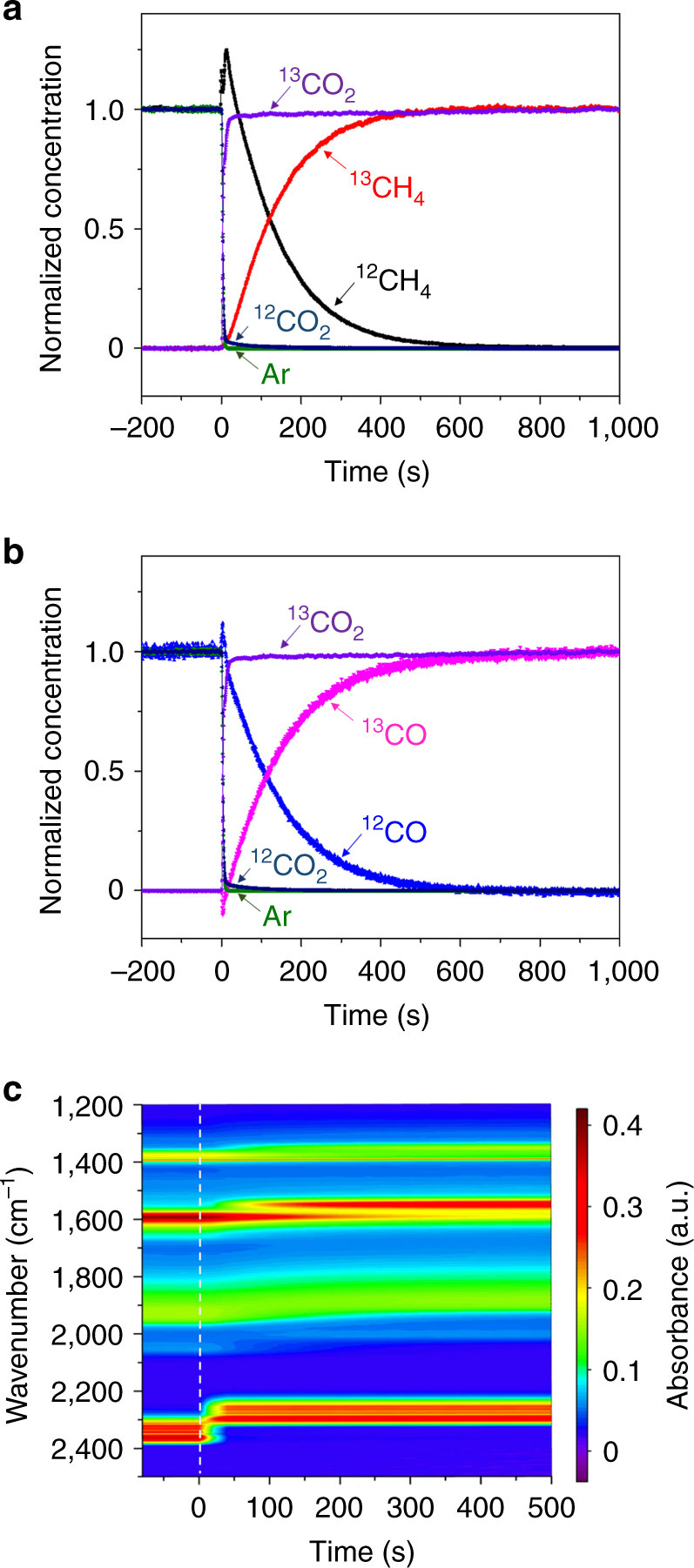
The responses of reactants, products and surface intermediates after the steady-state isotopic switch. a, b Normalized mass spectrometry signals of gases in the effluent and c FTIR contour plot collected as a function of time. The feed gas was switched at 0 s from 12CO2/H2/Ar to 13CO2/H2 over 5% Pd/Al2O3 at 533 K. Ar gas was used as an inert tracer
Figure 1c shows the corresponding operando FTIR contour plot. Before the switch, the absorption band between 2,300 and 2,400 cm−1 (red range) is attributed to gaseous 12CO2. The spectral range between 1,800 and 2,100 cm−1 (blue and green range) is assigned to chemisorbed carbonyls (∗ 12CO) on the surface of Pd metal particles22. The absorption bands around 1,400 (yellow range) and 1,600 cm−1 (red range) arise from adsorbed formates (H12COO∗) on the surface of the Al2O3 support22. After the switch, the bands due to 12CO2 quickly disappeared and a new band developed at lower wavenumbers (red range between 2,200 and 2,300 cm−1) attributable to 13CO2, consistent with their rapid responses in MS. The IR signal decay of surface species might be slightly affected by the location of IR beam on the sample wafer25. However, the shift of the IR features of 12CO∗ and H12COO∗ to those of 13CO∗ and H13COO∗ at lower wavenumbers were much slower compared with the shift of 12CO2 to 13CO2 bands. This observation suggests that neither H12COO∗ nor 12CO∗ undergoes reverse reactions to form 12CO2. The disappearance trends of the IR signatures of H12COO∗ and 12CO∗ are similar to those of the MS signals of 12CH4 and 12CO products, suggesting that both H12COO∗ and 12CO∗ are reactive intermediates rather than spectators and, the rate-determining steps for CH4 and CO formation are related to CO∗ and HCOO∗. It is noteworthy that no other species (e.g., bicarbonate) were observed under steady-state reaction conditions and no carbon deposition was identified in our previous study on Pd/Al2O3 during CO2 reduction24. Therefore, CO∗ and HCOO∗ are the two most abundant surface species and will be the focus of the following discussion.
To gain insight into the interaction between ∗CO and Pd on the 5% Pd/Al2O3, room temperature 12CO adsorption followed by temperature-programed ∗ 12CO desorption were studied by FTIR (Supplementary Fig. 1). At room temperature ∗ 12CO on Pd showed IR features at 2,098, 2,052, 1,960 and 1,928 cm−1. When the temperature was raised to 533 K, ∗ 12CO with features at 2,098 and 1,960 cm−1 desorbed, whereas ∗ 12CO with bands at 2,052 and 1,928 cm−1 did not desorb but was hydrogenated to CH4 once H2 was introduced. This reveals the existence of two types of adsorption sites on the Pd particles that bind CO weakly (∗ 12COw) and strongly (∗ 12COs). The stability of ∗COs at high temperatures is due to the dissociated ∗H, which adsorbs near to ∗COs on the Pd surface and transfers electron density to Pd to strengthen the Pd-CO bond22, 26. The FTIR spectrum of ∗ 12COs is very similar to that of ∗ 12CO obtained in CO2 reduction at 533 K, indicating that all ∗ 12CO observed under reaction conditions were ∗ 12COs. However, ∗ 12COw could not be observed during CO2 reduction at ≥ 533 K due to its rapid desorption. As we observe no ∗ 12COw under steady-state reaction conditions, the 12CO product generated after the switch was mainly from the conversion of the other abundant species, H12COO∗.
Normalized MS responses of 12CH4 and 12CO with increasing temperature are shown in Supplementary Fig. 2, whereas those for 13CH4 and 13CO are not displayed. The faster decay in both 12CH4 and 12CO signals at higher temperatures indicates the higher reactivities of intermediates. In contrast, the decay curves of 12CO2 and Ar did not change with temperature, further indicating that any elementary step that directly involves CO2 is not the rate-limiting step for the formation of CH4 and CO. The real mean surface residence times (Supplementary Fig. 3) of intermediates leading to 12CH4 and 12CO (denoted as ICH and ICO) together with the -derived activation energies E for 12CH4 and 12CO formation (Supplementary Fig. 4) are summarized in Table 1. The at 533 K was 134 s and gradually decreases with increasing temperature, reaching 41 s at 573 K. Similarly, the decreased from 107 to 55 s with increasing temperature from 533 to 573 K. The activation energies are 75 and 53 kJ mol−1 for the formation of CH4 and CO, respectively, in line with the values we obtained previously in a plug-flow reactor24. Series of corresponding operando FTIR spectra of ∗CO and HCOO∗ collected at 533–573 K are shown in Supplementary Figs. 5, 6, respectively. The shift of IR bands representing ∗ 12COs and H12COO∗ to those representing ∗ 13COs and H13COO∗ became faster with increasing temperature, but still remained much slower than the shift of the IR band of 12CO2 to that of 13CO2. The decay trends of ∗ 12COs and H12COO∗ FTIR intensity with increasing temperature was very similar to the decay trends of MS signals of 12CH4 and 12CO products (Supplementary Fig. 2), further indicating that HCOO∗ and CO∗ are related to the rate determining steps for CO and CH4 formation. In addition, the disappearances of ∗ 12COs and H12COO∗ follow an apparent first-order kinetics. The activation energies based on the Arrhenius plots of the two first-order rate constants (Supplementary Figs. 5, 6) are listed in Table 1. The activation energy for the conversion of ∗ 12COs was 73 kJ mol−1, an identical value to that determined for 12CH4 formation from the MS data. This result suggests that the rate-determining step along the CH4 formation path is the conversion of ∗COs. The activation energy of H12COO∗ conversion was 52 kJ mol−1, very similar to the 53 kJ mol−1 determined for the 12CO formation by MS, indicating that the conversion of HCOO∗ is the rate-determining step for CO formation.
Table 1.
Real mean surface residence times for ICH and ICO , and activation energies for CH4 (E CH4) and CO (E CO) formation and adsorbed ∗COs (E∗COs) and HCOO∗ (E HCOO∗) conversions in CO2 reduction at 533–573 K
| Temp. (K) | (s) | (s) | E CH4 (kJ mol −1) | E CO (kJ mol −1) | E∗COs (kJ mol −1) | E HCOO∗(kJ mol −1) |
|---|---|---|---|---|---|---|
| 533 | 134 | 107 | ||||
| 543 | 92 | 85 | 75 | 53 | 73 | 52 |
| 563 | 49 | 57 | ||||
| 573 | 41 | 55 |
Uncertainties in activation energies are ±2 kJ mol−1
Abundance of surface intermediates
Figure 2 shows the surface abundance of ICH and ICO from SSITKA at 533–573 K. The was 1.86 μmol at 533 K, and decreased to 1.01 μmol as the temperature was increased to 573 K. As demonstrated above, 12CO was produced from the existing H12COO∗. Thus, the amount of H12COO∗ that was converted to 12CO was less at higher temperatures than at lower temperatures. In contrast, the increased from 1.95 μmol at 533 K to 2.37 μmol at 573 K. This increase could be interpreted as being caused by an increased concentration of ∗ 12COs at higher temperatures. However, this is not the case, as no difference in intensity of ∗ 12COs was observed as the temperature was increased from 533 to 573 K (Supplementary Fig. 7). The constant intensity of ∗ 12COs implies that the amount of 12CH4 produced from ∗ 12COs, which have already existed at the moment of the switch, should be the same at 533–573 K. Therefore, the significant increase in the amount of 12CH4 at higher temperatures is attributed to the conversion of the other abundant species, H12COO∗. These results suggest that with increasing temperature, for a given amount of surface HCOO∗ at steady state, a progressively larger portion enters the CO2 methanation pathway, while a smaller portion enters the RWGS pathway (Fig. 2b). This explains the decreased at higher temperature. This also agrees well with the above SSITKA results, which show that the became shorter than with increasing temperature. Therefore, formates prefer to enter the faster pathway to form CH4 rather than enter the slower pathway to form CO at higher temperature. Similarly, Goodwin et al. discussed a case where two products share an intermediate: if > , then it must be that <, because more of this intermediate will form product 2 than product 1 due to the faster formation of product 2 (smaller )27. Therefore, the temperature effect on these parameters, , , and , further demonstrates that HCOO∗ is the intermediate shared by the processes of both CO2 methanation and RWGS.
Fig. 2.
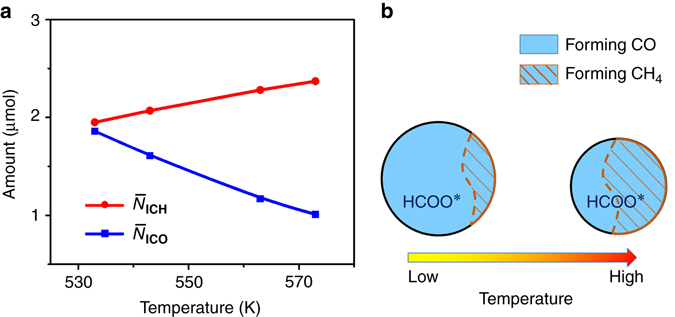
The distribution of surface intermediates in the pathways under steady state. a The amount of surface intermediates leading to 12CH4 and 12CO on the catalyst (41 mg) during CO2 reduction at 533–573 K. b Schematic representation of the proportion of surface coverage of HCOO∗ that is eventually converted to CH4 and CO with increasing temperature concluded from a
Discussion
Based on above results and SSITKA theory16, 17, a scheme relating HCOO∗, ∗COs and ∗COw species in CO2 reduction is proposed in Fig. 3. The reduction of formate produces ∗CO that first occupies strong adsorption sites on the Pd particles. Once the strong adsorption sites of Pd particles are saturated (the pool of ∗COs is completely filled), the excess ∗CO can only occupy the weak adsorption sites of Pd particles to form ∗COw. The ∗COs will be further hydrogenated to CH4 while the ∗COw will desorb.
Fig. 3.
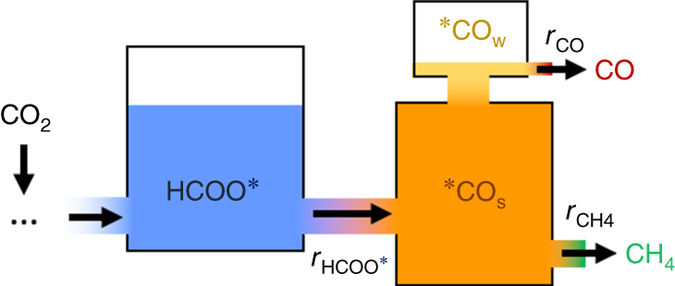
Factors governing the selectivities in CO2 reduction. Scheme for the pathways from formate to products in CO2 methanation and RWGS over Pd/Al2O3 catalysts. The filled pool of ∗COs and the unfilled pool of HCOO∗ are supported by the unchanged coverage of ∗ 12COs and the changed coverage of H12COO∗ at 533–573 K (Supplementary Fig. 7)
The scheme shows that the rate of CO formation (r CO) is determined and equal to the rate of HCOO∗ conversion () subtracted by the rate of CH4 formation (r CH4):
| 1 |
The rate of CH4 formation is equal to the rate of ∗COs conversion (, the rate-determining step for CH4 formation), which is proportional to the concentration of ∗COs. The completely filled pool of ∗COs in the 533–573 K temperature range suggests that has reached its maximum, , at each given temperature between 533–573 K. Therefore, it is the balance between the rate of HCOO∗ reduction to ∗CO () and the maximum rate of ∗COs conversion () that determines whether CO is observed in the gas phase or not. If > , both CH4 and CO will be observed (e.g., the case in Fig. 3). If, however, ≤ , which means that all the ∗CO produced from HCOO∗ are not sufficient to saturate all strong adsorption sites on the Pd metal for CH4 formation (i.e., the pool of ∗COs in Fig. 3 is not full or just full), then all ∗CO will be ∗COs and will be further hydrogenated to CH4. In this case, CO2 reduction will only be methanation and the rate of CH4 formation r CH4 () will not reach its maximum.
As the conversions of both HCOO∗ and ∗COs require the presence of ∗H (absorbed hydrogen) involved24, r CH4 () is determined by the concentrations of ∗COs ([∗COs]) and ∗H ([∗H]), as well as the rate constant of ∗COs conversion (k 1):
| 2 |
and is determined by the concentration of HCOO∗ ([HCOO∗]) and ∗H ([∗H]), and rate constant of HCOO∗ reduction (k 2):
| 3 |
so
| 4 |
It is known that ∗COs and HCOO∗ do not share and compete for active sites, as they are located on Pd metal and Al2O3 support, respectively22. Therefore, [∗COs] and [HCOO∗] can be independently changed to tune the rate of CO formation, r CO, as well as the reaction product distribution.
In the case of a completely filled pool of ∗COs (e.g., the case in Fig. 3), if aiming at a higher CH4 selectivity, [∗COs] should be increased or [HCOO∗] should be decreased. One method, e.g., is to add more metal sites onto the Al2O3 support. The increased metal loading will not only result in an increased number of metal sites for forming ∗COs but also lead to a decreased number of support sites that can accommodate HCOO∗. It means that the capacity of ∗COs pool (Fig. 3) is enlarged, meanwhile that of HCOO∗ is decreased. This, consequently, may lead to a situation where the result of Eq. (4) decreases even to 0, showing less or complete absence of CO in the gas phase. This hypothesis was tested on Pd/Al2O3 catalysts with different Pd loadings but similar Pd particle size distributions (Fig. 4), which minimized the potential effects of metal particle size on k 1 and k 2 influencing and . If the scheme and hypothesis are correct, Pd/Al2O3 catalysts with higher Pd loadings should exhibit higher CH4 selectivity than those with lower Pd loadings. Furthermore, this is exactly what the reactivity data of Fig. 4 shows: CH4 selectivity increased from 45 to 90% as the Pd loading was increased from 2.5 to 10% (Fig. 4). Previous studies on Ru/Al2O3 and Ni/SiO2 catalysts have also shown that CH4 selectivity in CO2 reduction reaction increased with the metal loading12, 28, 29, consistent with the findings reported in this work.
Fig. 4.
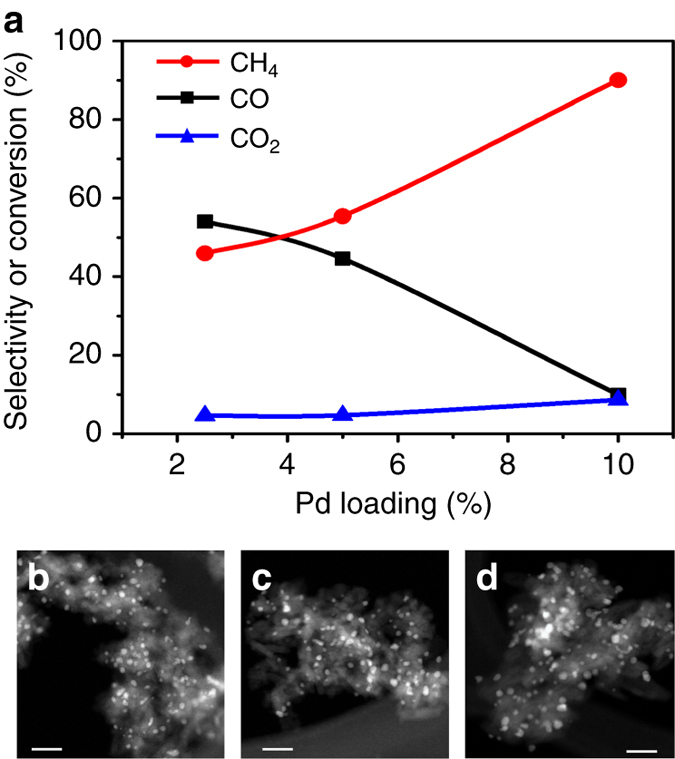
Controlled selectivities in CO2 reduction by tailor-made catalysts through mechanistic understanding. a CH4 and CO selectivities and CO2 conversion as a function of Pd loading at 573 K. b–d STEM images of 2.5, 5 and 10% Pd/Al2O3. STEM scale bars represent 20 nm. (Particle size distributions and temperature-programed ∗ 12CO desorption for the catalysts are displayed in Supplementary Fig. 8)
In the case of an incompletely filled pool of ∗COs, when CH4 selectivity is always 100%, if one aims at a higher CH4 formation rate, [∗COs] needs to be increased. For instance, Karelovic et al.30 reported that the rate of CO2 methanation over Rh/Al2O3 was greatly increased by adding Pd/Al2O3, which has no activity towards CO2 methanation at 473 K. They attributed the synergistic effect to the supply of dissociated H∗ from Rh/Al2O3 to Pd/Al2O3. It is noteworthy that although CO2 methanation cannot proceed on Pd/Al2O3 at 473 K, this temperature is high enough for RWGS reaction to occur producing CO22, 31. We propose that CO or ∗CO produced by/on Pd/Al2O3 could diffuse onto the Rh domains, increasing the concentration of ∗COs on Rh. The total amount of ∗COs produced by both Pd/Al2O3 and Rh/Al2O3 is still not enough to saturate all the active Rh sites for CH4 formation (to fill up the ∗COs pool of Rh, Fig. 3). Therefore, the CH4 selectivity remained ~100% but the methanation rate was higher on the Pd/Al2O3-Rh/Al2O3 mixture than on Rh/Al2O3 alone. The role of the Pd/Al2O3 component was to provide extra CO (∗CO) to the empty sites of Rh. The synergistic effect at lower catalyst temperature (423 K) was found to be negligible. Our study showed that RWGS cannot occur over Pd/Al2O3 at 423 K31, so the added Pd/Al2O3 cannot supply additional CO to Rh/Al2O3 catalyst for the further ∗CO hydrogenation to CH4 on Rh/Al2O3. Therefore, the above analysis of their results show that for an incompletely filled pool of ∗COs with 100% CH4 selectivity ( ≤ ) on a catalyst, adding a component (promoter) which can produce CO (∗CO) is a strategy for increasing the rate of CO2 methanation.
In conclusion, CO2 methanation and RWGS are not two parallel reactions during the CO2 reduction over Pd/Al2O3 catalysts. Instead, they share the initial steps and intermediates from bicarbonates to formates until after formate decomposition. The rate of formate decomposition to CO∗ is larger than the rate of ∗CO hydrogenation to CH4 and the excess CO∗ desorbs. The rate-determining step for CO2 reduction and for RWGS is the conversion of HCOO∗, whereas that for CH4 formation is the hydrogenation of ∗CO. The balance of the hydrogenation kinetics between HCOO∗ and ∗CO governs the selectivities to CH4 and CO. Given that ∗CO and HCOO∗ are mainly on metal (Pd) and support (Al2O3), respectively, the balance could be tuned to achieve the desired CH4 and CO selectivities by optimizing the loading of the metal and the surface area of the support. This work has important implications for other bifunctional systems where the balance between different catalytic functions determines the rates and product distribution.
Methods
Catalyst synthesis and SSITKA experiments
The Pd/Al2O3 catalysts were prepared on a γ-Al2O3 powder (Sasol, Puralox SBA-200) by the incipient wetness method using Pd(NH3)4(NO3)2 as the precursor. After impregnation, the samples were dried at 373 K for 24 h and then calcined at 773 K for 2 h in air (flow rate = 60 mL min−1) and followed by reduction at 773 K for 1 h in 10% H2/He (flow rate = 60 mL min−1) to obtain the Pd/Al2O3 catalysts31. Forty-one milligrams of 5 wt% Pd/Al2O3 was pressed onto a tungsten mesh and loaded into a home-made operando transmission IR cell32. The cell has a very small internal dead volume (~0.2 cm3 after the catalyst loading), resulting in a short gas hold-up time. This renders the system suitable for obtaining accurate kinetic information about intermediates and products during the SSITKA experiments. Before experiments, the catalyst was pretreated by calcination at 673 K for 1 h under air with a flow rate of 10 mL min−1, followed by reduction at 673 K for 1 h under 20% H2 in He with a flow rate of 10 mL min−1. The reactant gas mixture was composed of 4 mL min−1 H2, 1 mL min−1 12CO2 or 13CO2, 1 mL min−1 Ar and He as the diluent with a total gas flow of 10 mL min−1 at atmospheric pressure. Ar gas was used as an inert tracer to correct for gas hold-up of the system and for gas re-adsorption. After the reaction reached steady state at the reaction temperature of 533 K, the reactant was switched from H2/12CO2/Ar to H2/13CO2. During the switch, the gaseous effluent from the cell and the species on the catalyst surface were monitored by MS and FTIR, respectively. The switch was performed in the temperature range of 533–573 K, where the CO2 conversion was <8%. In order to obtain the real mean surface residence times () of intermediates leading to CH4 and CO, space velocity experiments were conducted by varying the total flow rate from 10 to 25 mL min−1 at constant partial pressures of CO2 and H2 to exclude gas hold-up and re-adsorption effects.
Data analysis
The SSITKA theory has earlier been described extensively16, 33. The integration of the normalized step-decay or step-input response always yields the overall mean surface-residence time, , of all adsorbed surface intermediates, which lead to CH4 and CO products . The error <5% caused by gas phase hold-up (~3 s) could be ignored due to the fast decay of standard Ar gas and CO2 gas. The rate constant is simply the reciprocal of average surface residence time for the active surface intermediates (k = = TOF∗ θ −1). The number of adsorbed species and CO can be obtained from integration of decay rates (r) of CH4 and CO products . In addition, is usually closely related to the number of active sites on the catalysts for product formation. In this study of CO2 reduction over 41 mg of 5% Pd/Al2O3, the completely originates from H12COO∗, which was demonstrated to be located on the Al2O3 support. As not all H12COO∗ was converted to 12CO, the amount of sites for formates on Al2O3 is estimated to be at least 1.86 μmol, i.e., obtained at 533 K. For CH4 formation via ∗ 12COs and a portion of H12COO∗, the upper limit of the amount of active Pd sites for ∗COs (forming CH4) is 1.95 μmol, i.e., at 533 K. This value accounts for 90% of the surface Pd atoms. The total amount of intermediates ( + ) on the surface of the catalyst slightly decreased from 3.81 μmol at 533 K to 3.38 μmol at 573 K, which was caused by the decreased amount of H12COO∗ on the surface shown in Supplementary Fig. 7.
Data availability
The data that support the findings of this study are available from the corresponding author on request.
Electronic supplementary material
Acknowledgements
We gratefully acknowledge the financial support of this work by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division.
Author contributions
X.W. designed and built up the SSITKA/Operando-FTIR/MS/GC system and performed all the experiments, analyzed the data and wrote the paper. H.S. revised the paper. J.S. managed the project and revised the paper.
Competing interests
The authors declare no competing financial interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-00558-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aresta M, Dibenedetto A, Angelini A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. technological use of CO2. Chem. Rev. 2014;114:1709–1742. doi: 10.1021/cr4002758. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Wang SP, Ma XB, Gong JL. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011;40:3703–3727. doi: 10.1039/c1cs15008a. [DOI] [PubMed] [Google Scholar]
- 3.Graciani J, et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science. 2014;345:546–550. doi: 10.1126/science.1253057. [DOI] [PubMed] [Google Scholar]
- 4.Sun W, et al. Heterogeneous reduction of carbon dioxide by hydride-terminated silicon nanocrystals. Nat. Commun. 2016;7:12553. doi: 10.1038/ncomms12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thampi KR, Kiwi J, Gratzel M. Methanation and photo-methanation of carbon-dioxide at room-temperature and atmospheric-pressure. Nature. 1987;327:506–508. doi: 10.1038/327506a0. [DOI] [Google Scholar]
- 6.Wang F, et al. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 2016;138:6298–6305. doi: 10.1021/jacs.6b02762. [DOI] [PubMed] [Google Scholar]
- 7.Sun FM, Yan CF, Wang ZD, Guo CQ, Huang SL. Ni/Ce-Zr-O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGS) Int. J. Hydrogen. Energ. 2015;40:15985–15993. doi: 10.1016/j.ijhydene.2015.10.004. [DOI] [Google Scholar]
- 8.Yeung CMY, et al. Engineering Pt in ceria for a maximum metal-support interaction in catalysis. J. Am. Chem. Soc. 2005;127:18010–18011. doi: 10.1021/ja056102c. [DOI] [PubMed] [Google Scholar]
- 9.Meng XG, et al. Photothermal conversion of CO2 into CH4 with H2 over Group VIII Nanocatalysts: an alternative approach for solar fuel production. Angew. Chem. Int. Ed. 2014;53:11478–11482. doi: 10.1002/anie.201404953. [DOI] [PubMed] [Google Scholar]
- 10.Roiaz M, et al. Reverse water-gas shift or sabatier methanation on Ni(110)? Stable surface species at near-ambient pressure. J. Am. Chem. Soc. 2016;138:4146–4154. doi: 10.1021/jacs.5b13366. [DOI] [PubMed] [Google Scholar]
- 11.Carrasquillo-Flores R, et al. Reverse water-gas shift on interfacial sites formed by deposition of oxidized molybdenum moieties onto gold nanoparticles. J. Am. Chem. Soc. 2015;137:10317–10325. doi: 10.1021/jacs.5b05945. [DOI] [PubMed] [Google Scholar]
- 12.Wu HC, et al. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: the influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015;5:4154–4163. doi: 10.1039/C5CY00667H. [DOI] [Google Scholar]
- 13.Matsubu JC, Yang VN, Christopher P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015;137:3076–3084. doi: 10.1021/ja5128133. [DOI] [PubMed] [Google Scholar]
- 14.Kattel S, et al. CO2 hydrogenation over oxide-supported PtCo catalysts: The role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 2016;55:7968–7973. doi: 10.1002/anie.201601661. [DOI] [PubMed] [Google Scholar]
- 15.Weatherbee GD, Bartholomew CH. Hydrogenation of CO2 on group-VIII metals .4. specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal. 1984;87:352–362. doi: 10.1016/0021-9517(84)90196-9. [DOI] [Google Scholar]
- 16.Shannon SL, Goodwin JG. Characterization of catalytic surfaces by isotopic-transient kinetics during steady-state reaction. Chem. Rev. 1995;95:677–695. doi: 10.1021/cr00035a011. [DOI] [Google Scholar]
- 17.Ledesma C, Yang J, Chen D, Holmen A. Recent approaches in mechanistic and kinetic studies of catalytic reactions using SSITKA technique. ACS Catal. 2014;4:4527–4547. doi: 10.1021/cs501264f. [DOI] [Google Scholar]
- 18.Meunier FC, et al. Quantitative analysis of the reactivity of formate species seen by DRIFTS over a Au/Ce(La)O2 water-gas shift catalyst: first unambiguous evidence of the minority role of formates as reaction intermediates. J. Catal. 2007;247:277–287. doi: 10.1016/j.jcat.2007.02.013. [DOI] [Google Scholar]
- 19.Meunier FC. The power of quantitative kinetic studies of adsorbate reactivity by operando FTIR spectroscopy carried out at chemical potential steady-state. Catal. Today. 2010;155:164–171. doi: 10.1016/j.cattod.2009.11.017. [DOI] [Google Scholar]
- 20.Wang J, Kispersky VF, Delgass WN, Ribeiro FH. Determination of the Au active site and surface active species via operando transmission FTIR and isotopic transient experiments on 2.3 wt.% Au/TiO2 for the WGS reaction. J. Catal. 2012;289:171–178. doi: 10.1016/j.jcat.2012.02.008. [DOI] [Google Scholar]
- 21.Hanspal S, Young ZD, Shou H, Davis RJ. Multiproduct steady-state isotopic transient kinetic analysis of the ethanol coupling reaction over hydroxyapatite and magnesia. ACS Catal. 2015;5:1737–1746. doi: 10.1021/cs502023g. [DOI] [Google Scholar]
- 22.Erdohelyi A, Pasztor M, Solymosi F. Catalytic-hydrogenation of CO2 over supported palladium. J. Catal. 1986;98:166–177. doi: 10.1016/0021-9517(86)90306-4. [DOI] [Google Scholar]
- 23.Solymosi F, Erdohelyi A, Lancz M. Surface interaction between H2 and CO2 over palladium on various supports. J. Catal. 1985;95:567–577. doi: 10.1016/0021-9517(85)90135-6. [DOI] [Google Scholar]
- 24.Wang X, Shi H, Kwak JH, Szanyi J. Mechanism of CO2 hydrogenation on Pd/Al2O3 catalysts: kinetics and transient DRIFTS-MS studies. ACS Catal. 2015;5:6337–6349. doi: 10.1021/acscatal.5b01464. [DOI] [Google Scholar]
- 25.Thomas S, et al. Modelling a reactor cell for operando IR studies: From qualitative to fully quantitative kinetic investigations. Catal. Today. 2017;283:176–184. doi: 10.1016/j.cattod.2016.07.008. [DOI] [Google Scholar]
- 26.Solymosi F, Erdohelyi A, Bansagi T. Infrared study of the surface interaction between H2 and CO2 over rhodium on various supports. J. Chem. Soc. Farad. T. 1. 1981;77:2645–2657. doi: 10.1039/f19817702645. [DOI] [Google Scholar]
- 27.McClaine BC, Davis RJ. Importance of product readsorption during isotopic transient analysis of ammonia synthesis on Ba-promoted Ru/BaX catalyst. J. Catal. 2002;211:379–386. doi: 10.1016/S0021-9517(02)93757-7. [DOI] [Google Scholar]
- 28.Wang X, Hong YC, Shi H, Szanyi J. Kinetic modeling and transient DRIFTS-MS studies of CO2 methanation over Ru/Al2O3 catalysts. J. Catal. 2016;343:185–195. doi: 10.1016/j.jcat.2016.02.001. [DOI] [Google Scholar]
- 29.Kwak JH, Kovarik L, Szanyi J. CO2 reduction on supported Ru/Al2O3 catalysts: Cluster size dependence of product selectivity. ACS Catal. 2013;3:2449–2455. doi: 10.1021/cs400381f. [DOI] [Google Scholar]
- 30.Karelovic A, Ruiz P. Improving the hydrogenation function of Pd/gamma-Al2O3 catalyst by Rh/gamma-Al2O3 addition in CO2 methanation at low temperature. ACS Catal. 2013;3:2799–2812. doi: 10.1021/cs400576w. [DOI] [Google Scholar]
- 31.Kwak JH, Kovarik L, Szanyi J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013;3:2094–2100. doi: 10.1021/cs4001392. [DOI] [Google Scholar]
- 32.Yang, Y. et al. Design and operating characteristics of a transient kinetic analysis catalysis reactor system employing in situ transmission Fourier transform infrared. Rev. Sci. Instrum. 77 094104 (2006).
- 33.Bertole CJ, Mims CA, Kiss G. Support and rhenium effects on the intrinsic site activity and methane selectivity of cobalt Fischer-Tropsch catalysts. J. Catal. 2004;221:191–203. doi: 10.1016/j.jcat.2003.08.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on request.