

Summary
Brca2 deficiency causes Mre11-dependent degradation of nascent DNA at stalled forks, leading to cell lethality. To understand the molecular mechanisms underlying this process, we isolated Xenopus laevis Brca2. We demonstrated that Brca2 protein prevents single-stranded DNA gap accumulation at replication fork junctions and behind them by promoting Rad51 binding to replicating DNA. Without Brca2, forks with persistent gaps are converted by Smarcal1 into reversed forks, triggering extensive Mre11-dependent nascent DNA degradation. Stable Rad51 nucleofilaments, but not RPA or Rad51T131P mutant proteins, directly prevent Mre11-dependent DNA degradation. Mre11 inhibition instead promotes reversed fork accumulation in the absence of Brca2. Rad51 directly interacts with the Pol α N-terminal domain, promoting Pol α and δ binding to stalled replication forks. This interaction likely promotes replication fork restart and gap avoidance. These results indicate that Brca2 and Rad51 prevent formation of abnormal DNA replication intermediates, whose processing by Smarcal1 and Mre11 predisposes to genome instability.
Keywords: Brca2, Rad51, Mre11, DNA replication, fork protection, Xenopus laevis
Graphical Abstract
Highlights
-
•
Brca2 promotes Rad51 binding to replicating DNA, preventing fork gaps
-
•
Stable Rad51 nucleofilaments directly protect DNA from Mre11-dependent degradation
-
•
Smarcal1-dependent fork reversal triggers extensive Mre11-dependent DNA degradation
-
•
Rad51 directly interacts with Pol α, promoting its function at stalled forks
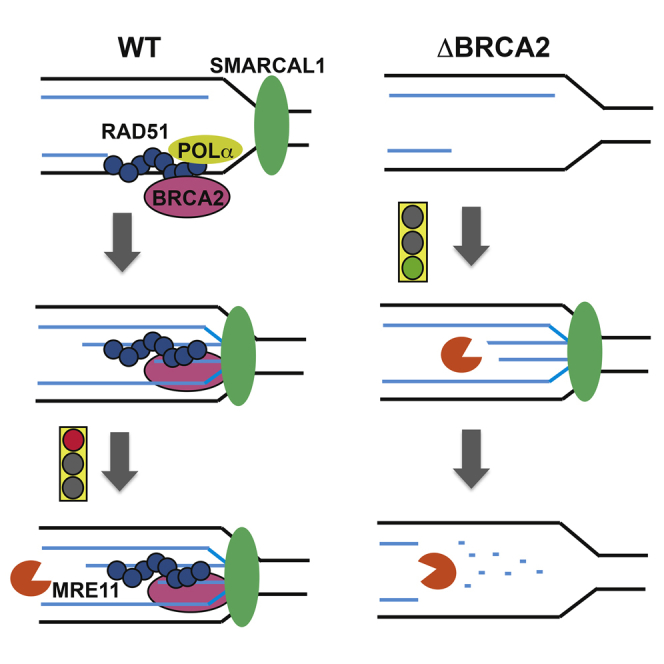
Kolinjivadi et al. show that Brca2 protects replication forks by promoting Rad51 binding to replicating DNA. Rad51 then directly interacts with Pol α, suppressing the occurrence of ssDNA gaps during replication. When forks stall, Brca2-dependent Rad51 nucleofilament formation prevents extensive Mre11-dependent nascent DNA degradation triggered by Smarcal1-mediated fork reversal.
Introduction
Maintenance of genome integrity during DNA replication relies on the ability to respond to DNA lesions and structures impairing replication fork progression (Flynn and Zou, 2011, Nam and Cortez, 2011, Toledo et al., 2011). A central role in preventing genome instability commonly associated with human cancer is provided by the homologous recombination (HR) Brca1 and Brca2 genes, which are often mutated in sporadic and familial cases of cancers (Holloman, 2011, Jackson et al., 2011).
HR proteins’ roles in chromosome breakage repair are well established (Kowalczykowski, 2015, Thorslund and West, 2007). However, their functions in DNA replication are poorly understood. This is in part due to the essential role of many HR genes in higher eukaryotes (Jasin, 2002, Thompson and Schild, 2001).
Cell-free systems based on vertebrate Xenopus laevis egg extract have been helpful to study essential proteins in DNA repair, cell-cycle checkpoints, and DNA replication (Byun et al., 2005, Hashimoto and Costanzo, 2011, Hashimoto et al., 2010, Hashimoto et al., 2011, Räschle et al., 2008, Sannino et al., 2016). Using electron microscopy (EM) to visualize replication intermediates (RIs) isolated from egg extracts, we uncovered a role for Rad51 in preventing Mre11-dependent degradation of nascent DNA during DNA replication (Hashimoto et al., 2010). Consistent with this, Brca2, which loads Rad51 onto DNA breaks and gaps, and Rad51 itself were shown to prevent extensive Mre11-dependent nascent DNA degradation at stalled forks in mammalian cells (Schlacher et al., 2011, Spies et al., 2016). Extensive DNA degradation was also observed after loss of the Rad51 stabilizing factor BOD1L (Higgs et al., 2015). More recently, inhibition of Mre11 recruitment to stalled forks by loss of the MLL3/4 complex protein PTIP and PARP1 has been shown to bypass the Brca2 requirement for survival (Ding et al., 2016, Ray Chaudhuri et al., 2016).
A crucial role in replication fork metabolism is played by Smarcal1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like1), which travels with the elongating replication fork and is regulated by ATR (Bétous et al., 2012, Couch et al., 2013). In vitro, Smarcal1 promotes branch migration and fork reversal on artificial DNA substrates (Bétous et al., 2012, Ciccia et al., 2012). Smarcal1 translocase-dependent fork remodeling could promote reversed fork (RVF) formation in vivo, triggering nascent DNA degradation in Brca2-defective cells.
RVFs are induced by replication fork-stalling agents (Zellweger et al., 2015) under DNA damage checkpoint-deficient conditions (Couch et al., 2013, Lopes et al., 2001). RVFs can also form when polymerase alpha (Pol α) function is compromised in primase mutants (Fumasoni et al., 2015) and in the absence of Tipin-Tim1, which promotes Pol α chromatin binding (Errico et al., 2014), suggesting the existence of a critical link between RVFs and defects in Pol α-dependent DNA polymerization.
Here we isolated Xenopus laevis Brca2 and investigated its molecular role at unperturbed and stalled replication forks using egg extract. We show that Brca2 licenses Rad51 binding to replication forks. Its absence leads to the accumulation of gapped RIs remodeled by Smarcal1 into RVFs, which act as entry points for extensive Mre11-dependent degradation of nascent DNA. Stable Rad51 nucleofilaments protect DNA from Mre11-dependent degradation, whereas Rad51 interaction with Pol α contributes to ensure continuous ungapped DNA replication. These results uncover the molecular mechanisms underlying Brca2-mediated replication fork protection.
Results
Brca2 and Rad51 Binding to Replicating Chromatin
To understand the role of Brca2 in DNA replication, we cloned full-length Xenopus Brca2 cDNA. Brca2 protein revealed significant homology to human Brca2, with all major domains conserved except for BRC5 (Figures 1A, S1, and S2). We then raised antibodies recognizing Xenopus Brca2 in egg extract (Figures S3A and S3B). In co-immunoprecipitations (coIP), Rad51 antibodies pulled down Brca2 protein. Reciprocal coIP with anti Brca2 antibodies confirmed Rad51 binding and revealed that only a fraction of total Rad51 in the cytoplasm was bound to Brca2 (Figure 1B). Furthermore, depletion of more than 99% of Brca2 reduced Rad51 levels in extract only slightly (Figure 1C). These results suggested that Brca2 and Rad51 exist as separate protein pools in egg cytoplasm, although a small fraction of total Brca2 forms protein complexes with Rad51. Consistently, depletion of Rad51 did not significantly reduce Brca2 levels in egg extract (Figure 1C).
Figure 1.

Brca2 and Rad51 Function in DNA Replication
(A) The Xenopus Brca2 protein. See also Figures S1 and S2.
(B) Brca2 and Rad51 immunoprecipitations (IPs) and western blots (WBs).
(C) Brca2 and Rad51 depletion from egg extract.
(D) Chromatin binding time course with or without recombinant geminin. NS, no sperm nuclei.
(E) Brca2 and Rad51 chromatin binding normalized to histone H2B. Mean optical density ± SD of three experiments (n = 3) is shown.
(F) Chromatin binding time course in mock and Brca2-depleted extracts.
(G) Relative incorporation of α-32P-dCTP over time in mock and Brca2-depleted extracts. Counts per minute for mock-treated extracts at 150 min were considered as 100%.
(H) DNA replication in extracts depleted as indicated and reconstituted with 50 ng/μL recombinant Brca2c or Brca2d proteins. L-APH (3 μM) or L-APH plus S1 nuclease (0.1 U/μL) was added at the start where indicated. Nuclei were pre-treated with MMS (1% v/v) or UV (1,000 J/m2) where indicated. Counts per minute in mock-treated extracts at 150 min were considered as 100%. Mean values ± SD (n = 3) are shown.
See also Figures S1–S3.
Under unperturbed conditions, Brca2 was loaded onto chromatin together with major replication factors, and its loading persisted after their dissociation (Figure 1D). Notably, Brca2 loading onto chromatin preceded and accompanied loading of Rad51, suggesting that, similar to Rad51 (Hashimoto et al., 2010), Brca2 binds to DNA during unperturbed replication. To verify whether Brca2 chromatin binding was dependent on active DNA replication, we used geminin to inhibit pre-replication complex assembly (Hashimoto et al., 2010). Geminin induced significant but incomplete inhibition of Brca2 chromatin binding, accompanied by a Rad51 chromatin loading decrease (Figures 1D and 1E). In contrast, the DNA polymerase inhibitor aphidicolin (M-APH, 20 μM), which induces stalled forks, increased Brca2 binding to chromatin (Figure S3C). Consistent with previous results (Jeyasekharan et al., 2010, Reuter et al., 2014) and under conditions in which DNA replication was inhibited, as shown by lack of DNA-bound Cdc45 because of activation of the ataxia telangiectasia mutated (ATM) checkpoint (Costanzo et al., 2000), induction of DNA double-strand breaks (DSBs) onto otherwise DSB-free sperm chromatin also triggered prolonged Brca2 chromatin binding (Figure S3D). These results indicated the existence of at least three binding modes of Brca2 to chromatin, one dependent on DNA replication and stimulated by stalled forks, a second independent of DNA replication, and a third induced by DSBs.
To understand the role of Brca2 during DNA replication and to elucidate its links with Rad51, we depleted endogenous Brca2 from egg extract. Depletion of Brca2 did not significantly affect chromatin binding of major replication factors. However, it almost completely prevented Rad51 assembly onto chromatin (Figures 1F and S3E), indicating that Brca2 was required for active loading of Rad51 onto replicating DNA, except for a small fraction of Rad51 associating with chromatin independently of Brca2.
Similar to Rad51 (Hashimoto et al., 2011), Brca2 depletion did not significantly affect chromosomal DNA replication under unchallenged conditions (Figure 1G). However, Brca2 was required to support restart of forks uncoupled by low amounts of aphidicolin (L-APH, 3 μM) and collapsed by S1 nuclease-induced DNA breakage (Figure 1H). These conditions do not activate the checkpoint and, in contrast to UV and methyl methanesulfonate (MMS) treatment, do not affect replication initiation (Hashimoto et al., 2011), allowing the analysis of Brca2 function during ongoing DNA replication.
To verify whether Brca2’s role in fork restart was linked to Rad51 chromatin binding stability, we cloned and expressed the Xenopus Brca2 C terminus (Brca2c; Figures S3F and S3G), which has been shown to stabilize Rad51 nucleofilaments through the Rad51 binding domain (Davies and Pellegrini, 2007, Esashi et al., 2007) and promote DNA strand exchange (Yang et al., 2002). Notably, addition of Brca2c to Brca2-depleted extracts rescued DNA replication in the presence of S1 nuclease and L-APH (Figure 1H) by stabilizing RAD51 binding to chromatin (Figure S3H). On the contrary, a Rad51 binding domain-deficient Brca2c protein (Brca2d; Figures S3F and S3G), was unable to restore DNA replication (Figure 1H) and Rad51 chromatin binding under similar conditions (Figure S3H). These experiments indicated that stabilization of Rad51 binding to chromatin was required for collapsed fork restart.
Brca2 and ssDNA Gap Formation during DNA Replication
Because bulk incorporation of labeled nucleotide could not detect replication impairment, we employed EM analysis of RIs to identify more subtle defects in the absence of Brca2, using methods established previously (Hashimoto and Costanzo, 2011, Hashimoto et al., 2010). This approach provided snapshot images of RIs stabilized by DNA-psoralen crosslinking prior to their isolation. For the analysis, we considered replication bubbles and Y-shaped RIs, obtained by restriction digestion, which helped to distinguish parental strands from the replicated ones (Lopes, 2009). We observed 70- to 500-nt-long ssDNA gaps on the replicated strands of replication fork junctions isolated from Brca2-depleted, but not from mock-treated extracts (Figures 2A and 2B). Similar fork junction gaps were found in Rad51-depleted extracts (Figures 2A and 2B; Hashimoto et al., 2010). Fork junction gaps were mostly asymmetric, with only one arm being affected (Figure 2A), and only rarely symmetric (Figure 2C).
Figure 2.

ssDNA Gaps and Nascent DNA Degradation in the Absence of Brca2
(A) EM micrographs showing RIs isolated from mock-treated (left) and Brca2- (center) and Rad51-depleted extracts (right). Empty arrows indicate newly replicated strands. Arrowheads indicate ssDNA gaps at fork junctions.
(B) Graph showing the distribution of fork gaps with different lengths. Bars indicate the percentage of RIs with different gap lengths in mock-treated and Brca2- or Rad51-depleted extracts. Mean values ± SEM relative to 150 RIs counted in three experiments (n = 3) are shown.
(C) EM micrograph showing RIs isolated from Brca2-depleted extract. The full black arrow indicates an internal ssDNA fork gap.
(D) Distribution of RIs with the indicated number of internal gaps in mock-treated and Brca2- or Rad51-depleted extracts. Mean values ± SEM (n = 3) are shown.
(E) EM micrograph showing an RI isolated from an undepleted extract incubated with H-APH (1.5 mM) as shown.
(F) EM micrograph showing RIs isolated from mock-treated (top) or Brca2-depleted (bottom) extracts treated with H-APH as in (E). Insets show a higher magnification of replication bubbles. Empty arrows indicate double-stranded DNA. Arrowheads show ssDNA.
See also Figure S4.
Fork junction gaps formed in Brca2- or Rad51-depleted extracts were not suppressed by Mre11 inhibition with mirin (Figure S4A), which has been shown to inhibit Mre11 activity during DNA replication in extracts (Hashimoto et al., 2010). This suggested that fork junction gaps were due to lack of DNA polymerization on one or both strands and not due to Mre11-dependent DNA degradation. In Brca2-depleted extracts, we also observed internal ssDNA gaps, situated at a distance from fork junctions (Figures 2C and 2D). Internal gaps were similar to the ones observed in Rad51-depleted extracts (Figure 2D), which were suppressed by mirin (Hashimoto et al., 2010). Because no major differences were observed in bulk nucleotide incorporation between mock- and Brca2-depleted extracts (Figure 1G), it was likely that internal and fork junction gaps were subsequently filled in by post-replicative repair.
Regulation of Nascent DNA Stability
Collectively, these observations revealed the presence of abnormal RIs formed in the absence of Brca2 during unchallenged replication. Because Brca2 protects nascent DNA from Mre11-dependent DNA degradation at stalled forks (Schlacher et al., 2011), we aimed to reproduce this process in Brca2-depleted egg extract to understand whether abnormal RIs were linked to this phenomenon. To induce fork stalling, we supplemented egg extract with high doses of aphidicolin (H-APH, 1.5 mM) 60 min after DNA addition (Figure 2E) to completely inhibit DNA elongation, as shown previously (Byun et al., 2005). Following 60 min of incubation with H-APH, large asymmetric fork junction gaps could be observed in Y-shaped RIs (Figure 2E) and in replication bubbles (Figure 2F, top). H-APH-induced fork junction gaps were present in more than 70% of RIs in both mock- and Brca2-depleted extracts (Figures S4B and S4C). Similar to spontaneous fork junction gaps found in Brca2- or Rad51-depleted extracts (Figure S4A), mirin did not affect H-APH-induced fork junction gap frequency and size (Figures S4B and S4C). H-APH-induced fork gaps isolated from mid S phase were on average ≈750 nt long, with some gaps reaching ≈1,600 nt in length (Figure S4C).
Significantly, in Brca2-depleted extracts treated with H-APH, we detected numerous replication bubbles in which nascent DNA was no longer present (Figure 2F, bottom; Figure S4D). Complete loss of nascent DNA resulting in extended regions of ssDNA on both replicated strands could be observed only in replication bubbles and not in Y-shaped RIs because single-stranded DNA bubbles were resistant to restriction digestion.
To verify whether the absence of nascent DNA in Brca2-depleted extracts was due to degradation of stalled nascent DNA, we monitored the stability of nascent DNA at stalled forks over time. To this end, nascent DNA was labeled with a short pulse of biotin-dUTP (deoxyuridine triphosphate) (Figure 3A), and biotin-dUTP remaining in nascent DNA after fork stalling was then quantified. In Brca2-depleted samples, we detected nascent DNA loss, which was inhibited by mirin or recombinant Brca2c, but not Brca2d proteins (Figure 3A), suggesting a critical role for Rad51 nucleofilament stabilization in nascent DNA protection against Mre11. Consistent with this, depletion of Rad51 also induced extensive Mre11-dependent degradation of nascent DNA (Figure S4E).
Figure 3.

Brca2- and Rad51-Mediated Protection from Mre11
(A) Top: experimental scheme. Bottom: relative percentage of residual biotin-dUTP in sperm nuclei quantified using a fluorescence method. The fluorescence intensity of mock at 0 min was considered as 100%. Extracts were treated as indicated and supplemented with 100 μM mirin or recombinant Brca2c or Brca2d. Mean values ± SD (n = 3) are shown.
(B and C) Gel showing the effect of Rad51WT and Rad51T131P (B) or RPA complex (C) pre-incubation with 5′ fluorescently labeled DNA substrate (20 nM), shown in the scheme containing phosphorothioate bonds (s), and subsequent incubation with Mre11 (30 nM). Reactions were resolved on denaturing 30% polyacrylamide gel.
(D) Mre11-dependent DNA degradation rates in the presence of Rad51WT, Rad51T131P, or RPA relative to the amount of substrate shown in (B), lane (−), which was considered as 100%. Mean values ± SD (n = 3) are shown.
(E) Electrophoretic mobility shift assay showing binding of Rad51WT, Rad51T131P, or RPA to the same fluorescently labeled DNA substrate (20 nM) resolved on 0.8% agarose gel.
See also Figures S4 and S5.
To verify whether formation of Rad51 nucleofilaments directly prevented Mre11-mediated DNA degradation, we reconstituted DNA protection in vitro using recombinant yeast Mre11, human Rad51, and heterotrimeric replication protein A (RPA) (Figure S4F). We observed extensive Mre11-mediated degradation of linear DNA containing a 35-nt-long 5′ ssDNA overhang (Figure 3B). As expected, no Mre11-dependent degradation was observed on substrates containing 3′ overhangs (Figure S4G). Strikingly, Rad51 pre-assembled on 5′ overhangs completely protected DNA from Mre11-mediated degradation of the 3′ end in the 3′ to 5′ direction (Figure 3B). Similar Rad51-mediated protection was also observed against human Mre11 protein (Figures S4F and S4H).
Interestingly, the Rad51T131P mutant protein identified in Fanconi anemia (FA) patient cells, which has a reduced ability to form stable Rad51 nucleofilaments (Wang et al., 2015), was much less efficient in protecting DNA from Mre11 nuclease at identical concentrations (Figure 3B). To confirm the specific effect of Rad51, we also tested ssDNA binding protein complex RPA. In contrast to Rad51, DNA-saturating concentrations of RPA were unable to protect DNA from Mre11-mediated degradation (Figure 3C). Relative quantification of undegraded DNA substrate at increasing protein concentrations further validated these observations (Figure 3D). Of note, higher concentrations of Rad51 (1,000 nM) were needed to saturate all binding sites on the DNA substrate (Figure 3E) compared with RPA, which could bind all available sites at 200 nM, as shown by the disappearance of free DNA into a higher protein-DNA complex in a mobility shift assay (Figure 3E). This reflected the smaller ssDNA binding site of Rad51, which occupies 3 nt (Qiu et al., 2013) compared with 30 nt occupied by RPA (Kim et al., 1994). Significantly, in contrast to Rad51, although RPA was able to saturate the DNA substrate at 200 nM, it was unable to protect it from degradation (Figure 3C). DNA protection by RPA was not seen even at RPA concentrations ranging from 400–800 nM and when using human Mre11 (Figures 3C and S4I). These results were largely confirmed by the observation of similar protection and DNA binding patterns when using substrates containing single-stranded DNA (ssDNA) gaps (Figures S5A–S5D).
Smarcal1-Dependent Fork Reversal
Because fork junction gaps were not directly susceptible to Mre11-dependent degradation in vivo (Figures S4A–S4C), we looked for other abnormal RIs that could trigger extensive nascent DNA degradation. We noticed the presence of numerous RVFs containing classical four-way junctions (Figure 4A) in RIs isolated from extracts treated with H-APH. RVF reversed branches were 1,250 ± 200 nt long, and most of them contained long ssDNA tails (Figure 4A), suggesting that they originated from remodeling of forks containing asymmetric gaps (Figure 2E).
Figure 4.

Smarcal1-Dependent Regulation of RVFs
(A) EM micrograph showing RVF intermediate isolated from extracts treated with H-APH (1.5 mM). The arrow indicates a double-stranded reversed branch. The arrowhead indicates the single-stranded tail of the reversed branch. The inset shows a high magnification of the RVF junction.
(B) Chromatin binding of the indicated factors at the indicated times following buffer or H-APH supplementation. Buffer and H-APH were added to egg extract 45 min after sperm nuclei. NS, absence of sperm nuclei.
(C) RVF frequency in extracts treated as indicated. Mean values ± SEM (n = 3) are shown. ∗,∗∗,∗∗∗p < 0.01, obtained by unpaired t test between the marked couples.
(D) Scheme showing the assay to quantify RVFs with ssDNA tails (STAR Methods).
(E) ELISA detection of BrdU in nascent ssDNA in nuclei incubated in extracts treated as shown. Where indicated, extracts were supplemented with 5 ng/μL recombinant human Smarcal1WT or catalytically dead Smarcal1HD. Mean intensity values ± SD (n = 3) are shown.
See also Figure S6.
To identify RVFs formation mechanism, we produced antibodies against the Xenopus ortholog of the Smarcal1 translocase, which has been shown to promote fork reversal in vitro on synthetic gapped substrates (Bétous et al., 2012, Ciccia et al., 2012). Anti-Smarcal1 antibodies could completely deplete egg extract of the endogenous protein (Figure S6A). Depletion of Smarcal1 did not affect the cytoplasm levels of other proteins involved in DNA replication and resection (Figures S6A and S6B) or DNA replication efficiency (Figure S6C). Similar to Rad51, high levels of Smarcal1 accumulated onto chromatin following H-APH treatment (Figure 4B). Strikingly, depletion of Smarcal1 significantly impaired fork reversal induced by H-APH (Figure 4C) and moderately increased H-APH-induced fork gaps (Figures S6D and S6E). Although depletion of Rad51 also mildly impaired RVF formation, consistent with previous findings (Zellweger et al., 2015), co-depletion of both Smarcal1 and Rad51 further suppressed RVFs, indicating that the two proteins act additively to sustain RVF levels (Figure 4C).
We then tested whether Smarcal1 was sufficient to promote conversion of gapped forks into RVFs. To this end, we added increasing amounts of recombinant Smarcal1 protein to un-depleted egg extract treated with H-APH, which could stimulate RVF formation (Figure S6F). Interestingly, high amounts of Smarcal1 (25 ng/μL) induced RVFs in which reversed branches were present at both junctions of the replication bubble (Figure S6G). This intermediate was found only at high doses of recombinant Smarcal1 and likely reflected the higher frequency of RVFs induced by hyperaccumulation of recombinant Smarcal1 onto chromatin.
To quantify RVFs independently of EM analysis, we exploited the presence of a high number of RIs with ssDNA tails in RVF branches that could be isolated from egg extract. To this end, we adapted an assay based on the ability of anti-bromodeoxyuridine (BrdU) antibodies to recognize incorporated BrdU only in the context of ssDNA (Couch et al., 2013) and used it to quantify RVFs because EM detected no other major source of extended unpaired nascent ssDNA on RIs (Figure 4A). We confirmed the existence of a linear relationship between the BrdU signal and the number of RVFs visualized by EM because the signal was strongly induced by H-APH and was significantly inhibited by Smarcal1 depletion (Figure 4E). The BrdU signal strength in Smarcal1-depleted extracts was restored by Smarcal1WT, but not by its previously characterized catalytically inactive version Smarcal1HD (Ciccia et al., 2012; Figure 4E), although both proteins were able to bind DNA (Figure S6H). These experiments indicated that Smarcal1 translocase activity was required to promote the formation of RVFs. The BrdU assay also confirmed a Rad51 and Smarcal1 additive role on RVFs observed by EM (Figure 4E).
Fork Reversal and Nascent DNA Degradation
To determine whether RVFs were responsible for the extensive nascent degradation observed in Brca2-depleted extracts, we co-depleted Smarcal1 and determined nascent DNA stability. Strikingly, Smarcal1 depletion significantly prevented the nascent DNA degradation observed in Brca2-depleted extracts treated with H-APH (Figure 5A). Supplementation of depleted extracts with recombinant Smarcal1WT, but not with Smarcal1HD partially restored nascent DNA degradation (Figure 5A). These experiments suggested that RVFs induced by active Smarcal1 triggered nascent DNA degradation in the absence of Rad51 bound to DNA.
Figure 5.

RVFs and Nascent DNA Degradation
(A) Residual biotin-dUTP in nuclei replicated in extracts treated as shown. Where indicated, extracts were Smarcal1-depleted and supplemented with buffer or 5 ng/μL recombinant Smarcal1WT or Smarcal1HD proteins. The fluorescence intensity of mock at 0 min was considered as 100%.
(B) ELISA detection of BrdU in nascent ssDNA in nuclei incubated in extracts treated as shown. Mean intensity values ± SD (n = 3) are shown.
(C and D) Gel showing the effect of Rad51WT and Rad51T131P (C) or RPA (D) pre-incubation with 5′ fluorescently labeled RVF, shown in the scheme, and subsequent incubation with Mre11. Reactions were resolved on denaturing 30% polyacrylamide gel.
(E) Mre11-dependent DNA degradation rates in the presence of Rad51WT, Rad51T131P, or RPA relative to the amount of substrate (20 nM) shown in (C), lane (−), which was considered as 100%. Mean values ± SD (n = 3) are shown.
(F) DNA binding of Rad51 WT or RPA to the same fluorescently labeled RVF DNA substrate (20 nM) resolved on 0.8% agarose gel.
See also Figure S6.
To test whether Brca2 was also required for the stability of RVFs formed by Smarcal1, we quantified RVFs induced by H-APH in the absence of Brca2 with the method shown in Figure 4D. This experiment revealed a significant reduction in the BrdU signal in extracts depleted of Brca2, suggesting that RVFs were decreased in the absence of Brca2 (Figure 5B). Significantly, the BrdU signal could be restored by addition of mirin (Figure 5B), indicating that Brca2 was required for the protection of RVFs from Mre11-mediated degradation but was dispensable for RVF formation.
Importantly, Smarcal1 depletion reduced the BrdU signal in Brca2-depleted extracts, and this inhibition was only partially rescued by mirin (Figure 5B), consistent with a role for Smarcal1 in promoting RVF formation.
Interestingly, low levels of BrdU signal were also detected in Brca2-depleted extracts treated with mirin in the absence of H-APH. This signal possibly reflected spontaneous RVFs arising in the absence of Brca2 and degraded by Mre11 (Figure S6I).
To test whether Brca2’s role in preventing RVFs degradation was directly related to its ability to stabilize Rad51 binding to these structures, we performed Mre11 nuclease protection assays using synthetic RVF substrates. Rad51WT, but not RAD51T131P protein, was able to protect synthetic RVFs from Mre11 nuclease-mediated degradation, whereas RPA was unable to protect RVFs (Figures 5C–5E), despite the fact that it could fully saturate all binding sites on synthetic RVFs in mobility shift assays, similar to Rad51 (Figure 5F). These observations confirmed the critical role of stable Rad51 nucleofilaments in protecting RVFs.
Rad51-Pol α Interaction
Our results indicated that forks with persistent gaps induced by DNA polymerization inhibition obtained with H-APH could be converted by Smarcal1 into RVFs that were susceptible to Mre11-dependent degradation in the absence of Rad51 bound to DNA. However, spontaneous fork junction gaps because of lack of DNA polymerization were also present in Rad51- or Brca2-depleted extracts under unchallenged conditions (Figures 2A, 2B, and S4A–S4C), potentially predisposing to RVF formation (Figure S6I). Given their relevance, we tested how Rad51 prevented spontaneous fork gap formation. We hypothesized that Rad51 directly influenced replicative polymerases function. To verify this hypothesis, we immunoprecipitated Rad51 from egg extracts and performed western blot experiments, which highlighted the presence of Pol α, but not Pol δ, in Rad51 immunoprecipitates (Figure 6A). To determine the presence of a direct interaction between Rad51 and Pol α, we produced a recombinant human Pol α-primase complex, which is made of four subunits: the catalytic α and non-catalytic B subunits and the large (PriL) and small subunits (PriS) of the primase (Pellegrini, 2012). We also produced a version of the Pol α-primase complex in which the α and B subunits lacked their N-terminal domains (ΔNTD). The Pol α-primase complex, containing a His10 tag present at the N terminus of Pri-S, was able to pull down recombinant Rad51 (Figure 6B). Conversely, the version of the complex missing the NTDs was not able to bind Rad51 protein (Figure 6B). The NTD of the catalytic α subunit containing the first 109 amino acids was instead able to directly interact with Rad51 (Figure 6C). This NTD region of the Pol α catalytic subunit contains a pattern of negatively charged and aromatic amino acids that is highly conserved in eukaryotes (Figure S7A) and might play a role in binding Rad51. Importantly, this interaction was inhibited by stoichiometric amounts of Brca2 BRC4 peptide (Figure 6D), which prevents Rad51 polymerization (Thorslund and West, 2007). This observation suggested that the interaction with Pol α required an oligomeric form of Rad51.
Figure 6.

Rad51-Pol α Interaction at Stalled Forks
(A) WB of egg extract IPs using the indicated antibodies. Flowthrough (FT) and extract input (XE) are also shown.
(B) Top: Coomassie-stained gel showing pull-down experiments using Rad51 and hPol α complexes made of the Pol α catalytic subunit, B subunit, Pri-S, and Pri-L. The Pol α catalytic and B subunits were either full-length or missing the first 333 and 148 amino acids, respectively (ΔNTD). His-tagged Pri-S was used for the pull-down. Bottom: WB with anti-Rad51 antibodies of samples shown at the top.
(C) Top: Coomassie-stained gel showing pull-down experiments using Rad51 and the first 109 amino acids of Pol α (MBP-Pol α NTD) bound to amylose resin. Bottom: WB with anti-Rad51 antibodies of the samples shown at the top.
(D) Coomassie-stained gel showing pull-down experiments using Rad51 and the first 109 amino acids of Pol α (MBP-Pol α NTD) bound to amylose resin in the presence of increasing amounts of BRC4 peptide.
(E) Chromatin binding time course following addition of recombinant human full-length (hPol α) and NTD truncated (ΔNTD) complexes shown in (B) in the presence of DMSO or M-APH. Anti-Xenopus Pol α p180 and anti-human Pol α p180 were used for the WB shown at the top and center, respectively.
(F) IPOND showing proteins bound to chromatin containing nascent DNA following biotin pull- down with streptavidin beads. Extracts were supplemented with M-APH 45 min after nuclei addition and pulse-labeled for 10 min with biotin-dUTP as indicated. Pol α, δ, and ε inputs are also shown.
(G) DNA replication in mock-treated or Rad51-depleted restarting extracts (Figure S7F). Values in mock-treated stalling extracts transferred to mock-treated restarting extracts were considered as 100%. Mean values ± SD (n = 3) are shown. See also Figure S7.
To assess the functional relevance of this interaction in the context of replication forks, we adopted a previously established approach based on the displacement of endogenous replication factors by their recombinant versions, which are preferentially incorporated into replicating DNA (Simon et al., 2016). Wild-type (WT) and ΔNTD Pol α-primase complexes were added to egg extract and shown to bind chromatin under unchallenged and challenged conditions induced by intermediate doses of aphidicolin (M-APH, 20 μM), which produced stalled forks but still allowed partial DNA polymerization (Figure 6E). However, in contrast to the WT, the ΔNTD Pol α-primase complex was unable to efficiently displace endogenous Pol α bound to chromatin. These findings suggested that the NTD was involved in the localization of the Pol α-primase complex to replication forks, possibly through its interaction with Rad51 bound to forks, although participation of other replisome components in this process could not be excluded.
To verify the role of Rad51 in promoting stable DNA binding of replicative polymerases, we tested the effect of Rad51 depletion using an assay based on isolation of proteins on nascent DNA (IPOND), in which biotin-dUTP pulses were used to label and pull down nascent chromatin (Sirbu et al., 2013), omitting the formaldehyde crosslinking step to isolate high-affinity nascent DNA binding proteins present at replication forks. Depletion of Rad51 did not affect DNA binding of replication factors such as Cdc45 or Psf3 to stalled forks (Figure 6F) or Pol α concentration in egg extract (Figure S7B). However, it strongly inhibited Pol α association with nascent DNA in the presence of stalled forks induced by M-APH, and it mildly inhibited its DNA binding under unchallenged conditions (Figures 6F and S7C). Rad51 depletion also impaired Pol δ binding to stalled replication forks (Figures 6F and S7D), although no interaction was observed with Pol δ (Figure 6A). Because Pol δ binds to and elongates the primers synthesized by Pol α-primase, reduced Pol α levels could indirectly prevent optimal Pol δ loading onto DNA (Pellegrini and Costa, 2016). In contrast to Pol α/δ, Pol ε binding to replication forks in the presence of M-APH was not significantly affected by Rad51 depletion, although it was partially inhibited compared with un-depleted, untreated extracts (Figures 6F and S7E). Rad51-mediated Pol α DNA binding stabilization might also indirectly facilitate Pol ε association with DNA. However, this hypothesis will need further investigation.
We then studied Rad51 involvement in DNA replication restart at stalled forks. To this end, replication forks were stalled by M-APH in Rad51-proficient and -deficient extracts (stalling extracts) and allowed to restart in Rad51-proficient and -deficient extracts (restarting extracts) (Figure S7F) treated with geminin and roscovitine, a CDK2 inhibitor, to prevent de novo origin assembly and firing (Figure S7F; Errico et al., 2007, Trenz et al., 2006). Replication could efficiently resume only in the presence of Rad51 because depletion of Rad51 from both stalling and restarting extracts severely impaired fork restart (Figure 6G). These findings confirmed that Rad51-mediated DNA binding stabilization of replicative polymerases and nascent DNA was critical to ensure efficient resumption of DNA replication at stalled forks.
Discussion
Brca2 and Rad51 at Replication Forks
Here, using Xenopus egg extract and EM-based approaches to analyze replication intermediates, we provided an extensive characterization of the Brca2 and Rad51 role in DNA replication. We showed the following: Brca2 is loaded onto chromatin during DNA replication; Rad51 replicative function requires Brca2, which loads most of Rad51 onto replicating chromatin; the Brca2 C-terminal segment promotes replication fork restart following template breakage; Brca2, like Rad51, prevents the formation of ssDNA gaps at fork junctions and behind them under unchallenged conditions; persistent fork gaps are remodeled into RVFs by Smarcal1; Smarcal1-mediated RVFs trigger extensive Mre11-dependent nascent DNA degradation in the absence of Brca2; stable Rad51 nucleofilaments directly protect DNA from Mre11-dependent degradation; Brca2 protects RVFs from Mre11-mediated degradation and prevents their spontaneous formation; and Rad51 interacts with the Pol α NTD domain. These results provide molecular insights into the Brca2- and Rad51-dependent fork protection mechanisms, revealing surprising links between the HR and DNA replication proteins.
The unexpected interaction between Rad51 and Pol α could explain the spontaneous accumulation of fork junction gaps in the absence of Brca2/Rad51. ssDNA gaps might occur on DNA sequences, lesions, or structures prone to create transient blocks to DNA polymerization (Figure 7A). Compatibly with its ability to load Rad51 onto gapped DNA in vitro (Jensen et al., 2010), Brca2 might promote Rad51 nucleofilament formation ahead of stalled polymerases, facilitating their progression through DNA segments difficult to replicate (Figure 7B). Rad51-dependent modification of the DNA template structure and stabilization of replicative polymerase binding to forks could facilitate nascent DNA synthesis (Figure 7C), avoiding gap formation. Rad51 polymerization is likely required for the interaction with Pol α because this is prevented by the BRC4 peptide. Pol α progression on the DNA could then displace the nucleofilament with its NTD domain, which, being negatively charged, might weaken Rad51 binding to DNA as it progresses.
Figure 7.

General Model
See Discussion for an explanation.
This process might happen on lagging strands given the presence of extended ssDNA regions that might fold into secondary structures obstructing DNA polymerization, although Brca2/Rad51 action on stalled leading strands cannot be excluded. These findings might also explain the Rad51 requirement for stalled fork restart (Figure 6F; Petermann et al., 2010).
In the absence of Brca2/Rad51, fork gaps transiently accumulate during replication (Figure 7D). DNA synthesis resuming downstream of the stalled polymerase could partially fill the fork junction gap, leading to the formation of internal gaps behind forks (Figure 7E), which could be further enlarged by the action of Mre11 in the absence of Rad51-mediated protection (Figure 7F; Hashimoto et al., 2010). If unrepaired, fork junction gaps might give rise to spontaneous RVFs in the absence of active Mre11.
Lower-fidelity non-replicative polymerases (McVey et al., 2016) might participate in ssDNA gap filling (Figure 7E). This process could contribute to developing the base substitution signature typical of Brca2-defective cancer cells (Nik-Zainal et al., 2016).
Smarcal1-Dependent Fork Remodeling
When left unrepaired, persistent fork junction gaps, such as the ones induced by aphidicolin (Figure 7G), become a substrate of Smarcal1, which promotes the formation of RVFs (Figure 7H). Smarcal1 has been linked to RVF formation in vitro and to fork restart in vivo (Bétous et al., 2012, Ciccia et al., 2009, Ciccia et al., 2012). We showed that Smarcal1 plays a major role in promoting RVFs following replication fork arrest in vivo because RVFs are severely decreased in the absence of Smarcal1 and stimulated by its overexpression. Smarcal1 might be recruited by fork gaps formed on the leading strand in the presence of RPA or on the lagging strand when RPA molecules become limiting (Bétous et al., 2013).
Unfilled fork junction gaps likely play a major role in promoting RVFs as also shown by the increased formation of RVFs caused by the absence of a functional primase gene (Fumasoni et al., 2015) or Pol α-stabilizing factors such as Tipin-Timeless (Errico et al., 2014).
Similar to previous findings (Zellweger et al., 2015), Rad51 depletion decreased RVFs. Interestingly, depletion of both Smarcal1 and Rad51 further suppressed RVFs, suggesting that both proteins act additively to promote them. Other translocases, such as FBH1, ZRANB3, HLTF, or FANCM, might also contribute to this process (Ciccia and Symington, 2016) because depletion of both Rad51 and Smarcal1 did not completely prevent fork regression (Figures 4C and 4E).
The RVFs observed here were induced by high doses of aphidicolin, which compromise ATR activation by suppressing DNA primer synthesis (Byun et al., 2005, Van et al., 2010); lower doses trigger robust ATR activation that prevents Smarcal1-mediated fork remodeling (Couch et al., 2013). Because the ATR phosphorylation site responsible for human Smarcal1 inhibition is conserved, a similar mechanism is likely to regulate Xenopus Smarcal1.
Given the inhibitory effect of ATR on Smarcal1, RVFs might occur more frequently in genomic regions in which ATR activation is suppressed, such as centromeres (Aze et al., 2016), or in any other situation in which the ATR checkpoint is compromised (Lopes et al., 2001).
Rad51-, Brca2-, and Mre11-Dependent Regulation of Nascent DNA Stability
So far, Rad51 has only been indirectly linked to nascent DNA protection. Here we provide biochemical evidence that Rad51 nucleofilaments directly protect ssDNA from Mre11-mediated resection. The formation of a stable Rad51 nucleofilament is critical for this function because Rad51T131P, which cannot form stable Rad51 nucleofilaments (Wang et al., 2015), is unable to protect DNA. The Rad51T131P mutation was identified in FA cells (Wang et al., 2015), suggesting that protection of nascent DNA is important to prevent genome instability associated with FA. This is in agreement with the role of other FA proteins at replication forks (Schlacher et al., 2012).
Brca2-dependent Rad51 DNA binding is important to protect RVFs from Mre11-mediated degradation (Figures 7H and 7I). Without Brca2, RVFs formed by Smarcal1-dependent remodeling of persistent fork gaps induced by aphidicolin (Figure 7J) function as entry points for Mre11 and, possibly, other nucleases (Figure 7K), triggering extensive degradation of both leading and lagging strands (Figure 7L). RVF formation might make DNA ends available for nuclease action by locally displacing replisome components or disrupting local chromatin organization, which could normally prevent nascent DNA degradation at fork gaps.
Because nascent DNA degradation is not completely suppressed by Smarcal1 depletion in Brca2-depleted extracts, it is likely that degradation at gaps behind forks continues independent of Smarcal1. This could be due to Smarcal1-independent RVFs and Mre11-dependent resection of internal gaps behind forks.
Because of its limited processivity (Cejka, 2015) Mre11 might only initiate DNA degradation at RVFs, which could then be completed by nucleases such as ExoI, DNA2, and others in combination with WRN or BLM helicases (Berti and Vindigni, 2016, Cejka, 2015). The presence of 5′ or 3′ ssDNA tails of different lengths, modified bases, or proteins bound to the end of the reversed branch, such as Ku70/80, might influence the rate of resection and the type of nuclease recruited. In these cases, similar to the processing of DSBs with blocked ends (Anand et al., 2016), endonuclease activity of Mre11 stimulated by CtIP phosphorylated by cyclin dependent kinases (CDKs) might be particularly useful to initiate RVF degradation.
Restoration of RVFs by suppression of Mre11 activity in Brca2-depleted extracts indicates that Brca2 is mainly required for protection of RVFs but dispensable for their formation. Residual chromatin binding of Rad51 in the absence of Brca2, although insufficient to protect DNA, might contribute to RVF formation together with Smarcal1. Alternatively, Rad51 function could be limited to RVF protection. Rad51 involvement in RVF formation or protection would be compatible with the observed Smarcal1 and Rad51 additive roles in sustaining RVF levels.
Nevertheless, Smarcal1-dependent RVFs, which can still be observed in the absence of Rad51 (Figures 4C and 4E), are sufficient to the trigger extensive nascent DNA degradation that occurs when Rad51 is completely depleted (Figure S4E) or prevented from binding DNA (Figures 3A and 5A; Schlacher et al., 2011).
Overall, these findings are consistent with a major role for Brca2 and Rad51 in preventing deregulated nascent DNA degradation and promoting continuous and accurate DNA replication across replication blocks. Because the Rad51 ortholog RecA in prokaryotes protects nascent DNA from nuclease-mediated degradation in the presence of DNA damage and promotes loading of DNA polymerases to restart replication forks (Courcelle and Hanawalt, 2003), it is possible that both of these functions have been conserved in eukaryotes.
The characterization of the biochemical mechanisms underlying Brca2/Rad51-mediated fork protection described here could be useful to identify novel strategies to target Brca2-defective cancer cells.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti BrdU B44 | BD Bioscience | N/A |
| Anti-Xenopus Pol α p180 (Mouse monoclonal); peptide antigen used: VKRLPAVTKPGH | This study | Abmart: clone 13026-1-3/C199 |
| Anti human Pol α p180 | Abcam | ab31777; RRID: AB_731976 |
| Anti-Xenopus Pol δ 125 kDa (Mouse Monoclonal); peptide antigen used: SSQTKKLRGDWDDD | This study | Abmart: clone 19570-1-1/C316 |
| Anti-Xenopus Pol ε p60 (Rabbit polylconal) | Shou Waga, Japan Women’s University | N/A |
| Anti-Orc1 (Rabbit polyclonal) | (Aze et al., 2016) | N/A |
| Anti Cdc45 (Rabbit polyclonal) | (Aze et al., 2016) | N/A |
| Anti Psf3 (Rabbit polyclonal) | (Hashimoto et al., 2010) | N/A |
| Anti-Mre11 (Rabbit polyclonal) | Jean Gautier, Columbia University | N/A |
| Anti-RPA70(Rabbit polyclonal) | Jean Gautier, Columbia University | N/A |
| Anti-Rad51 (Rabbit polyclonal); peptide antigen used: CAEAMFAINADGVGDAKD | This study | N/A |
| Anti Xenopus Smarcal1 (Xenopus full length protein used as antigen) | This study | N/A |
| Anti Xenopus Brca2 (Rabbit polyclonal); peptide antigen used: KPHIKEDQNEPESNSEYCS | This study | N/A |
| Anti-Histone H2B | Millipore | clone 07-371; RRID: AB_310561 |
| Anti-Mcm7 | Santa Cruz | clone sc-9966; RRID: AB_627235 |
| Anti-Rad51 goat antibodies | Santa Cruz | sc6862; RRID: AB_2177096 |
| Anti-Flag | Cogentech | N/A |
| Anti-Histone H3 | Abcam | clone ab1791; RRID: AB_302613 |
| Anti-Rad51 mouse antibodies | Abcam | clone ab123 |
| Bacterial and Virus Strains | ||
| WT and catalytic dead (HD, D549A E550A) mutant Smarcal1 baculovirus vectors | (Ciccia et al., 2012) | N/A |
| Mre11-6xHis baculovirus vector | Peter Cejka, University of Zurich | N/A |
| Biological Samples | ||
| Xenopus egg extract | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Xenopus Brca2 protein | This study | N/A |
| Xenopus and human Smarcal1WT and Smarcal1HD proteins | This study | N/A |
| Rad51 protein | This study | N/A |
| Yeast Mre11 protein | This study | N/A |
| Human Mre11-6xHis protein | Peter Cejka, University of Zurich | N/A |
| Human Polymerase α1-1462 and B1-598 subunit proteins | This study | N/A |
| Human Primase His10-tagged human Primase PriS and PriL subunit proteins | This study | N/A |
| RPA protein complex | This study | N/A |
| MBP tagged human Polymerase α1-109 NTD | This study | N/A |
| Geminin | (Aze et al., 2016) | N/A |
| BRC4 peptide: LLGFHTASGKKVKIAKESLDKVKNLFDE | Sigma | N/A |
| Xenopus 6xHis-TEV-SMARCAL1(FL) | This study | N/A |
| 3,5,8-Trimethylpsoralen TMP | Sigma | T6137 |
| Biotin-16-dUTP | Roche | 11093070910 |
| Mirin | Sigma | M9948 |
| BrdU | Sigma | B5002 |
| Dynabeads-ProteinA | Thermo Fisher | 10002D |
| Roscovitine | Calbiochem | 557360 |
| Aphidicolin | Sigma | A0781 |
| Corionic Ganadotropin | Sigma | CL10 |
| Spermine | Sigma | S3256 |
| Methyl Methanesulfonate (MMS) | Sigma | 129925 |
| Spermidine | Sigma | S2626 |
| Lysolecithin | Sigma | L1381 |
| Maltose | Sigma | M5885 |
| Imidazole | Sigma | I5513 |
| BSA | Sigma | A2058 |
| Calcium ionophore | Sigma | A23187 |
| Proteinase K | Sigma | 3115887001 |
| RNase A | Thermo Fisher | EN0531 |
| Benzonase | Sigma | E1014 |
| Critical Commercial Assays | ||
| ECL | Amerhasm | • RPN2232 |
| pCRBluntIITOPO vector | Thermo Fisher | K28002 |
| SuperscriptIII reverse transcriptase | Thermo Fisher | 18080093 |
| QuickChange Site-Directed Mutagenesis kit | Agilent Genomics | 200515 |
| SensoLyte Biotin Quantitation Kit | Anaspec | AS-72163 |
| Terminal Transferase and 10X buffer | NEB | M0315 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | M0530 |
| Deposited Data | ||
| Xenopus Brca2 cDNA | This study | GenBank: KY024483 |
| Raw images | This study | http://dx.doi.org/10.17632/xf5bvrr7gh.1 |
| Experimental Models: Cell Lines | ||
| High Five insect cells | Invitrogen | B85502 |
| Experimental Models: Organisms/Strains | ||
| Xenopus laevis females | Nasco | LM00535MX |
| Xenopus laevis males | Nasco | LM00715MX |
| Oligonucleotides | ||
| xBrca2-N1 GAGACATGGCTACATCTCAACTTGG | Sigma | N/A |
| xBrca2-C2 TCACTTTACCTGCCATTCTGCTGG | Sigma | N/A |
| xBrca2-Xho-rev(3177) CCGCTCGAGTCACTTTACCTGCCATTCTGCTGG | Sigma | N/A |
| Brca2-Bgl-forw(2295) GGAAGATCTACCCAAATGACTTCAAATCTTCGCTGTAGC | Sigma | N/A |
| Brca2-Xho-rev(3020) CCGCTCGAGTCAGTTCTTACTGCACCCAGTTGTTC | Sigma | N/A |
| Rad51T131P _for GATCTGGGTCTTCCCAGGTCGGAATTCTCCAAACA | Sigma | N/A |
| Rad51T131P _rev TGTTTGGAGAATTCCGACCTGGGAAGACCCAGATC | Sigma | N/A |
| Recombinant DNA | ||
| pFastbac-HTb Xenopus laevis Smarcal1 | GenScript | GenBank: NM_001096199 |
| ADA400-Xenopus laevis BRCA2 | This study | N/A |
| ADA411-GST-Brca22295-3177 (Brca2c) | This study | N/A |
| ADA417-GST-Brca22295-3020 (Brca2d) | This study | N/A |
| pET11d-human RPA protein complex | This study | N/A |
| pET-11c-human Rad51 | (Sigurdsson et al., 2001) | N/A |
| pET-11c-human Rad51T131P | This study | N/A |
| pFBDM-StrepII-tagged human Polymerase α1-1462 and B1-598 subunit | This study | N/A |
| pFBDM-His10-tagged full length human Primase subunits PriL and PriS | This study | N/A |
| pMAT11-MBP- human Polymerase α1-109 NTD | N/A | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| ImageJ | Freeware | N/A |
| Gatan Micrograph software | Gatan | N/A |
| Multi Gauge V3.2 software | Fuji | N/A |
| Other | ||
| FEI Tecnai 12 EM microscope | FEI | N/A |
| GATAN high-resolution camera | Gatan | N/A |
| Cary Eclipse fluorescence spectrophotometer | Varian | N/A |
| Bioruptor | Diagenode | N/A |
| Stratalinker equipped with 254 and 365 nm ultraviolet light bulbs | Stratagene | N/A |
Contact for Reagent and Resource Sharing
Resource and reagent requests should be directed to and will be fulfilled by the Lead Contact, Vincenzo Costanzo (vincenzo.costanzo@ifom.eu).
Experimental Model
Eggs derived from Xenopus laevis frogs were used as experimental model system. Collection of eggs from the female frogs was performed in a non-invasive way following chorionic gonadotropin (Sigma, CG10) injections. Occasional surgical procedures were performed on the male frogs to harvest sperm nuclei. Experimental protocols were approved by IFOM Animal Welfare committee and the Italian Ministry of Health. The number of animals used was kept to a minimum and was calculated taking into account the number eggs required to obtain the cytoplasmic extract needed for the experiments described.
The animals were kept in highly regulated and monitored conditions with room and water temperature at 19 C. Basic husbandry requirements were provided by the IFOM Xenopus facility.
Method Details
Protein expression and purification
6xHis-TEV-SMARCAL1(FL) from Xenopus was expressed in High Five insect cells (Invitrogen, B85502) infected with the respective recombinant baculoviruses by Silvia Monzani and Sebastiano Pasqualato, Cogentech, Milan Proteins were isolated according to standard procedures. Briefly, cell pellets were resuspended in lysis buffer-1 (50 mM HEPES pH7.6, 300 mM NaCl, 10% glycerol, 2 mM 2-mercaptoethanol) supplemented with protease inhibitors cocktail setIII (Calbiochem), lysed by sonication and cleared by centrifugation. The cleared lysate was incubated with Talon metal affinity resin (Clontech) for 2 hr at 4°C. The resins were then washed with 30 vol. of lysis buffer-1 and the bound proteins were eluted with either 10 mM maltose (Sigma, M5885) or 250 mM imidazole (Sigma, I5513). The eluate was further purified by SEC on Superdex-200 column (GE Healthcare) pre-equilibrated in SEC buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol). Relevant fractions were concentrated in 50 kDa molecular mass cut-off Amicon ultra centrifugal filters (Millipore). GST-Brca22295-3177 (Brca2c) and GST-Brca22295-3020 (Brca2d) were expressed in E. coli BL21(C41) transformed with the respective expressing plasmids. Protein expression was induced by the addition of 0.2 mM IPTG at 20°C and cells were incubated overnight. Cell pellets were resuspended in lysis buffer-2 (50 mM Tris pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM EDTA and 1 mM DTT) supplemented with protease inhibitors cocktail setIII (Calbiochem), lysed by sonication and cleared by centrifugation. The cleared lysate was incubated with glutathione Sepharose 4B (GE Healthcare) for 2 hr at 4°C. The resin was then washed with 30 vol. lysis buffer-2 and the bound proteins were eluted with 10 mM reduced Glutathione in (50 mM Tris pH 8.0 150 mM NaCl). To cleave the GST-tag the beads were instead washed with PreScission buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM DTT, 1 mM EDTA), resuspend in an equal volume of PreScission buffer, and incubated overnight at 4°C with 0.1 mg/ml PreScission protease (produced in Cogentech, Milan). The eluate was further purified by SEC on Superdex-200 column (GE Healthcare) pre-equilibrated in SEC buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol). Relevant fractions were concentrated in 30 kDa molecular mass cut-off Amicon ultra centrifugal filters (Millipore). Double StrepII-tagged human Polymerase α (1-1462) and B subunit (1-598) were cloned into the pFBDM vector, which was subsequently used to generate recombinant baculovirus using the MultiBac system. The same procedure was followed to produce a baculovirus for the expression of full-length human Primase (His10-tagged PriS and PriL). Protein expression was performed by co-infecting Sf9 insect cells at a density of 2x106 cells/ml with baculoviruses encoding Pol α – B and Primase, and shaking the cells at 120 rpm for 72 hr at 27°C. An N-terminally truncated version of the complex, comprising double StrepII-tagged Polymerase α (334-1462) and B subunit (149-598), was also cloned into pFBDM and expressed with full-length Primase in the same way. Pol α complexes were purified using Strep-Tactin Superflow resin (IBA). The N-terminal region of human Polymerase α (1-109) was cloned into the pMAT11 vector (http://camelot.bioc.cam.ac.uk/∼marko/vector/data/pmat.pdf), which encodes an N-terminal, TEV-cleavable His6-MBP tag. The protein was expressed in Rosetta2 (DE3) cells, and purified by Ni-NTA affinity chromatography and size exclusion chromatography (25 mM Na-HEPES pH 7.2, 160 mM KCl).
Human Rad51 was overexpressed in bacterial cells from pET-11c plasmid. Rad51T131P was generated by site-directed mutagenesis (QuickChange, Agilent genomics) using primers listed in the Key Resources Table. The correct sequence was verified by sequencing. All purification steps were carried out at 4°C. Cells were lysed by sonication in cell-breakage buffer (50 mM Tris pH 7.5, 10% sucrose, 10 mM EDTA, 1 mM DTT and 0.01% NP40) containing 1M KCl and protease inhibitors (2 μg/ml aprotinin, 5 μg/ml benzamidine, 10 μM chymostatin, 10 μM leupeptin and 1 μM pepstatin A). The crude extract was clarified by ultracentrifugation (100 000 x g, 1 h) and the supernatant was subjected to ammonium sulfate precipitation at 0.242 g/ml. The precipitate was resuspended in buffer K (20 mM K2HPO4, 10% glycerol, 0.5 mM EDTA) supplemented with protease inhibitors and loaded on 35 mL Q-Sepharose column. Proteins were eluted with a 350 mL gradient of 100–700 mM KCl in buffer K and then loaded onto a 5 mL hydroxyapatite (HAP, Bio-Rad) column and eluted with a 50 mL gradient of 0–600 mM KH2PO4 in buffer T (20 mM Tris-HCl (pH 7.5), 10% glycerol, 1 mM EDTA). Fractions containing Rad51 were dialyzed for 1 hr against buffer T supplemented with 50 mM KCl and then loaded on 1 mL MonoQ column (GE Healthcare) and eluted with 10 mL gradient of 100-600 mL KCl in buffer T. Fractions containing Rad51 were diluted in buffer T, loaded on 1 mL MonoS column (GE Healthcare) and eluted with 10 mL gradient of 100-500 mL KCl in buffer T. The peak fractions were pooled, concentrated and stored at −80°C in small aliquots.
Human RPA was overexpressed in bacterial cells from pET11d plasmid and purified by the same protocol described previously for yeast RPA (Sigurdsson et al., 2001). Briefly, soluble cell extract was loaded on 20 mL Affi-Gel Blue agarose column and the protein was eluted with 120 mL gradient of 0.5-2.5 M NaSCN in buffer T. Fractions containing RPA were dialyzed against 2 l of buffer T for 3 hr and loaded on 5 mL HAP column. RPA was eluted with 50 mL gradient of 10-140 mM KH2PO4 in buffer T. Eluate fractions were loaded on 1 mL MonoQ column and eluted with 10 mL gradient of 50-300 mM KCl in buffer T. Fractions containing RPA were pooled, concentrated and stored at −80°C.
Yeast Mre11 purification was performed as previously shown (Chavdarova et al., 2015). Briefly, Saccaromyces cerevisiae cells expressing Mre11 were lysed and subjected to ammonium sulfate precipitation at 0.28 g/ml. The precipitate was dissolved in buffer K supplemented with protease inhibitors and loaded on 7 mL Q-Sepharose column. Mre11 was eluted with a 200 mL gradient of 100–1000 mM KCl in buffer K and loaded onto a 1 mL HAP column. Proteins were eluted with a 10 mL gradient of 0–400 mM KH2PO4 in buffer K. Fractions containing Mre11 were loaded on a 1 mL MonoS column and eluted with a 10 mL gradient of 100–1000 mM KCl in buffer K. Peak fractions containing Mre11 were pooled, concentrated and stored at −80°C.
Human Mre11-6xHis protein was produced by overexpression in Sf9 insect cells infected by recombinant Mre11-6xHis baculovirus provided by Peter Cejka (University of Zurich, Switzerland). Cell pellet was resuspended in buffer P (50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 1 mM β-mercaptoethanol, 1 mM EDTA) supplemented with protease inhibitors, stirred for 30 min and clarified by ultracentrifugation (1 hr at 100 000 x g). Soluble extract was incubated with pre-equilibrated Ni-NTA agarose resin (QIAGEN) for 1 hr with continuous mixing. Resin was washed with buffer P and bound proteins were eluted with buffer P supplemented with 250 mM imidazole. The fractions containing MRE11 were diluted in buffer T, loaded on 1 mL MonoQ column and the protein was eluted with 10 mL of 100-1000 mM KCl in buffer T. The peak fractions were stored at −80°C in small aliquots.
Egg extract and chromatin binding
Xenopus interphase egg extracts and sperm nuclei were prepared as previously described (Aze et al., 2016). Briefly, Xenopus eggs were collected in MMR buffer (5 mM K-HEPES pH7.5, 100 mM NaCl, 0.5 mM KCl, 0.25 mM MgSO4, 0.5 mM CaCl2, 25 μM EDTA) from chorionic gonadotropin injected female frogs. The eggs were de-jellied in 10 mM Tris pH8.0, 110 mM NaCl and 5 mM DTT and rinsed three times in MMR. De-jellied eggs were released in interphase in presence of 5 μM Calcium Ionophore (Sigma) for 5-6 min, washed three times with MMR and rinsed twice in ice cold S-buffer (50 mM K-HEPES pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 2 mM β-mercaptoethanol). Activated eggs were then packed by centrifugation at 1200 x rpm for one minute and the excess of buffer was discarded. Eggs were crushed at 13000 x rpm for twelve minutes at 4°C. The crude extract was collected and centrifuged at 70.000 x rpm for 12 min at 4°C in a TLA100 rotor (Beckman). The interphase extract was obtained by collecting and mixing the cleared cytoplasmic fraction together with the nuclear membranes. For sperm nuclei preparation 4 testis were removed from 2 male frogs and placed in petri dishes containing 10 mL EB buffer (50 mM KCl, 50 mM HEPES KOH pH7.6, 5 mM MgCl2, 2 mM DTT). Testis were finely chopped with razor blade. The material was then transferred to 15 mL Falcon tube and spun at 2,000 x g, in a swinging bucket rotor for 5 min at 4°C. The pellet was resuspended in a total volume of 2 mL of room temperature SuNaSp buffer (0.25 M sucrose, 75 mM NaCl, 0.5 mM spermidine, 0.15 mM spermine). To remove membranes 100 μL of 2 mg/ml lysolecithin (Sigma) were added and incubated for 10 min at room temperature. Reaction was stopped by adding 3% BSA (Sigma). The pellet was resuspended again in 2 mL EB and spun at 2,000 x g for 5 min at 4°C. The final pellet was resuspended in 400 μL of EB + 30% glycerol. Sperm nuclei were tested for absence of DNA breaks with TUNEL assay as previously described (Aze et al., 2016). Briefly, 20 μL of different sperm nuclei preparations (4000 n/μl) were incubated at 37°C for 4 hr in 170 μL H2O supplemented with 20 μL 10 x TdT buffer (NEB), 90 U Terminal transferase (NEB) and 1 μL 32P-dCTP. Aliquots of the reaction were then precipitated with 5% TCA, 2% pyrophosphate solution and spotted on Whatman GF-C glass fiber filter. After ethanol washes, filters were dried and the incorporated TCA precipitable radioactivity was counted in scintillation counter. Sperm nuclei preparations with the lowest counts were used for all the experiments. For DNA replication assays sperm nuclei (4000 n/μl) were added to interphase egg extract treated as shown in Figure legend. Extracts were supplemented with 32P-dCTP, incubated at 23°C for the times indicated in figure legend and then stopped with Stop buffer (1% SDS, 8 mM EDTA, 80 mM Tris-HCl pH 8 and 1 mg/ml proteinase K). The mixture was then incubated at 50°C for 2 hr. Aliquots of replication reactions precipitated with 5% TCA, 2% pyrophosphate solution and spotted on Whatman GF-C glass fiber filter. After ethanol washes, filters were dried and the incorporated TCA precipitable radioactivity was counted in scintillation counter and quantified as previously shown (Aze et al., 2016).
For replication fork restart sperm DNA (4000 n/μl) were incubated in 50 μL mock-treated or Rad51-depleted extracts. After nuclei addition (60 min) extracts were supplemented with H-APH and incubated for additional 60 min. Chromatin was then isolated and transferred to mock-treated or Rad51-depleted restarting extracts supplemented with geminin and roscovitine. DNA replication was quantified by α-32P-dCTP DNA incorporation for 120 as described above.
For chromatin binding 40 μL egg extract containing sperm DNA were isolated from master reactions treated as shown in Figure legends at the indicated time points. For immunoblotting, samples were diluted with 10 volumes of EB (100 mM KCl, 2.5 mM MgCl2, and 50 mM HEPES–KOH pH 7.5) containing 0.25% NP-40 and centrifuged through a 0.5 M sucrose layer at 10000 x g at 4°C for 5 min. Pellets were washed once with EB and suspended in Laemmli loading buffer. Proteins were then resolved on a SDS-PAGE and monitored by WB.
Nuclease protection
Fluorescently labeled DNA substrates were prepared by annealing as described elsewhere (Marini and Krejci, 2012). Briefly, equimolar amounts of the corresponding oligonucleotides were mixed in hybridizing buffer (50 mM Tris, 100 mM NaCl, and 10 mM MgCl2), heated to 75°C for 3 min and cooled slowly to room temperature for annealing. The substrates were then purified by HPLC using a 1 mL Mono Q column (GE Healthcare Life Sciences) and a 20 mL gradient in 10 mM Tris buffer containing 1 M NaCl. The purity was checked on native PAGE. The corresponding fractions were then concentrated on a concentrator (Vivaspin) and washed with buffer W (25 mM Tris and 3 mM MgCl2). The concentrations were determined using the absorbance at 260 nm and the corresponding molar extinction coefficients. For the nuclease protection assays, DNA substrate (20 nM) was incubated with Rad51WT, Rad51T131P or RPA in the P buffer (80 mM KCl, 10 mM Tris-HCl pH 7.5, 1 mM DTT, 10 μg/ml BSA, 2 mM MnCl2, 2 mM MgCl2 and 2 mM ATP) for 5 min at 37°C followed by incubation with 30-50 nM recombinant yeast or human Mre11 as indicated in figure legend for 30 min at 30°C. Reactions were deproteinized by incubation with 0.1% SDS and 500 μg/ml of proteinase K at 37°C for 5 min, heat-denatured, analyzed on 30% denaturing PAGE gels (acrylamide:bisacrylamide, 19:1) and scanned using a Fuji FLA 9000 imager. Where indicated, gels were quantified using Multi Gauge V3.2 software (Fuji).
Mobility shift
Fluorescently labeled DNA substrate (20 nM) was incubated with the amounts of Rad51WT, Rad51T131P or RPA indicated in figure legend in the P buffer for 5 min at 37°C. Reactions were resolved on 0.8% agarose gel and scanned using a Fuji FLA 9000 imager.
Electron microscopy
DNA for electron microscopy analysis was processed as previously described with some modifications (Hashimoto et al., 2010). Briefly, for replication intermediate visualization sperm nuclei (4000 n/μl) were incubated at 23°C in 200 μl egg extract for 60 min, diluted with 400 μL of EB buffer, layered onto 800 μL EB-EDTA (EB buffer + 1 mM EDTA) + 30% (w/v) sucrose and centrifuged at 3,000xg for 10 min at 4°C. Pellets were resuspended in 100 μl EB-EDTA and transferred to a 96-well plate. 4,5′,8-Trimethylpsoralen (TMP) was added at 10 μg/ml to each well. Samples were incubated on ice for 5 min in the dark and irradiated with 365 nm ultraviolet light for 7 min on a precooled metal block. The procedure from TMP addition to irradiation with ultraviolet light was repeated three more times. Samples were then supplemented with 0.1% (w/v) SDS to lysate nuclei and treated with 100 μg/ml RNase A for 1h at 37°C. For complete protein digestion, psoralen-crosslinked chromatin was incubated with proteinase K (1 mg/ml) for 2 h at 50°C. Genomic DNA was extracted by adding one volume of 1:1 (v/v) phenol–chloroform mixture, precipitated with isopropanol, washed with 70% ethanol and processed for electron microscopy as previously shown (Hashimoto et al., 2010). Electron microscopy grid shadowing was done with a Leica MED20, and image acquisition with an FEI Tecnai 20 EM microscope equipped with a GATAN high-resolution camera at the IFOM electron microscopy facility. Blind analysis of EM images was performed by EM specialists.
Reagents
Aphidicolin was used at a concentration of 1.5 mM (H-APH), 20 μM (M-APH) or 3 μM (L-APH) as indicated in Figure legends. Roscovitin and geminin were used as previously described (Hashimoto et al., 2010). Briefly, interphase egg extracts were supplemented with 0.5 mM Roscovitine and 60 nM recombinant geminin to obtain restarting egg extracts, which were used to test the ability of partially replicated chromatin to resume replication as described above. Recombinant geminin was previously described (Aze et al., 2016). Brca2c and Brac2d were used at 50 ng/μl. Smarcal1 WT and HD protein were used at doses indicated in Figure legend. Mirin was used at 100 μM. MMS and UV pre-treatment of sperm nuclei has been previously described (Hashimoto et al., 2010). Briefly, sperm nuclei (40000 n/μl) were incubated with 1% (v/v) methyl methane sulfonate (MMS, Sigma) at 4°C for 2 hr or irradiated with 1,000 J/m2 ultraviolet (UV) light using a Stratalinker UV irradiator equipped with 254 nm UV light bulbs.
Nascent ssDNA detection
Nascent unpaired ssDNA in replicating egg extract was detected as previously shown with some modifications (Couch et al., 2013). Briefly, for each sample 40 μM BrdU (Sigma) and 10 μM biotin-16-dUTP (Roche) were added to 200 μL interphase egg extracts 10 min after addition of sperm nuclei (4000 n/μl). Extracts were mock-treated, Brca2- depleted, Rad51- or Smarcal1- depleted and supplemented with recombinant proteins, APH or mirin as described in Figure legends. To isolate nuclei reactions were stopped by dilution with 900 μL ice cold EB, layered onto 500 μl of EB plus 30% (w/v) sucrose and centrifuged at 3,000 x g for 10 min at 4°C. To stabilize replication structures and prevent branch migration DNA was psoralen-crosslinked. To this end pellets were resuspended in 100 μl EB and transferred to a 96-well plate (100 μl per well). 4,5,8-Trimethylpsoralen (TMP) was added at 10 μg/μl to each well. Samples were incubated for 5 min at 4°C in the dark and irradiated with 365 nm UV light for 7 min on a precooled metal block using a Stratalinker UV irradiator equipped with 365 nm light bulbs. The procedure from TMP addition to irradiation with ultraviolet light was repeated three more times. For complete protein digestion, psoralen-crosslinked chromatin was incubated with proteinase K (1 mg/ml) and RNase A (167 μg/ml1) for 2 h at 37°C in EB. Genomic DNA was extracted with one volume of 1:1 (v/v) phenol–chloroform mixture, precipitated with isopropanol and partially digested with restriction enzyme StuI, which generates genomic fragments with blunt ends in the context of AGG’CCT sequence. DNA was plated into 96 well streptavidin coated plates (Thermo Fisher, 15500), which were incubated with mouse monoclonal antibodies against BrdU (1:500) (Anti BrdU B44 BD Bioscience) in PBS-Triton 0.05% for 2 hr at 23°C to selectively detect nascent-strand ssDNA. Plates were washed three times with PBS-Triton 0.05% and probed with HRP labeled anti mouse IgGs (Dako). After washing chemoluminescence was detected by standard ECL using a plate reader.
Nascent DNA degradation
Sperm nuclei (4000 n/μl) were incubated in 250 μl egg extracts at 23°C. Extracts were mock-treated, Smarcal1-, Brca2-, Rad51- or Smarcal1 and Rad51 depleted as described in Figure legends. 45 min after nuclei addition extracts were supplemented with 40 μM biotin dUTP. 15 min after dUTP addition 1.5 mM aphidicolin was added. 50 μL samples were taken immediately after APH addition (0) and at 60, 120, 180 and 240 min after aphidicolin addition and supplemented with 250 μL Stop buffer. Samples were incubated with proteinase K (1 mg/ml) and RNase A (167 μg/ml) for 2 h at 37°C. Genomic DNA was extracted with phenol–chloroform, ethanol precipitated and further separated from free nucleotides by gel filtration on G50 columns (Amersham). DNA was fragmented by sonication, heat denatured, digested with 50 U Benzonase (Sigma) and processed for SensoLyte fluorescence biotin quantification assay (AnaSpec) according to the manufacturer’s instructions. Fluorescence was monitored using a Cary Eclipse fluorescence spectrophotometer.
Depletions and pull downs
To immuno-deplete Brca2, Rad51 and Smarcal1 0.5-1 mL egg extract were incubated with affinity purified IgGs (35-50 μg) at RT for 1 hr with 250 μl Dynabeads-ProteinA (Thermo Fisher, 10002D) for one to three subsequent depletion rounds lasting about 1 hr each. For Brca2 four rounds of depletion were required. For co-immunoprecipitations beads used for depletions were extensively washed and probed with antibodies as indicated in Figure legends.
To obtain mock-treated extract a parallel depletion was carried out using the same protocol with Dynabeads-ProteinA conjugated with affinity purified rabbit pre-immune IgGs.
For Pol α co-precipitation experiments shown in Figure 6B saturating quantities of full-length and truncated Polymerase α complexes were added to 30 μL Dynabeads-ProteinA pre-equilibrated in PD buffer (1xPBS, 5% glycerol, 0.5 mM TCEP, 0.2% Igepal). 2 × 1 mL washes in PD buffer supplemented with 350 mM NaCl, followed by 1 × 1 mL wash in PD buffer, were performed, after which 50 U Benzonase (EMD Millipore) was added. 400 μL 20 μM purified Rad51 in PD buffer was added, along with BSA (Sigma Aldrich) to a final concentration of 2.5 mg/ml. Samples were incubated at 4°C for 90 min with rolling, after which 4 × 1 mL washes were performed, followed by elution with 100 μL PD buffer supplemented with 300 mM imidazole. Samples were analyzed by SDS-PAGE with Coomassie blue staining. Western blotting was performed using the goat polyclonal anti-Rad51 antibody sc-6862 (Santa Cruz Biotechnology). For experiments in Figure 6C saturating quantities of purified, DNA-free His6-MBP-Polymerase α (1-109), or His6-MBP, were added to 100 μL Amylose resin (NEB) pre-equilibrated in PD buffer, and incubated at 4°C for 20 min. 2 × 1 mL washes in PD buffer were performed, after which 400 μL 20 μM purified Rad51 in PD buffer was added, along with BSA (Sigma Aldrich) to a final concentration of 2.5 mg/ml. Samples were incubated at 4°C for 90 min with rolling, after which 4 × 1 mL washes were performed, followed by elution with 100 μL PD buffer supplemented with 20 mM maltose. Samples were analyzed by SDS-PAGE with Coomassie blue staining.
For experiments in Figure 6D 2, 5, 10, 20 and 40 μM BRC4 peptide (Sigma), whose sequence has been described in the Key Resources Table, and 10 μM Rad51 were incubated at 37°C for 15 min in PBS, 0.2% Igepal, 1mM TCEP and 5% glycerol (Pull down buffer). Amylose resin was washed with PDB and incubated with 500 μL of 40 μM His-MBP-Polα NTD for an hour at 4°C. The resin was washed with pull down buffer and BRC4-Rad51 complex was added for another hour of incubation at 4°C. The resin was then washed and eluted in 100 μL of 50mM maltose in pull down buffer. Samples were then analyzed by SDS-PAGE with Coomassie blue staining.
Ipond
100 μl extracts were used for each sample. Sperm nuclei were then added to reach a final concentration of 4000 nuclei/μl. 45 min after sperm nuclei addition 10 min DNA labeling pulses were carried out supplementing the extracts with 40 μM Biotin-16-dUTP (Roche) and either 20 μM aphidicolin or DMSO as control. DNA replication was stopped by diluting 100 μL reactions with 200 μL cold EB-EDTA buffer (50 mM HEPES-KOH pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 1 mM EDTA). Samples were homogenized by using a cut p1000 tip and overlaid on 600 μL EB-EDTA-Sucrose buffer (EB-EDTA buffer + 30% w/v sucrose). Nuclei were collected by centrifugation at 8300 x g at 4°C for 10 min in a swinging-bucket rotor (TLA 100.3, Beckman). The supernatant and the dense sucrose layer were carefully removed and the nuclear pellet resuspended with 400 μL EB-NP40 buffer (50 mM HEPES-KOH pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.25% NP40) to lysate nuclei. Samples were then subjected twice to a 10 min sonication step (30 s ON / 40 s OFF cycle and Max Power with a Bioruptor device, Diagenode). After the sonication step 20 μL from each sample were kept apart (5% Input to be loaded as control for SDS-PAGE). Biotinylated DNA fragments were then pulled-down by incubation with 40 μL Dynabeads M-280 Streptavidin (Thermo Fisher, 11205D) for 30 min at 4°C. Dynabeads M-280 Streptavidin + the pull-down fractions were then washed three times with 200 μL EB-EDTA buffer and eventually resuspended with 30 μL of 1X denaturing loading buffer. The entire volume was eventually loaded on for SDS-PAGE and WB analysis.
Cloning
The cDNA sequences encoding Xenopus laevis Brca2 was obtained by RT-PCR, from RNA derived from Xenopus eggs with Trizol reagent (ThermoFisher). SuperscriptIII reverse transcriptase (Thermo Fisher) and an oligo(dT)20 was used for the first strand DNA synthesis. The full-length Brca2 sequence was amplified by PCR using a Phusion High-Fidelity DNA Polymerase (New England Biolabs) and Brca2-N1 and Brca2-C2 reverse primers (Key Resources Table) derived from full-length oocyte cDNA next generation sequence (unpublished data). The PCR product was cloned into pCRBluntIITOPO vector (Invitrogen) obtaining the ADA400 plasmid. Xenopus Brca2 cDNA was sequenced and the sequence was deposited in GenBank: KY024483. The sequences encoding the C-terminal Brca2 fragments Brca22295-3177 (Brca2c) and Brca22295-3020 (Brca2d) were amplified by PCR using the primers Brca2-Bgl-forw(2295), Brca2-Xho-rev(3177), Brca2-Xho-rev(3020) and sub-cloned in the first cassette of the dicistronic vector pGEX6p-2rbs (GenBank: KM817768), obtaining the plasmids ADA411 (for the expression of GST-Brca22295-3177) and ADA417 (for the expression of GST-Brca22295-3020). The cDNA encoding for Xenopus laevis Smarcal1 (GenBank: NM_001096199) cloned into pFastbac-HTb, was obtained from GenScript. WT and catalytic dead (HD, D549A E550A) mutant SMARCAL1 baculovirus vectors to express human Smarcal1 and expression protocols were previously described (Ciccia et al., 2012). All sequences were checked by DNA sequencing. All primers were purchased from Sigma.
Quantification and Statistical Analysis
Statistical analysis was performed with PRISM software and indicated in figure legend. Images shown represent typical results of experiments repeated at least three times.
Data and Software Availability
The accession number for the Xenopus BRCA2 sequence reported in this paper is NCBI GenBank: KY024483.
Raw images have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/xf5bvrr7gh.1.
Author Contributions
A.M.K., V.S., A.D.A., H.T., and G.B. performed most of the experiments. K.Z. produced the recombinant Rad51, RPA, and Mre11 proteins and performed the in vitro assays. M.K. and R.S. produced the polymerase complexes and performed the pull-down assays. A.C. provided essential reagents. L.P., L.K., and V.C. analyzed the data, coordinated the experiments, and wrote the manuscript.
Acknowledgments
We thank F. Pezzimenti for technical support with Xenopus laevis, J. Gannon for antibody production, M. Lopes for helpful discussions, and Giulia Rota for technical help. This work was funded by the Associazione Italiana per Ricerca sul Cancro (AIRC), European Research Council (ERC) Consolidator Grant 614541, the Armenise-Harvard Foundation career development award, Epigen Progetto Bandiera (4.7), AICR-Worldwide Cancer Research (13-0026), and Fondazione Telethon Grant GGP13-071 (to V.C.); by Wellcome Trust Investigator Award 104641/Z/14/Z (to L.P.); by the Czech Science Foundation (GACR 17-17720S and 13-26629S) and Project LQ1605 from the National Program of Sustainability II (MEYS CR) (to L.K.); and by NIH grant R01CA197774 and Susan G. Komen CCR grant CCR16377030 (to A.C.). V.S. is funded by a Fondazione Veronesi (FUV) personal postdoctoral fellowship.
Published: July 27, 2017
Footnotes
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.07.001.
Contributor Information
Luca Pellegrini, Email: lp212@cam.ac.uk.
Lumir Krejci, Email: lkrejci@chemi.muni.cz.
Vincenzo Costanzo, Email: vincenzo.costanzo@ifom.eu.
Supplemental Information
References
- Anand R., Ranjha L., Cannavo E., Cejka P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell. 2016;64:940–950. doi: 10.1016/j.molcel.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Aze A., Sannino V., Soffientini P., Bachi A., Costanzo V. Centromeric DNA replication reconstitution reveals DNA loops and ATR checkpoint suppression. Nat. Cell Biol. 2016;18:684–691. doi: 10.1038/ncb3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M., Vindigni A. Replication stress: getting back on track. Nat. Struct. Mol. Biol. 2016;23:103–109. doi: 10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Couch F.B., Mason A.C., Eichman B.F., Manosas M., Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun T.S., Pacek M., Yee M.C., Walter J.C., Cimprich K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J. Biol. Chem. 2015;290:22931–22938. doi: 10.1074/jbc.R115.675942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavdarova M., Marini V., Sisakova A., Sedlackova H., Vigasova D., Brill S.J., Lisby M., Krejci L. Srs2 promotes Mus81-Mms4-mediated resolution of recombination intermediates. Nucleic Acids Res. 2015;43:3626–3642. doi: 10.1093/nar/gkv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Symington L.S. Stressing out about RAD52. Mol. Cell. 2016;64:1017–1019. doi: 10.1016/j.molcel.2016.11.036. [DOI] [PubMed] [Google Scholar]
- Ciccia A., Bredemeyer A.L., Sowa M.E., Terret M.E., Jallepalli P.V., Harper J.W., Elledge S.J. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V., Robertson K., Ying C.Y., Kim E., Avvedimento E., Gottesman M., Grieco D., Gautier J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell. 2000;6:649–659. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- Couch F.B., Bansbach C.E., Driscoll R., Luzwick J.W., Glick G.G., Bétous R., Carroll C.M., Jung S.Y., Qin J., Cimprich K.A., Cortez D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J., Hanawalt P.C. RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 2003;37:611–646. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- Davies O.R., Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007;14:475–483. doi: 10.1038/nsmb1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X., Ray Chaudhuri A., Callen E., Pang Y., Biswas K., Klarmann K.D., Martin B.K., Burkett S., Cleveland L., Stauffer S. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016;7:12425. doi: 10.1038/ncomms12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A., Costanzo V., Hunt T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc. Natl. Acad. Sci. USA. 2007;104:14929–14934. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A., Aze A., Costanzo V. Mta2 promotes Tipin-dependent maintenance of replication fork integrity. Cell Cycle. 2014;13:2120–2128. doi: 10.4161/cc.29157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F., Galkin V.E., Yu X., Egelman E.H., West S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- Flynn R.L., Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011;36:133–140. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M., Zwicky K., Vanoli F., Lopes M., Branzei D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 Complex. Mol. Cell. 2015;57:812–823. doi: 10.1016/j.molcel.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Costanzo V. Studying DNA replication fork stability in Xenopus egg extract. Methods Mol. Biol. 2011;745:437–445. doi: 10.1007/978-1-61779-129-1_25. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Puddu F., Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2011;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs M.R., Reynolds J.J., Winczura A., Blackford A.N., Borel V., Miller E.S., Zlatanou A., Nieminuszczy J., Ryan E.L., Davies N.J. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell. 2015;59:462–477. doi: 10.1016/j.molcel.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Holloman W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011;18:748–754. doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M., Mattair D., Lin H., Gutierrez-Barrera A.M., Elsayegh N., Litton J.K., Hortobagyi G.N., Arun B. Identifying genomic rearrangements in BRCA1 and BRCA2 in high-risk individuals for hereditary breast and ovarian cancer. J. Clin. Oncol. 2011;29:163. [Google Scholar]
- Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21:8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- Jensen R.B., Carreira A., Kowalczykowski S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyasekharan A.D., Ayoub N., Mahen R., Ries J., Esposito A., Rajendra E., Hattori H., Kulkarni R.P., Venkitaraman A.R. DNA damage regulates the mobility of Brca2 within the nucleoplasm of living cells. Proc. Natl. Acad. Sci. USA. 2010;107:21937–21942. doi: 10.1073/pnas.1009577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Paulus B.F., Wold M.S. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski S.C. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2015;7:a016410. doi: 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M. Electron microscopy methods for studying in vivo DNA replication intermediates. Methods Mol. Biol. 2009;521:605–631. doi: 10.1007/978-1-60327-815-7_34. [DOI] [PubMed] [Google Scholar]
- Lopes M., Cotta-Ramusino C., Pellicioli A., Liberi G., Plevani P., Muzi-Falconi M., Newlon C.S., Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- Marini V., Krejci L. Unwinding of synthetic replication and recombination substrates by Srs2. DNA Repair (Amst.) 2012;11:789–798. doi: 10.1016/j.dnarep.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M., Khodaverdian V.Y., Meyer D., Cerqueira P.G., Heyer W.D. Eukaryotic DNA polymerases in homologous recombination. Annu. Rev. Genet. 2016;50:393–421. doi: 10.1146/annurev-genet-120215-035243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam E.A., Cortez D. ATR signalling: more than meeting at the fork. Biochem. J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L. The Pol α-primase complex. Subcell. Biochem. 2012;62:157–169. doi: 10.1007/978-94-007-4572-8_9. [DOI] [PubMed] [Google Scholar]
- Pellegrini L., Costa A. New insights into the mechanism of DNA duplication by the eukaryotic replisome. Trends Biochem. Sci. 2016;41:859–871. doi: 10.1016/j.tibs.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y., Antony E., Doganay S., Koh H.R., Lohman T.M., Myong S. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat. Commun. 2013;4:2281. doi: 10.1038/ncomms3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Schärer O.D., Walter J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A., Callen E., Ding X., Gogola E., Duarte A.A., Lee J.E., Wong N., Lafarga V., Calvo J.A., Panzarino N.J. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M., Zelensky A., Smal I., Meijering E., van Cappellen W.A., de Gruiter H.M., van Belle G.J., van Royen M.E., Houtsmuller A.B., Essers J. BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J. Cell Biol. 2014;207:599–613. doi: 10.1083/jcb.201405014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannino V., Kolinjivadi A.M., Baldi G., Costanzo V. Studying essential DNA metabolism proteins in Xenopus egg extract. Int. J. Dev. Biol. 2016;60:221–227. doi: 10.1387/ijdb.160103vc. [DOI] [PubMed] [Google Scholar]
- Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K., Wu H., Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S., Trujillo K., Song B., Stratton S., Sung P. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J. Biol. Chem. 2001;276:8798–8806. doi: 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- Simon A.C., Sannino V., Costanzo V., Pellegrini L. Structure of human Cdc45 and implications for CMG helicase function. Nat. Commun. 2016;7:11638. doi: 10.1038/ncomms11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu B.M., McDonald W.H., Dungrawala H., Badu-Nkansah A., Kavanaugh G.M., Chen Y., Tabb D.L., Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013;288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies J., Waizenegger A., Barton O., Sürder M., Wright W.D., Heyer W.D., Löbrich M. Nek1 regulates Rad54 to orchestrate homologous recombination and replication fork stability. Mol. Cell. 2016;62:903–917. doi: 10.1016/j.molcel.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L.H., Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001;477:131–153. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- Thorslund T., West S.C. BRCA2: a universal recombinase regulator. Oncogene. 2007;26:7720–7730. doi: 10.1038/sj.onc.1210870. [DOI] [PubMed] [Google Scholar]
- Toledo L.I., Murga M., Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol. Oncol. 2011;5:368–373. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenz K., Smith E., Smith S., Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van C., Yan S., Michael W.M., Waga S., Cimprich K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010;189:233–246. doi: 10.1083/jcb.200909105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.T., Kim T., Wagner J.E., Conti B.A., Lach F.P., Huang A.L., Molina H., Sanborn E.M., Zierhut H., Cornes B.K. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol. Cell. 2015;59:478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Jeffrey P.D., Miller J., Kinnucan E., Sun Y., Thoma N.H., Zheng N., Chen P.L., Lee W.H., Pavletich N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.