

Summary
DNA damage tolerance during eukaryotic replication is orchestrated by PCNA ubiquitination. While monoubiquitination activates mutagenic translesion synthesis, polyubiquitination activates an error-free pathway, elusive in mammals, enabling damage bypass by template switching. Fork reversal is driven in vitro by multiple enzymes, including the DNA translocase ZRANB3, shown to bind polyubiquitinated PCNA. However, whether this interaction promotes fork remodeling and template switching in vivo was unknown. Here we show that damage-induced fork reversal in mammalian cells requires PCNA ubiquitination, UBC13, and K63-linked polyubiquitin chains, previously involved in error-free damage tolerance. Fork reversal in vivo also requires ZRANB3 translocase activity and its interaction with polyubiquitinated PCNA, pinpointing ZRANB3 as a key effector of error-free DNA damage tolerance. Mutations affecting fork reversal also induced unrestrained fork progression and chromosomal breakage, suggesting fork remodeling as a global fork slowing and protection mechanism. Targeting these fork protection systems represents a promising strategy to potentiate cancer chemotherapy.
Keywords: DNA damage tolerance, replication fork reversal, replication fork progression, cancer chemotherapeutics, PCNA ubiquitination, postreplication repair, ZRANB3 DNA translocase, electron microscopy in vivo, single-molecule approaches
Graphical Abstract
Highlights
-
•
Fork slowing and reversal upon damage require K63-linked PCNA polyubiquitination
-
•
ZRANB3 mediates fork slowing/reversal in vivo via binding to polyubiquitinated PCNA
-
•
ZRANB3 DNA translocase—not nuclease—activity mediates fork slowing and reversal
-
•
Mammalian error-free postreplication repair entails global fork slowing and reversal
Vujanovic et al. show that UBC13-mediated, K63-linked PCNA polyubiquitination mediates DNA damage-induced replication fork slowing and reversal, via recruitment to forks of ZRANB3 DNA translocase. These data link the postreplication repair pathway, yet elusive in mammals, to the regulation of fork progression and remodeling.
Introduction
Replicating cells react to genotoxic stress activating different molecular pathways, devoted to regulate origin firing and to protect the stability of ongoing replication forks (Berti and Vindigni, 2016). Replication completion in the presence of DNA lesions is assisted by the activation of the so-called “post-replication repair” (PRR) pathway, which is modulated in eukaryotic cells by controlled ubiquitination of the DNA polymerase clamp, i.e., proliferating cellular nuclear antigen (PCNA) (Branzei and Psakhye, 2016, García-Rodríguez et al., 2016). Accumulation of single-stranded DNA (ssDNA) at replication forks facing DNA lesions triggers recruitment of the E2-E3 pair RAD6-RAD18, mediating PCNA mono-ubiquitination on the K164 residue (Hoege et al., 2002, Niimi et al., 2008). This modification has been linked to the recruitment of translesion synthesis (TLS) polymerases, promoting error-prone DNA damage bypass at the expense of increased mutations rates (Bienko et al., 2005, Plosky et al., 2006). In yeast, further modification of the same PCNA residue via K63-linked polyubiquitination—which requires the dimeric E2 MMS2/UBC13 and the Rad5 E3 ligase (Hoege et al., 2002)—promotes error-free PRR, an alternative DNA damage tolerance pathway that fills postreplicative ssDNA gaps via template-switching (TS) and recombinational mechanisms, involving sister chromatid junctions (Branzei and Psakhye, 2016, Giannattasio et al., 2014). Alternative models for error-free PRR and TS entail remodeling of the replication fork in a four-way junction—a process known as replication fork reversal—to allow TS to occur directly at the elongating fork (Higgins et al., 1976). However, replication fork reversal in yeast cells has only been observed upon fork stalling in replication or checkpoint mutants (Fumasoni et al., 2015, Sogo et al., 2002) or upon topological stress induced by Topoisomerase I (Top1) poisons (Ray Chaudhuri et al., 2012), questioning the physiological role of fork reversal in this organism.
In mammals, detection of PCNA polyubiquitination proved more difficult and has so far required acute genotoxic treatments and/or overexpression of the responsible enzymes (Brun et al., 2010, Motegi et al., 2008). Rad5 has two related proteins in human cells—HLTF and SHPRH—both contributing to PCNA polyubiquitination (Motegi et al., 2008, Unk et al., 2010), possibly assisting the response to different types of DNA damage (Lin et al., 2011). A third E3 ligase has also been invoked (Krijger et al., 2011). Whether PCNA polyubiquitination directly promotes TS and/or regulates TLS has long been controversial (García-Rodríguez et al., 2016). Also, no direct data are currently available on whether error-free PRR in higher eukaryotes mostly entails post-replicative junctions or fork reversal. Intriguingly, besides their E3 ligase activity, both Rad5 in yeast and HLTF in human cells possess specific domains capable of reversing replication forks in vitro (Blastyák et al., 2007, Kile et al., 2015), although their contribution to fork reversal in vivo is currently uncertain.
Recent visual inspection of mammalian replication intermediates in vivo has uncovered replication fork reversal as a global and genetically controlled response to various challenges to the replication process. These include oncogene activation, unstable repetitive sequences, and treatments with various genotoxins (Follonier et al., 2013, Neelsen et al., 2013, Ray Chaudhuri et al., 2012, Zellweger et al., 2015). These transient structures were proposed to exert a protective role upon replication stress, but to date only a few factors have been directly implicated in their formation, stabilization, and restart (Neelsen and Lopes, 2015). Importantly, genetic defects in reversed fork formation or stabilization upon genotoxic treatments were also shown to impair active replication fork slowing, thus linking controlled fork progression and fork remodeling upon replication stress (Ray Chaudhuri et al., 2012, Zellweger et al., 2015). PCNA ubiquitination was shown to be dispensable for continued fork progression upon UV damage in DT40 cells (Edmunds et al., 2008), but its potential contribution to actively remodel and slow down replication forks in mammalian cells has not been thoroughly investigated.
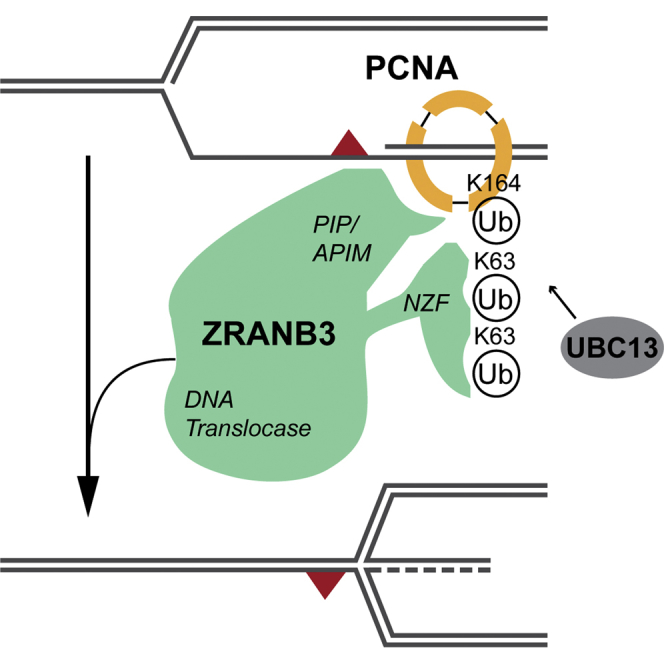
A number of proteins, mostly belonging to RECQ helicase or SWI/SNF protein families, are able to reverse forks in biochemical assays (Neelsen and Lopes, 2015). HARP/SMARCAL1 and AH2/ZRANB3 DNA translocases can re-anneal RPA-coated DNA strands (Yusufzai and Kadonaga, 2008, Yusufzai and Kadonaga, 2010) and reverse synthetic replication forks (Bétous et al., 2012, Bétous et al., 2013, Ciccia et al., 2012), but their contribution to fork reversal in vivo is elusive. ZRANB3 was shown to associate with replication factories, to assist restart of stalled forks and to mildly contribute to genotoxin resistance (Ciccia et al., 2012, Yuan et al., 2012). These functions require the DNA translocase activity of ZRANB3, as well as its binding to PCNA—via PIP and APIM domains—and to polyubiquitinated PCNA—via its NZF domain (Ciccia et al., 2012, Yuan et al., 2012). ZRANB3 carries also a structure-specific endonuclease activity in the HNH domain (Weston et al., 2012), but its functional relevance is currently unclear.
Here, combining DNA fiber analysis and electron microscopy (EM) visualization of replication intermediates in vivo, we provide evidence that PCNA polyubiquitination mediates active fork slowing and reversal upon genotoxic treatments. Moreover, we report that a known “reader” of this modification—the translocase ZRANB3—regulates fork speed, fork remodeling, and chromosome integrity in vivo, via its DNA translocase activity and its ability to interact with polyubiquitinated PCNA.
Results
PCNA Ubiquitination Mediates Active Fork Slowing and Reversal upon Genotoxic Stress
In order to assess whether PCNA ubiquitination is required to actively reduce replication fork speed upon replication stress, we investigated by DNA fiber spreading replication fork progression in PCNA-K164R mouse embryonic fibroblasts (MEFs) and their wild-type counterparts (Langerak et al., 2007). We combined incorporation of halogenated nucleotides and optional treatments with nanomolar doses of the Top1 inhibitor camptothecin (CPT) or the DNA crosslinking agent mitomycin C (MMC) (Zellweger et al., 2015). Both treatments induced a significant slowdown of replication fork progression, but this response was abolished by the K164R PCNA mutation (Figures 1A and 1B). Thus, PCNA ubiquitination is strictly required to mediate active fork slowing upon these genotoxic treatments.
Figure 1.

PCNA Ubiquitination Is Required for Replication Fork Slowing and Reversal upon Genotoxic Stress
(A) Labeling scheme of DNA fiber experiments: cells were provided with chlorodeoxyuridine (CldU, red). 30 min later, cells were washed and supplemented with iododeoxyuridine (IdU, green) and optionally treated with camptothecin (CPT) 50 nM and/or mitomycin C (MMC) 200 nM for 30 min. Green tracks were measured to assess fork speed.
(B) Control and PCNA-K164R mouse embryonic fibroblasts (MEFs) were subjected to the DNA fiber protocol described in (A). At least one hundred tracts were scored per sample. Whiskers: 10th–90th percentile (∗∗∗p < 0.001; ns, non-significant; Mann-Whitney test). Similar results were obtained in at least two biological replicates.
(C) Representative electron microscopy images of reversed (left) or normal (right) replication forks. P, parental strand; D, daughter strand; R, regressed arm.
(D) Frequency of reversed forks in the indicated MEFs, upon optional 1 hr treatment with CPT 50 nM or MMC 200 nM, assessed by EM visualization. Similar results were obtained in two biological replicates and in independent MEF clones (Tables S1A and S1B).
We next used psoralen crosslinking coupled to EM (Zellweger and Lopes, 2017) to investigate in vivo replication fork architecture and to reveal the possible conversion of standard replication forks into four-way junctions—known as reversed forks (Figure 1C)—previously associated with a variety of genotoxic treatments (Ray Chaudhuri et al., 2012, Zellweger et al., 2015). As expected, reversed fork frequency in wild-type MEFs was markedly increased by CPT and MMC treatments. Remarkably, PCNA-K164R MEFs did not significantly increase reversed fork levels upon both genotoxic treatments (Figure 1D, Table S1A), showing that PCNA ubiquitination is required for effective replication fork reversal. Similar DNA fiber and EM results were obtained upon CPT treatment in independent lines of wild-type and PCNA-K164R MEFs (Figures S1A and S1B, Table S1B).
In order to visualize endogenous PCNA modifications occurring upon different genotoxic treatments, we performed cell fractionation to enrich for polyubiquitinated species of PCNA (Figure S1C). This approach enabled us to detect endogenous levels of polyubiquitinated PCNA, which were induced upon acute UV irradiation (Figure S1D), as previously reported (Motegi et al., 2008). Albeit technically challenging, using this protocol we detected low levels of polyubiquitinated PCNA also upon nanomolar doses of CPT or MMC, as those typically used for our DNA fiber and EM experiments (Figure S1D).
K63-Linked, UBC13-Dependent Polyubiquitination Is Required for Fork Slowing and Reversal upon Genotoxic Stress
In order to distinguish whether PCNA mono- or poly-ubiquitination mediates the observed fork slowing and reversal upon genotoxic stress, we took advantage of a previously characterized ubiquitin replacement system in U2OS cells (Xu et al., 2009). Doxycycline addition allows the replacement of endogenous ubiquitin with similar levels of exogenous wild-type or K63R mutant ubiquitin and does not impair overall cell-cycle progression (Figures S2A and S2B). DNA fiber experiments revealed that impairing K63-linked ubiquitin chain formation by the K63R mutation significantly affects CPT-induced replication fork slowing (Figure 2A). Moreover, replacement of endogenous ubiquitin with the K63R mutant markedly reduced the frequency of fork reversal upon CPT and MMC treatment (Figure 2B, Table S2A). Thus, K63-linked polyubiquitination is essential for efficient fork remodeling upon genotoxic stress.
Figure 2.

K63-Linked, UBC13-Dependent Polyubiquitination Is Required for Drug-Induced Fork Slowing and Reversal
(A) Cells conditionally (+Dox) replacing endogenous ubiquitin with a K63R ubiquitin mutant were subjected to the DNA fiber protocol as in Figure 1A. The ratio between green and red tracts is plotted, to display drug-induced fork slowing.
(B) Frequency of replication fork reversal in cells replacing endogenous ubiquitin with WT or K63R-ubiquitin, upon optional 1 hr treatment with CPT 50 nM or MMC 200 nM, assessed by EM visualization. In brackets, the number of analyzed molecules. Similar results were obtained in two biological replicates (Table S2A).
(C) Wild-type (WT) or UBC13-knockout (UBC13-KO) HCT116 cells were subjected to the DNA fiber protocol in Figure 1A.
(D) The same cell lines as in (C) were used for DNA fiber analysis, upon optional 5 J/m2 UV-C irradiation in between the two labeling periods. Top right: the western blot shows the absence of UBC13 in UBC13-KO HCT116 cells. GAPDH, loading control. In (A), (C), and (D) at least one hundred tracts were scored per sample. Whiskers: 10th–90th percentile (∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ns, non-significant; Mann-Whitney test). Similar results were obtained in at least two biological replicates.
(E) Frequency of replication fork reversal in WT and UBC13-KO HCT116 cells, assessed by EM visualization, upon optional 1 hr treatment with CPT 50 nM or MMC 200 nM, or 1 hr after 5 J/m2 UV-C irradiation. In brackets, the number of analyzed molecules. Similar results were obtained in two biological replicates (Table S2B).
In light of the reported redundancy of different E3 ligases mediating PCNA polyubiquitination (Krijger et al., 2011, Motegi et al., 2008), we next assessed the relevance of PCNA polyubiquitination by inactivation of the E2 enzyme required for this modification, i.e., UBC13 (García-Rodríguez et al., 2016). UBC13 knockout (KO) in HCT116 cells did not affect cell-cycle progression (Figure S2C) but abolished the reduction in replication fork speed upon CPT, MMC, and UV-C treatments (Figures 2C and 2D). Moreover, UBC13-KO cells failed to appreciably induce replication fork reversal, monitored by EM, upon all tested genotoxic treatments (Figure 2E; Table S2B). Similar results were obtained by siRNA-mediated downregulation of UBC13 in CPT- or MMC-treated U2OS cells (Figures S2D–S2F, Table S2C). Taken together, the data presented so far strongly suggest that PCNA polyubiquitination is required to mediate active fork slowing and fork reversal upon different genotoxic treatments.
The ZRANB3 Translocase Is Required for Fork Slowing and Reversal upon Different Genotoxic Treatments
As the DNA translocase ZRANB3 was reported as a specific interactor of polyubiquitinated PCNA and was shown to mediate replication fork reversal in biochemical assays (Ciccia et al., 2012), we directly tested its requirement for replication fork slowing and reversal in vivo. Therefore, a ZRANB3 knockout U2OS cell line generated by CRISPR/Cas9 technology—which did not display altered cell-cycle progression (Figure S3A)—was compared to its wild-type counterpart for replication fork slowing by DNA fiber assays and for fork reversal by EM, upon treatment with CPT, MMC, and UV-C. As observed upon impairment of UBC13-dependent K63-linked polyubiquitination (Figure 2), ZRANB3-KO cells displayed unrestrained fork progression in response to all tested treatments, with fork slowing being completely abolished upon CPT, MMC, and UV treatments (Figures 3A and 3B). Moreover, ZRANB3-KO cells displayed unaffected frequencies of fork reversal in unperturbed conditions but were unable to efficiently promote replication fork reversal upon all tested genotoxic treatments (Figure 3C, Table S3A). Similar effects were observed by DNA fiber spreading and EM analysis of another ZRANB3-KO clone and upon siRNA-mediated downregulation of ZRANB3 in U2OS cells (Figures S3A–S3F, Tables S3B and S3C).
Figure 3.

ZRANB3 Is Required for Efficient Replication Fork Slowdown and Fork Reversal upon Different Genotoxic Treatments
(A) Wild-type (WT) or ZRANB3-knockout (ZRANB3-KO) U2OS cells were subjected to the DNA fiber protocol as in Figure 1A.
(B) The same cell lines as in (A) were used for DNA fiber analysis, upon optional 5 J/m2 UV-C irradiation in between the two labelings. Top right: the western blot shows the absence of ZRANB3 in ZRANB3-KO U2OS cells. β tubulin, loading control. In (A) and (B), at least one hundred tracts were scored per sample. Whiskers: 10th–90th percentile (∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.1; ns, non-significant; Mann-Whitney test). Similar results were obtained in at least two biological replicates.
(C) Frequency of replication fork reversal in WT and ZRANB3-KO U2OS cells, assessed by EM visualization, upon optional 1 hr treatment with CPT 50 nM or MMC 200 nM, or 1 hr after 5 J/m2 UV-C irradiation irradiation. In brackets, the number of analyzed molecules are shown. Similar results were obtained in two biological replicates and in two independent ZRANB3-KO clones (Tables S3A and S3B).
Damage-Induced Fork Slowing and Reversal Protect Chromosome Integrity and Require ZRANB3-PCNA Interaction and ZRANB3 DNA Translocase Activity
ZRANB3 contains multiple domains and motifs, which mediate its enzymatic activities or the interaction with PCNA and its ubiquitinated forms (Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012). To assess the relevance of these activities and interactions in replication fork slowing and reversal in vivo, we analyzed specific point mutations in ZRANB3 (Figure 4A), affecting respectively its interaction with PCNA (PIP+APIM domains), its interaction with polyubiquitinated PCNA (NZF-zinc finger), its DNA translocase activity (DEXDc domain; helicase dead [HD]) (Ciccia et al., 2012), or a crucial residue of the HNH domain, which was shown to provide ZRANB3 nuclease activity (Weston et al., 2012). We obtained stable cell lines by viral transduction of ZRANB3-KO U2OS cells, re-expressing FLAG/HA-tagged wild-type ZRANB3 or one of these mutant forms. We ensured that all tagged proteins were expressed at approximately the level of endogenous ZRANB3 in the original U2OS cell line (Figure 4A) and that none of the mutant cell lines had marked delays in cell-cycle progression (Figure S4A). Using these cell lines, we assessed whether wild-type and mutant forms of ZRANB3 could complement the defects in fork slowing and reversal observed in ZRANB3-KO cells (Figure 3), focusing on CPT treatments. Expression of WT ZRANB3 in ZRANB3-KO cells restored effective CPT-induced fork slowing and reversal (Figures 4B and 4C, Table S4), showing that the tagged protein is functional upon genotoxic treatment. Expression of the PIP+APIM mutant failed to restore fork slowing and reversal, suggesting that ZRANB3-PCNA interaction is essential to regulate fork progression and remodeling upon damage. Similarly, mutations destabilizing the NZF zinc finger impaired CPT-induced fork slowing and reversal (Figures 4B and 4C, Table S4). A complete defect in fork slowing and reversal was also observed upon expression of the helicase dead (HD) mutant that impairs ZRANB3 DNA translocation activity, while a mutant that inactivates the HNH nuclease motif restored efficient control of fork progression and remodeling (Figures 4B and 4C, Table S4). Thus, effective interaction of ZRANB3 with both unmodified and polyubiquitinated PCNA and its DNA translocase activity, but not an intact HNH nuclease domain, are required for replication fork slowing and reversal upon genotoxic stress. In order to test whether ZRANB3-mediated fork slowing and reversal limit DNA damage-induced chromosomal instability, we assessed chromosomal abnormalities by metaphase spreads after CPT treatment. To exclude selection of compensatory mutations in ZRANB3-KO cells, we chose to transiently inactivate ZRANB3 by siRNA in cell lines stably expressing siRNA-resistant HA-tagged ZRANB3 variants that retain (WT, HNH) or impair (PIP-APIM) fork slowing and reversal activities (Figures 4A–4C; Figure S4B). ZRANB3 downregulation led to increased chromosomal abnormalities upon CPT treatment (Figure 4D). Notably, this increase was suppressed by the presence of WT and HNH mutant forms of ZRANB3, while the PIP-APIM ZRANB3 mutant—specifically defective in fork slowing and reversal—failed to restore chromosome integrity after CPT treatment. These data strongly suggest that ZRANB3-mediated fork slowing and reversal prevent chromosomal instability upon genotoxic treatments.
Figure 4.

Fork Progression, Fork Remodeling and Chromosomal Integrity Defects upon Inactivation of Different ZRANB3 Domains
(A) Left: schematic representation of ZRANB3 domain organization and of mutations analyzed in this study. Right: western blot analysis of ZRANB3 in the indicated cell lines.
(B) The indicated stable cell lines, expressing WT or mutant ZRANB3, were used for DNA fiber analysis as in Figure 1A, upon optional CPT 50 nM treatment. At least one hundred tracts were scored per sample. Whiskers: 10th–90th percentile (∗∗∗∗p < 0.0001; ns, non-significant; Mann-Whitney test). Similar results were obtained in at least two biological replicates.
(C) Frequency of replication fork reversal in the indicated cell lines, assessed by EM visualization, upon optional 1 hr treatment with CPT 50 nM. In brackets, the number of analyzed molecules. Similar results were obtained in two biological replicates (Table S4).
(D) Left: number of chromosomal abnormalities per indicated cell line, as determined by metaphase spreading upon optional 8 hr CPT treatment (50 nM) and 16 hr nocodazole treatment (200 ng/mL). Error bars, standard deviations. Right: representative DAPI stained metaphase; the arrow points to a chromosome break. Scale bar, 5 μm. Western blot analysis of ZRANB3 protein levels in U2OS cell lines used in (D, left). In (A) and (D), the expression level of HA-tagged ZRANB3 WT and mutant proteins (retarded mobility) is close to endogenous ZRANB3 levels in U2OS cells. β tubulin, loading control.
Discussion
Our data establish the genetic dependency of replication fork slowing and reversal on PCNA ubiquitination, UBC13, and K63-linked polyubiquitination—all of which are known to mediate the error-free PRR pathway in mammalian cells (García-Rodríguez et al., 2016)—thus showing that activation of this pathway entails global fork slowing and reversal in response to a variety of genotoxic treatments. In agreement with the prevalent occurrence of error-free PRR at replication forks in higher eukaryotes, in human cells we failed to detect significant accumulation of postreplicative junctions—under similar experimental conditions that were previously successfully used to visualize and characterize TS postreplicative intermediates in S. cerevisiae cells (Giannattasio et al., 2014)—even upon acute treatments and genetic stabilization of these structures (Figures S4C–S4E). We thus propose that—differently from yeast cells—activation of the error-free PRR pathway in human cells leads to extensive fork remodeling, transiently limiting fork progression on the damaged template. In turn, genetic inactivation of fork remodeling in error-free PRR mutants causes unrestrained fork progression, occasionally leading to chromosomal breaks, visible in the next mitosis. The choice between fork reversal and postreplicative error-free PRR likely reflects differences in the efficiency of re-priming in higher versus lower eukaryotes, the abundance of repetitive DNA in human cells and the different control of several key players in these pathways (Branzei and Psakhye, 2016, Sale, 2012).
By cell fractionation, we were able to enrich and detect endogenous levels of polyubiquitinated PCNA upon acute UV treatment, as previously reported (Motegi et al., 2008). Albeit technically challenging (Niimi et al., 2008), in the same experimental conditions PCNA polyubiquitination could also be detected upon sublethal (nanomolar) MMC and CPT treatments. Importantly, while extensive TS is expected at UV-induced lesions, CPT- and MMC-induced DNA lesions are expected to delay template unwinding and should not extensively involve DNA damage bypass by TS. We propose that activation of this branch of the PRR likely results from a common molecular feature detected at replication forks upon all tested genotoxic treatments—i.e., ssDNA accumulation (Zellweger et al., 2015)—which reportedly promotes the recruitment of the E3 ligases for PCNA ubiquitination (Niimi et al., 2008) and may thus mediate replication fork remodeling even in the absence of a DNA lesion specifically requiring TS. This scenario is in agreement with the surprising evidence that reversed fork frequency in human cells is not significantly dependent on the type and dose of genotoxic treatments (Ray Chaudhuri et al., 2012, Zellweger et al., 2015). In this context, fork reversal should be considered as a general fork protection mechanism, which actively delays global fork progression and promotes DNA damage tolerance when required. The mechanisms underlying the global remodeling of replication forks from local DNA damage at a subset of forks certainly deserve further investigation.
In addition, our data uncover the key role in active fork slowing and reversal of a known interaction partner of polyubiquitinated PCNA, i.e., the DNA translocase ZRANB3. This protein was shown to contribute in human cells to genome maintenance upon genotoxic treatments and to the restart of stalled replication forks, via its DNA translocase activity and its multiple interactions with unmodified and polyubiquitinated PCNA (Ciccia et al., 2012, Yuan et al., 2012). Our data reveal the dependency on the same activity and domains for DNA damage-induced replication fork slowing and reversal, strongly suggesting that the key role of ZRANB3 in the replication stress response entails recruitment to replication forks to assist fork remodeling, in keeping with its known biochemical properties (Ciccia et al., 2012). Besides formation of reversed forks, ZRANB3 may also mediate reversed fork accumulation by controlling their stability, preventing unscheduled restart or processing. Interestingly, binding to polyubiquitinated PCNA is not required for ZRANB3 recruitment, but rather for its retention at replication factories (Ciccia et al., 2012), which may suggest its involvement in modulating fork restart. Our DNA fiber, EM, and chromosomal analyses of different ZRANB3 mutants reinforce the tight association between fork reversal, active fork slowing, and chromosome stability in human cells. Inactivation of the ZRANB3 nuclease domain (HNH) had no visible impact on fork slowing or reversal and, in keeping with our model, did not increase chromosomal breakage upon CPT treatment. Alternative mechanistic roles for ZRANB3 nuclease activity in response to replication stress will require further investigation.
We noted that the low levels of replication fork reversal consistently observed in unperturbed cells were not significantly affected by ZRANB3 depletion or inactivation, possibly suggesting that endogenous impediments to fork progression lead to fork remodeling via ZRANB3-independent mechanisms. However, this may also reflect the functional redundancy of other members of the same family—such as SMARCAL1 or RAD54—possibly providing fork reversal activities when ZRANB3 is inactive. Similarly, despite the marked reduction in fork reversal that we observed in ZRANB3-defective cells upon all tested genotoxic treatments, it is well possible that other translocases play a role in fork remodeling in response to specific types of replication interference, as suggested by additive contributions to cell survival and fork restart upon genotoxic treatments (Ciccia et al., 2012, Yuan et al., 2012). The functional analysis of this redundancy and/or damage specificity would require extensive EM analysis, upon simultaneous inactivation of several members of this protein family.
As the E3 ligases responsible for PCNA polyubiquitination—Rad5 in yeast and HLTF, among others, in human cells—were shown to possess fork reversal activities in biochemical assays (Blastyák et al., 2007, Kile et al., 2015), it will be important to clarify whether these activities stimulate fork reversal in vivo and how they are possibly coordinated with additional enzymatic activities recruited to replication forks via binding to polyubiquitinated PCNA (i.e., ZRANB3). Similarly, the central recombinase RAD51 was shown to mediate fork reversal in vivo (Zellweger et al., 2015). Understanding the mechanistic cross-talk between translocase and strand exchange activities in driving fork reversal will require complex biochemical reconstitution of this transaction, possibly including chromatinized substrates. Moreover, as additional factors have been proposed to bind polyubiquitinated PCNA in yeast and human cells (Saugar et al., 2012), it will be important to test their potential contribution to fork slowing and reversal upon different genotoxic treatments.
Overall, this study contributes to our mechanistic understanding of active fork slowing and remodeling upon genotoxic treatments. As these fork protection mechanisms are expected to provide resistance to cancer chemotherapeutic treatments acting via DNA damage, further elucidation of the underlying mechanisms will be required to identify promising targets to potentiate cancer chemotherapy.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ZRANB3 | Proteintech | Cat# 23111-1-AP |
| UBC13 | Invitrogen | Cat# 371100; RRID: AB_2533298 |
| Rabbit polyclonal anti-B tubulin | Santa Cruz Biotechnology | Cat# sc-9104; RRID: AB_2241191 |
| Rabbit monoclonal anti-Ub K164 PCNA | Cell Signaling | Cat# 13439S |
| Mouse monoclonal anti-PCNA | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_628110 |
| Mouse monoclonal anti-GAPDH | Millipore | Cat# MAB374; RRID: AB_2107445 |
| Rabbit polyclonal anti-BLM | Abcam | Cat# ab476; RRID: AB_304596 |
| Rabbit polyclonal anti-TFIIH p89 (S-19) | Santa Cruz Biotechnology | Cat# sc-293; RRID: AB_2262177 |
| Donkey anti-rabbit IgG-HRP | GE HealthCare | Cat# NA934V |
| Donkey anti-mouse IgG-HRP | GE HealthCare | Cat# NA931V |
| Mouse anti-BrdU/IdU | Becton Dickinson | Cat# 347580; RRID: AB_400326 |
| Rat anti-BrdU/CldU | Abcam | Cat# ab6326; RRID: AB_305426 |
| Goat anti-mouse Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11001; RRID: AB_2534069 |
| Donkey Anti-rat Cy3 | Jackson ImmunoResearch | Cat# 712-166-153; RRID: AB_2340669 |
| Rabbit anti-Lamin A (C-terminal) | Sigma-Aldrich | Cat# L1293 RRID: AB_532254 |
| Mouse anti-ubiquitin (clone P4D1) | Santa Cruz Biotechnology | Cat# sc-8017 RRID: AB_628423 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Camptothecin | Sigma-Aldrich | Cat# C991 |
| Mitomycin C | Sigma-Aldrich | Cat# M0503 |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| Methyl methanesulfonate | Sigma-Aldrich | Cat# M4016 |
| Aphidicolin | Sigma-Aldrich | Cat# A0781 |
| Nocodazole | Sigma-Aldrich | Cat# M1404 |
| Chloroquine diphosphate salt | Sigma-Aldrich | Cat# C6628 |
| Hexadimethrine bromide - Polybrene | Sigma-Aldrich | Cat# 107689 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| N-Ethylmaleimide | Sigma-Aldrich | Cat# E1271 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat# 13778-500 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Cat# H-1200 |
| cis-Diammineplatinum(II) dichloride | Sigma-Aldrich | Cat# P4934 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8340 |
| Proteinase K, recombinant, PCR Grade | Sigma-Aldrich | Cat# 03115852001 |
| ECL Advance Blocking Reagent | GE HealthCare | Cat# RPN418V |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| EcoRI | NEB | Cat# R0101S |
| 5-Chloro-2′-deoxyuridine | Sigma-Aldrich | Cat# C6891 |
| Etoposide | Sigma-Aldrich | Cat# E1383 |
| Hydrogen peroxide solution | Sigma-Aldrich | Cat# 349887 |
| Doxorubicin | Sigma-Aldrich | Cat# D1515 |
| 5-Iodo-2′-deoxyuridine | Sigma-Aldrich | Cat# I7125 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10425 |
| Deposited Data | ||
| Raw imaging data | This paper | http://dx.doi.org/10.17632/bj4ggp8b65.1 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | HTB-96 |
| HEK293T | ATCC | CRL-11268 |
| Phoenix-AMPHO | ATCC | CRL-3213 |
| U2OS ZRANB3 knock out (clone 35) | David Cortez lab | N/A |
| U2OS ZRANB3 HNH mutant (from ZRANB3 KO clone 35) | This paper | N/A |
| U2OS ZRANB3 HD mutant (from ZRANB3 KO clone 35) | This paper | N/A |
| U2OS ZRANB3 PIP+APIM mutant (from ZRANB3 KO clone 35) | This paper | N/A |
| U2OS ZRANB3 NZF mutant (from ZRANB3 KO clone 35) | This paper | N/A |
| U2OS ZRANB3 wild type (from ZRANB3 KO clone 35) | This paper | N/A |
| HCT116 | ATCC | CCL-247 |
| HCT116 Ubc13 knock out | Niels Mailand lab | Thorslund et al., 2015 |
| MEFs PCNA wild type (clone 2976) | Heinz Jacobs lab | N/A |
| MEFs PCNA wild type (clone 2977) | Heinz Jacobs lab | N/A |
| MEFs PCNA 164K164R/K164R mutant (clone 2978) | Heinz Jacobs lab | N/A |
| MEFs PCNA 164K164R/K164R mutant (clone 2979) | Heinz Jacobs lab | N/A |
| Oligonucleotides | ||
| siCtrl: CGUACGCGGAAUACUUCGAdTdT | Microsynth | N/A |
| UBC13 siRNA: AAUGGCAGCCCCUAAAGUAdTdT | Microsynth | N/A |
| BLM siRNA: CCGAAUCUCAAUGUACAUAGA dTdT | Microsynth | N/A |
| ZRANB3 siRNA: siGENOME siRNA D-010025-03-005 | Dharmacon | Cat# 84083 |
| Recombinant DNA | ||
| pML113 | This lab | Follonier et al., 2013 |
| pMSCV-FLAG-HA-ZRANB3 WT | Alberto Ciccia lab | N/A |
| pMSCV-FLAG-HA-ZRANB3 PIP+APIM (Q519A, I522A, F525A, F526A, T1071X) | Alberto Ciccia lab | N/A |
| pMSCV-FLAG-HA-ZRANB3 NZF-zinc (C644A, C641A) | Alberto Ciccia lab | N/A |
| pMSCV-FLAG-HA-ZRANB3 HD (D157A, E158A) | Alberto Ciccia lab | N/A |
| pMSCV-FLAG-HA-ZRANB3 HNH (H1043L) | Alberto Ciccia lab | N/A |
| Software and Algorithms | ||
| GraphPad Prism6 for Mac OS X | GraphPad Software | https://www.graphpad.com/ |
| ImageJ (used for DNA fibers and EM data) | ImageJ Software | https://imagej.nih.gov/ij/ |
| FlowJo (FACS data analysis) | FlowJo Software | https://www.flowjo.com/ |
| Attune NxT (FACS data analysis) | Attune NxT Software | https://www.thermofisher.com/ |
| FusionCapt Advance Solo 7 17.02 control and analysis software for chemiluminescence detection (used for western blot) | Vilber Lourmat | http://www.vilber.de/ |
| Other | ||
| Digital Radiometer (used for UV irradiation measurements) | UVP, Upland, CA | Model: UVX Digital Radiometer. Seral No.: E 29127 |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact, Prof. Massimo Lopes (lopes@imcr.uzh.ch).
Experimental Model and Subject Details
Source of cell lines used in the study is reported in the Key Resources Table.
Method Details
Cell culture and cell lines
Human osteosarcoma U2OS and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS, GIBCO), 100 U/ml penicillin and 100 μg/ml streptomycin in an atmosphere containing 6% CO2 at 37°C. Cells were treated with different cancer chemotherapeutics and DNA damaging agents as indicated, trypsinized, and processed for cell cycle analysis, western blots, and EM DNA extraction.
U2OS ZRANB3 knock out (KO) cells were generated in David Cortez’ lab using CRISPR-Cas9 technology. Cells were transfected with pSpCas9(BB)-2A-Puro (Addgene plasmid #48139) containing guide RNAs targeting the first exon (AGCTTTGCTCTTAGTCTGTCAGG, TTTTTTATGTTATGAACCCTAGG) and selected with 2 μg/ml puromycin for two days prior to plating for individual clones. Gene editing was confirmed by PCR (primers: TTGCTTTCAAACTCAGTGCTTT and TGGATAAAGCTAACTTGGTCACAAT) and immunoblotting.
U2OS wild-type or ZRANB3-KO cells were cultured at 37°C (6% CO2) in DMEM supplemented with 7.5% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin.
Human HCT116 cell lines were cultured at 37°C (6% CO2) in McCoy’s (5A) Modified Medium (26600-023, GIBCO) supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin.
U2OS cells carrying different ubiquitin mutants were kept in media without tetracycline until the expression was needed. Then 2 μg/ml of doxycycline was added to the growth media to induce expression.
The mutant ZRANB3 constructs were generated by site directed mutagenesis of pMSCV-FLAG-HA-ZRANB3 (Ciccia et al., 2012). The following mutants were utilized in this study: (1) PIP+APIM (Q519A, I522A, F525A, F526A, T1071X); (2) NZF-zinc (C644A, C641A); (3) HD (D157A, E158A); (4) HNH (H1043L).
Generation of stable cell lines expressing exogenous ZRANB3
Phoenix retrovirus producer cells were transfected (grown in DMEM media with 10% FBS and 1% Glutamine) with retroviral vectors containing HA-tagged WT or mutant ZRANB3 (PIP+APIM, NZF-zinc, HD or HNH). 1 h before transfection we added Chloroquine (final concentration of 20 μm) and then added the transfection mixture (H2O, 10 μg plasmid DNA and 2 M CaCl2). 8 h post transfection fresh media was added. 48 h post transfection supernatant from Phoenix cells containing viruses was collected. Polybrene (8 mg/ml) was added to the supernatant, and it was used as a media for target cells to be infected. 3 h later the infection procedure was repeated. After the second infection procedure, the media was changed to usual growth media (DMEM, 7.5% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin). 48 h post infection cells were kept in selection media containing puromycin (3 μg/ml) for few days until cells from an uninfected control group had died. Successful integration in the infected cells lead to survival, while the cells with no integration died. Surviving cells were collected and amplified in selection media to obtain the stable cell lines. Exogenous ZRANB3 protein levels were assessed by western blot using an antibody directed against ZRANB3 and compared to endogenous ZRANB3 levels in wild-type U2OS.
Transfections
For siRNA experiments, cells were transfected with the indicated siRNAs using RNAiMAX (Invitrogen) according to manufacturer’s instruction. Experiments were performed 60 hours post transfection.
Drugs and reagents
Camptothecin was dissolved in dimethyl sulfoxide (DMSO) to yield a 20mM stock (7mg/ml). Mitomycin C (M0503, Sigma-Aldrich, St. Louis, MO, USA) was prepared in ddH2O to obtain a 3mM stock (1mg/ml). UV irradiation was administered using an ultraviolet 254 nm lamp. The intensity of irradiation was measured with digital radiometer (UVP. Upland. CA).
Western blotting
Intracellular protein levels were determined by western blot analysis of whole cell extracts. Mammalian cell extracts were prepared in Laemmli sample buffer (4% SDS, 20% glycerol, 120 mM Tris-HCl pH 6.8). 20-60 μg of total protein from cell isolates were loaded onto 6%–12% casted SDS-gels, or gradient 4%–15% SDS-gels. Proteins were separated electrophoretically at 12 mA (for 1 gel; 2 gels at 24 mA) for 15-30 min and then at 18 mA until the end (for 1 gel; 2 gels at 36 mA) followed by transferring (wet-transfer) of the proteins on nitrocellulose blotting membranes (Immobilon-P membrane, RPN303D, Fisher Scientific) in buffer containing 20% methanol and 80% 1x transfer buffer (transfer buffer 10x: 25 mM Tris, 192 mM glycine, 10% methanol), either for 2 h (100 V, 4°C) or overnight (30 V, 4°C). Prior to addition of primary antibodies, membranes were blocked for 1 h in 1x Tris-buffered saline (TBS) containing Tween20 (0.1%) and 2% ECL blocking solution (GE Healthcare). Membranes were incubated with appropriate primary antibodies either overnight at 4°C or for 3-4 h at RT. Secondary antibodies were added in blocking solution for 1 h at RT. Membranes were washed 3 times with 0.1% TBST (10 min each) after incubation with primary and secondary antibodies. The membrane was then exposed to an enhanced chemiluminescent system (detection reagent final volume equivalent to 0.125 ml/cm2 membrane, GE Healthcare), and a charge-coupled device image analyzer was used to visualize immunoreactive bands (Fusion SOLO chemiluminescence imaging system, Vilber Lourmat). For ubiquitin immunoblotting, U2OS cells were lysed in RIPA buffer (50 mM Tris PH 7.4, 150 mM NaCl, 1% TRITON x 100, 1% deoxycholate, 0.1% SDS) containing protease inhibitors (cat. No. P8340 Sigma), 1 mM PMSF, 10 mM NaF, 2 mM N-ethyl maleimide (NEM), sodium pyrophosphate. 10 μg of the protein extracts were separated onto gradient 4%–15% casted SDS-gel and wet-transferred on PVDF membrane (Immobilon-PSQ membrane, 0.2 μm, cat. no. ISEQ00010). After transfer, the membrane was immediately incubated in a denaturing solution containing 6 M guanidinium chloride, 20 mM Tris-HCl pH 7.4, 10 mM PMSF and 5 mM 2-mercaptoethanol (30 min at 4°C). The membrane was then washed extensively with TBS-Tween20 (0.1%), saturated minimum 6 hr in TBS-BSA (5%) and incubated with anti-ubiquitin antibody (i.e., clone P4D1, diluted 1:1000 in TBS-BSA 5%) for 1 hr. After extensive washes in TBS-Tween, the membrane was incubated with secondary antibody as usual.
Chromatin fractionation to detection endogenous ubiquitinated PCNA
Treated cells were solubilized in SB1 buffer [150 mM NaCl, 50 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.1% Triton X-100, protease inhibitors, 1 mM PMSF, 1 mM sodium orthovanadate, 10 mM NaF, 2 mM N-ethyl maleimide (NEM)], cell extracts (fraction S1) were collected by centrifugation. This extraction was repeated twice. The pellet was then resuspended in SB2 buffer (50 mM Tris-HCl pH 8.0, 1 mM MgCl2, 1 mM PMSF, 1 mM sodium orthovanadate, 10 mM NaF, 2 mM NEM), sonicated and incubated with benzonase and MNase for 30 min at 30°C. This extraction was repeated twice. Cell extracts were then collected by centrifugation (fraction S2) and the pellet (P) was solubilized in 2.5% SDS at 95°C.
Antibodies
The following primary antibodies were used for western blotting: ZRANB3 (23111-1-AP, Proteintech, 1:600 dilution), Beta tubulin (sc-9104, Santa Cruz Biotechnology, 1:1000 dilution), GAPDH (MAB374, Millipore, 1:20000 dilution, provided by A. Sartori, IMCR, Zurich), Ub K164 PCNA (13439S, Cell Signaling, 1:1000 dilution), UBC13 (#4919, Cell Signaling Technology, 1:1000 dilution; 371100, Invitrogen, 1:1000 dilution), PCNA (sc-56; Santa Cruz Biotechnology, 1:1000 dilution), Lamin A (L1293, Sigma, 1:1000 dilution). Secondary antibodies used for western blotting were anti-rabbit and anti-mouse ECL (GE Healthcare, 1:2500 dilution).
FACS analysis of cell cycle progression
Asynchronously growing U2OS cells (40%–60% confluency) were labeled for 30 min with 10 μM EdU. Cells were trypsinized, harvested, and fixed for 15 min with 4% formaldehyde/PBS. Then the cells were washed with 1% BSA/PBS, pH 7.4, permeabilized with 1% saponin/1% BSA/PBS. Incorporated EdU was labeled according to the manufacturer’s instructions (#C35002; Invitrogen). DNA was stained with 1 μg/ml DAPI. Samples were measured using Attune NxT flow cytometer (ThermoFisher Scientific) and analyzed with FlowJo software.
Replication fork progression by DNA fiber analysis
The procedure was essentially carried out as previously described (Ray Chaudhuri et al., 2012). Briefly, asynchronously growing U2OS cells were labeled with 30 μM chlorodeoxyuridine (CldU, Sigma-Aldrich), a thymidine analog, for 30 min, washed twice with PBS, treated with appropriate dosage with any of the genotoxic agents (or non-treated as control) and exposed to 250 μM IdU. The cells were quickly trypsinized and resuspended in PBS at 2.5 × 105 cells per ml. The labeled cells were diluted 1:5 with unlabeled cells, and 2.5 μL of cells were mixed with 7.5 μL of lysis buffer (200 mM Tris-HCl (pH 7.5), 50 mM EDTA, 0.5% (w/v) SDS) on a glass slide. After 9 min, the slides were tilted at 15°-45°, and the resulting DNA spreads were air-dried, fixed in 3:1 methanol/acetic acid and refrigerated overnight. The DNA fibers were denatured with 2.5 M HCl for 1 h, washed with PBS and blocked with 2% BSA in PBST for 40 min. The newly replicated CldU and IdU tracks were labeled (for 2.5 h in the dark, at RT) with anti-BrdU/CldU antibodies recognizing CldU (ab6326, Abcam, rat) and BrdU/IdU (347580, Becton Dickinson, mouse), respectively. After washing 5x3min in PBST (0.2%) the following secondary antibodies were used (incubated for 1h in the dark, at RT): anti-mouse Alexa 488 (Molecular Probes), anti-rat Cy3 (Jackson Immunoresearch). After washing 5x3 min in PBST (0.2%) the slides were air-dried completely, mounted with 20 uL/slide Antifade Gold (Invitrogen), and sealed to a coverslip by transparent nail polish. Microscopy was carried out with an Olympus IX81 fluorescence microscope and acquired with a CCD camera (Orca AG, Hamamatsu) or Leica DMRB equipped with a camera (model DFC360 FX, Leica). The images were processed with CellR software (Olympus) or Leica Application Suite 3.3.0. Statistical analysis of tract length was performed using GraphPad Prism. The significance of the difference between the means was determined by Student’s t test.
Electron microscopic analysis of genomic DNA
The procedure was performed as recently described (Zellweger and Lopes, 2017), with minor modifications described below. Following the depletion of the protein of interest, asynchronous subconfluent cells were treated with 25 nM CPT for 1 h or 4 mM HU for 5 h. Where indicated, cells were pretreated with 50 μM Mirin for 1h. Cells were collected, resuspended in PBS and crosslinked with 4,5′, 8-trimethylpsoralen (10 μg/ml final concentration), followed by irradiation pulses with UV 365 nm monochromatic light (UV Stratalinker 1800; Agilent Technologies). For DNA extraction, cells were lysed (1.28 M sucrose, 40 mM Tris-HCl [pH 7.5], 20 mM MgCl2, and 4% Triton X-100; QIAGEN) and digested (800 mM guanidine–HCl, 30 mM Tris–HCl [pH 8.0], 30 mM EDTA [pH 8.0], 5% Tween-20, and 0.5% Triton X-100) at 50°C for 2 h in presence of 1 mg/ml proteinase K. The DNA was purified using chloroform/isoamylalcohol (24:1) and precipitated in 0.7 volume of isopropanol. Finally, the DNA was washed with 70% EtOH and resuspended in 200 μL TE (Tris-EDTA) buffer. 100 U of restriction enzyme (PvuII high fidelity, New England Biolabs) were used to digest 12 μg of mammalian genomic DNA for 4-5 h. Replication intermediates enrichment was performed by QIAGEN Plasmid Mini Kit columns. The QIAGEN-tip 20 surface tension was reduced by applying 1 mL QBT buffer. The columns were washed and equilibrated with 10 mM Tris-HCl (pH 8.0), 1 M NaCl, followed by 10 mM Tris-HCl (pH 8.0), 300 mM NaCl, respectively. DNA was then loaded onto the columns. The columns were then washed with high NaCl solution (10 mM Tris-HCl [pH 8.0] and 900 mM NaCl) and eluted in caffeine solution (10 mM Tris-HCl [pH 8.0], 1 M NaCl, and 1.8% [w/v] caffeine). To purify and concentrate the DNA an Amicon size-exclusion column was used. DNA was then resuspended in TE buffer. The Benzyldimethylalkylammonium chloride (BAC) method was used to spread the DNA on the water surface and then load it on carbon-coated 400-mesh copper grids. Subsequently, DNA was coated with platinum using a High Vacuum Evaporator MED 020 (BalTec). Microscopy was performed with a transmission electron microscope (Tecnai G2 Spirit; FEI; LaB6 filament; high tension ≤ 120 kV) and picture acquisition with a side mount charge-coupled device camera (2,600 × 4,000 pixels; Orius 1000; Gatan). For each experimental condition at least 70 replication fork molecules were analyzed. DigitalMicrograph version 1.83.842 (Gatan) and ImageJ (National Institutes of Health) were used to process and analyze the images.
Chromosomal breakage and abnormalities by metaphase spreading
After transfection with the indicated siRNAs, cells were treated for 8 h with 50 nM CPT. The compound was washed off three times with 1 × PBS, upon which cells were released into fresh medium containing 200 ng/ml nocodazole for 16 h. Cells were harvested and swollen with 75 mM KCl for 20 min at 37°C. Swollen mitotic cells were collected and fixed with methanol:acetic acid (3:1). The fixing step was repeated two times. Fixed cells were dropped onto pre-hydrated glass slides and air-dried overnight. The following day, slides were mounted with Vectashield medium containing DAPI. Images were acquired with a microscope (model DMRB; Leica) equipped with a camera (model DFC360 FX; Leica) and visible chromatid breaks/ gaps were counted.
Quantification and Statistical Analysis
For DNA fiber experiments at least one hundred tracts were scored per sample. Every experiment was repeated at least twice. The results were analyzed using GraphPad Prism6 for Mac OS X, using Mann – Whitney test. Whiskers: 10-90th percentile (∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.1; ns, non-significant). Every EM experiment was repeated twice and at least 70 molecules per sample were analyzed (see Tables S1–S4).
Data and Software Availability
Original imaging data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/bj4ggp8b65.1.
Author Contributions
M.V. performed and analyzed all EM, DNA fibers, western blot, and FACS experiments, with technical assistance of J.K., R.Z., and R.H. N.T. generated important preliminary data. M.C.R. produced the PCNA ubiquitination data and assisted J.K. for the metaphase spreads. J.A.S. performed DNA fiber experiments and FACS analyses in the ubiquitin replacement systems. K.Z. performed all 2D gel experiments on postreplicative junctions. H.J. produced and provided PCNA-K164R MEFs. C.L.H and D.C. produced and provided the ZRANB3-KO U2OS cells. A.T., J.-W.H., and A.C. designed, produced, and characterized biochemically the ZRANB3 mutants. L.P. supervised the detection of ubiquitinated PCNA. M.L. designed and supervised the project and wrote the manuscript, assisted by L.P.
Acknowledgments
We thank the Center for Microscopy and Image Analysis of the University of Zurich for technical assistance with electron microscopy. We are grateful to Z.J. Chen for sharing the ubiquitin replacement system, to M. Gatti and K. Mutreja for technical assistance, and to all members of the Lopes group for discussions. This work was supported by the SNF grants 31003A_146924 and 31003A_169959 and the ERC Consolidator Grant 617102 to M.L., by the SNF grant 31003A_166370 and the Helmut Horten grant to L.P., by the NIH grant R01CA197774 to A.C., by the NIH grant GM116616 to D.C. and by the KWF grant NKI-2012-5243 and the ZonMW Top grant 91213018 to H.J.
Published: September 7, 2017
Footnotes
Supplemental Information includes four figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.08.010.
Supporting Citations
The following references appear in the Supplemental Information: Follonier and Lopes, 2014, Wu and Hickson, 2003.
Supplemental Information
References
- Berti M., Vindigni A. Replication stress: getting back on track. Nat. Struct. Mol. Biol. 2016;23:103–109. doi: 10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Couch F.B., Mason A.C., Eichman B.F., Manosas M., Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M., Green C.M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A.R. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Blastyák A., Pintér L., Unk I., Prakash L., Prakash S., Haracska L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell. 2007;28:167–175. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D., Psakhye I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016;40:137–144. doi: 10.1016/j.ceb.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Brun J., Chiu R.K., Wouters B.G., Gray D.A. Regulation of PCNA polyubiquitination in human cells. BMC Res. Notes. 2010;3:85. doi: 10.1186/1756-0500-3-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds C.E., Simpson L.J., Sale J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Follonier C., Lopes M. Combined bidimensional electrophoresis and electron microscopy to study specific plasmid DNA replication intermediates in human cells. Methods Mol. Biol. 2014;1094:209–219. doi: 10.1007/978-1-62703-706-8_16. [DOI] [PubMed] [Google Scholar]
- Follonier C., Oehler J., Herrador R., Lopes M. Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat. Struct. Mol. Biol. 2013;20:486–494. doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- Fumasoni M., Zwicky K., Vanoli F., Lopes M., Branzei D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 Complex. Mol. Cell. 2015;57:812–823. doi: 10.1016/j.molcel.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez N., Wong R.P., Ulrich H.D. Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front. Genet. 2016;7:87. doi: 10.3389/fgene.2016.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M., Zwicky K., Follonier C., Foiani M., Lopes M., Branzei D. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014;21:884–892. doi: 10.1038/nsmb.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins N.P., Kato K., Strauss B. A model for replication repair in mammalian cells. J. Mol. Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Kile A.C., Chavez D.A., Bacal J., Eldirany S., Korzhnev D.M., Bezsonova I., Eichman B.F., Cimprich K.A. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol. Cell. 2015;58:1090–1100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger P.H.L., Lee K.-Y., Wit N., van den Berk P.C.M., Wu X., Roest H.P., Maas A., Ding H., Hoeijmakers J.H.J., Myung K., Jacobs H. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: existence of an alternative E3 ligase. DNA Repair (Amst.) 2011;10:438–444. doi: 10.1016/j.dnarep.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak P., Nygren A.O.H., Krijger P.H.L., van den Berk P.C.M., Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.R., Zeman M.K., Chen J.Y., Yee M.C., Cimprich K.A. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell. 2011;42:237–249. doi: 10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A., Liaw H.-J., Lee K.-Y., Roest H.P., Maas A., Wu X., Moinova H., Markowitz S.D., Ding H., Hoeijmakers J.H.J., Myung K. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA. 2008;105:12411–12416. doi: 10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen K.J., Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Neelsen K.J., Zanini I.M.Y., Herrador R., Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A., Brown S., Sabbioneda S., Kannouche P.L., Scott A., Yasui A., Green C.M., Lehmann A.R. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. USA. 2008;105:16125–16130. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plosky B.S., Vidal A.E., Fernández de Henestrosa A.R., McLenigan M.P., McDonald J.P., Mead S., Woodgate R. Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin. EMBO J. 2006;25:2847–2855. doi: 10.1038/sj.emboj.7601178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- Sale J.E. Competition, collaboration and coordination--determining how cells bypass DNA damage. J. Cell Sci. 2012;125:1633–1643. doi: 10.1242/jcs.094748. [DOI] [PubMed] [Google Scholar]
- Saugar I., Parker J.L., Zhao S., Ulrich H.D. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012;40:245–257. doi: 10.1093/nar/gkr738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo J.M., Lopes M., Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C., Bekker-Jensen S., Mailand N. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527:389–393. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- Unk I., Hajdú I., Blastyák A., Haracska L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst.) 2010;9:257–267. doi: 10.1016/j.dnarep.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Weston R., Peeters H., Ahel D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012;26:1558–1572. doi: 10.1101/gad.193516.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Hickson I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- Xu M., Skaug B., Zeng W., Chen Z.J. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell. 2009;36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Ghosal G., Chen J. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell. 2012;47:410–421. doi: 10.1016/j.molcel.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T., Kadonaga J.T. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T., Kadonaga J.T. Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proc. Natl. Acad. Sci. USA. 2010;107:20970–20973. doi: 10.1073/pnas.1011196107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R., Lopes M. Dynamic architecture of eukaryotic DNA replication forks in vivo, visualized by electron microscopy. In: Muzi-Falconi M., Brown G., editors. Genome Instability: Methods and Protocols. Springer Science+Business Media; 2017. [Google Scholar]
- Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.