

Abstract
Glutamine (Gln) is considered the preferred precursor for the neurotransmitter pool of glutamate (Glu), the major excitatory transmitter in the mammalian CNS. Here, an activity-regulated, high-affinity Gln transport system is described in developing and mature neuron-enriched hippocampal cultures that is potently inhibited by riluzole (IC50 1.3 +/− 0.5µM), an anti-glutamatergic drug, and is blocked by low concentrations of 2-(methylamino)isobutyrate (MeAIB), a system A transport inhibitor. K+-stimulated MeAIB transport displays an affinity (Km) for MeAIB of 37 +/− 1.2 µM, saturates at ~200 µM, is dependent on extracellular Ca2+, and is blocked by inhibition of voltage gated Ca2+-channels (VGCCs). Spontaneous MeAIB transport is also dependent on extracellullar Ca2+ and VGCCs, but is also blocked by the Na+ channel blocker tetrodotoxin, by Glu receptor antagonists, and by GABA indicating its dependence on intact neural circuits driven by endogenous glutamatergic activity. The transport of MeAIB itself does not rely on Ca2+, but on Na+ ions, and is pH-sensitive. Activity-regulated, riluzole-sensitive spontaneous and K+-stimulated transport is minimal at 7–8 days in vitro (DIV), coordinantly induced during the next two weeks, and is maximally expressed by DIV>20; the known period for maturation of the Glu/Gln cycle and regulated presynaptic Glu release. Competition analyses with various amino acids indicate that Gln is the most likely physiological substrate. Activity-regulated Gln/MeAIB transport is not observed in astrocytes. The functional identification of activity-regulated, high-affinity, riluzole-sensitive Gln/MeAIB transport in hippocampal neurons may have important ramifications in the neurobiology of activity stimulated presynaptic Glu release, the Glu/Gln cycle between astrocytes and neurons, and neuronal Glu-induced excitotoxicity.
Keywords: glutamate/glutamine cycle, activity-dependent regulation, neurotransmitter cycling, neuronal glutamine transporter, excitotoxicity, neuroprotection
Graphical abstract
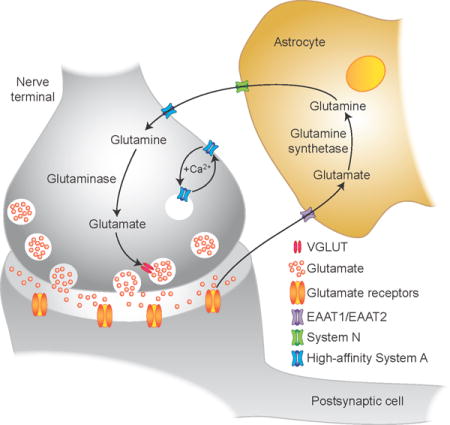
This report describes a Ca2+-regulated ‘system A’ glutamine transport system in hippocampal neuron-enriched primary cultures that is dependent on neural activity in mature synapses, potently inhibited by riluzole (a blocker of synaptic glutamate release), and that is up-regulated during the critical postnatal period of functional maturation of the glutamate/glutamine cycle between astrocytes and neurons and synaptic glutamate release. The novel high-affinity system A transporter described here may have physiological and pathological implications in understanding the neurobiology of excitotoxic synaptic glutamate release in acute and chronic neurodegenerative diseases.
INTRODUCTION
Excessive presynaptic Glu release is involved in several acute and chronic neurodegenerative diseases (Benveniste et al. 1984, Obrenovitch & Urenjak 1997, Puel et al. 1998, Dudek & Sutula 2007, Kostandy 2012, Dorsett et al. 2016). Riluzole is a benzothiazole anticonvulsant that was shown many years ago to be neuroprotective in numerous conditions of excessive synaptic Glu release including global and focal cerebral ischemia (Pratt et al. 1992, Doble 1996, Stutzmann 1997, Kanthasamy et al. 1999). Riluzole has received renewed attention as an anti-glutamatergic agent because of novel methods that increase its brain penetration (Verma et al. 2016), the reported beneficial effects of riluzole in animal models of Alzheimer’s disease (Hunsberger et al. 2015, Mokhtari et al. 2016, Pereira et al. 2016), and riluzole-derivatives with increased anti-convulsant properties (Coleman et al. 2015). Today, riluzole is registered under the trade name Rilutek for the treatment of amyotrophic lateral sclerosis (ALS). Riluzole exhibits various pharmacological activities, the most potent of which is inhibition neuronal Na+ channels at concentrations of 1–50 µM (Benoit & Escande 1991, Herbert et al. 1994, Song et al. 1997, Stefani et al. 1997, Prakriya & Mennerick 2000, Spadoni et al. 2002), which would reduce neuronal circuit activity and Ca2+-dependent synaptic Glu release (Cheramy et al. 1992, Martin et al. 1993, Wang et al. 2004). Na+-dependent Gln import into synapses from astrocytes to replenish synaptic Glu stores released is a potential novel target to reduce excessive synaptic Glu release (Kanamori & Ross 2013, Tani et al. 2014, Marx et al. 2015). Direct functional evidence for activity-regulated Ca2+-dependent Gln transport in hippocampal neurons is, however, lacking.
The concept of Glu/Gln cycling for replenishment of neurotransmitter Glu pools originated from 13C- and 14C-NMR determinations in vivo (Sibson et al. 2001, Rae et al. 2003) and ‘classic’ biochemical experiments in vitro where it was shown that 3H-Gln was a preferred precursor for synaptic Glu released in various neuronal preparations including the hippocampus (Hamberger et al. 1979, Reubi 1980). Activity-induced modulation of synaptic efficacy (Bacci et al. 2002, Armano et al. 2002) and glutamatergic epileptiform activity (Tani et al. 2010, Kanamori & Ross 2011) requires transport of Gln that is significantly reduced by inhibition of Gln synthesis in astrocytes or by application of MeAIB that blocks Gln import into neurons. MeAIB is a competitive and reversible inhibitor of the low affinity (Km ~0.5 mM) Na+-coupled neuronal Gln transporters SNAT1 and SNAT2 (Mackenzie & Erickson 2004). An activity-induced electrogenic Gln transporter has recently been reported that is inhibited by high concentrations (20 mM) of MeAIB (Billups et al. 2013) suggesting that activity-regulated Gln/MeAIB transport in the specialized calyx of Held synapse is mediated by SNAT1 or SNAT2 (Blot et al. 2009, Marx et al. 2015). While SNAT1 and SNAT2 are localized to CNS neurons they are not expressed in axon terminals in the cerebral cortex or hippocampus (Mackenzie et al. 2003, Conti & Melone 2006, Grewal et al. 2009, Jenstad et al. 2009), suggesting that an unidentified neuronal Gln/MeAIB transporter supports Glu transmission in the hippocampus.
Here, the hypothesis that a novel, high-affinity, Ca2+-regulated Gln/MeAIB transport system exists in hippocampal neurons is tested; such activity should be optimally regulatable by neuronal activity under physiologic levels of Gln (~30 µM) reported in extrasynaptic space (Kanamori & Ross 2013). I report that high-affinity, activity-regulated, Ca2+-dependent Gln/MeAIB transport exists in hippocampal neurons and that it is inhibited by riluzole, a drug that reduces synaptic release of Glu and Glu-induced excitotoxicity in vitro and in vivo. I also report that both spontaneous and K+ depolarization-stimulated Gln/MeAIB transport are dramatically induced between 8 and 20 days in vitro (DIV), which is coordinately regulated with the postnatal maturation of the Glu/Gln cycle (Chowdhury et al. 2007, Hertz 2013) and synaptic vesicular Glu release in cortical and hippocampal neurons in vitro and in vivo (Bolshakov & Siegelbaum 1995, Wasling et al. 2004, Wilson et al. 2005, De Gois et al. 2005). The identification of activity-regulated, riluzole-sensitive, high-affinity neuronal Gln/MeAIB transport emphasizes the importance of this potential therapeutic target in conditions of excessive synaptic release of Glu in hippocampal neurons during development, in mature synapses, and in the aged. A preliminary report of these results has recently appeared (Erickson 2016)
Materials & Methods
Cell culture
Primary hippocampal neurons were prepared from embryonic day 19 Sprague Dawley rats (Envigo, Indianapolis, Indiana.RRID:RGD_5508397) of both sexes (Brewer et al. 1993). All animal procedures were approved by the Institutional Animal Care and Use Committee at LSUHSC, and complied with the NIH guidelines. Pregnant dams were subjected to an overdose of inhalation of compressed CO2 administered in an anesthesia chamber and then decapitated using a guillotine. All measures were taken to minimize pain, discomfort, or suffering of animals. Briefly, during dissection the hippocampi were collected in Hanks Balanced Salt Solution (without Ca2+) containing 10 mM MgCl2, 10 mM HEPES (pH 7.2), 0.2 mM glutamine, penicillin (100 U/ml) and streptomycin (100 mg/ml) (pen/strep) at 4°C. Hippocampi were then incubated in oxygenated Neurobasal medium (NB) without B27 (Invitrogen, Gaithersburg, MD) containing 0.1 mg/ml papain (Sigma, St. Louis, MO) and 0.2 mg/ml cysteine for 15 min at 37°C with shaking. The tissue was transferred to a 15 ml conical tube and allowed to settle and the medium then replaced twice with DMEM containing 10% (v/v) FBS. The tissue was then dissociated by trituration with fire-polished Pasteur pipettes in DMEM containing FBS, the cells were counted, diluted in DMEM containing 10% (v/v) FBS, 0.5 mM glutamax, and 25 µM Glu, and then seeded in six-well dishes (4 X 105 cells/well) precoated with 25 µg/ml poly-D-lysine (>300,000 MW, Sigma). After 2 h at 37° C the medium was removed and replaced with NB medium supplemented with B27, 1% (v/v) FBS, 0.5 mM glutamax, 25 µM Glu, and pen/strep. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 with the addition of 1 ml of NB medium supplemented with B27, 0.5 mM glutamax and pen/strep every third day of the first week, and every other day thereafter.
Astrocyte-enriched cultures were also prepared as secondary cultures from same plating of hippocampal cells as above, except that the cells were maintained in DMEM containing 10% FBS, instead of NB medium. In addition, after one week the cells were trypsinized to remove them from the bottom of the flask, and then split 1/10 in DMEM containing 10% FBS and transferred to a new flask. Once confluent, these cells were again split 1/10 in DMEM containing FBS and allowed to reach confluency. Cells were then dispersed (4 X 105/well) in poly-D-lysine coated 6-well clusters for analysis. Astrocytes were treated with high K+ depolarization (+/− Ca2+) and transport of 14C-MeAIB (20 µM, 5 min) at 37°C was measured.
Uptake studies
Uptake studies were performed using primary neuron-enriched hippocampal cultures at various days of maturation in vitro (DIV). Briefly, cells (most studies were performed at DIV 14) were first rinsed in buffer (3 ml) containing 125 mM NaCl, 4.8 mM KCl, 25 mM HEPES (pH 7.4), 1.2 mM KH2PO4, 2.5 mM MgSO4, 5.6 mM glucose, and 1 mM EGTA (N buffer), and then K+-stimulated (5 min) or spontaneous uptake experiments were performed. Following K+-stimulated or spontaneous Gln/MeAIB uptake the dishes were removed from the 37°C incubator and placed on ice, and the cells were then washed with ice-cold N buffer (2.5 ml). SDS (1%, 2 ml) was then added to solubilize the cells and the amount of radioactivity determined by liquid scintillation counting (EcoScint, Nat. Diag.). Various controls (4°C, K+ 4.8 mM, no Ca2+ +EGTA, +MeAIB 5 mM) were appropriately included with each experiment as indicated.
For K+-stimulated uptake experiments, N buffer-rinsed cells were bathed (37° C, 5 min) in a) N buffer, b) N buffer (without EGTA) containing 1.2 mM CaCl2, c) high K+ buffer containing 69.8 mM NaCl, 60 mM KCl, 25 mM HEPES (pH 7.4), 1.2 mM KH2PO4, 2.5 mM MgSO4, 5.6 mM glucose, and 1 mM EGTA, or d) high K+-buffer (without EGTA) containing 1.2 mM CaCl2. The solutions were removed and the cells rinsed in N buffer to chelate Ca2+. Uptake studies were then performed immediately in N buffer (37° C, 5 min) that contains 20 µM unlabeled Gln or MeAIB spiked with 3H-Gln or 14C-MeAIB (Perkin-Elmer; 2 µl/well), respectively. For kinetic analysis, various concentrations of unlabeled MeAIB (3.9 µM – 500 µM) were added and uptake was terminated at 3 min. Various amino acid substrate competitors (2 mM) were added directly to the radiolabeled 14C-MeAIB solution and uptake was performed for 5 min. Amino acids tested included: alanine (ala), proline (pro), sarcosine, histidine (his), MeAIB, Gln, leucine (leu), glycine (gly), serine (ser), N,N’dimethylglcine (N’N-DMG), betaine, aminoisobutyric acid (AIB), 2-amino-2-norbornane-carboxylic acid (BCH), D-serine, arginine (arg), cyclo-D-serine, D-aspartate (D-asp), taurine, and γ-aminobutyric acid (GABA). In some experiments verapamil (ver, 20 µM) or tetrodotoxin (TTX, 1 µM) were present during K+-stimulation, but they were not present during the 5 min uptake period.
For spontaneous uptake experiments N buffer-rinsed cells were bathed (37° C, 5 or 15 min) in a) N buffer or b) N buffer (without EGTA, with 1.2 mM CaCl2) containing 20 µM unlabeled MeAIB spiked with 14C-MeAIB (2 µL/well). Cells were pretreated with vehicle (DMSO or water) for control values or with verapamil (20 µM), α-methl-4-carboxyphenylglycine (MCPG, 1 mM), 2-amino-5-phosphonopentanoic acid (AP5, 50 µM) together with 2,3-dioxo-6nitro-1,2,3,4-tetrahydrobenzo[f]-quinoxaline-7-sulfonamide (NBQX, 10 µM), or TTX (1 µM) 2 min prior to addition of 14C-MeAIB and these drugs were present during the uptake period. Riluzole (0.039 – 20 µM) and phenytoin (1.56 – 200 µM) dose-response inhibition experiments were performed by pre-incubating cells in Ca2+-containing medium with various concentrations of inhibitors (2 min) and then replacing the medium with the same medium (+ drug) containing 20 µM MeAIB.
Data analysis
Uptake of radioactive amino acids was independently measured in culture wells and the final values were derived from at least three independent hippocampal neuronal preparations. Scintillation counts were converted into molar quantities by calculation of specific activity (dpm/pmol) of the radiolabeled/unlabeled Gln or MeAIB mixture used. Error bars represent the means +/− S.E. or means +/− S.D. (for kinetics) of the average values from independent experiments. Comparisons of mean values from 3–6 independent neuronal preparations were done by one-way ANOVA with pairwise comparisons between groups adjusted with Tukey-Kramer posthoc analysis. All nonlinear regression analyses were performed in Prism7 (GraphPad Software Inc. La Jolla, CA).
RESULTS
K+-stimulated, Ca2+-dependent Gln transport exists in hippocampal neurons
High K+-depolarization (56 mM) of axon terminals was the original biochemical method used to show that Gln was a preferred precursor for the Ca2+-dependent releasable pool of Glu from hippocampal neurons. An important question that still remains is whether such synaptic activation induces Ca2+-dependent Gln transport activity in hippocampal neurons? To examine this question neuron-enriched hippocampal primary cultures were subjected to brief (5 min) high K+ depolarization (60 mM) in Ca2+-containing Krebs-Ringer solution, washed in Ca2+-free Krebs-Ringer solution containing EGTA (N buffer), and then 3H-Gln (20 µM) transport was measured in N buffer over 5 min. Uptake in the absence of Ca2+ or at 4°C was also examined for specificity and to control for non-specific effects. Low concentrations of Gln were used to minimize transport from low affinity neuronal Gln transporters, such as SNAT1 and SNAT2. We also examined a selective subpopulation of Gln transporters sensitive to inhibition by MeAIB (5 mM). MeAIB-inhibitable 3H-Gln transport at low concentrations of Gln (20 µM) in the absence of Ca2+ is low (<5% of total 3H-Gln uptake) under Ca2+-free spontaneous conditions. However, MeAIB inhibitable and Ca2+-dependent 3H-Gln uptake increases approximately 25% following a brief exposure to K+-depolarization (Fig. 1). MeAIB is a transported substrate of the ‘classic’ system A transporters (Christensen 1990) and is not metabolized, which renders it more stable than Gln. Therefore, 14C-MeAIB (20 µM) was used in all subsequent experiments to identify, and characterize, a subpopulation of Gln/MeAIB transporters that relies on extracellular Ca2+ ions for functional expression in a whole cell assay and that is stimulated by K+-depolarization.
Fig. 1. Ca2+-dependent 3H-Gln transport in hippocampal neurons is stimulated by brief high K+-induced depolarization.

A relatively low concentration of 3H-Gln (20 µM) in the uptake medium is used to isolate a high affinity K+-stimulated Gln transport system in cultured hippocampal neurons. Abundant uptake of 3H-Gln by neurons is observed following 5 min incubation in normal Krebs buffer without Ca2+ (N buffer) but 3H-Gln transport is not significantly affected by inclusion of MeAIB (5 mM). The uptake of 3H-Gln increases ~25% by brief exposure (5 min) to high K+-depolarization (plus Ca2+) of axon terminals (K), which is blocked by inclusion of MeAIB (5 mM) in the uptake buffer (K-M). The MeAIB-sensitive portion of 3H-Gln transport increases ~8X fold following K+-depolarization (M sen, +Ca2+). The transport of 3H-Gln is conducted in N buffer at DIV 14. Uptake values obtained at 4°C were less than 5% and were subtracted. N, normal Krebs buffer without Ca2+ and with EGTA (1 mM). N-M, Same buffer with MeAIB (5 mM). K, Krebs buffer containing Ca2+ (1.2 mM) and with Na+ replaced with high K+ (60 mM, 5 min). K-M, Same K+-buffer with MeAIB (5 mM). M-sen, MeAIB-sensitive transport with same buffers. Data are the mean +/− S.E. values for n=3 independent experiments. F5,18 = 59.4, p < 0.01 (main effect). *p = 0.04 (K vs K-M), ** p = 0.003 (Msen+Ca vs Msen-Ca), N.S. not significant.
K+-stimulated, Ca2+-dependent MeAIB transport exists in hippocampal neurons
Here, the identification and characterization of K+-stimulated, Ca2+-dependent 14C-MeAIB transport activity in primary cultures of hippocampal neurons is reported. K+-stimulated 14C-MeAIB transport was measured in Ca2+-free Krebs buffer (N buffer, pH 7.4) in cells that have had brief (5 min) prior exposure to K+-depolarizing conditions (60 mM) in the presence or absence of Ca2+, and the cells are then rinsed in N buffer to remove Ca2+, which is also required for endocytosis (Hosoi et al. 2009). Under these conditions Ca2+-dependent 14C-MeAIB transport increased ~10-fold by K+-depolarization (Fig. 2). K+-stimulated 14C-MeAIB transport is eliminated by omission of exogenous Ca2+ (+EGTA) or by inclusion of verapamil (20 µM), an inhibitor of P-type voltage-gated Ca2+ channels, during the period of K+-depolarization (Fig. 2). The transport process itself is not dependent on Ca2+ because 14C-MeAIB uptake occurs in N buffer, which does not contain Ca2+ (N buffer also contains EGTA to chelate residual Ca2+ ions). Direct high K+-depolarization of axon terminals bypasses any dependence on intact neuronal circuitry; hence, K+-induced 14C-MeAIB transport is not affected by tetrodotoxin (TTX, 1 µM) present during K+-stimulation. However, the transport process itself is dependent on the presence of Na+, as K+-stimulated Ca2+-dependent 14C-MeAIB transport is not observed when Li+ ions are substituted for Na+ in the uptake buffer (Fig. 3). The dependence of 14C-MeAIB transport on the pH of the uptake buffer was also examined and found to be modestly increased at pH 8.2 and decreased at pH 6.0 (Fig. 3).
Fig. 2. Ca2+-dependent 14C-MeAIB transport is stimulated by brief high K+-induced depolarization and exists spontaneously in neurons, but not in astrocytes.

Hippocampal neurons (DIV 14) were first rinsed in N buffer that does not contain Ca2+, but EGTA (1 mM). For K+-stimulated uptake, cells are exposed to high K+ (60 mM) (with or without 1.2 mM CaCl2) for 5 min at 37°C. Cells were then rinsed in N buffer and 14C-MeAIB uptake is performed for 5 min in N buffer (37°C). For spontaneous uptake, cells are first rinsed in N buffer and then incubated (5 min, 37°C) in N buffer or in normal Krebs medium with Ca2+ (1.2 mM) containing MeAIB (20 µM). Uptake values obtained at 4°C were less than 5% and were subtracted. K+, high K+ depolarization. N, Krebs buffer without Ca2+ and with EGTA. –Ca, 5 min pre-treatment in same buffers without Ca2+ (with 1 mM EGTA). Ver, Verapamil (20 µM) is included during the 5 min K+-depolarization period in Ca2+ containing high K+ buffer. Astrocytes pretreated with high K+ depolarization (plus Ca2+) do not transport significant amounts of 14C-MeAIB (20 µM, 5 min, 37°C). Data are the mean +/− S.E. for n=3 independent experiments. F7,24 = 25.5, p < 0.01 (main effect) ** p < 0.001.
Fig. 3. Ca2+-dependent, high affinity 14C-MeAIB transport in hippocampal neurons is dependent on Na+ ions and is pH-sensitive.

Cultures are exposed to high K+ (60 mM) (plus 1.2 mM CaCl2) for 5 min at 37°C, then rinsed in N buffer, and 14C-MeAIB (20 µM) uptake is performed (5 min, 37°C) in: A, N buffer or N buffer that contains 145 mM LiCl, instead of Na+, or B, N buffer at pH 8.2, 7.4, or 6.0. Uptake values obtained at 4°C were less than 5% and were subtracted. Data are the mean +/− S.E. values for n=3 independent experiments. F4,18 = 73.1, p < 0.01 (main effect). ** p < 0.01.
Kinetics of K+-induced, Ca2+-dependent 14C-MeAIB transport activity differentiate this neuronal transport system from SNAT1- or SNAT2-mediated activity, which display much lower affinity (Km ~ 0.5 mM). K+-induced, Ca2+-dependent MeAIB transport in neurons displays an affinity (Km) for MeAIB of 37 +/− 1.2 µM and saturates at 200 µM (Fig. 4). The relative ability of various amino acid substrates at 2 mM to block K+-induced/Ca2+-dependent 14C-MeAIB transport, which is indicative of relative substrate affinity of the transporter, is shown in Fig. 5. Amino acids tested include alanine, proline, sarcosine, histidine, MeAIB, Gln, leucine, glycine, serine, N’N dimethylglycine (N’N-DMG), betaine, aminoisobutyric acid (AIB), 2-amino-2-norbornanecarboxylic acid (BCH), D-serine, arginine, cyclo-D-serine, D-aspartic acid, taurine, and γ-aminobutyric acid (GABA). The preferred substrate competitors alanine, proline, sarcosine, histidine, MeAIB and Gln displayed greater than 90% inhibition of 14C-MeAIB transport when present at 2 mM concentration (Fig. 5). Taurine, GABA, cyclo-D-serine, and D-aspartic acid do not interfere with K+-stimulated transport of MeAIB. Activity-induced, Ca2+-dependent MeAIB transport is not observed in cultured hippocampal astrocytes (Fig. 2). High K+ was used here to maximally depolarize axon terminals, which is reasonable for the initial screen to identify the substrates and inhibitors and kinetically characterize this novel high-affinity Ca2+-dependent Gln/MeAIB transport system in primary hippocampal neurons.
Fig. 4. Kinetic analysis of Ca2+-dependent K+-stimulated 14C-MeAIB transport in hippocampal neurons.

A, Saturation isotherm of initial uptake velocity (3 min) of 14C-MeAIB (3.9 – 500 µM) in hippocampal neuron-enriched primary cultures (DIV 14). B, Lineweaver-Burk analysis of initial uptake velocity. Cells are exposed to high K+ (60 mM) (with 1.2 mM CaCl2) for 5 min at 37°C, then rinsed in N buffer, and 14C-MeAIB uptake is performed in N buffer (37°C). Uptake values obtained at 4°C were less than 5% and were subtracted. The Km and Vmax value for MeAIB was calculated from the nonlinear regression of specific MeAIB uptake velocity versus the extracellular concentration of MeAIB. Data are from a representative experiment performed in duplicate, and Km values are derived from n=4 independent experiments.
Fig. 5. Substrate competition profiles for Ca2+-dependent, high affinity 14C-MeAIB transport.

Cultures are exposed to high K+ (60 mM plus 1.2 mM CaCl2) for 5 min at 37°C, then are rinsed in N buffer, and 14C-MeAIB (20 µM) uptake is performed in N buffer (5 min, 37°C). Transport is conducted in the absence or presence of various amino acid competitive substrates (2 mM). Uptake values obtained at 4°C were less than 5% and were subtracted. Subtracted data were normalized to 14C-MeAIB uptake in the absence of unlabeled amino acids. N’N-DMG, N’N dimethylglycine. AIB, aminoisobutyric acid. BCH, 2-amino-2-norbornane-carboxylic acid. Cyclo-D-ser, cyclo-D-serine. Data are the mean +/− S.E. values from n=3–5 independent experiments. **p < 0.01, indicates significant differences from uptake in the absence of unlabeled substrate. ***p < 0.01) indicate amino acids (ala, pro, sarcosine, his, MeAIB, Gln) that reduce 14C-MeAIB transport by greater than 90%. N.S., not significant.
Spontaneous Ca2+-regulated MeAIB transport exists in hippocampal neurons
High K+-stimulation (60mM) of axon terminals is not physiological. Does activity-regulated, Ca2+-dependent Gln/MeAIB transport spontaneously exist under physiologic conditions in neuronal hippocampal cultures? To examine this question, neurons were incubated under physiological conditions in Krebs buffer with 14C-MeAIB (20 µM; with or without Ca2+). Spontaneous transport of low concentrations of MeAIB requires exogenous Ca2+ (1.2 mM) and is blocked by inhibitors of VCGGs (verapamil 20 µM) (Fig. 2, Fig. 6), like K+ stimulated Gln/MeAIB transport (Fig 1, 2). Spontaneous, Ca2+-dependent 14C-MeAIB transport is blocked by TTX (1 µM), an inhibitor of Na+ channels that are required for action-potential mediated Ca2+-stimulated exocytosis of Glu and neural circuit activity (Fig. 6), unlike K+ stimulated transport. Spontaneous transport does not occur with Li+ ions substituted for Na+ (not shown). Transport is dependent on excitatory-driven neural circuit activity because it is blocked by inhibitors of AMPA and NMDA Glu receptors that mediate postsynaptic Glu transmission (Fig. 6). Spontaneous transport is not affected by MCPG (Fig. 6), a group I/II metabotropic Glu receptor inhibitor. Spontaneous MeAIB transport is inhibited by high concentrations (2 mM) of GABA (Fig. 6), which activates GABA receptors to increase inhibitory transmission and shunt Glu-driven neural circuit activity. Together, these results indicate that a Ca2+-dependent Gln/MeAIB transporter exists spontaneously in hippocampal neuronal cultures and requires Na+- and Ca2+-dependent Glu transmission for functional activity.
Fig. 6. Spontaneous 14C-MeAIB transport requires extracellular Ca2+ and is blocked by inhibitors of VGCCs, Na+ channel activity, glutamatergic transmission via AMPA and NMDA receptors, and by GABA.

Cultures (DIV 14) were rinsed in N buffer and then preincubated in Krebs buffer with Ca2+ with or without inhibitors (2 min) and then in the same buffers containing 14C-MeAIB (20 µM) at 37°C (5 min). Uptake values obtained at 4°C were less than 5% and were subtracted. Con, control. Ver, verapamil 20 µM, A/N, AP5, 50 µM / NBQX 10 µM, TTX, tetrodotoxin 1 µM; MCPG, α-methyl-4-carboxyphenylglycine, 1 mM. Selected amino acids (2 mM) included with 14C-MeAIB during transport reveal that spontaneous 14C-MeAIB transport exhibits that same substrate preference to K+-stimulated transport, except for GABA, which is inhibitory due to the blockade of excitatory glutamatergic transmission. Data are the mean +/− S.E. values from n=3 independent experiments. F12,39 = 571 (main effect), p < 0.0001. **p < 0.0001.
Alanine, proline, sarcosine, histidine, MeAIB and Gln display greater than 90% inhibition of 14C-MeAIB transport when present at 2 mM concentration (Fig. 6), which indicates that they are also preferred potential substrates for spontaneous Ca2+-dependent 14C-MeAIB transport, like they are for K+-stimulated transport. Gln is present at much greater levels in extrasynaptic space in vivo, compared to all other amino acids. Taurine, an osmolyte and regulator of Ca2+ homeostasis in neurons, does not interfere with 14C-MeAIB transport (Fig. 6). Together these results indicate that a Ca2+-regulated, Gln-sensitive MeAIB transporter exists spontaneously in primary neuron-enriched hippocampal cultures.
Spontaneous, Ca2+-regulated MeAIB transport is potently inhibited by riluzole, a neuroprotective agent
Riluzole is neuroprotective in conditions of excessive synaptic Glu release because it inhibits Glu release from synapses by unclear mechanisms. Does riluzole inhibit activity-regulated Gln/MeAIB import into hippocampal neurons? To examine this question, neurons were pre-incubated with riluzole or other potential anti-glutamatergic agents for 2 min in Ca2+-containing Krebs buffer and then with same medium containing 14C-MeAIB (20 µM) for 15 min. Drugs examined are anti-glutamatergic agents used in epilepsy, global and focal ischemic stroke, ALS, Glu-induced hearing loss, that include riluzole, lamotrigine, phenytoin, carbazepam, topiramate, ethosuximide, valproic acid, Gabapentin, zonasamide, and levetiracetam. At 20 µM concentration, riluzole completely blocks spontaneous Ca2+-dependent MeAIB transport while lamatrogine, phenytoin and toperimate were much less effective inhibiting approximately 54%, 25%, and 20% of control values, respectively. The relative inhibitory constant (IC50) for riluzole to inhibit spontaneous Ca2+-dependent MeAIB transport is 1.3 +/− 0.1 µM (Fig. 7). Phenytoin displays an IC50 value of 57 +/− 4 µM to block activity-regulated, Ca2+-dependent, high-affinity Gln/MeAIB transport in hippocampal neuronal cultures (Fig. 7), which is 50X less potent than riluzole. All other agents tested are ineffective at 20 µM concentration. The relative potency values for riluzole and phenytoin described here are consistent with the reported differences between riluzole and phenytoin to provide neuroprotection against presynaptic Glu-induced excitotoxicity in acute and chronic neurodegenerative conditions.
Fig. 7. Spontaneous Ca2+-regulated 14C-MeAIB transport is potently and preferentially blocked by riluzole, an anti-glutamatergic drug.

Spontaneous Ca2+-dependent 14C-MeAIB transport (20 µM) is measured (15 min, 37°C) with and without various anti-glutamatergic drugs, following their brief (2 min) pre-incubation at 37°C. Spontaneous transport in Ca2+-free Krebs buffer was measured in all independent experiments and is generally less than 15% of Ca2+-containing control samples, was not affected by any of the drugs tested, and was subtracted as the negative background control. The IC50 value for riluzole (black circles) and phenytoin (open squares) inhibition of 14C-MeAIB transport was calculated from dose response curves. Data shown are from a representative experiment, and the experiment was repeated n=4 times.
K+-stimulated, and spontaneous Ca2+-regulated MeAIB transport is endogenously up-regulated during the 2nd and 3rd weeks of development in hippocampal neurons
The Glu/Gln cycle and synaptic VGLUT1-mediated glutamatergic transmission develops in hippocampal and cortical pyramidal neurons during the 2nd and 3rd postnatal weeks in vivo and in vitro. Does K+-stimulated and Ca2+-dependent spontaneous transport of 14C-MeAIB develop in neuronal cultures along a similar time-line? To examine this question, neurons were incubated in Krebs buffer with Ca2+ for spontaneous conditions, or in Ca2+-free conditions after brief K+-depolarization with Ca2+ (see Experimental Procedures) across several postnatal developmental time points (DIV 8, 12, 14, 16, and 20). Spontaneous and K+ depolarization-induced, Ca2+-dependent 14C-MeAIB transport is not present in DIV 8 cultures (Fig. 8). Both K+-stimulated and spontaneously active 14C-MeAIB transport are dramatically and coordinately up-regulated during the 2nd and 3rd postnatal weeks (DIV 8–20). High-affinity, Ca2+-dependent 14C-MeAIB transport is K+-depolarization stimulated, spontaneously active, and is completely blocked by the presence of 5 mM MeAIB at all developmental ages. Collectively, these results demonstrate that activity dependent, Ca2+-regulated Gln/MeAIB transport is developmentally up-regulated during the critical period of maturation of glutamatergic presynaptic strength and is maximally expressed in fully-differentiated, mature hippocampal neurons.
Fig. 8. Developmental up-regulation of the expression of activity-dependent spontaneous and K+-stimulated Ca2+ sensitive 14C-MeAIB transport.

Spontaneous and K+-stimulated 14C-MeAIB transport (20 µM) is measured (5 min, 37°C) in hippocampal neuron-enriched cultures at different days in vitro (DIV 8, 12, 14, 16, and 20). N, normal Krebs buffer without Ca2+ and with EGTA. N+C, same buffer with Ca2+ (1.2 mM) and without EGTA. K, Krebs buffer Na+ replaced with high K+ (60 mM) but without Ca2+ and with EGTA. K+C, Same K+ buffer with Ca2+ and without EGTA. Ca2+-dependent spontaneous and K+-stimulated transport are controlled by inclusion of MeAIB (5 mM). Background levels at 4°C are consistently at <100 pmol/well. MeAIB (5 mM) subtracted data are the mean +/− S.E. values from n=3 independent experiments. F9,72 = 481, p < 0.0001 (main effect). * p < 0.05, ** p < 0.001.
DISCUSSION
This report describes, for the first time, a high-affinity, Ca2+-regulated, K+-stimulated, and spontaneously active Gln/MeAIB transport system in hippocampal neuron-enriched primary cultures that is regulated endogenously by neuronal activity in mature synapses, potently inhibited by riluzole (a blocker of Glu release), and that is up-regulated during the critical postnatal period of functional maturation of the Glu/Gln cycle between astrocytes and neurons and synaptic Glu release. The novel Gln/MeAIB transport system described here may have physiological and pathological implications in understanding the neurobiology of excitotoxic synaptic Glu release in the hippocampus in acute and chronic neurodegenerative diseases.
Gln transport is robust in hippocampal neurons due to the multitude of Gln transporter subtypes (at least 14) that exist in the CNS that subserve multiple physiologic functions in many cellular compartments (Schioth et al. 2013, Albrecht et al. 2007). However, only a small percentage (<5%) of the total amount of Gln transported in hippocampal neuronal culture (5 min at 20 µM 3H-Gln concentration) is sensitive to inhibition by MeAIB (5 mM), a selective blocker of system A transport (Christensen 1990, McGivan & Pastor-Anglada 1994). Yet, Ca2+-dependent MeAIB-sensitive Gln transport activity can be stimulated by brief high K+-depolarization of axon terminals suggesting that an activity-regulated Ca2+-dependent Gln/MeAIB transporter exists in hippocampal neurons. The transport of MeAIB was therefore used here as a specific substrate to isolate and to further characterize this Gln transporter subpopulation, and because MeAIB is not metabolized, and so is more stable than Gln. As surmised, Ca2+-dependent transport of MeAIB is greatly stimulated following brief K+-depolarization. K+-stimulated MeAIB transport is dependent on extracellular Ca2+, and in the presence of 1.2 mM Ca2+, MeAIB transport is blocked by inhibition of VGCC’s (verapamil, 20 µM). This K+ depolarization-induced Gln/MeAIB transport system displays relatively high affinity for MeAIB (37 +/− 1.2 µM) and is maximally active at MeAIB concentrations of 200 µM, unlike low-affinity (Km ~ 0.5 mM) neuronal Gln/MeAIB transporters SNAT1 or SNAT2 (Mackenzie & Erickson 2004). The novel, high affinity Gln transporter in hippocampal neurons described here would operate efficiently at low extrasynaptic levels of Gln (~30 µM) reported in the hippocampus in vivo (Kanamori & Ross 2013), which would be ideal for activity-dependent regulation of Gln cycling into synapses. While other precursors could contribute to regulating cytoplasmic Glu levels and vesicular Glu stores in cortical and hippocampal axon terminals (Hassel & Brathe 2000, Lieth et al. 2001, Kam & Nicoll 2007, Takeda et al. 2012), Gln is still considered the preferred precursor for loading the Ca2+-dependent releasable pool of vesicular Glu (Leke & Schousboe 2016).
The present study challenges existing research paradigms that the known low-affinity neuronal Gln/MeAIB transporters SNAT1 or SNAT2 are involved in supplying Gln at synapses for Glu transmission (Chaudhry et al. 2002, Mackenzie & Erickson 2004, Conti & Melone 2006). A higher affinity Gln/MeAIB transport system that is regulated by neuronal activity, and inhibited by riluzole, is a potential new target to affect presynaptic Glu transmission in hippocampal neurons. MeAIB is a substrate-competitor for several other amino acid transporters as well, such as the lysosomal amino acid transporter LYAAT-1 (Sagne et al. 2001), the electrogenic H+-dependent amino acid cotransporter PAT2 (Boll et al. 2002), the synaptic vesicle enriched amino acid transporter NTT4/Rxt1 (Zaia & Reimer 2009), and a broad spectrum amino acid transporter SNAT8 (Hagglund et al. 2015). Gln uptake by neuronal SNAT7 and SNAT8 isoforms is weak in oocytes and they may also transport cationic and anionic amino acids (Hagglund et al. 2011, Hagglund et al. 2015). NTT4/Rxt1 is found in preparations enriched in synaptic vesicles (Masson et al. 1999) and recently was described to be a leu/pro/his/Gln/MeAIB transporter when molecularly engineered for expression at the plasma membrane (Zaia & Reimer 2009). The transporter responsible for activity-dependent, spontaneous and K+-stimulated, Ca2+-regulated neuronal 14C-MeAIB transport may also, of course, be still an orphan or a misidentified Slc member.
Low levels of Ca2+-regulated Gln/MeAIB transport described in the present study could be a result of the intracellular sequestration of an inducible, Ca2+-dependent recycling of a high-affinity Gln/MeAIB transporter protein in excitatory synapses providing an important regulatory mechanism for activity regulated Glu transmission. Indeed, regulated cycling of transporters involved in the recycling of other neurotransmitters, such as acetylcholine and GABA, rely on cycling of intracellular pools of high-affinity choline (CHT1) and GABA (GAT1) transporters to the plasma membrane in nerve terminals (Deken et al. 2003, Ribeiro et al. 2007, Ferguson et al. 2003). SNAT2 is also regulated at the level of recycling of an intracellular pool of SNAT2 vesicles in many cell types as an osmolyte (Franchi-Gazzola et al. 2006) including neurons, but is not required for presynaptic Glu transmission in cortical neurons in vitro (Grewal et al. 2009). The dependence of spontaneous Ca2+-regulated MeAIB transport on Na+ (TTX-inhibitable, Li+ intolerant), Ca2+ (VGCC-inhibitable), and postsynaptic Glu receptor function (NBQX/AP5-inhibitable) described here indicates the requirement for action potential stimulated and Glu-driven neural circuit activity. Blockade of endogenous synaptic Glu transmission by inclusion of GABA, to increase synaptic inhibition, also blocks spontaneous Ca2+-regulated MeAIB transport. Collectively these results support the presence of intracellular stores of an activity-regulated, high affinity Gln/MeAIB transporter protein in hippocampal neurons that exhibits Ca2+-dependent cycling to the plasma membrane for functional expression and that is inducible by neuronal activity.
Biochemical studies and 13C-NMR determinations have similarly identified the time period during which the Glu/Gln cycle activity develops in the rat to between postnatal days 10 and 30 (Chowdhury et al. 2007, Hertz 2013). Presynaptic Glu release and Glu transmission also increases dramatically in pyramidal neurons during this time period in vivo and in vitro (Bolshakov & Siegelbaum 1995, Wasling et al. 2004, Wilson et al. 2005, De Gois et al. 2005). In the present study, spontaneous and K+-stimulated Gln/MeAIB transport is negligible in DIV 8 cultures but is maximal (>10-fold increase) by DIV 20. The developmental period selected for neuronal experiments to investigate the role for regulated Gln/MeAIB transport in excitatory transmission is therefore critical. Gln transport during this ‘critical’ period of postnatal development may contribute in mechanisms of synaptic growth and differentiation. Synaptic growth, re-organization and plasticity may require Gln because activity-dependent Gln transport could facilitate presynaptic Glu release that strengthens selective excitatory synaptic pathways in the brain. For instance, Glu-dependent secretion of BDNF sculpts cortical and hippocampal neural circuits during development (Lu 2003, Kuczewski et al. 2010, Park & Poo 2013), stimulates energy metabolism (Burkhalter et al. 2003) and regulates Gln/MeAIB-sensitive dendritic development (Burkhalter et al. 2007). Thus, activity-regulated synaptic Gln transport during development may facilitate proper establishment of synaptic Glu release in neural circuits for the development of stable neural networks.
The inhibition of spontaneous Gln/MeAIB transport by riluzole suggests that neural activity regulated Gln/MeAIB transport may be linked to Na+-dependent/action-potential driven and Ca2+-regulated synaptic release of Glu. Riluzole has been shown to inhibit synaptic Glu release (Martin et al. 1993, Kretschmer et al. 1998, Lingamaneni & Hemmings 1999, Wang et al. 2004). Neuroprotection by riluzole may occur by inhibiting Glu release in neurons via mechanisms that include inhibition of Na+ channels (Benoit & Escande 1991, Martin et al. 1993, Herbert et al. 1994, Stefani et al. 1997, Prakriya & Mennerick 2000, Spadoni et al. 2002). Riluzole inhibition of Na+ channels would reduce action-potential driven neuronal circuit activity and thus, synaptic Glu release that drives AMPA receptor mediated Glu activity and subsequent NMDA-mediated excitotoxicity. Phenytoin, an established Na+ channel blocker, also blocks activity-regulated Gln/MeAIB transport, and both riluzole and phenytoin are neuroprotective in ischemia-induced neuronal death in vivo (Malgouris et al. 1989, Pratt et al. 1992, Taft et al. 1989, Ates et al. 2013). However, riluzole is more than 50X more potent than phenytoin to inhibit activity-regulated Gln/MeAIB transport, which agrees with its improved potency to prevent excitotoxic Glu-induced neuronal damage in the hippocampus (Ates et al. 2013). These results support the development of selective riluzole-derived drugs to target neural-regulated Gln/MeAIB transport activity for neuroprotection.
A recent quantitative analysis of the rates of release and resupply of vesicular Glu required to sustain synaptic Glu release confirms that maintaining the synaptic cytoplasmic Glu supply via Gln (or tricarboxylic acid cycle intermediates) released from glia is essential for continued Ca2+-dependent synaptic Glu release under spontaneous conditions, and under conditions of increased neural activity (Marx et al. 2015). Excessive and sustained synaptic release of Glu is an initial event that triggers Ca2+-dependent Glu-mediated cell death in postsynaptic neurons in various deleterious conditions including traumatic brain injury, peri-operative neuronal and cardiac stress, epilepsy, global and focal cerebral ischemia, mild Alzheimer’s disease, noise-induced hearing loss, and in several in vitro models of neurodegenerative diseases including oxygen-glucose reperfusion injury (Bittigau & Ikonomidou 1997, Dodd 2002, Holmes 2002, Kostandy 2012, Dorsett et al. 2016, Mancini et al. 2016, Pereira et al. 2016). Riluzole is neuroprotective in all of these acute and chronic neurodegenerative conditions (Malgouris et al. 1989, Pratt et al. 1992, Bae et al. 2000, Ruel et al. 2005, Heurteaux et al. 2006, Hunsberger et al. 2015, Pereira et al. 2016, Verma et al. 2016) by uncertain mechanisms. Riluzole is more effective as a neuroprotective agent when given prophylactically, which is in line with its presumed role to reduce synaptic Glu release and resultant Glu-induced excitotoxicity postsynaptically. The identification of Ca2+-regulated, riluzole-sensitive, high-affinity Gln/MeAIB transport in hippocampal neurons supports a role for an activity-regulated Gln transporter in regulating Glu release from synapses and resulting synaptic Glu-induced excitotoxicity.
Acknowledgments
This work was directly supported by LSUHSC Research Enhancement Fund 5497500049 and, in part, by National Institutes of Health Grant R21 14975051.
The abbreviations used are
- Gln
glutamine
- MeAIB
2-(methylamino)isobutyrate
- VGCCs
voltage gated calcium channels
- SNAT
sodium-coupled neutral amino acid transporter
- BCH
2-amino-2-norbornanecarboxylic acid
- AIB
aminoisobutyric acid
- N’N-DMG
N’N-dimethylglycine
- TTX
tetrodotoxin
- Ver
verapamil
- NBQX
2,3-dioxo-6nitro-1,2,3,4-tetrahydrobenzo[f]-quinoaliine-7-sulfonamide
- AP5
2-amino-5-phosphonopentanoic acid
- MCPG
α-methyl-4-carboxyyphenylglycine
- DIV
days in vitro
- NTT4/Rxt1
synaptic vesicle enriched amino acid transporter
- LYAAT-1
lysosomal amino acid transporter
- EAAT
excitatory amino acid transporter
- PAT2
proton-dependent amino acid cotransporter
- CHT1
high-affinity choline transporter
- GAT1
GABA transporter
- VGLUT1
vesicular Glu transporter
- NB
Neurobasal medium
Footnotes
All experiments were conducted in compliance with the ARRIVE guidelines.
Conflict of interest: The author declares no competing financial interests.
Author contribution: J.D.E. conceived the study, performed the experiments, analyzed the data, and wrote the paper.
References
- Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332–343. doi: 10.2741/2067. [DOI] [PubMed] [Google Scholar]
- Armano S, Coco S, Bacci A, Pravettoni E, Schenk U, Verderio C, Varoqui H, Erickson JD, Matteoli M. Localization and functional relevance of system A neutral amino acid transporters in cultured hippocampal neurons. J Biol Chem. 2002;277:10467–10473. doi: 10.1074/jbc.M110942200. [DOI] [PubMed] [Google Scholar]
- Ates O, Cayli SR, Gurses I, Karabulut AB, Yucel N, Kocak A, Cakir CO, Yologlu S. Do sodium channel blockers have neuroprotective effect after onset of ischemic insult? Neurol. Res. 2013;29:317–323. doi: 10.1179/016164107X159225. [DOI] [PubMed] [Google Scholar]
- Bacci A, Sancini G, Verderio C, Armano S, Pravettoni E, Fesce R, Franceschetti S, Matteoli M. Block of glutamate-glutamine cycle between astrocytes and neurons inhibits epileptiform activity in hippocampus. J Neurophysiol. 2002;88:2302–2310. doi: 10.1152/jn.00665.2001. [DOI] [PubMed] [Google Scholar]
- Bae H-J, Lee Y-S, Kang D-W, Gu J-S, Yoon B-W, Roh J-K. Neuroprotective effect of low dose riluzole in gerbil model of transient global ischemia. Neurosci Lett. 2000;294:29–32. doi: 10.1016/s0304-3940(00)01536-6. [DOI] [PubMed] [Google Scholar]
- Benoit E, Escande D. Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflugers Arch. 1991;419:603–609. doi: 10.1007/BF00370302. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Billups D, Marx M-C, Mela I, Billups B. Inducible presynaptic glutamatergic transport supports glutamatergic transmission at the calyx of Held synapse. J Neurosci. 2013;33:17429–17434. doi: 10.1523/JNEUROSCI.1466-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Ikonomidou C. Glutamate in neurologic diseases. J. Child Neurol. 1997;12:471–485. doi: 10.1177/088307389701200802. [DOI] [PubMed] [Google Scholar]
- Blot A, Billups D, Bjorkmo M, Quazi AZ, Uwechue NM, Chaudhry FA, Billups B. Functional expression of two system A glutamine transporter isoforms in rat auditory brainstem neurons. Neuroscience. 2009;164:998–1008. doi: 10.1016/j.neuroscience.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boll M, Foltz M, Rubio-Aliaga I, Kottra G, Daniel H. Functional characterization of two novel mammalian electrogenic proton-dependent amino acid cotransporters. J. Biol. Chem. 2002;277:22966–22973. doi: 10.1074/jbc.M200374200. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Burkhalter J, Fiumelli H, Allaman I, Chatton Y-Y, Martin J-L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J. Neurosci. 2003;23:8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhalter J, Fiumelli H, Erickson JD, Martin JL. A critical role for system A amino acid transport in the regulation of dendritic development by brain-derived neurotrophic factor (BDNF) J Biol Chem. 2007;282:5152–5159. doi: 10.1074/jbc.M608548200. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Edwards RH. The glutamine commute: take the N line and transfer to the A. J. Cell Biol. 2002;157:349–355. doi: 10.1083/jcb.200201070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheramy A, Barbeito L, Godeheu G, Glowinski J. Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci. Lett. 1992;147:209–212. doi: 10.1016/0304-3940(92)90597-z. [DOI] [PubMed] [Google Scholar]
- Chowdhury GM, Pater AB, Mason GF, Rothman DL, Behar KL. Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J. Cereb. Blood Flow Metab. 2007;27:1895–1907. doi: 10.1038/sj.jcbfm.9600490. [DOI] [PubMed] [Google Scholar]
- Christensen HN. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev. 1990;70:43–77. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- Coleman N, Nguyen HM, Cao Z, et al. The riluzole derivative 2-amino-6-trifluoromethylthio-benzothiazole (SKA-19), a mixed KCa2 activator and Na V blocker, is a potent novel anticonvulsant. Neurotherapeutics. 2015;12:234–249. doi: 10.1007/s13311-014-0305-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Melone M. The glutamine commute: lost in the tube? Neurochem Int. 2006;48:459–464. doi: 10.1016/j.neuint.2005.11.016. [DOI] [PubMed] [Google Scholar]
- De Gois S, Schafer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deken SL, Wang D, Quick MW. Plasma membrane GABA transporters reside on distinct vesicles and undergo rapid regulated recycling. J. Neurosci. 2003;23:1563–1568. doi: 10.1523/JNEUROSCI.23-05-01563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47(6 Suppl 4):S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- Dodd PR. Excited to death: different ways to lose your neurons. Biogerontology. 2002;3:51–56. doi: 10.1023/a:1015255312948. [DOI] [PubMed] [Google Scholar]
- Dorsett CR, McGuire JL, DePasuale EA, Gardner AE, Floyd CL, McCullumsmith RE. Glutamate neurotransmission in rodent models of traumatic brain injury. J. Neurotrauma. 2016 doi: 10.1089/neu.2015.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Sutula TP. Epileptogenesis in the dentate gyrus: a critical perspective. Prog. Brain Res. 2007;163:755–773. doi: 10.1016/S0079-6123(07)63041-6. [DOI] [PubMed] [Google Scholar]
- Erickson JD. Functional identification of an activity-induced, Ca2+-dependent glutamine transporter in hippocampal neurons. American Society for Neurochemistry Annual Meeting; Denver, Colorado. 2016. [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD. Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J. Neurosci. 2003;23:9697–9709. doi: 10.1523/JNEUROSCI.23-30-09697.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi-Gazzola R, Dall'Asta V, Sala R, Visigalli R, Bevilacqua E, Gaccioli F, Gazzola GC, Bussolati O. The role of the neutral amino acid transporter SNAT2 in cell volume regulation. Acta Physiol (Oxf) 2006;187:273–283. doi: 10.1111/j.1748-1716.2006.01552.x. [DOI] [PubMed] [Google Scholar]
- Grewal S, Defamie N, Zhang X, De Gois S, Shawki A, Mackenzie B, Chen C, Varoqui H, Erickson JD. SNAT2 amino acid transporter is regulated by amino acids of the SLC6 gamma-aminobutyric acid transporter subfamily in neocortical neurons and may play no role in delivering glutamine for glutamatergic transmission. J. Biol. Chem. 2009;284:11224–11236. doi: 10.1074/jbc.M806470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagglund MG, Hellsten SV, Bagchi S, et al. Transport of L-glutamine, L-alanine, L-arginine and L-histidine by the neuron-specific Slc38a8 in CNS. J. Mol. Biol. 2015;427:1495–1512. doi: 10.1016/j.jmb.2014.10.016. [DOI] [PubMed] [Google Scholar]
- Hagglund MG, Sreedharan S, Nilsson VC, Shaik JH, Almkvist I, Backlin S, Wrange O, Fredriksson R. Identification of SLC38A7 (SNAT7) protein as a glutamine transporter expressed in neurons. J. Biol. Chem. 2011;286:20500–20511. doi: 10.1074/jbc.M110.162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberger AC, Chiang GH, Sandoval E, Cotman CW. Glutamate as a CNS transmitter. II. Regulation of synthesis in the releasable pool. Brain Res. 1979;168:531–541. doi: 10.1016/0006-8993(79)90307-x. [DOI] [PubMed] [Google Scholar]
- Hassel B, Brathe A. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J. Neurosci. 2000;20:1342–1347. doi: 10.1523/JNEUROSCI.20-04-01342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert T, Drapeau P, Pradier L, Dunn RJ. Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol. 1994;45:1055–1060. [PubMed] [Google Scholar]
- Hertz L. The glutamate-glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol. 2013;4:1–16. doi: 10.3389/fendo.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C, Laigle C, Blondeau N, Jarretou G, Lazdunski M. Alpha-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience. 2006;137:241–251. doi: 10.1016/j.neuroscience.2005.08.083. [DOI] [PubMed] [Google Scholar]
- Holmes GL. Seizure-induced neuronal injury: animal data. Neurology. 2002;59:S3–6. doi: 10.1212/wnl.59.9_suppl_5.s3. [DOI] [PubMed] [Google Scholar]
- Hosoi N, Holt M, Sakaba T. Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron. 2009;63:216–229. doi: 10.1016/j.neuron.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Hunsberger HC, Seitzner DS, Rudy CC, Hickman JE, Libell EM, Speer RR, Gerhardt GA, Reed MN. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015;135:381–394. doi: 10.1111/jnc.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenstad M, Quazi AZ, Zilberter M, et al. System A transporter SNAT2 mediates replenishment of dendritic pools controlling retrograde signaling by glutamate. Cereb Cortex. 2009;19:1092–1106. doi: 10.1093/cercor/bhn151. [DOI] [PubMed] [Google Scholar]
- Kam K, Nicoll R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci. 2007;27:9192–9200. doi: 10.1523/JNEUROSCI.1198-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori K, Ross BD. Chronic electrographic seizure reduces glutamine and elevates glutamate in the extracellular fluid of rat brain. Brain Res. 2011;1371:180–191. doi: 10.1016/j.brainres.2010.11.064. [DOI] [PubMed] [Google Scholar]
- Kanamori K, Ross BD. Electrographic seizures are significantly reduced by in vivo inhibition of neuronal uptake of extracellular glutamine in rat hippocampus. Epilepsy Res. 2013;107:20–36. doi: 10.1016/j.eplepsyres.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthasamy AG, Yun RJ, Njuyen B, Truong DD. Effect of riluzole on the neurological and neuropathological changes in an animal model of cardiac arrest-induced movement disorder. J. Pharmacol. Exp. Ther. 1999;288:1340–1348. [PubMed] [Google Scholar]
- Kostandy BB. The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol Sci. 2012;33:223–237. doi: 10.1007/s10072-011-0828-5. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD, Kratzer U, Schmidt WJ. Riluzole, a glutamate release inhibitor, and motor behavior. N.S. Arch. Pharm. 1998;358:181–190. doi: 10.1007/pl00005241. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Gaiarsa JL. Acivity-dependent dendritic secretion of brain-derived neurotrophic factor modulates synaptic plasticity. Eur. J. Neurosci. 2010;32:1239–1244. doi: 10.1111/j.1460-9568.2010.07378.x. [DOI] [PubMed] [Google Scholar]
- Leke R, Schousboe A. The glutamine transporters and their role in the glutamate/GABA-glutamine cycle. Adv. Neurobiol. 2016;13:223–257. doi: 10.1007/978-3-319-45096-4_8. [DOI] [PubMed] [Google Scholar]
- Lieth E, LaNoue KF, Berkich DA, Xu B, Ratz M, Taylor C, Hutson SM. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001;76:1712–1723. doi: 10.1046/j.1471-4159.2001.00156.x. [DOI] [PubMed] [Google Scholar]
- Lingamaneni R, Hemmings HC. Effects of anticonvulsants on veratridine- and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci Lett. 1999;276:127–130. doi: 10.1016/s0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Schafer MK, Erickson JD, Hediger MA, Weihe E, Varoqui H. Functional properties and cellular distribution of the system A glutamine transporter SNAT1 support specialized roles in central neurons. J Biol Chem. 2003;278:23720–23730. doi: 10.1074/jbc.M212718200. [DOI] [PubMed] [Google Scholar]
- Malgouris C, Bardot F, Daniel M, Pellis F, Rataud J, Uzan A, Blanchard J-C, Laduron PM. Riluzole, a novel antiglutamate, prevents memory loss and hippocampal neuronal damage in ischemic gerbils. J. Neurosci. 1989;9:3720–3727. doi: 10.1523/JNEUROSCI.09-11-03720.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini A, Chelini A, Di Capua A, et al. Synthesis and biological evaluation of a new class of benzothiazines as neuroprotective agents. Eur J Med Chem. 2016;126:614–630. doi: 10.1016/j.ejmech.2016.11.053. [DOI] [PubMed] [Google Scholar]
- Martin D, Thompson MA, Nadler JD. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- Marx M-C, Billups D, Billups B. Maintaining the presynaptic glutamate supply for excitatory neurotransmission. J. Neurosci. Res. 2015;93:1031–1044. doi: 10.1002/jnr.23561. [DOI] [PubMed] [Google Scholar]
- Masson J, Riad M, Chaudhry FA, et al. Unexpected localization of the Na+/Cl−dependent-like orphan transporter, Rxt1, on synaptic vesicles in the rat central nervous system. Eur. J. Neurosci. 1999;11:1349–1361. doi: 10.1046/j.1460-9568.1999.00540.x. [DOI] [PubMed] [Google Scholar]
- McGivan JD, Pastor-Anglada M. Regulatory and molecular aspects of mammalian amino acid transport. Biochem J. 1994;299(Pt 2):321–334. doi: 10.1042/bj2990321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari Z, Baluchnejadmojarad T, Nikbakht F, Mansouri M, Roghani M. Riluzole ameliorates learning and memory deficits in Aβ25-35-induced rat model of Alzheimer's disease and is independent of cholinoceptor activation. Biomed Pharmacother. 2016;87:135–144. doi: 10.1016/j.biopha.2016.12.067. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J. Altered glutamatergic transmission in neurological disorders: From high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol. 1997;51:39–87. doi: 10.1016/s0301-0082(96)00049-4. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Pereira AC, Gray JD, Kogan JF, Davidson RL, Rubin RG, Okamoto M, Morrison JH, McEwen BS. Age and Alzheimer's disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry. 2016;22:296–305. doi: 10.1038/mp.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Mennerick S. Selective depression of low-release probablity excitatory synapses by sodium channel blockers. Neuron. 2000;26:671–682. doi: 10.1016/s0896-6273(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Pratt J, Rataud J, Bardot F, Roux M, Blanchard J-P, Laduron PM, Stutzmann JM. Neuroprotective actions of riluzole in rodent models of global and focal cerebral ischaemia. Neurosci Lett. 1992;140:225–230. doi: 10.1016/0304-3940(92)90108-j. [DOI] [PubMed] [Google Scholar]
- Puel JL, Ruel J, Gervais d'Aldin C, Pujol R. Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport. 1998;9:2109–2114. doi: 10.1097/00001756-199806220-00037. [DOI] [PubMed] [Google Scholar]
- Rae C, Hare N, Bubb WA, McEwan SR, Broer A, McQuillan JA, Balcar VJ, Conigrav AD, Broer S. Inhibition of glutamine transport depletes glutamate and GABA neurotransmitter pools: further evidence for metabolic compartmentation. J. Neurochem. 2003;85:503–514. doi: 10.1046/j.1471-4159.2003.01713.x. [DOI] [PubMed] [Google Scholar]
- Reubi JC. Comparative study of the release of glutamate and GABA, newly synthesized from glutamine, in various regions of the central nervous system. Neuroscience. 1980;5:2145–2150. doi: 10.1016/0306-4522(80)90130-x. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Pinthong M, Black SA, Gordon AC, Prado VF, Prado AM, Rylett RJ, Ferguson SS. Regulated recycling and plasma membrane recruitment of the high-affinity choline transporter. Eur. J. Neurosci. 2007;26:3437–3448. doi: 10.1111/j.1460-9568.2007.05967.x. [DOI] [PubMed] [Google Scholar]
- Ruel J, Wang J, Pujol R, Hameg A, Dib M, Puel JL. Neuroprotective effect of riluzole in acute noise-induced hearing loss. Neuroreport. 2005;16:1087–1090. doi: 10.1097/00001756-200507130-00011. [DOI] [PubMed] [Google Scholar]
- Sagne C, Agulhon C, Ravassard P, Darmon M, Hamon M, El Mestikawy S, Gasnier B, Giros B. Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc Natl Acad Sci. 2001;98:7206–7211. doi: 10.1073/pnas.121183498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioth HB, Roshanbin S, Hagglund MG, Fredriksson R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol. Aspects Med. 2013;34:571–585. doi: 10.1016/j.mam.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shuulman RG. In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during [2-13C]glucose infusion. J. Neurochem. 2001;76:975–989. doi: 10.1046/j.1471-4159.2001.00074.x. [DOI] [PubMed] [Google Scholar]
- Song JH, Huang CS, Nagata K, Yeh JZ, Narahashi T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 1997;282:707–714. [PubMed] [Google Scholar]
- Spadoni F, Hainsworth AH, Mercuri NB, Caputi L, Martella G, Lavaroni F, Bermardi G, Stefani A. Lamotrigine derivatives and riluzole inhibit INa2P in cortical neurons. Neuroreport. 2002;13:1167–1170. doi: 10.1097/00001756-200207020-00019. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Bernardi G. Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp Neurol. 1997;147:115–122. doi: 10.1006/exnr.1997.6554. [DOI] [PubMed] [Google Scholar]
- Stutzmann JM, Wahl F, Pratt J, Mary V, Reibaud M, Tecoult E, Rataud J. Neuroprotective profile of riluzole in in vivo models of acute neurodegenerative diseases. CNS Drug Reviews. 1997;3:83–101. [Google Scholar]
- Taft WC, Clifton GL, Blair RE, DeLorenzo RJ. Phenytoin protects against ischemia-produced neuronal cell death. Brain Res. 1989;483:143–148. doi: 10.1016/0006-8993(89)90045-0. [DOI] [PubMed] [Google Scholar]
- Takeda K, Ishida A, Takahashi K, Ueda T. Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from α-ketoglutarate for vesicular loading. J. Neurochem. 2012;121:184–196. doi: 10.1111/j.1471-4159.2012.07684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron. 2014;81:888–900. doi: 10.1016/j.neuron.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Huguenard JR, Reimer RJ. Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J. Neurosci. 2010;30:1288–1300. doi: 10.1523/JNEUROSCI.0106-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Arora I, Javed K, Akhtar M, Samim M. Enhancement in the neuroprotective power of riluzole against cerebral ischemia using a brain targeted drug delivery vehicle. ACS Appl Mater Interfaces. 2016;8:19716–19723. doi: 10.1021/acsami.6b01776. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes) Neuroscience. 2004;125:191–201. doi: 10.1016/j.neuroscience.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Wasling P, Hanse E, Gustafsson B. Developmental changes in release properties of the CA3-CA1 glutamate synapse in rat hippocampus. J Neurophysiol. 2004;92:2714–2724. doi: 10.1152/jn.00464.2004. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia KA, Reimer RJ. Synaptic vesicle protein NTT4/XT1 (SLC6A17) catalyzes Na+-coupled neutral amino acid transport. J. Biol. Chem. 2009;284:8439–8448. doi: 10.1074/jbc.M806407200. [DOI] [PMC free article] [PubMed] [Google Scholar]