

Abstract
Background
Male breast cancer (MBC) represents a poorly characterised group of tumours, the management of which is largely based on practices established for female breast cancer. However, recent studies demonstrate biological and molecular differences likely to impact on tumour behaviour and therefore patient outcome. The aim of this study was to investigate methylation of a panel of commonly methylated breast cancer genes in familial MBCs.
Methods
60 tumours from 3 BRCA1 and 25 BRCA2 male mutation carriers and 32 males from BRCAX families were assessed for promoter methylation by methylation-sensitive high resolution melting in a panel of 10 genes (RASSF1A, TWIST1, APC, WIF1, MAL, RARβ, CDH1, RUNX3, FOXC1 and GSTP1). An average methylation index (AMI) was calculated for each case comprising the average of the methylation of the 10 genes tested as an indicator of overall tumour promoter region methylation. Promoter hypermethylation and AMI were correlated with BRCA carrier mutation status and clinicopathological parameters including tumour stage, grade, histological subtype and disease specific survival.
Results
Tumours arising in BRCA2 mutation carriers showed significantly higher methylation of candidate genes, than those arising in non-BRCA2 familial MBCs (average AMI 23.6 vs 16.6, p = 0.01, 45% of genes hypermethylated vs 34%, p < 0.01). RARβ methylation and AMI-high status were significantly associated with tumour size (p = 0.01 and p = 0.02 respectively), RUNX3 methylation with invasive carcinoma of no special type (94% vs 69%, p = 0.046) and RASSF1A methylation with coexistence of high grade ductal carcinoma in situ (33% vs 6%, p = 0.02). Cluster analysis showed MBCs arising in BRCA2 mutation carriers were characterised by RASSF1A, WIF1, RARβ and GTSP1 methylation (p = 0.02) whereas methylation in BRCAX tumours showed no clear clustering to particular genes. TWIST1 methylation (p = 0.001) and AMI (p = 0.01) were prognostic for disease specific survival.
Conclusions
Increased methylation defines a subset of familial MBC and with AMI may be a useful prognostic marker. Methylation might be predictive of response to novel therapeutics that are currently under investigation in other cancer types.
Electronic supplementary material
The online version of this article (10.1186/s12885-017-3632-7) contains supplementary material, which is available to authorized users.
Keywords: Male breast cancer, Familial breast cancer, Methylation, BRCA1, BRCA2, Promoter methylation
Background
Male breast cancer (MBC) is a poorly studied disease. Indeed, MBC accounts for ~1% of all breast cancers but it contributes to a higher proportion of breast cancer-related deaths [1, 2]. As a significant proportion of MBCs arise within breast/ovarian families, the majority of MBC research has focused on cancer predisposition. However, differences in genotype-phenotype between female and male breast cancers suggest that MBCs have alternate and novel drivers [3–5].
It is now well recognised that aberrant modification of gene expression by promoter methylation is often pathogenic and not an inconsequential contributor to oncogenesis: indeed epigenomic changes are often more commonly observed than gene mutations and chromosomal instability in many cancers [6]. In cancer, aberrant methylation is frequently seen within CpG islands in promoter regions often resulting in transcriptional silencing [7] often occurring early in cancer development. From a clinical perspective, gene methylation may not only contribute to the biological understanding of cancer subsets, but may also be utilised in screening, staging and monitoring of disease activity, as methylation is stable in formalin-fixed paraffin-embedded pathology material and in plasma. Methylated genes may also be attractive treatment targets in MBC using therapies in trials in other tumour types [8].
To date only three MBC studies, composed of a total of 182 male breast cancers, have evaluated methylation in MBCs, which showed that promoter gene methylation in MBC, as compared to normal male breast tissue, is a common event and associated with a more aggressive phenotype [9–11]. However, the methodologies used are prone to give false positive results and/or are non-quantitative. To address the paucity of data we have performed methylation profiling in a well-characterised series of MBC. Our aims were to 1) determine the frequency and level of methylation of important breast cancer genes in a large cohort of familial MBCs, 2) identify clinicopathological associations, including patient outcome, that may define a biological effect of gene methylation and 3) identify and characterise potential molecular subgroups defined by their methylation patterns with clinicopathological correlation.
Methods
Patient samples
Primary male breast cancers examined in this study were obtained from the Kathleen Cunningham Foundation Consortium (kConFab) breast/ovarian familial cancer repository (Table 1). Cases are accepted into the registry based on a strong family history of breast and ovarian cancer with criteria for admission to the kConFab study as outlined previously [12], with all participants providing informed consent to participate in research studies. Patients were from Australia and New Zealand and diagnosed between 1980 and 2009.
Table 1.
Clinicopathological description of male breast cancers in this study
| Feature | ||
|---|---|---|
| Age (years) | Median = 62.5 | Range: 30–85 |
| Mutation carrier status | ||
| BRCA1 | 3 | 5.0% |
| BRCA2 | 25 | 41.7% |
| BRCAX | 32 | 53.3% |
| Size (mm) | Median = 17 | Range: 2–50 |
| Histological subtype | ||
| Invasive carcinoma - no special type (IC-NST) | 46 | 76.7% |
| Invasive papillary carcinoma | 8 | 13.3% |
| IC-NST with areas of micropapillary | 4 | 6.7% |
| Invasive lobular carcinoma | 2 | 3.3% |
| Grade | ||
| 1 | 2 | 3.3% |
| 2 | 30 | 50.0% |
| 3 | 28 | 46.7% |
| DCIS | ||
| Present | 41 | 68.3% |
| Absent | 15 | 25.0% |
| Unknown | 4 | 6.7% |
| Nodal Status | ||
| N0 | 28 | 46.7% |
| N1 | 20 | 33.3% |
| Nx | 12 | 20.0% |
| Paget’s Disease | ||
| Present | 8 | 13.3% |
| Absent | 44 | 73.3% |
| Unknown | 8 | 13.3% |
| ER status (Allred score) | ||
| Negative (0–4/8) | 2 | 3.3% |
| Positive (5–8/8) | 58 | 96.7% |
| PgR status (allred score) | ||
| Negative (0–4/8) | 8 | 13.3% |
| Positive (5–8/8) | 52 | 86.7% |
| HER2 (SISH) | ||
| Amplified | 5 | 8.3% |
| Non-amplified | 55 | 91.7% |
| Phenotype | ||
| Luminal | 54 | 90.0% |
| HER2 | 5 | 8.3% |
| Basal | 1 | 1.7% |
The flow of patients through the study was according to the REMARK criteria outlined in Additional file 1 [13]. Of the 118 cases within the kConFab registry, 58 cases were excluded due to unavailability of tissue. Sixty cases had sufficient material at an appropriate DNA concentration for methylation testing as outlined below. These cases belonged to three groups: 3 MBCs that arose in BRCA1 mutation carriers, 25 that arose in BRCA2 mutation carriers and 32 that occurred in males from BRCAX families (i.e. where an underlying germline mutation had not been identified).
Clinical parameters, including disease specific survival (DSS) were obtained from referring clinical centres, kConFab questionnaires and state death registries [14, 15]. Information on pedigrees, mutational status and testing were available from the kConFab central registry. Histological classification was based on criteria set by the World Health Organisation 2012 [16] and all slides and pathological records from all cases were reviewed centrally. Immunohistochemistry for estrogen receptor (ERα), progesterone receptor (PgR), basal markers (cytokeratin 5 (CK5), EGFR) and HER2 silver in-situ hybridisation (SISH) was performed as previously reported [4]. Stratification of intrinsic phenotypes was based on Nielsen et al. [17], and placed into luminal (ERα/PgR positive, HER2 negative, CK5 and/or EGFR negative), basal (ER α/PgR and HER2 negative; CK5 and/or EGFR positive), HER2 (HER2 positive) and null/negative (HER2, ERα, PgR, CK5 and EGFR negative) phenotypes. Permission to access the kConFab samples and data was granted by the kConFab Executive Committee (Project #115/07–17). This work was carried out with approval from the Peter MacCallum Cancer Centre Ethics Committee (Project No: 11/61).
Germline BRCA1/2 testing
Mutation testing for BRCA1 and BRCA2 mutations was performed as previously reported [18, 19]. Once the family mutation had been identified, all pathogenic (including splice site) variants of BRCA1 and BRCA2 were genotyped by kConFab in all available family members’ DNA.
DNA extraction
Genomic DNA was extracted from formalin-fixed, paraffin embedded (FFPE) samples. A 3 μM haematoxylin and eosin (H&E) stained slide was cut from FFPE blocks and stained to identify for tumour enriched areas showing >80% tumour purity. From the relevant area on the FFPE block, one to two 2 mm punch biopsy cores were taken. The cores were then dewaxed and hydrated through a decreasing alcohol series. Genomic DNA was then extracted using the DNeasy Tissue kit (Qiagen, Hilden, Germany) following proteinase K digestion at 56 °C for 3 days.
Bisulfite modification
Genomic DNA (600 ng) was bisulfite modified using the MethylEasy™ Xceed kit (Genetic Signatures, North Ryde, Australia) according to the manufacturer’s instructions. The bisulfite modified DNA was eluted into 50 μL of EB buffer. CpGenome™ Universal Methylated DNA (Chemicon/Millipore, Billerica, MA) and whole-genome amplified DNA [20] were used as the fully methylated and unmethylated controls, respectively. DNA methylation standards (10, 25 and 50%) were made by mixing the fully methylated control with the unmethylated DNA control.
Methylation-sensitive high resolution melting (MS-HRM)
Methylation screening was performed using MS-HRM to quantitate methylation in bisulfite-modified samples according to the sequence-dependent thermostability in which the level and presence of homogenous and heterogeneous methylation can be detected [21, 22]. MS-HRM primers were specifically designed to generate short amplicons enabling use in formalin-fixed paraffin embedded (FFPE) samples and are summarised in Additional file 2.
PCR amplification and HRM analysis were performed on the Rotor-Gene 6000 (Corbett, Sydney). Samples were run in duplicate. Conditions for each gene are described in Additional file 2. The reaction was performed using a final volume of 20 μL and the mixture consisted of 1 × PCR buffer (Qiagen, Hilden, Germany), 2.5–4.0 mmol/L of MgCl2, 200 μmol/L of each dNTP, forward and reverse primers, 5 μmol/L of SYTO9 intercalating dye (Invitrogen, Carlsbad, CA), 0.5 U of HotStarTaq DNA polymerase (Qiagen, Hilden, Germany) and 10 ng of bisulfite modified DNA. The methylation level of each DNA sample was determined visually by comparing it against the standard curves. Heterogeneous DNA methylation was defined by melting profiles that did not directly conform to any of the methylation controls due to the formation of heteroduplexes between closely but not identically related single complementary DNA strands. Complexes that complete melting slightly after the unmethylated controls were indicative of low levels of DNA methylation. In contrast, complexes with a late melting profile typically contained more heavily methylated epialleles (Fig. 1).
Fig. 1.
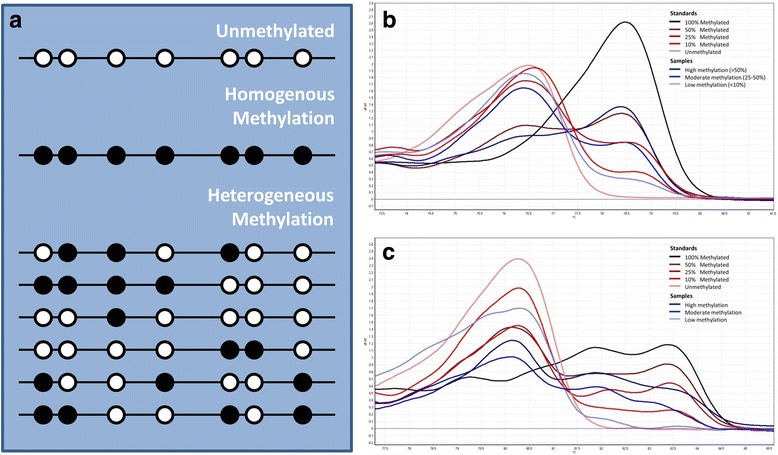
a Schematic representation of an unmethylated sample, homogenously methylated sample and heterogeneously methylated sample (circles represent CpG islands with white indicating unmethylated and black indicating methylated sites), b quantitation of homogenous methylation (RARβ), c quantitation of heterogeneous methylation (RUNX3)
Methylation scoring
A cut-off of 10% methylation was used to primarily exclude low level methylation of uncertain biological significance. The remaining samples were further grouped into moderate methylation (10–50% fully methylated, or moderate heterogenous methylation) and high methylation (>50% fully methylated, or high-level heterogenous methylation) (Fig. 1). Positive methylation (hypermethylation) for each gene was thus considered when duplicate samples showed >10% or moderate to high heterogeneous methylation The samples were also given a percentage methylation for each gene by comparing the methylation to the curves of the standard, which was then averaged across all the genes to give a average methylation index (AMI) scored between 0 and 100% for each tumour sample [23]. The AMI measurement is based on the cumulative methylation index [24], which is the sum of the percentages of methylation of the individual genes, but corrects for the number of genes tested. Using the AMI scores, groups were dichotomised into low and high based on the median AMI as a cut-off point. This analysis does not make assumptions as to the effect of any particular level of methylation.
Cluster analysis
Unsupervised complete linkage clustering was performed with Euclidean metric distance. Unsupervised hierarchical cluster analysis of methylation at each gene was used to detect possible distinct molecular signatures. Analysis was performed using Cluster and Tree View software written by Michael Eisen (Stanford University) as previously published [25–27].
Statistical analysis
Comparison of groups was made with using Mann-Whitney U for non-parametric continuous distributions and Fisher’s exact test for threshold data. Kaplan-Meier survival curves were plotted using breast cancer related death as the endpoint and compared using a log rank test. Pearson’s correlation coefficient was measured for the cluster analysis. Analysis was performed with GraphPad Prism 5 software (GraphPad Prism version 5.04 for Windows, GraphPad Software, La Jolla California USA). A two-tailed P-value test was used in all analyses and a p-value or less than 0.05 was considered statistically significant.
Results
Methylation analysis of MBCs finds associations with genotype and clinico-pathological characteristics
We performed methylation analysis on 60 MBC (25 BRCA2, 3 BRCA1 and 32 BRCAX), whose clinical and pathological features are summarised in Table 1. The features of these cases are consistent with familial male breast cancers in the literature [28], primarily being invasive carcinomas of no special type (76%), ER and PR positive (97% and 87% respectively) and HER2 unamplified (92%). Fifty four (90%), five (8%) and one (2%) tumour(s) were luminal, HER2 and basal phenotypes respectively.
We selected 10 genes for analysis based on their frequency of methylation and/or association with prognosis in previous studies of breast cancer, as follows. Methylation of GSTP1 and RASSF1A is common in MBC [10, 11]. Methylation of WIF1, TWIST, FOXC1, APC, RARb and MAL have also been associated with patient outcome in FBC [29–33]. CDH1, RARB and RUNX3 are frequently methylated in 22–72% [34–36], 20–45% [35, 37, 38] and 50–90% of FBC respectively [39, 40].
GSTP1 was the most commonly methylated gene (82%), followed by RASSF1A (68%), with both showing a pattern of predominantly high level methylation (Table 2). Other genes were more varied: RARβ, APC and RUNX3 had moderate levels of methylation, while heterogeneous methylation was observed in TWIST1, MAL and WIF1, with a mix of moderate and high heterogeneous methylation. Only low level methylation was observed in CDH1 with no cases showing hypermethylation. There were no statistically significant associations of specific gene methylation with patient genotype, however, there were trends for higher methylation frequency of RARβ (44% vs 20%, p = 0.08) and TWIST1 (52% vs 26%, p = 0.06) in BRCA2 carriers. Overall, the BRCA2 group also showed a higher rate of gene hypermethylation (45% vs 34%, p < 0.01) in our target suppressor gene panel than the other groups.
Table 2.
Percentage of cases with hypermethylation
| GSTP1 | RASSF1A | MAL | TWIST | RUNX3 | RARβ | APC | FOXC1 | CDH1 | WIF1 | TOTAL HYPERMETHYLATED GENES | AMI (mean) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BRCA1 (n = 3) | 2 (66%) | 1 (33%) | 1 (33%) | 0 | 1 (33%) | 1 (33%) | 1 (33%) | 1 (33%) | 0 | 1 (33%) | 9 (30%) | 13.4 |
| BRCA2 (n = 25) | 22 (88%) | 20 (80%) | 14 (56%) | 13 (52%) | 8 (32%) | 11 (44%) | 8 (32%) | 6 (24%) | 0 | 11 (44%) | 113 (45%) | 23.6 |
| BRCAX (n = 32) | 25 (78%) | 20 (63%) | 12 (38%) | 9 (28%) | 9 (28%) | 6 (19%) | 7 (22%) | 8 (25%) | 0 | 14 (56%) | 110 (34%) | 17.0 |
| All (n = 60) | 49 (82%) | 41 (68%) | 27 (45%) | 22 (37%) | 18 (30%) | 18 (30%) | 16 (27%) | 15 (25%) | 0 | 26 (43%) | 232 (39%) | 14.0 |
| p = 0.06 | p = 0.08 | P < 0.01 | p = 0.01 |
We examined the association of specific gene methylation with patient and tumour characteristics (Table 3). APC hypermethylation was significantly associated with older age (69.1 years vs 60.4 years, p = 0.01, Table 2) whereas MAL hypermethylation was significantly inversely associated with age (59.1 years vs 65.7 years, p = 0.04). Significantly larger tumour size was noted for cases with RARβ hypermethylation (median 22.3 mm vs 16.5 mm; p = 0.01). RARβ hypermethylation was also associated with a higher percentage of Paget’s disease (31% vs 8%, p = 0.04). RUNX3 hypermethylation was associated with increased frequency of IC-NST histological type (94% vs 69%, p = 0.046) and RASSF1A hypermethylation associated with the coexistence of high grade DCIS (33% vs 6% (p = 0.02).
Table 3.
Correlation of hypermethylation with clinicopathological variables (associations approaching significance, p < 0.05 in bold)
| GSTP1 | RASSF1A | MAL | RUNX3 | RARβ | APC | FOXC1 | AMI (median) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypermethylation | + | - | + | - | + | - | + | - | + | - | + | - | + | - | > | < |
| Age (years) | 59.1 | 65.7 | 67.2 | 60.9 | 69.1 | 60.4 | ||||||||||
| p-value | 0.04 | 0.07 | 0.01 | |||||||||||||
| Tumour size (mm) | 22.3 | 16.5 | 21.4 | 17.1 | 20.8 | 15.8 | ||||||||||
| p-value | 0.01 | 0.08 | 0.02 | |||||||||||||
| IC-NST Histology | 94% | 69% | ||||||||||||||
| p-value | 0.046 | |||||||||||||||
| Grade 3 | 51% | 18% | ||||||||||||||
| p-value | 0.09 | |||||||||||||||
| Paget’s Disease | 31% | 8% | ||||||||||||||
| p-value | 0.04 | |||||||||||||||
| DCIS present | 33% | 6% | ||||||||||||||
| p-value | 0.02 | |||||||||||||||
| Lymphovascular invasion | 49% | 18% | 20% | 51% | 53% | 40% | ||||||||||
| p-value | 0.09 | 0.07 | 0.09 | |||||||||||||
| Perineural invasion | 63% | 36% | ||||||||||||||
| p-value | 0.07 | |||||||||||||||
| Node positive | 52% | 24% | 62% | 34% | ||||||||||||
| p-value | 0.08 | 0.07 | ||||||||||||||
| HER2 positive | 13% | 0 | ||||||||||||||
| p-value | 0.11 | |||||||||||||||
High overall levels of methylation have been associated with aggressive tumour features such as mitotic count, grade and poor patient outcome in MBC [10] and FBC [30, 41]. Therefore, we calculated a measure of overall methylation for each sample, the AMI. There was a significant increase in AMI in BRCA2 mutation carriers compared with other MBCs (23.6 vs 16.6, p = 0.01, Fig. 2). In addition, the AMI was positively correlated with tumour size (median 22.4 mm vs 15.4 mm, p = 0.02).
Fig. 2.
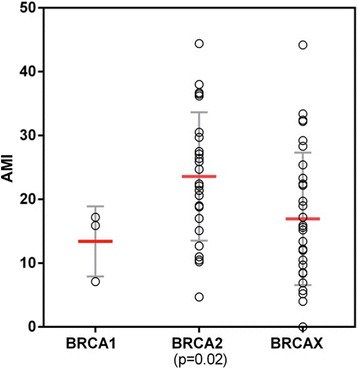
Average methylation index (AMI) for samples stratified by BRCA status (Central bar – median, error bars = 1 standard deviation)
Cluster analysis identifies subgroups of MBC
In order to evaluate whether methylation profiles could discover novel subgroups in MBC, as has been seen for FBC [42, 43] and colorectal cancer [44], we performed an unsupervised clustering analysis. Four main clusters with at least 7 samples in each group were identified (Fig. 3). MBCs arising in BRCA2 carriers showed a significantly greater frequency (6/7 vs 19/53, p = 0.02) of Cluster 3 membership (characterised by RASSF1A, WIF1, GSTP1 and RARβ methylation). No other clinicopathological association or prognostic differences were seen between the clusters.
Fig. 3.
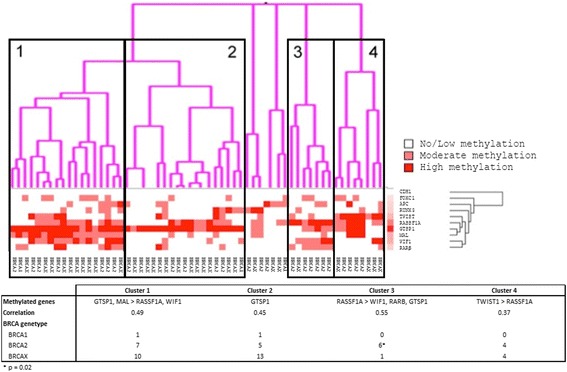
Unsupervised cluster analysis of methylation amongst male breast cancers (gradation is seen in the shading between white (no methylation) and red (high methylation))
Analysis of methylation patterns within the BRCA2 subgroup of tumours showed two clusters with correlation coefficients >0.8) (Additional file 3). Cluster A contained 12 tumours and was characterised by high GSTP1 methylation and MAL methylation and relatively lower RASSF1A methylation. Cluster B contained 8 tumours and showed primarily high RASSF1A methylation. Cluster A tumours showed an earlier age at diagnosis than other BRCA2 tumours. Other variables did not align to one or the other cluster. Analysis of BRCAX tumours by cluster analysis showed only very small clusters of 6 or less patients with a correlation coefficient above 0.8 (Additional file 3).
A high average methylation index and TWIST1 hypermethylation associated with worse disease specific survival
Both a high AMI (HR:3.3, 95% CI:1.3–7.0, p = 0.01) and hypermethylation of TWIST1 (HR:3.7, 95% CI:2.0–12.9, p = 0.001) were adverse features for disease specific survival (Fig. 4) with TWIST1 methylation (HR:4.7, 95% CI:2.0–27.5, p = 0.01) also being associated with a significantly shorter survival in the BRCA2 MBC subgroup. Because BRCA2 tumours have higher methylation overall and also worse survival than other MBC cohorts [45, 46], we also evaluated survival within the BRCA2 carriers, and observed a trend towards worse outcome with higher AMI in this sub-group (HR:3.3, 95% CI: 0.8–9.7, p = 0.1). Hypermethylation of FOXC1 (HR:2.3, 95% CI:0.99–8.1, p = 0.053) showed a strong trend towards worse DSS; hypermethylation of other genes showed no prognostic information. No significant association with progression-free survival was detected for any gene or AMI. Multivariate analysis was not performed due to inadequate numbers of cases.
Fig. 4.
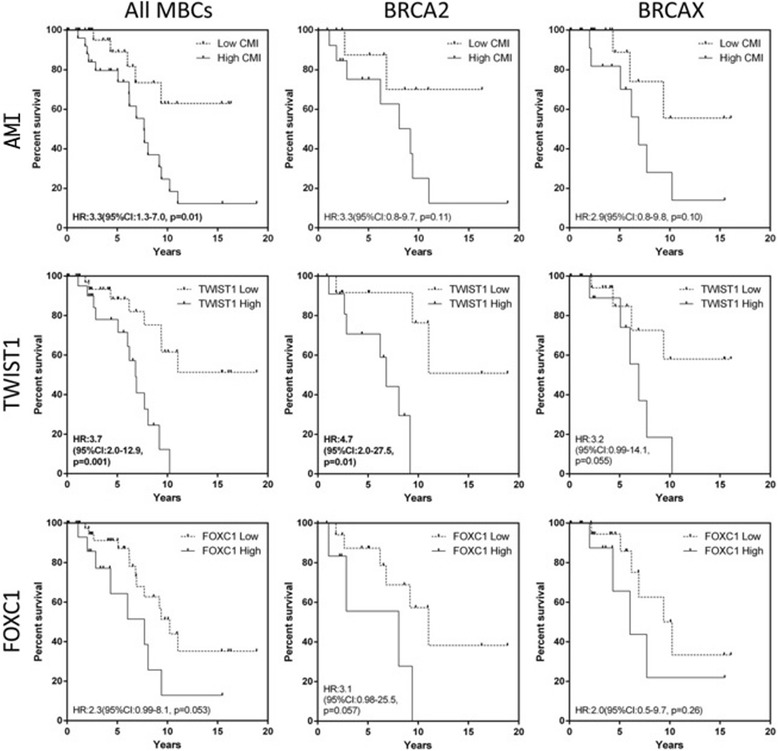
Disease specific survival of average methylation index (AMI), TWIST1 and FOXC1 in all male breast cancers and within the BRCA2 and BRCAX subgroups
Discussion
Aberrant methylation of promoter regions of tumour suppressor genes has been shown to be a frequent mechanism of gene silencing in most cancers, including breast cancers [47–49]. In many instances, this is observed in adjacent normal tissues or in pre-invasive lesions [50]. Perhaps best seen in colorectal cancer [51], subsets may demonstrate methylation patterns with clinical relevance.
We have used methylation sensitive high-resolution melting analysis of methylation as it has been demonstrated to be highly sensitive, robust and effective in evaluating FFPE tissue, able to differentiate and semi-quantitate homogenous and heterogeneous methylation [22, 52]. This current comparative study is the largest to examine methylation using a robust technology of well characterised and acknowledged tumour suppressor genes shown to be methylated and important in the pathogenesis FBC, in a clinically well annotated cohort of familial male breast cancers with known mutation status. We have identified frequent promoter hypermethylation (≥30%) in GSTP1, RASSF1A, MAL, TWIST, RUNX3, and RARβ, and identified significant associations with clinico-pathological features in five of the genes assayed. One caveat to some of these associations is that the small sample size and their level of statistical significance close to the p < 0.05 threshold may mean that false positive results are included due to the multiple tests performed.
Currently there are only three published methylation studies in a total of 182 male breast cancers. Of the genes we investigated only methylation at GSTP1, RARβ and RASSF1A have been individually assessed, The largest study by Kornegoor et al. [10] examined candidate methylation of 25 genes in 108 MBCs by methylation specific multiplex ligation dependent probe amplification (MS-MLPA), detecting methylation in RARβ (5%) and GSTP1 (44%), somewhat lower than our results. This study did not segregate MBC into sporadic and familial groups, which have been shown to contain distinct geno-phenotypic characteristics and may explain the difference in frequency observed. The second study by Pinto et al. [11] evaluated RASSF1A (76%) and RARβ (8%) in 27 familial MBCs using quantitative methyl-specific PCR. The lower frequency of RARβ hypermethylation observed may be explained by the lower proportion of BRCA2 cases included (3/27 compared to 25/60 in our cohort). Consistent with this possibility we observed a trend for RARβ methylation to be higher in BRCA2 cases. Finally, Johanssen et al. [9] performed genome-wide methylation profiling in 47 MBCs, and identified two clusters of cases; unfortunately germline mutation status was only available for 8 cases.
One of the most striking findings in this study is the high frequency of GSTP1 methylation (82%), which has not been noted before. GSTP1 encodes for glutathionine S transferase P [53] and may be a critical gene in the development of familial MBCs. Very high levels of GSTP1 methylation are also seen in prostate cancer, which is another male cancer that can be associated with BRCA2 mutation [54, 55]. We noted high levels of GSTP1 methylation in both BRCA2 (88%) and BRCAX tumours (78%), well above that noted by Kornegoor et al. (44%) and that reported in FBCs (generally <60%) [56, 57]. The reason for this result is unlikely to be assay related, as using the same methodology we have shown similar levels of methylation in FBC to that reported in the literature. There are two other possibilities. Firstly, GSTP1 methylation may be ERβ mediated as studies of prostate cancer lines show that the ERβ/eNOS complex causes GSTP1 repression by local chromatin remodelling following recruitment to estrogen responsive elements [58]. Secondly, GSTP1 functions as a caretaker gene [53, 58, 59] with its loss resulting in increased oxidative DNA damage and mutagenesis, thus, in BRCA2 deficient cancers already sensitive to oxidative stress [60], any loss of GSTP1 may have a more pronounced effect and be integral in tumour development.
We also noted overall methylation differences between the BRCA2 and BRCAX subgroups further supporting previous studies showing a possible BRCA2 MBC subset. In female BRCA2 carriers, promoter hypermethylation has also been shown to be elevated compared to non-familial and BRCA1 carriers [49, 61]. Methylation profiling of FBC was able to discriminate BRCA1, BRCA2 and two subsets of BRCAX tumours [61]. This study is the first to report on methylation of male breast cancers arising in BRCA1 mutation carriers. These tumours are rare, and while we only have three cases within our cohort, this is a novel group. We were unable to see a significant correlation between gene hypermethylation and BRCA1 status but did observe the lowest levels of methylation of all the groups, mirroring the findings seen in BRCA1 associated female breast cancer. Further investigation of this rare subgroup is warranted.
This high level of methylation could potentially be used for screening in BRCA2 male carriers as methylation is not seen in normal tissues, serum or plasma of normal individuals but can be detected in blood. GSTP1 may be the prime candidate as studies evaluating its use as a biomarker for prostate cancer are well advanced.
To aid the above possible screening strategies we have developed an index of methylation (AMI) to investigate the quanta of methylation. We observed that AMI correlated with larger tumour size and shorter disease specific survival suggesting that either a stochastic accumulation of methylation and/or a methylator phenotype leads to a more aggressive tumour, as observed in the study of Kornegoor et al. [10]. Similarly, Johansson et al. [9] found that a highly methylated MBC subgroup was more proliferative and showed a trend towards worse patient outcome. In sporadic FBC conflicting results regarding methylation and survival have been found, with higher methylation subgroups showing either improved prognosis [43] or poor overall survival [62]. These differences are perhaps explained by the influence of the intrinsic subtypes, which show distinct methylation patterns and patient outcome [49]. The association between multi-gene hypermethylation and outcome in familial FBC does not appear to have been evaluated. Notably, in our cohort a high AMI maintained a trend towards prognostic significance in BRCA2 tumours further suggesting that as above, methylation has particular biological importance in this subset of tumours.
Conclusions
We have shown that tumour promoter methylation within our target suppressor gene panel is commonly observed in familial and particularly BRCA2 male breast cancers suggesting aberrant hypermethylation may be a significant driver in MBCs carrying prognostic information. In addition, the presence of specific methylation patterns particular to MBC subtypes such as BRCA2 carriers further supports emerging evidence suggesting the presence of unique and distinct MBC subsets that differ from other MBC subgroups and from FBC.
Additional files
REMARK patient flow through study (XLSX 34 kb)
Methylation specific high resolution melting condition and primers (XLSX 36 kb)
a) BRCA2 subgroup cluster analysis, b) BRCAX subgroup cluster analysis, c) Numbers and sizes of clusters within BRCA2 and BRCAX subgroups using various correlation coefficient cut-offs (listed on the x-axis), d) age of diagnosis of patient within Cluster A, B and other BRCA2 tumours (DOCX 234 kb)
Acknowledgements
We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow up Study (National Breast Cancer Foundation and Cancer Australia #628333) for their contributions to this resource, and the many families who contribute to kConFab.
Funding
kConFab is supported by grants from the National Breast Cancer Foundation, the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Siddhartha Deb received a postgraduate scholarship from the NHMRC. Funding from the National Breast Cancer Foundation (AD), Victorian Cancer Agency (KG), the Cancer Council of Victoria (AD) and Cancer Australia also supported this study.
Abbreviations
- AMI
Average methylation index
- CK5
Cytokeratin 5
- DCIS
Ductal carcinoma in situ
- DSS
Disease specific survival
- ERα
Estrogen receptor
- FBC
Female breast cancer
- FFPE
Formalin-fixed, paraffin embedded
- H&E
Haematoxylin and eosin
- IC-NST
Invasive carcinomas of no special type
- kConFab
Kathleen Cuningham Foundation Consortium
- MBC
Male breast cancer
- MS-HRM
Methylation-sensitive high resolution melting
- MS-MLPA
Methylation-specific multiplex ligation dependent probe amplification
- PgR
Progesterone receptor
- QMSP
Quantitative methyl-specific PCR
Authors’ contributions
SD – Project conceptualization, DNA extraction and performing methylation assays, data analysis, preparation of manuscript, KLG – data interpretation, preparation of manuscript, JMP - Preparation of standards and performing methylation assays, ET – Performing methylation assay, kConFab Investigators – preparation of clinical data, AD – Project and assay design, technical supervision, manuscript review, SBF – Project conceptualization, manuscript preparation and review. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This work was carried out with approval from the Peter MacCallum Cancer Centre Ethics Committee (Project No: 11/61). All patients provided written informed consent for the use of their tissue and data.
Consent for publication
not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-017-3632-7) contains supplementary material, which is available to authorized users.
Contributor Information
Siddhartha Deb, Email: deb_siddhartha@hotmail.com.
Kylie L. Gorringe, Email: kylie.gorringe@petermac.org
Jia-Min B. Pang, Email: jiaminpang@gmail.com
David J. Byrne, Email: david.byrne@petermac.org
Elena A. Takano, Email: elena.takano@petermac.org
kConFab Investigators, Email: heather.thorne@petermac.org.
Alexander Dobrovic, Email: alex.dobrovic@onjcri.org.au.
Stephen B. Fox, Phone: +613 855 98422, Email: stephen.fox@petermac.org
References
- 1.Korde LA, Zujewski JA, Kamin L, Giordano S, Domchek S, Anderson WF, Bartlett JM, Gelmon K, Nahleh Z, Bergh J, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol. 2010;28(12):2114–2122. doi: 10.1200/JCO.2009.25.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss JR, Moysich KB, Swede H. Epidemiology of male breast cancer. Cancer Epidemiol Biomark Prev. 2005;14(1):20–26. [PubMed] [Google Scholar]
- 3.Deb S, Do H, Byrne D, Jene N, kConFab I, Dobrovic A, Fox SB. PIK3CA mutations are frequently observed in BRCAX but not BRCA2-associated male breast cancer. Breast Cancer Res. 2013;15(4):R69. doi: 10.1186/bcr3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deb S, Jene N, Fox SB. Genotypic and phenotypic analysis of familial male breast cancer shows under representation of the HER2 and basal subtypes in BRCA-associated carcinomas. BMC Cancer. 2012;12:510. doi: 10.1186/1471-2407-12-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deb S, Johansson I, Byrne D, Nilsson C, Investigators K, Constable L, Fjallskog ML, Dobrovic A, Hedenfalk I, Fox SB. nuclear HIF1A expression is strongly prognostic in sporadic but not familial male breast cancer. Mod Pathol. 2014;27(9):1223–1230. doi: 10.1038/modpathol.2013.231. [DOI] [PubMed] [Google Scholar]
- 6.Heichman KA, Warren JD. DNA methylation biomarkers and their utility for solid cancer diagnostics. Clin Chem Lab Med. 2012;50(10):1707–1721. doi: 10.1515/cclm-2011-0935. [DOI] [PubMed] [Google Scholar]
- 7.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 8.Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res. 2007;13(6):1634–1637. doi: 10.1158/1078-0432.CCR-06-2076. [DOI] [PubMed] [Google Scholar]
- 9.Johansson I, Lauss M, Holm K, Staaf J, Nilsson C, Fjallskog ML, Ringner M, Hedenfalk I. Genome methylation patterns in male breast cancer - identification of an epitype with hypermethylation of polycomb target genes. Mol Oncol. 2015;9(8):1565–1579. doi: 10.1016/j.molonc.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kornegoor R, Moelans CB, Verschuur-Maes AH, Hogenes M, de Bruin PC, Oudejans JJ, van Diest PJ. Promoter hypermethylation in male breast cancer: analysis by multiplex ligation-dependent probe amplification. Breast Cancer Res. 2012;14(4):R101. doi: 10.1186/bcr3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto R, Pilato B, Ottini L, Lambo R, Simone G, Paradiso A, Tommasi S. Different methylation and microRNA expression pattern in male and female familial breast cancer. J Cell Physiol. 2013;228(6):1264–1269. doi: 10.1002/jcp.24281. [DOI] [PubMed] [Google Scholar]
- 12.Mann GJ, Thorne H, Balleine RL, Butow PN, Clarke CL, Edkins E, Evans GM, Fereday S, Haan E, Gattas M, et al. Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Res. 2006;8(1):R12. doi: 10.1186/bcr1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Reporting recommendations for tumor marker prognostic studies. J Clin Oncol. 2005;23(36):9067–9072. doi: 10.1200/JCO.2004.01.0454. [DOI] [PubMed] [Google Scholar]
- 14.Suter CM, Martin DI, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. 2004;36(5):497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
- 15.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 16.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJE. WHO classification of Tumours of the breast: IARC: Lyon. 2012. [Google Scholar]
- 17.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 18.Hogervorst FB, Nederlof PM, Gille JJ, McElgunn CJ, Grippeling M, Pruntel R, Regnerus R, van Welsem T, van Spaendonk R, Menko FH, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63(7):1449–1453. [PubMed] [Google Scholar]
- 19.Lum A, Le Marchand L. A simple mouthwash method for obtaining genomic DNA in molecular epidemiological studies. Cancer Epidemiol Biomark Prev. 1998;7(8):719–724. [PubMed] [Google Scholar]
- 20.Kristensen LS, Mikeska T, Krypuy M, Dobrovic A. Sensitive melting analysis after real time- methylation specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. Nucleic Acids Res. 2008;36(7):e42. doi: 10.1093/nar/gkn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikeska T, Candiloro IL, Dobrovic A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics. 2010;2(4):561–573. doi: 10.2217/epi.10.32. [DOI] [PubMed] [Google Scholar]
- 22.Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007;35(6):e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang JM, Deb S, Takano EA, Byrne DJ, Jene N, Boulghourjian A, Holliday A, Millar E, Lee CS, O'Toole SA, et al. Methylation profiling of ductal carcinoma in situ and its relationship to histopathological features. Breast Cancer Res. 2014;16(5):423. doi: 10.1186/s13058-014-0423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fackler MJ, Malone K, Zhang Z, Schilling E, Garrett-Mayer E, Swift-Scanlan T, Lange J, Nayar R, Davidson NE, Khan SA, et al. Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clin Cancer Res. 2006;12(11 Pt 1):3306–3310. doi: 10.1158/1078-0432.CCR-05-2733. [DOI] [PubMed] [Google Scholar]
- 25.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makretsov NA, Huntsman DG, Nielsen TO. Yorida E, Peacock M, Cheang MC, Dunn SE, Hayes M, van de Rijn M, Bajdik C, et al. Hierarchical clustering analysis of tissue microarray immunostaining data identifies prognostically significant groups of breast carcinoma. Clin Cancer Res. 2004;10(18 Pt 1):6143–6151. doi: 10.1158/1078-0432.CCR-04-0429. [DOI] [PubMed] [Google Scholar]
- 27.van de Rijn M, Gilks CB. Applications of microarrays to histopathology. Histopathology. 2004;44(2):97–108. doi: 10.1111/j.1365-2559.2004.01766.x. [DOI] [PubMed] [Google Scholar]
- 28.Deb S, Lakhani SR, Ottini L, Fox SB. The cancer genetics and pathology of male breast cancer. Histopathology. 2016;68(1):110–118. doi: 10.1111/his.12862. [DOI] [PubMed] [Google Scholar]
- 29.Cheol Kim D, Thorat MA, Lee MR, Cho SH, Vasiljevic N, Scibior-Bentkowska D, Wu K, Ahmad AS, Duffy S, Cuzick JM, et al. Quantitative DNA methylation and recurrence of breast cancer: a study of 30 candidate genes. Cancer Biomark. 2012;11(2–3):75–88. doi: 10.3233/CBM-2012-0266. [DOI] [PubMed] [Google Scholar]
- 30.Cho YH, Shen J, Gammon MD, Zhang YJ, Wang Q, Gonzalez K, Xu X, Bradshaw PT, Teitelbaum SL, Garbowski G, et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res Treat. 2012;131(1):197–205. doi: 10.1007/s10549-011-1712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejeux E, Ronneberg JA, Solvang H, Bukholm I, Geisler S, Aas T, Gut IG, Borresen-Dale AL, Lonning PE, Kristensen VN, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horne HN, Lee PS, Murphy SK, Alonso MA, Olson JA, Jr., Marks JR: Inactivation of the MAL gene in breast cancer is a common event that predicts benefit from adjuvant chemotherapy. Mol Cancer Res 2009, 7(2):199–209. [DOI] [PMC free article] [PubMed]
- 33.Trifa F, Karray-Chouayekh S, Jmal E, Jmaa ZB, Khabir A, Sellami-Boudawara T, Frikha M, Daoud J, Mokdad-Gargouri R. Loss of WIF-1 and Wnt5a expression is related to aggressiveness of sporadic breast cancer in Tunisian patients. Tumour Biol. 2013;34(3):1625–1633. doi: 10.1007/s13277-013-0694-2. [DOI] [PubMed] [Google Scholar]
- 34.Caldeira JR, Prando EC, Quevedo FC, Neto FA, Rainho CA, Rogatto SR. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006;6:48. doi: 10.1186/1471-2407-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parrella P, Poeta ML, Gallo AP, Prencipe M, Scintu M, Apicella A, Rossiello R, Liguoro G, Seripa D, Gravina C, et al. Nonrandom distribution of aberrant promoter methylation of cancer-related genes in sporadic breast tumors. Clin Cancer Res. 2004;10(16):5349–5354. doi: 10.1158/1078-0432.CCR-04-0555. [DOI] [PubMed] [Google Scholar]
- 36.Sebova K, Zmetakova I, Bella V, Kajo K, Stankovicova I, Kajabova V, Krivulcik T, Lasabova Z, Tomka M, Galbavy S, et al. RASSF1A and CDH1 hypermethylation as potential epimarkers in breast cancer. Cancer Biomark. 2011;10(1):13–26. doi: 10.3233/CBM-2012-0230. [DOI] [PubMed] [Google Scholar]
- 37.Park SY, Kwon HJ, Choi Y, Lee HE, Kim SW, Kim JH, Kim IA, Jung N, Cho NY, Kang GH. Distinct patterns of promoter CpG island methylation of breast cancer subtypes are associated with stem cell phenotypes. Mod Pathol. 2012;25(2):185–196. doi: 10.1038/modpathol.2011.160. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Xu X, Liu J, Liu H, Fu L, Gu L. Epigenetic regulation of retinoic acid receptor beta2 gene in the initiation of breast cancer. Med Oncol. 2011;28(4):1311–1318. doi: 10.1007/s12032-010-9685-y. [DOI] [PubMed] [Google Scholar]
- 39.Lau QC, Raja E, Salto-Tellez M, Liu Q, Ito K, Inoue M, Putti TC, Loh M, Ko TK, Huang C, et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006;66(13):6512–6520. doi: 10.1158/0008-5472.CAN-06-0369. [DOI] [PubMed] [Google Scholar]
- 40.Subramaniam MM, Chan JY, Soong R, Ito K, Ito Y, Yeoh KG, Salto-Tellez M, Putti TC. RUNX3 inactivation by frequent promoter hypermethylation and protein mislocalization constitute an early event in breast cancer progression. Breast Cancer Res Treat. 2009;113(1):113–121. doi: 10.1007/s10549-008-9917-4. [DOI] [PubMed] [Google Scholar]
- 41.Sharma G, Mirza S, Yang YH, Parshad R, Hazrah P, Datta Gupta S, Ralhan R. Prognostic relevance of promoter hypermethylation of multiple genes in breast cancer patients. Cell Oncol. 2009;31(6):487–500. doi: 10.3233/CLO-2009-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3(75):75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 45.Gargiulo P, Pensabene M, Milano M, Arpino G, Giuliano M, Forestieri V, Condello C, Lauria R, De Placido S. Long-term survival and BRCA status in male breast cancer: a retrospective single-center analysis. BMC Cancer. 2016;16:375. doi: 10.1186/s12885-016-2414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwiatkowska E, Teresiak M, Filas V, Karczewska A, Breborowicz D, Mackiewicz A. BRCA2 mutations and androgen receptor expression as independent predictors of outcome of male breast cancer patients. Clin Cancer Res. 2003;9(12):4452–4459. [PubMed] [Google Scholar]
- 47.Bardowell SA, Parker J, Fan C, Crandell J, Perou CM, Swift-Scanlan T. Differential methylation relative to breast cancer subtype and matched normal tissue reveals distinct patterns. Breast Cancer Res Treat. 2013;142(2):365–380. doi: 10.1007/s10549-013-2738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng W, Shen L, Wen S, Rosen DG, Jelinek J, Hu X, Huan S, Huang M, Liu J, Sahin AA, et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007;9(4):R57. doi: 10.1186/bcr1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jonsson G, Olsson H, Borg A, Ringner M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12(3):R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pang JMB, Dobrovic A, Fox SB. DNA methylation in ductal carcinoma in situ of the breast. Breast Cancer Res. 2013;15(3):206. doi: 10.1186/bcr3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deb S, Fox SB. Molecular profiling in colorectal cancer: current state of play and future directions. Colorectal Cancer. 2014;3(1):41–56. doi: 10.2217/crc.13.82. [DOI] [Google Scholar]
- 52.Dobrovic A, Kristensen LS. DNA methylation, epimutations and cancer predisposition. Int J Biochem Cell Biol. 2009;41(1):34–39. doi: 10.1016/j.biocel.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Nebert DW, Vasiliou V. Analysis of the glutathione S-transferase (GST) gene family. Hum Genomics. 2004;1(6):460–464. doi: 10.1186/1479-7364-1-6-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91(24):11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Millar DS, Ow KK, Paul CL, Russell PJ, Molloy PL, Clark SJ. Detailed methylation analysis of the glutathione S-transferase pi (GSTP1) gene in prostate cancer. Oncogene. 1999;18(6):1313–1324. doi: 10.1038/sj.onc.1202415. [DOI] [PubMed] [Google Scholar]
- 56.Buyru N, Altinisik J, Ozdemir F, Demokan S, Dalay N. Methylation profiles in breast cancer. Cancer Investig. 2009;27(3):307–312. doi: 10.1080/07357900802350814. [DOI] [PubMed] [Google Scholar]
- 57.Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998;58(20):4515–4518. [PubMed] [Google Scholar]
- 58.Re A, Aiello A, Nanni S, Grasselli A, Benvenuti V, Pantisano V, Strigari L, Colussi C, Ciccone S, Mazzetti AP, et al. Silencing of GSTP1, a prostate cancer prognostic gene, by the estrogen receptor-beta and endothelial nitric oxide synthase complex. Mol Endocrinol. 2011;25(12):2003–2016. doi: 10.1210/me.2011-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione-S-transferase gene (GSTP1) in prostate cancer cells. Oncogene. 2002;21(7):1048–1061. doi: 10.1038/sj.onc.1205153. [DOI] [PubMed] [Google Scholar]
- 60.Fridlich R, Annamalai D, Roy R, Bernheim G, Powell SN. BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair (Amst) 2015;30:11–20. doi: 10.1016/j.dnarep.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flanagan JM, Cocciardi S, Waddell N, Johnstone CN, Marsh A, Henderson S, Simpson P, da Silva L, kConFab I, Khanna K, et al. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Am J Hum Genet. 2010;86(3):420–433. doi: 10.1016/j.ajhg.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, Tse CK, Swift-Scanlan T, Geradts J, Troester MA, et al. DNA methylation profiling in the Carolina breast cancer study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res. 2014;16(5):450. doi: 10.1186/s13058-014-0450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
REMARK patient flow through study (XLSX 34 kb)
Methylation specific high resolution melting condition and primers (XLSX 36 kb)
a) BRCA2 subgroup cluster analysis, b) BRCAX subgroup cluster analysis, c) Numbers and sizes of clusters within BRCA2 and BRCAX subgroups using various correlation coefficient cut-offs (listed on the x-axis), d) age of diagnosis of patient within Cluster A, B and other BRCA2 tumours (DOCX 234 kb)