

Abstract
Brugada syndrome (BrS) is an autosomal dominant inherited channelopathy. It is associated with a typical pattern of ST-segment elevation in the precordial leads V1–V3 and potentially lethal ventricular arrhythmias in otherwise healthy patients. It is frequently seen in young Asian males, in whom it has previously been described as sudden unexplained nocturnal death syndrome. Although it typically presents in young adults, it is also known to present in children and infants, especially in the presence of fever. Our understanding of the genetic pathogenesis and management of BrS has grown substantially considering that it has only been 24 years since its first description as a unique clinical entity. However, there remains much to be learned, especially in the pediatric population. This review aims to discuss the epidemiology, genetics, and pathogenesis of BrS. We will also discuss established standards and new innovations in the diagnosis, prognostication, risk stratification, and management of BrS. Literature search was run on the National Center for Biotechnology Information's website, using the Medical Subject Headings (MeSH) database with the search term “Brugada Syndrome” (MeSH), and was run on the PubMed database using the age filter (birth–18 years), yielding 334 results. The abstracts of all these articles were studied, and the articles were categorized and organized. Articles of relevance were read in full. As and where applicable, relevant references and citations from the primary articles were further explored and read in full.
Keywords: Brugada syndrome, Brugada pattern, children
INTRODUCTION AND HISTORY
In 1987 in Maastricht, Netherlands, Professor Pedro Brugada and his brother Josep encountered a 3-year-old Polish patient with recurrent cardiac arrest whose sister had died suddenly at the same age. The peculiar electrocardiographic (ECG) findings of these children were unique and heretofore undescribed. In 1992, the brothers published a seminal case series of eight patients with recurrent aborted sudden death due to polymorphic ventricular tachycardia (VT), who had structurally normal hearts and distinct ECG findings that we now recognize as the Brugada syndrome (BrS).[1]
BrS is an autosomal dominant inherited channelopathy. It is associated with a typical pattern of ST-segment elevation in the precordial leads V1–V3 and potentially lethal ventricular arrhythmias (VA) in otherwise healthy patients. It is frequently seen in young Asian males, in whom it has previously been described as sudden unexplained nocturnal death syndrome.[2] Although it typically presents in young adults, it is also known to present in children and infants, especially in the presence of fever.
This review aims to discuss the epidemiology, genetics, and pathogenesis of BrS. We will also discuss established standards and new innovations in the diagnosis, prognostication, risk stratification, and management of BrS.
DISEASE BURDEN
Since ECG findings are a requisite for the diagnosis of BrS, it is hard to know the true prevalence of BrS as presumably many patients are asymptomatic. Different studies propose varying prevalence rates ranging from 1 in 5000[3] to 1 in 2000.[4] BrS is thought to be responsible for 4–12% of sudden cardiac death in children and young athletes.[5] In some studies, it is implicated in 20% of sudden unexplained death in the young.[3] 10%–20% of sudden infant death syndrome is due to identifiable inherited channelopathies including BrS.[3,6]
There are now many population studies from different sources around the globe. Among adult studies, the incidence of Brugada pattern on ECG has ranged from 0.12% to 0.8%.[7,8,9,10] There is a strong male predominance, with BrS being about 8–10 times more prevalent in adult males.[4] In adults with BrS, there is a 10%–30% risk of cardiac arrest which is more likely in symptomatic patients.[11,12]
The rarity of this disease in the pediatric population can be surmised by the fact that the largest retrospective study in children conducted across 13 tertiary hospitals in three countries dating back 15 years (since the discovery of BrS) yielded only 30 patients.[13] In large studies of asymptomatic children from Japan, BrS ECG pattern was found in 0.01%–0.02%,[14,15] suggesting that BrS may exist in children but becomes clinically unmasked with increasing age. Indeed, 23% of asymptomatic children who were first-degree relatives of patients with BrS demonstrated abnormal ECG on ajmaline challenge after puberty despite having negative testing before puberty.[16] The strong male predominance seen in adults is not reflected in pediatric studies, where there is at most a mild male predominance.[13,17] The incidence of life-threatening arrhythmias in children with BrS is around 10%.[13]
GENETICS
From the initial descriptions of BrS with its occurrence in family members and certain ethnic groups, it seemed likely that BrS was an inheritable disorder. Just 6 years after the publication of the Brugada case series, Chen et al. uncovered loss of function mutations in the sodium channel gene SCN5A associated with idiopathic ventricular fibrillation (VF).[18] Since then, hundreds of mutations in the SCN5A gene (which encodes α subunit of the Nav1.5 voltage-gated sodium channel) has been described in association with BrS.[19] Eleven percent of adults and 21% of children have double mutations.[20,21] It is also well described that there are silent carriers of BrS mutations up to 40% in some cohorts.[21]
While mutations in SCN5A represent the largest proportion of identifiable mutations (15%–30%), there are other minor genes that have been implicated more recently. These include loss of function mutations in SCN1B (which encodes the β1 subunit of the Nav1.5 voltage-gated sodium channel),[22] glycerol-3-phosphate-dehydrogenase-1-like gene resulting in reduced inward sodium current,[23,24] and CACNA1C, CACNB2B, and CACNA2D1 (encoding subunits of cardiac L-type calcium channel).[25] Newer reports of gain of function mutations in KCNE3[26] and KCND3[27] (associated with outward potassium current channel), KCNJ8 causing increase in IKATP current, and HCN4 causing reduction of pacemaker If current have been associated with BrS.[28] There is recent evidence linking fibroblast growth factor gene 12 with the loss of function in Nav1.5.[29]
However, it is estimated that only 11%–30% of patients with BrS have identifiable genetic defects, suggesting that there is a vast expanse of genetic abnormality linked to BrS that we are yet to chart.[2,11,19,30]
CLINICAL PRESENTATION
Patients with BrS may present with palpitations, syncope, aborted or completed sudden death, including some cases of sudden infant death. VF or polymorphic VT may be documented. The most common initial presentation in children is a family history of BrS (47%), followed by incidental ECG finding (25%), syncope (14%), arrhythmias (13%), and aborted sudden death (1%).[31] This reflects the general proportion of presentation in adult studies.[32] Most syncopal episodes occur at rest and are often precipitated by fever (45%), including vaccination-related fever episodes.[13,20] Palpitations related to atrial fibrillation (AFib) may be the initial presentation in some cases.[33]
Other identifiable causes of sudden death or aborted sudden death including long QT syndrome, catecholaminergic polymorphic VT, short QT syndrome, idiopathic VT, and idiopathic VF should be considered in the differential diagnosis and excluded during the workup.
DIAGNOSIS
It is important to distinguish patients with BrS from those with the Brugada pattern. Diagnosis of BrS requires having the typical Brugada ECG pattern with clinical symptoms (usually suggesting VAs). Patients with typical ECG pattern of BrS without clinical symptoms are considered to have the Brugada pattern.
The Brugada pattern can also be induced by medications and toxins including sodium channel blockers, beta-blockers, tricyclic antidepressants, local anesthetics, alcohol, and cocaine. The clinical significance of drug-induced Brugada pattern in the absence of symptoms, family history of BrS, or family with Brugada ECG is undetermined.[34]
ELECTROCARDIOGRAPHIC FINDINGS
BrS is diagnosed based on characteristic ECG findings. There are three ECG patterns associated with BrS [Figure 1]. The so-called “coved” ST-elevation in the right precordial leads seen in Type 1 is most common and characteristic. The Type 1 pattern of ST-elevation on ECG is the cornerstone of diagnosis in BrS. The Brugada pattern can be highly variable with time and may be present only one-third of the time in patients who have demonstrated spontaneous Type 1 pattern.[35] Consensus criteria have defined Type 1 ECG pattern as ≥2 mm ST-segment elevation with typical coved appearance, Type 2 ECG pattern as ≥2 mm J-point elevation and ≥1 mm saddleback ST-segment elevation with positive T-wave, and Type 3 ECG pattern as ≥2 mm J-point elevation with <1 mm saddleback ST-segment elevation in the right precordial leads.[36] Other ECG abnormalities have also been noted. In a pediatric cohort, PR interval, QRS duration, and QTc interval were all significantly longer in BrS patients than in controls.[13]
Figure 1.
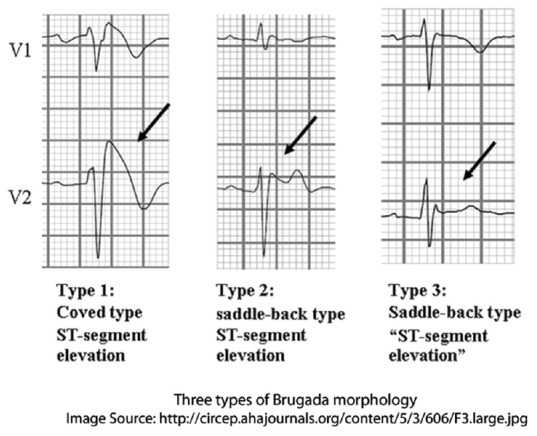
Electrocardiographic patterns in Brugada syndrome
An elegant study of body surface distribution of ECG findings in BrS demonstrated that maximal ST-elevation was in the region of the right ventricular outflow tract (RVOT). This would explain the typical distribution of the ST-segment elevation in V1–V3.[37] In an adult study, findings in V1 and V2 together were diagnostic in 99% of positive ECGs, and finding in lead V3 alone was not diagnostic.[38] Modified placement of leads V1 and V2 to higher intercostal spaces may demonstrate the Brugada pattern. Use of right-sided precordial lead placement V1R–V3R has also been used to detect the pattern.[39] In cases where it may be hard to differentiate the Type 2 Brugada pattern from r’ in V1 in healthy athletes, the duration of the base of the triangle of r’ in V1/V2 taken at 5 mm from the peak of r’ >160 ms is specific and sensitive for BrS [Figure 2].[40] Computer algorithms for recognizing Type 1, Type 2/3, and suggestive Brugada patterns have been developed with reasonable accuracy.[41]
Figure 2.
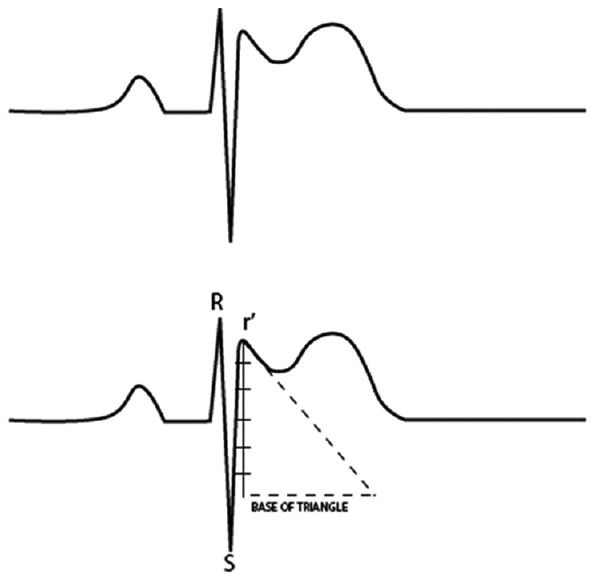
Measuring the duration of the base of the triangle of r’ in V1 V2 taken at 5 mm from the peak of r’
Drug challenge
In case of nondiagnostic baseline ECG, sodium channel blockers such as flecainide, procainamide, and ajmaline have been used to unmask the Brugada pattern.
In a pediatric case series using intravenous procainamide infusion (15 mg/kg up to a maximum dose of 1 g over 15 min), 31% had a positive response. There were no sustained arrhythmias.[42] However, the malignant response in the form of VAs have been reported during ajmaline challenge. The occurrence of VA during ajmaline challenge does not correlate with higher risk for future events.[43,44] Ajmaline dose of 1 mg/kg has been used, with infusion over 5 min in one study including pediatric patients and ranging from 1 mg/s to over 10 min in adult studies. The specific protocol of ajmaline infusion does not seem to matter, with no difference between rapid and slow infusion for diagnostic yield or sustained VA, though slower infusion rate allowing for early discontinuation has been recommended.[45] Repeat ajmaline challenge after the onset of puberty has unmasked the Brugada pattern in patients with negative result before puberty.[16] Flecainide testing (using infusion of 2 mg/kg over 10 min in a study including adolescents) has also been shown to be reproducible and powerful. However, sustained VA has been documented in up to 18% of patients. Deleterious effects appear to be more frequent in patients with identified SCN5A mutation.[46]
Approach to diagnosis
Diagnostic criteria for BrS in adults have been outlined by the recent consensus statement.[47]
ST-segment elevation with Type 1 morphology ≥2 mm in ≥1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd, or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs
Type 2 or Type 3 ST-segment elevation in ≥1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd, or 4th intercostal space when a provocative drug test with intravenous administration of Class I antiarrhythmic drugs induces a Type I ECG morphology.
A diagnostic algorithm [Figure 3] has been proposed for patients with idiopathic VF with normal routine investigations in adults, including the use of drug testing and possible genetic testing. This algorithm yielded a diagnosis of channelopathy in 51% patients, 40% of whom were diagnosed with BrS.[48] Similar guidelines for children do not exist, but this might be reasonably adapted for younger patients.
Figure 3.
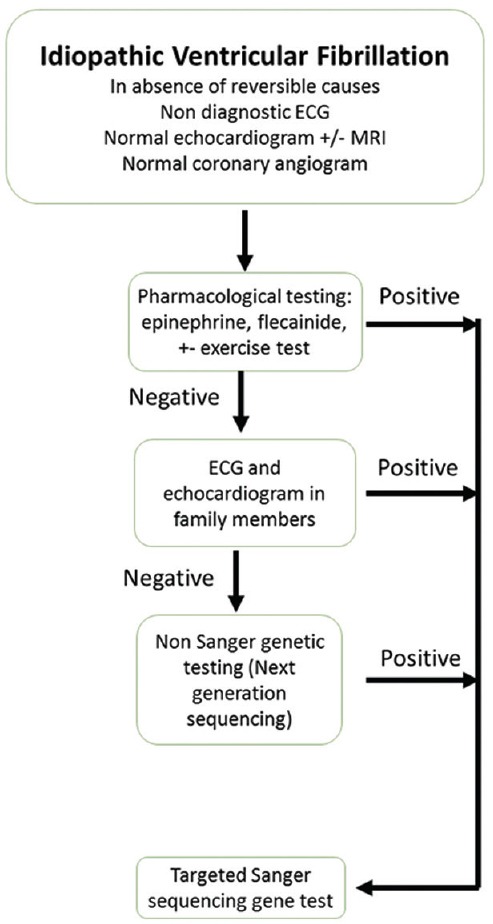
Proposed diagnostic algorithm for idiopathic ventricular fibrillation in adults
Risk stratification
In clinical practice, we attempt to apply our current knowledge of clinical, ECG, and genetic characteristics to determine risk for sudden death. Unfortunately, the literature on risk stratification in the pediatric population is scarce.
HISTORICAL FACTORS
A history of syncope or aborted sudden death is predictive of future event risk.[32,49,50] Studies involving young adults have demonstrated that the rate of cardiac events is highest in patients with documented VF as compared to patients presenting with syncope and lowest in asymptomatic patients.[49]
Electrocardiographic factors
Spontaneous Type 1 pattern on ECG at baseline is associated with risk for cardiac arrest.[11] There is no significant difference in the prognosis of patients, in whom placement in higher intercostal spaces is required to demonstrate the Type 1 pattern.[51] Although the Type 1 pattern is most commonly seen in the right precordial leads, 9% of patients with positive ECG have Type 1 ST-elevation pattern in one or more peripheral leads (augmented vector right and inferior leads), with a higher incidence of SCN5A mutations, inducibility on electrophysiological (EP) study, and sudden death.[52] QTc in lead V2 >420 ms was also associated with cardiac events.[49] AFib and atrial flutter (AFl) have been significantly correlated with malignant arrhythmias.[49,53]
Electrophysiological study
The prognostic value of programmed electrical stimulation in BrS remains controversial. Large studies of adults have demonstrated that VA inducibility was significantly associated with arrhythmic events on follow-up.[32,54] Ventricular effective refractory period <200 ms and QRS fragmentation were significant predictors of arrhythmias.[55] However, a meta-analysis of EP testing in adults with BrS suggested that this was not of prognostic value.[56] Not surprisingly, in a survey of pediatric electrophysiologists, 38% reported that none of their patients with BrS received EP studies and 11% reported that all their patients had received EP studies.[31]
Genetic factors
Missense mutations with >90% loss of function and mutations leading to premature truncation of proteins are more highly associated with cardiac events.[20]
OTHER CLINICAL ASPECTS
Special consideration for anesthesia
In patients with BrS requiring anesthesia for any reason, the choice of anesthetic agent requires caution. A full discussion on the anesthetic management for this population is beyond the scope of this article. Propofol and local anesthetics which have sodium channel blocking effects have a theoretical risk for precipitating arrhythmias in BrS. Some reviews of the literature have demonstrated that most patients with BrS tolerate anesthesia without complication.[57]
Clinical associations
Loss of function SCN5A mutations has been associated with significant atrial and ventricular lead capture issues.[58] SCN5A has also been associated with cardiac conduction disease, AFl, and familial sick sinus syndrome among others.[59,60,61] Ajmaline challenge has been demonstrated to unmask Brugada pattern in patients with myotonic dystrophy Type 1.[62,63] Familial studies have documented a link between BrS and epilepsy syndromes.[64]
Asymptomatic relatives
Asymptomatic patients with a family history of BrS are usually the most common form of presentation. In a study of 112 pediatric relatives with a family history of sudden arrhythmic death, two were diagnosed with BrS after ajmaline challenge.[65]
Proposed criteria of an isolated lead with coved type ECG (instead of >1 right precordial lead) have been shown to be sensitive and specific for identifying carriers of SCN5A mutation. Carriers had longer P-waves, PR intervals, and QRS durations; higher transmural dispersion of repolarization; and higher prevalence of late potentials.[66]
Pathogenesis
The pathogenesis of BrS appears to be multifaceted. Since the discovery of the first putative mutations associated with BrS, many more genes and mutations have been associated with BrS, and with these new discoveries, we continue to unravel more potential mechanisms of pathogenesis. Currently, there are two broad hypotheses to explain the Brugada pattern - the depolarization theory and the repolarization theory.
Repolarization theory
The early phase of the action potential (AP) in epicardial cells demonstrates spike and dome morphology (Phase 1 and Phase 2). This period of the AP is influenced by the fast inward sodium current INa, transient outward potassium current Ito, and L-type calcium current ICal. As the dome is depressed, the epicardial AP is shortened.[67]
The interplay of currents during Phase 1 of the AP can lead to depression of the AP dome. Normally, there is higher Ito in the epicardium. In certain situations (such as SCN5A or KCNE3 mutations), the Ito may become increased in a nonuniform manner in different regions of the epicardium, leading to epicardial dispersion of repolarization [Figure 4]. This creates a transmural gradient between epicardium and endocardium, which manifests as the typical ST-segment elevation pattern on surface ECG. This theory also may explain why potential reentry circuits for VA would exist.[4,68]
Figure 4.
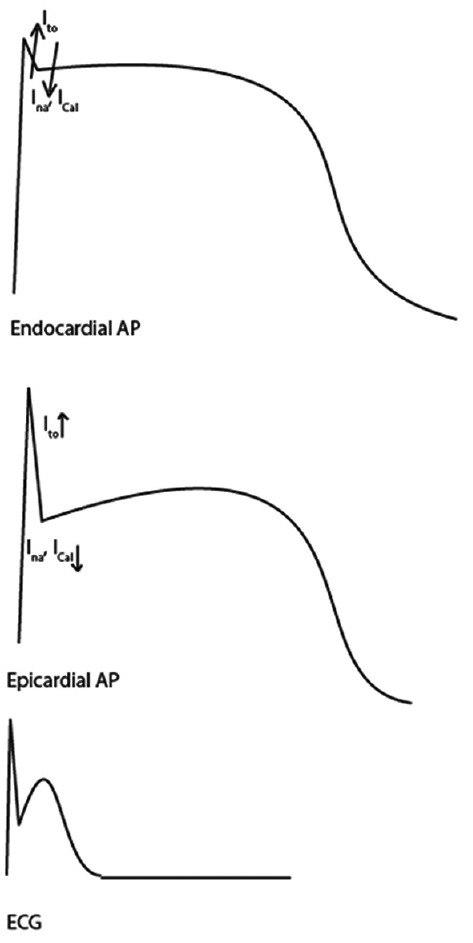
Repolarization theory of Brugada syndrome
Genetics as related to the pathogenesis of the repolarization theory
Our growing knowledge of the underlying genetics gives credence to the repolarization theory.
SCN5A encodes the alpha subunit of the fast sodium channel NaV1.5. Loss of function mutations in SCN5A leads to reduced INa during the AP, causing a reduction in the doming of the AP. Reduction in INa reduces the amplitude of AP and also impacts the Ito and Ical.[67]
The gene GPD1L encodes glycerol-phosphate-dehydrogenase-1-like protein (homologous to GPD1). BrS-associated mutations in this gene may be responsible for the loss of function of the sodium channel (and hence reduced the amplitude of sodium current INa). The enzyme GPD1L catalyzes the conversion of glycerol-3-phosphate to dihydroxyacetone phosphate. Mutations causing loss of function of this enzyme, in turn, cause increased levels of glycerol-3-phosphate, promoting the protein kinase C-dependent phosphorylation of the SCN5A.[69]
Kv4.3 (encoded by KCND3) is a major component of the α subunit of Ito. β subunits (encoded by KCNE3) can interact with potassium channels Kv4.3 such that the current is reduced. Mutations in KCNE3 reverse this suppression of function and increase Ito and thus may produce the BrS phenotype.[26]
Transcription factors Irx5, calcineurin, and NFATc3 contribute to the nonuniform distribution of Kv4 expression and Ito function.[26]
Mutations causing loss of function of genes encoding α and β subunits of the L-type calcium channels (CACNA1C, CACNB2B, and CACNA2D1) also cause reduction in the AP duration.[25]
Depolarization theory
According to the depolarization theory, a delay in conduction around the RVOT tissue drives current toward the RVOT and then back from the RVOT to the right ventricular (RV), resulting in the ST-segment elevation followed by inverted T-wave on ECG.[4] Some of the mild anatomic changes in the RV and RVOT may support the theory that there is an anatomic substrate for delayed conduction. In addition, the high prevalence of conduction abnormalities seen in BrS may also be contributory to this theory.
Early repolarization and late RV wall activation have been hypothesized as the cause for the typical ST-segment elevation in BrS. Although, in BrS, the typical ECG findings occur in the absence of structural heart disease, sodium channel blockers can provoke Brugada pattern in patients with arrhythmogenic RV cardiomyopathy and Chagas disease. Structural abnormalities in the RV such as subepicardial discontinuity can cause localized conduction block due to current to load mismatch.[70]
Gross electrical and anatomical features supporting depolarization theory
Studies of the gross electromechanical features of the heart in BrS have unraveled findings supportive of the depolarization theory.
Body mapping studies have shown that repolarization abnormalities in BrS such as ST-segment elevation and RTc prolongation are localized to the RVOT region, while depolarization abnormalities such as QRS prolongation are distributed more homogeneously.[71]
Biopsy specimens from patients with BrS demonstrate moderate abnormalities including fatty tissue deposition, fibrosis, and hypertrophy in the RV myocardium.[72] MRI studies have demonstrated a high prevalence of mild structural changes of the RV including wall motion abnormalities, reduced outflow tract ejection fraction, enlargement of inflow tract diameter, and global RV end systolic volume.[73]
Effect of hormones
There may be some hormonal influence of testosterone causing the high proportion of male cases in adults, which is not so prominent in the pediatric population. For instance, there are case reports of patients, in whom ST-elevation normalized after orchiectomy.[13]
Effect of temperature
The exact reason for increased susceptibility during fever is unclear. It is probable that in genetically predisposed individuals, the temperature dependence of the sodium channel is altered, which may explain the increased incidence of unmasking of Brugada pattern or arrhythmic events during fever.[13]
Diurnal and autonomic effects
In a large family carrying SCN5A mutations with features of LQT3 and BrS, there was a diurnal- and bradycardia-related variation of ventricular repolarization with QT interval prolonging during nighttime, which may help explain the prevalence of nocturnal sudden death in this family.[74]
Relationship with LQT3
Since both diseases represent dysfunction of the SCN5A gene, LQT3 and BrS may overlap or coexist in the same patients. Flecainide, which is used to treat LQT3 and unmask BrS, can cause patients with LQT3 to demonstrate ST-segment elevations such as BrS.[75]
TREATMENT
Lifestyle modification
Lifestyle modification is an important facet of management. This includes strict antipyresis during febrile illnesses, possible hospitalization during febrile illnesses, and avoidance of medications known to precipitate events. An updated list of relatively and absolutely contraindicated medications, along with relevant literature, as well as resources for medical professionals and patients, is available at www.brugadadrugs.org.
Medical therapy
Quinidine is a Type Ia antiarrhythmic which is known to reduce the Ito and has been used as therapy in BrS.[68] There are reports of successful treatment in the pediatric population with 16 months event-free period after initiating therapy. It appears to have a safe side effect profile in this population.[76] Quinidine has been successfully used to suppress electrical storm, recurrent VF and normalize the ECG in children and adolescents with recurrent VF after implantable cardioverter defibrillator (ICD) implantation.[77,78] A related molecule, hydroquinidine prevented inducible VT/VF on EPS in 76% of asymptomatic patients without ICD, as well as 100% of patients with ICD who had appropriate shocks. In cases where VT/VF remains inducible, ICD was placed.[79] There are currently no large-scale pediatric studies looking at efficacy and safety of quinidine.
There are interesting data regarding the use of propranolol as long-term therapy. Chockalingam et al. argue that it helps prevent tachycardia-related worsening of conduction defects in children, especially when febrile, which is a key difference in the clinical picture of BrS in children as compared to adults.[20]
Implantable cardioverter defibrillator
ICD placement for management of VT/VF is the treatment of choice for symptomatic adults and is the usual course of management for symptomatic children.
In adults, prophylactic ICD implantation has been used in patients with baseline spontaneous or drug-induced Type 1 ECG pattern, history of syncope, or family history of sudden death. While it is effective in providing appropriate shocks, the rate of inappropriate shocks is high.[80] Adult studies demonstrate 8%–16% appropriate shocks and 8%–27% inappropriate shocks, with complication rates as high as 32%.[81,82,83] Inappropriate shocks can occur due to lead failure/dislodgement, T-wave oversensing, device failure, sinus tachycardia, and supraventricular tachycardia. Other complications include pneumothorax, brachial plexus injury, RV perforation, infection, and psychiatric illness.
Along with the high rate of inappropriate shocks, children are more likely to have venous obstruction, tricuspid regurgitation (with intravenous leads), and lead disruption with age. Experts debate whether prophylactic ICD implantation in children is the best option and have proposed quinidine therapy be thought of as “Bridge to ICD” for symptomatic patients.”[84]
Interventional electrophysiology
Radiofrequency ablation of fibrillation-causing ventricular premature beats emanating from RVOT has been reported in an adolescent patient with BrS, and in fact, this patient did not have further ICD shocks in the 6-month follow-up period.[85] Pulmonary vein isolation has been utilized in drug-resistant AFib in BrS.[86] However, there is no consensus regarding the utility of interventional EP for the management of BrS.
Sports participation
Recent guidelines from the AHA/ACC regarding eligibility and disqualification for competitive athletes recommend that symptomatic athletes with suspected BrS are restricted from competitive sports until comprehensive workup is completed. For genotype-positive but phenotype-negative patients, sports participation is reasonable with appropriate precautionary measures including avoiding drugs that can cause BrS ECG changes or are associated with arrhythmias, electrolyte replenishment, avoidance of hyperthermia, availability of an automatic external defibrillator, and establishment of emergency action plan. Participation may also be considered for previously symptomatic patients with BrS, with precautionary measures, if asymptomatic on the treatment for 3 months or more.[87]
APPROACH TO MANAGEMENT
In adults, symptomatic patients are recommended to undergo ICD placement. It appears that children experience an even higher rate of ICD placement-related complications.[88] A survey of pediatric electrophysiologists reflected a lack of consensus regarding the approach to therapy. ICD implantation for symptomatic patients was recommended by 97% of providers. Thirty-one percent reported using pharmacological therapy including quinidine, beta-blockers, and mexiletine. Medications are also prescribed when ICD is refused or cannot be placed. Some providers also use a combination of ICD and medical therapy.[31]
APPROACH TO MANAGEMENT OF ASYMPTOMATIC RELATIVES
The management of genotype-positive asymptomatic patients is controversial. Previously, many of these patients would have undergone prophylactic ICD placement, but there is evidence that the potential harm of ICD placement outweighs the benefit in this group. Crosson and Nies recommend that asymptomatic offspring be screened by ECG with high precordial leads, repeating during febrile illness if initial ECG is negative, and avoiding medications that can cause BrS such as ECG changes while awaiting a febrile ECG. Currently, EP testing and provocative drug testing are not routinely recommended for asymptomatic relatives though this may change depending on the results of large adult studies looking at the clinical utility of quinidine.[89] For asymptomatic children, 27% of providers responding to a survey recommended ICD, and 11% recommended medical therapy.[31]
THE FUTURE
Despite rapid progress, there is much-uncharted territory to explore in BrS. Continuing advances in uncovering the underlying genetics allows us to understand the genetic mechanisms behind BrS. This in turn will help resolve the current debates regarding pathogenesis and create a unifying model of BrS pathogenesis. This will further guide optimal treatment. In the realm of pediatrics, more studies in children with BrS are needed. Specifically, we must try to improve our understanding of the progression of BrS with age and the influence of hormones and temperature regulation. Further insights into prognostication and risk stratification in children are also needed. Our current approach to therapy in children is rudimentary, and more studies are required to assess the role of medical therapy and creation of a consensus for managing BrS in children.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Triedman JK. Brugada and short QT syndromes. Pacing Clin Electrophysiol. 2009;32(Suppl 2):S58–62. doi: 10.1111/j.1540-8159.2009.02386.x. [DOI] [PubMed] [Google Scholar]
- 3.Chockalingam P, Wilde A. The multifaceted cardiac sodium channel and its clinical implications. Heart. 2012;98:1318–24. doi: 10.1136/heartjnl-2012-301784. [DOI] [PubMed] [Google Scholar]
- 4.Jellins J, Milanovic M, Taitz DJ, Wan SH, Yam PW. Brugada syndrome. Hong Kong Med J. 2013;19:159–67. [PubMed] [Google Scholar]
- 5.Sarquella-Brugada G, Campuzano O, Iglesias A, Sánchez-Malagón J, Guerra-Balic M, Brugada J, et al. Genetics of sudden cardiac death in children and young athletes. Cardiol Young. 2013;23:159–73. doi: 10.1017/S1047951112001138. [DOI] [PubMed] [Google Scholar]
- 6.Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. Cardiac channelopathies and sudden infant death syndrome. Cardiology. 2011;119:21–33. doi: 10.1159/000329047. [DOI] [PubMed] [Google Scholar]
- 7.Letsas KP, Gavrielatos G, Efremidis M, Kounas SP, Filippatos GS, Sideris A, et al. Prevalence of Brugada sign in a Greek tertiary hospital population. Europace. 2007;9:1077–80. doi: 10.1093/europace/eum221. [DOI] [PubMed] [Google Scholar]
- 8.Juang JM, Phan WL, Chen PC, Lai LP, Tsai MH, Lin JW, et al. Brugada-type electrocardiogram in the Taiwanese population – Is it a risk factor for sudden death? J Formos Med Assoc. 2011;110:230–8. doi: 10.1016/S0929-6646(11)60035-1. [DOI] [PubMed] [Google Scholar]
- 9.Bozkurt A, Yas D, Seydaoglu G, Acartürk E. Frequency of Brugada-type ECG pattern (Brugada sign) in Southern Turkey. Int Heart J. 2006;47:541–7. doi: 10.1536/ihj.47.541. [DOI] [PubMed] [Google Scholar]
- 10.Sidik NP, Quay CN, Loh FC, Chen LY. Prevalence of Brugada sign and syndrome in patients presenting with arrhythmic symptoms at a Heart Rhythm Clinic in Singapore. Europace. 2009;11:650–6. doi: 10.1093/europace/eup079. [DOI] [PubMed] [Google Scholar]
- 11.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation. 2002;105:1342–7. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 12.Kamakura S, Ohe T, Nakazawa K, Aizawa Y, Shimizu A, Horie M, et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ Arrhythm Electrophysiol. 2009;2:495–503. doi: 10.1161/CIRCEP.108.816892. [DOI] [PubMed] [Google Scholar]
- 13.Probst V, Denjoy I, Meregalli PG, Amirault JC, Sacher F, Mansourati J, et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation. 2007;115:2042–8. doi: 10.1161/CIRCULATIONAHA.106.664219. [DOI] [PubMed] [Google Scholar]
- 14.Yamakawa Y, Ishikawa T, Uchino K, Mochida Y, Ebina T, Sumita S, et al. Prevalence of right bundle-branch block and right precordial ST-segment elevation (Brugada-type electrocardiogram) in Japanese children. Circ J. 2004;68:275–9. doi: 10.1253/circj.68.275. [DOI] [PubMed] [Google Scholar]
- 15.Oe H, Takagi M, Tanaka A, Namba M, Nishibori Y, Nishida Y, et al. Prevalence and clinical course of the juveniles with Brugada-type ECG in Japanese population. Pacing Clin Electrophysiol. 2005;28:549–54. doi: 10.1111/j.1540-8159.2005.40020.x. [DOI] [PubMed] [Google Scholar]
- 16.Conte G, de Asmundis C, Ciconte G, Julià J, Sieira J, Chierchia GB, et al. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA. 2014;312:2039–41. doi: 10.1001/jama.2014.13752. [DOI] [PubMed] [Google Scholar]
- 17.Conte G, Dewals W, Sieira J, de Asmundis C, Ciconte G, Chierchia GB, et al. Drug-induced brugada syndrome in children: Clinical features, device-based management, and long-term follow-up. J Am Coll Cardiol. 2014;63:2272–9. doi: 10.1016/j.jacc.2014.02.574. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 19.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chockalingam P, Clur SA, Breur JM, Kriebel T, Paul T, Rammeloo LA, et al. The diagnostic and therapeutic aspects of loss-of-function cardiac sodium channelopathies in children. Heart Rhythm. 2012;9:1986–92. doi: 10.1016/j.hrthm.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Selga E, Campuzano O, Pinsach-Abuin ML, Pérez-Serra A, Mademont-Soler I, Riuró H, et al. Comprehensive Genetic Characterization of a Spanish Brugada Syndrome Cohort. PLoS One. 2015;10:e0132888. doi: 10.1371/journal.pone.0132888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricci MT, Menegon S, Vatrano S, Mandrile G, Cerrato N, Carvalho P, et al. SCN1B gene variants in Brugada Syndrome: A study of 145 SCN5A-negative patients. Sci Rep. 2014;4:6470. doi: 10.1038/srep06470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–9. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delpón E, Cordeiro JM, Núñez L, Thomsen PE, Guerchicoff A, Pollevick GD, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–18. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, et al. Transient outward current (I (to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–32. doi: 10.1016/j.hrthm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crotti L, Marcou CA, Tester DJ, Castelletti S, Giudicessi JR, Torchio M, et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: Implications for genetic testing. J Am Coll Cardiol. 2012;60:1410–8. doi: 10.1016/j.jacc.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm. 2013;10:1886–94. doi: 10.1016/j.hrthm.2013.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.García-Castro M, García C, Reguero JR, Miar A, Rubín JM, Alvarez V, et al. The spectrum of SCN5A gene mutations in Spanish Brugada syndrome patients. Rev Esp Cardiol. 2010;63:856–9. doi: 10.1016/s1885-5857(10)70171-7. [DOI] [PubMed] [Google Scholar]
- 31.Harris BU, Miyake CY, Motonaga KS, Dubin AM. Diagnosis and management of pediatric brugada syndrome: A survey of pediatric electrophysiologists. Pacing Clin Electrophysiol. 2014;37:638–42. doi: 10.1111/pace.12346. [DOI] [PubMed] [Google Scholar]
- 32.Giustetto C, Drago S, Demarchi PG, Dalmasso P, Bianchi F, Masi AS, et al. Risk stratification of the patients with Brugada type electrocardiogram: A community-based prospective study. Europace. 2009;11:507–13. doi: 10.1093/europace/eup006. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez-Mañero M, Namdar M, Sarkozy A, Casado-Arroyo R, Ricciardi D, de Asmundis C, et al. Prevalence, clinical characteristics and management of atrial fibrillation in patients with Brugada syndrome. Am J Cardiol. 2013;111:362–7. doi: 10.1016/j.amjcard.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Konigstein M, Rosso R, Topaz G, Postema PG, Friedensohn L, Heller K, et al. Drug-induced Brugada syndrome: Clinical characteristics and risk factors. Heart Rhythm. 2016;13:1083–7. doi: 10.1016/j.hrthm.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 35.Richter S, Sarkozy A, Veltmann C, Chierchia GB, Boussy T, Wolpert C, et al. Variability of the diagnostic ECG pattern in an ICD patient population with Brugada syndrome. J Cardiovasc Electrophysiol. 2009;20:69–75. doi: 10.1111/j.1540-8167.2008.01282.x. [DOI] [PubMed] [Google Scholar]
- 36.Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation. 2002;106:2514–9. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu W, Matsuo K, Takagi M, Tanabe Y, Aiba T, Taguchi A, et al. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: Clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms. J Cardiovasc Electrophysiol. 2000;11:396–404. doi: 10.1111/j.1540-8167.2000.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 38.Richter S, Sarkozy A, Paparella G, Henkens S, Boussy T, Chierchia GB, et al. Number of electrocardiogram leads displaying the diagnostic coved-type pattern in Brugada syndrome: A diagnostic consensus criterion to be revised. Eur Heart J. 2010;31:1357–64. doi: 10.1093/eurheartj/ehq049. [DOI] [PubMed] [Google Scholar]
- 39.Hashemi A, Shahrzad S, Shahrzad S, Saber S, Taban S, Aslani A, et al. Positive Brugada challenge test in V1 R-V3 R as a predictor of malignant prognosis in Brugada patients. Ann Noninvasive Electrocardiol. 2013;18:421–6. doi: 10.1111/anec.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serra G, Baranchuk A, Bayés-De-Luna A, Brugada J, Goldwasser D, Capulzini L, et al. New electrocardiographic criteria to differentiate the Type-2 Brugada pattern from electrocardiogram of healthy athletes with r’-wave in leads V1/V2. Europace. 2014;16:1639–45. [Google Scholar]
- 41.Nishizaki M, Sugi K, Izumida N, Kamakura S, Aihara N, Aonuma K, et al. Classification and assessment of computerized diagnostic criteria for Brugada-type electrocardiograms. Heart Rhythm. 2010;7:1660–6. doi: 10.1016/j.hrthm.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 42.Jordan CP, Berul CI, Moak JP, Greene EA. Universal ECG changes in pediatric patients undergoing procainamide challenge. Pacing Clin Electrophysiol. 2014;37:1106–10. doi: 10.1111/pace.12402. [DOI] [PubMed] [Google Scholar]
- 43.Conte G, Sieira J, Sarkozy A, de Asmundis C, Di Giovanni G, Chierchia GB, et al. Life-threatening ventricular arrhythmias during ajmaline challenge in patients with Brugada syndrome: Incidence, clinical features, and prognosis. Heart Rhythm. 2013;10:1869–74. doi: 10.1016/j.hrthm.2013.09.060. [DOI] [PubMed] [Google Scholar]
- 44.Gandjbakhch E, Fressart V, Duthoit G, Marquié C, Deharo JC, Pousset F, et al. Malignant response to ajmaline challenge in SCN5A mutation carriers: Experience from a large familial study. Int J Cardiol. 2014;172:256–8. doi: 10.1016/j.ijcard.2013.12.269. [DOI] [PubMed] [Google Scholar]
- 45.Arnalsteen-Dassonvalle E, Hermida JS, Kubala M, Six I, Quenum S, Leborgne L, et al. Ajmaline challenge for the diagnosis of Brugada syndrome: Which protocol? Arch Cardiovasc Dis. 2010;103:570–8. doi: 10.1016/j.acvd.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Gasparini M, Priori SG, Mantica M, Napolitano C, Galimberti P, Ceriotti C, et al. Flecainide test in Brugada syndrome: A reproducible but risky tool. Pacing Clin Electrophysiol. 2003;26(1 Pt 2):338–41. doi: 10.1046/j.1460-9592.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 47.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Jiménez-Jáimez J, Peinado R, Grima EZ, Segura F, Moriña P, Sánchez Muñoz JJ, et al. Diagnostic approach to unexplained cardiac arrest (from the FIVI-Gen Study) Am J Cardiol. 2015;116:894–9. doi: 10.1016/j.amjcard.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 49.Hiraoka M, Takagi M, Yokoyama Y, Sekiguchi Y, Aihara N, Aonuma K EJapan Idiopathic Ventricular Fibrillation Study Investigators. Prognosis and risk stratification of young adults with Brugada syndrome. J Electrocardiol. 2013;46:279–83. doi: 10.1016/j.jelectrocard.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092–6. doi: 10.1161/01.CIR.0000104568.13957.4F. [DOI] [PubMed] [Google Scholar]
- 51.Miyamoto K, Yokokawa M, Tanaka K, Nagai T, Okamura H, Noda T, et al. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol. 2007;99:53–7. doi: 10.1016/j.amjcard.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 52.Rollin A, Sacher F, Gourraud JB, Pasquié JL, Raczka F, Duparc A, et al. Prevalence, characteristics, and prognosis role of type 1 ST elevation in the peripheral ECG leads in patients with Brugada syndrome. Heart Rhythm. 2013;10:1012–8. doi: 10.1016/j.hrthm.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Abud A, Goyeneche R, Carlessi A, Strada B, Becker C. Possible prognostic value of atrial fibrillation and atrial flutter in Brugada syndrome. Arch Cardiol Mex. 2013;83:4–7. doi: 10.1016/j.acmx.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 54.Sieira J, Conte G, Ciconte G, de Asmundis C, Chierchia GB, Baltogiannis G, et al. Prognostic value of programmed electrical stimulation in Brugada syndrome: 20 years experience. Circ Arrhythm Electrophysiol. 2015;8:777–84. doi: 10.1161/CIRCEP.114.002647. [DOI] [PubMed] [Google Scholar]
- 55.Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, et al. Risk stratification in Brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59:37–45. doi: 10.1016/j.jacc.2011.08.064. [DOI] [PubMed] [Google Scholar]
- 56.Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AA, et al. Role of programmed ventricular stimulation in patients with Brugada syndrome: A meta-analysis of worldwide published data. Eur Heart J. 2007;28:2126–33. doi: 10.1093/eurheartj/ehm116. [DOI] [PubMed] [Google Scholar]
- 57.Kloesel B, Ackerman MJ, Sprung J, Narr BJ, Weingarten TN. Anesthetic management of patients with Brugada syndrome: A case series and literature review. Can J Anaesth. 2011;58:824–36. doi: 10.1007/s12630-011-9546-y. [DOI] [PubMed] [Google Scholar]
- 58.Chiang DY, Kim JJ, Valdes SO, de la Uz C, Fan Y, Orcutt J, et al. Loss-of-Function SCN5A Mutations Associated With Sinus Node Dysfunction, Atrial Arrhythmias, and Poor Pacemaker Capture. Circ Arrhythm Electrophysiol. 2015;8:1105–12. doi: 10.1161/CIRCEP.115.003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hothi SS, Ara F, Timperley J. p.Y1449C SCN5A mutation associated with overlap disorder comprising conduction disease, Brugada syndrome, and atrial flutter. J Cardiovasc Electrophysiol. 2015;26:93–7. doi: 10.1111/jce.12470. [DOI] [PubMed] [Google Scholar]
- 60.Nakajima S, Makiyama T, Hanazawa K, Kaitani K, Amano M, Hayama Y, et al. A novel SCN5A mutation demonstrating a variety of clinical phenotypes in familial sick sinus syndrome. Intern Med. 2013;52:1805–8. doi: 10.2169/internalmedicine.52.0085. [DOI] [PubMed] [Google Scholar]
- 61.Neu A, Eiselt M, Paul M, Sauter K, Stallmeyer B, Isbrandt D, et al. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Hum Mutat. 2010;31:E1609–21. doi: 10.1002/humu.21302. [DOI] [PubMed] [Google Scholar]
- 62.Pambrun T, Mercier A, Chatelier A, Patri S, Schott JJ, Le Scouarnec S, et al. Myotonic dystrophy type 1 mimics and exacerbates Brugada phenotype induced by Nav1.5 sodium channel loss-of-function mutation. Heart Rhythm. 2014;11:1393–400. doi: 10.1016/j.hrthm.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 63.Pambrun T, Bortone A, Bois P, Degand B, Patri S, Mercier A, et al. Unmasked Brugada pattern by ajmaline challenge in patients with myotonic dystrophy type 1. Ann Noninvasive Electrocardiol. 2015;20:28–36. doi: 10.1111/anec.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013;105:415–8. doi: 10.1016/j.eplepsyres.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 65.Wong LC, Roses-Noguer F, Till JA, Behr ER. Cardiac evaluation of pediatric relatives in sudden arrhythmic death syndrome: A 2-center experience. Circ Arrhythm Electrophysiol. 2014;7:800–6. doi: 10.1161/CIRCEP.114.001818. [DOI] [PubMed] [Google Scholar]
- 66.Santos LF, Rodrigues B, Moreira D, Correia E, Nunes L, Costa A, et al. Criteria to predict carriers of a novel SCN5A mutation in a large Portuguese family affected by the Brugada syndrome. Europace. 2012;14:882–8. doi: 10.1093/europace/eur421. [DOI] [PubMed] [Google Scholar]
- 67.Rook MB, Bezzina Alshinawi C, Groenewegen WA, van Gelder IC, van Ginneken AC, Jongsma HJ, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507–17. doi: 10.1016/s0008-6363(99)00350-8. [DOI] [PubMed] [Google Scholar]
- 68.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–6. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 69.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. 2009;297:H1446–52. doi: 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoogendijk MG, Potse M, Linnenbank AC, Verkerk AO, den Ruijter HM, van Amersfoorth SC, et al. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Heart Rhythm. 2010;7:238–48. doi: 10.1016/j.hrthm.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Yokokawa M, Takaki H, Noda T, Satomi K, Suyama K, Kurita T, et al. Spatial distribution of repolarization and depolarization abnormalities evaluated by body surface potential mapping in patients with Brugada syndrome. Pacing Clin Electrophysiol. 2006;29:1112–21. doi: 10.1111/j.1540-8159.2006.00505.x. [DOI] [PubMed] [Google Scholar]
- 72.Zumhagen S, Spieker T, Rolinck J, Baba HA, Breithardt G, Böcker W, et al. Absence of pathognomonic or inflammatory patterns in cardiac biopsies from patients with Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2:16–23. doi: 10.1161/CIRCEP.107.737882. [DOI] [PubMed] [Google Scholar]
- 73.Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M, et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J. 2009;30:2241–8. doi: 10.1093/eurheartj/ehp252. [DOI] [PubMed] [Google Scholar]
- 74.van den Berg MP, Haaksma J, Veeger NJ, Wilde AA. Diurnal variation of ventricular repolarization in a large family with LQT3-Brugada syndrome characterized by nocturnal sudden death. Heart Rhythm. 2006;3:290–5. doi: 10.1016/j.hrthm.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 75.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation. 2000;102:945–7. doi: 10.1161/01.cir.102.9.945. [DOI] [PubMed] [Google Scholar]
- 76.Baruteau AE, Mabo P, Probst V. Quinidine therapy in children affected by Brugada syndrome: Are we far from a safe alternative? Cardiol Young. 2009;19:652–4. doi: 10.1017/S104795110999093X. [DOI] [PubMed] [Google Scholar]
- 77.Mok NS, Chan NY, Chiu AC. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol. 2004;27(6 Pt 1):821–3. doi: 10.1111/j.1540-8159.2004.00537.x. [DOI] [PubMed] [Google Scholar]
- 78.Mehrotra S, Juneja R, Naik N, Pavri BB. Successful use of quinine in the treatment of electrical storm in a child with Brugada syndrome. J Cardiovasc Electrophysiol. 2011;22:594–7. doi: 10.1111/j.1540-8167.2010.01907.x. [DOI] [PubMed] [Google Scholar]
- 79.Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol. 2004;43:1853–60. doi: 10.1016/j.jacc.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 80.Sarkozy A, Boussy T, Kourgiannides G, Chierchia GB, Richter S, De Potter T, et al. Long-term follow-up of primary prophylactic implantable cardioverter-defibrillator therapy in Brugada syndrome. Eur Heart J. 2007;28:334–44. doi: 10.1093/eurheartj/ehl450. [DOI] [PubMed] [Google Scholar]
- 81.Rosso R, Glick A, Glikson M, Wagshal A, Swissa M, Rosenhek S, et al. Outcome after implantation of cardioverter defibrillator [corrected] in patients with Brugada syndrome: A multicenter Israeli study (ISRABRU) Isr Med Assoc J. 2008;10:435–9. [PubMed] [Google Scholar]
- 82.Daoulah A, Alsheikh-Ali AA, Ocheltree AH, Ocheltree S, Al-Kaabi S, Malik M, et al. Outcome after implantable cardioverter-defibrillator in patients with Brugada syndrome: The Gulf Brugada syndrome registry. J Electrocardiol. 2012;45:327–32. doi: 10.1016/j.jelectrocard.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 83.Conte G, Sieira J, Ciconte G, de Asmundis C, Chierchia GB, Baltogiannis G, et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: A 20-year single-center experience. J Am Coll Cardiol. 2015;65:879–88. doi: 10.1016/j.jacc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 84.Viskin S. Brugada syndrome in children: Don’t ask, don’t tell? Circulation. 2007;115:1970–2. doi: 10.1161/CIRCULATIONAHA.106.686758. [DOI] [PubMed] [Google Scholar]
- 85.Darmon JP, Bettouche S, Deswardt P, Tiger F, Ricard P, Bernasconi F, et al. Radiofrequency ablation of ventricular fibrillation and multiple right and left atrial tachycardia in a patient with Brugada syndrome. J Interv Card Electrophysiol. 2004;11:205–9. doi: 10.1023/B:JICE.0000048571.19462.54. [DOI] [PubMed] [Google Scholar]
- 86.Conte G, Chierchia GB, Wauters K, De Asmundis C, Sarkozy A, Levinstein M, et al. Pulmonary vein isolation in patients with Brugada syndrome and atrial fibrillation: A 2-year follow-up. Europace. 2014;16:528–32. doi: 10.1093/europace/eut309. [DOI] [PubMed] [Google Scholar]
- 87.Ackerman MJ, Zipes DP, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 10: The Cardiac Channelopathies: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol. 2015;66:2424–8. doi: 10.1016/j.jacc.2015.09.042. [DOI] [PubMed] [Google Scholar]
- 88.Shah MJ. Implantable cardioverter defibrillator-related complications in the pediatric population. Pacing Clin Electrophysiol. 2009;32(Suppl 2):S71–4. doi: 10.1111/j.1540-8159.2009.02389.x. [DOI] [PubMed] [Google Scholar]
- 89.Crosson JE, Nies M. Brugada syndrome in children. Expert Rev Cardiovasc Ther. 2015;13:173–81. doi: 10.1586/14779072.2015.999765. [DOI] [PubMed] [Google Scholar]