

Abstract
Angiosarcoma is a rare, aggressive subtype of soft-tissue sarcoma with a propensity for local recurrence and metastasis associated with a generally poor prognosis, unless diagnosed early. Given the vascular endothelial cell origin of angiosarcoma, tumours may develop in essentially any organ; however, there is a predilection for the skin where half of all tumours arise, increasing in prevalence with age. The most common risk factors are chronic lymphoedema and history of radiation. We review the most important radiological findings along the spectrum of angiosarcoma from head to toe throughout the body, including uncommon and rare locations. Key imaging features of angiosarcoma across multiple organ systems will be described, as well as the impact on management and prognosis.
INTRODUCTION
Angiosarcoma is an aggressive, malignant endothelial-cell tumour of lymphatic or vascular origin.1–3 It has a high rate of local recurrence and metastasis.4 Angiosarcoma represents <1% of all soft-tissue sarcomas, with a generally poor prognosis and overall survival ranging anywhere from 6 to 16 months.3,5–7
Angiosarcoma arises from vascular endothelial cells which show atypia and can grow along pre-existing vascular channels, sinusoidal or cavernous spaces. They can also form poorly organized vessels, solid masses or nodules.8 The pathological appearance of angiosarcoma varies according to the tumour grade. Low-grade angiosarcoma displays a small solid component with low-grade cytology and abundant open vascular lumina. High-grade lesions are densely cellular and infiltrative with a high mitotic rate and atypical cells. High-grade tumours have a variable amount of vascular lumen formation that can be focal in appearance.8,9
Histopathological identification of angiosarcoma is sometimes challenging because it can be mistaken for other vascular tumours. In the low-grade form, angiosarcoma may resemble a haemangioma, whereas the aggressive form may have overlapping features with anaplastic melanoma and epithelial carcinomas.4,9
Because angiosarcoma is such a rare entity, most knowledge is derived from case reports or small cohort studies that are limited by small sample sizes, incomplete data and diverse treatment approaches.3 In fact, only a few published reports include >100 patients. Nonetheless, useful cumulative information regarding this endothelial cell tumour has been observed.6
Owing to its origin from endothelial cells, angiosarcoma can develop throughout the body and can occur at any age.10 However, they are more common in older individuals, with a reported median age between 60 and 71 years;6 there is generally no gender predilection except for cutaneous lesions, which have been shown to be more prevalent among males.8 The cutaneous form is the most common, accounting for about half of all tumours, with the head and neck as the most frequently involved region.11 Specifically, the scalp accounts for approximately 50% of all angiosarcoma cases, yet this still only comprises <0.1% of all head and neck malignancies. Only 10% of all angiosarcomas occur in deep soft tissues, and the rest are found in parenchymal organs such as the breast, bone, spleen and liver.5,12 Angiosarcoma primarily spreads haematogenously, with the lungs as the most common site for metastases. Metastases also frequently affect the liver (Figure 8), bones (Figure 2), soft tissues and lymph nodes.3 There is usually multiorgan involvement at the time of diagnosis, often rendering it difficult to ascertain the site of origin and approach to therapy.10
Figure 8.

A 68-year-old female with cardiac angiosarcoma, hepatic and splenic metastases. Contrast-enhanced axial CT chest image (a) and axial bright-blood (b) MR images showing large discrete, soft-tissue mass that extends from the right atrial pericardium into the right atrium (black arrows). The mass occupies most of the right atrial cavity. Axial CT image of the upper abdomen (c) demonstrates multiple hypodense, peripherally enhancing lesions in the liver and spleen, consistent with metastatic disease (white arrows).
Figure 2.

A 47-year-old male with left chest wall angiosarcoma and left forearm metastases. Contrast-enhanced axial CT (a) image of the upper chest shows a large heterogeneous soft-tissue mass of the left anterior chest wall (white arrow). Forearm radiograph (b) demonstrates a lucent lytic lesion of the left proximal radius with soft-tissue swelling (black arrow). Axial T1 weighted MR image of the left forearm with intravenous contrast (c) shows an associated large heterogeneous enhancing soft-tissue mass of the flexor compartment (black-and-white arrow).
The classification of angiosarcoma is based on both the site of origin as well as the underlying risk factors involved. Angiosarcoma can be divided into primary cutaneous (in the absence of lymphoedema or radiation), parenchymal tissue/visceral (which includes primary breast lesions), deep soft tissue, and lymphoedema-associated and post-radiation angiosarcomas.3,5 Furthermore, as scientific advances are made in molecular and genetic biology, angiosarcoma can be genetically discriminated from other sarcomas with greater potential for improved therapeutic outcomes.5
Herein, this article provides a clinical and imaging review of angiosarcoma by anatomical site, summarizing the key clinical and imaging characteristics of this rare cancer.
AETIOLOGY
There are well-established risk factors linked to the development of angiosarcoma; the two most common are chronic lymphoedema and radiation therapy. Other factors include familial syndromes, exposure to environmental chemical toxins and foreign bodies.3,13,14
Chronic lymphoedema, irrespective of its cause (e.g. following breast cancer treatment, parasitic infections such as filariasis or Milroy's disease) is a definite risk factor for angiosarcoma, a phenomenon given the name Stewart–Treves syndrome.13 Some reports attribute radiation-induced angiosarcoma arising after the treatment of breast cancer as more likely related to radiation and/or lymphoedema.3,8
Radiation therapy is another independent risk factor for subsequent angiosarcoma development, contributing to about one-fourth of all cases.3,15 This applies to a variety of cancers that may be treated with radiation (e.g. cervical cancer, endometrial cancer and lymphoma) in addition to breast cancer. Large epidemiological studies have confirmed the increased risk of angiosarcoma following adjuvant radiotherapy for breast cancer.3,8 To differentiate it from lymphoedema-associated angiosarcoma, radiation-induced angiosarcoma must fulfil certain criteria including pathological confirmation, location within the radiation field and occurrence several years after treatment with no evidence of chronic lymphoedema. Post-radiation angiosarcoma arising in sites other than the breast tends to occur at least 10 years after the diagnosis of the primary cancer. Conversely, most post-radiation angiosarcoma has been reported to develop within 5 years after radiation treatment for breast cancer.8,16
Familial syndromes associated with angiosarcoma include neurofibromatosis type-1, Maffucci syndrome, bilateral retinoblastoma, von Recklinghausen syndrome, haemochromatosis and Klippel–Trenaunay syndrome. Studies have also documented the ability of angiosarcomas to originate within other lesions such as port wine stains, haemangiomas and lymphangiomas, albeit a rare occurrence.3,5,9,16
There is also a link between chemical exposure and angiosarcoma which is most evident with hepatic lesions. Some of the known chemical toxins include the occupational use of vinyl chloride, iatrogenic exposure to thorium dioxide (Thorotrast®) for radiological examinations in the past, arsenic containing insecticides used in agriculture and long-term use of anabolic steroids.5,17 Additionally, foreign body-associated angiosarcoma may in part be due to synthetic materials used in grafts or prostheses (e.g. Dacron®) vs chronic sequelae of an otherwise-normal foreign body reaction to items such as shrapnel, steel, surgical sponges and bone wax.5,18
The observation of angiosarcoma in patients with renal transplantation has raised suspicion that immunosuppression may be a risk factor involved in the development of this tumour. The exact pathogenesis, however, is unclear, including their predilection to occur at or near the site of arteriovenous fistulae.3,5,16,19,20 Despite the suggestion by some epidemiological studies that angiosarcoma occurs with increased frequency in patients with acquired immune deficiency syndrome, the exact role that immunosuppression plays in its pathogenesis is still not clear.3,8,19,21
CLINICAL FEATURES AND IMAGING FINDINGS
We illustrate an overview of angiosarcoma according to their anatomical sites. In each category, there is a concise review of the clinical presentation and prognosis, with a special emphasis on the imaging features and utility of various imaging modalities.
Angiosarcoma of the head and neck
Approximately 60% of angiosarcoma occurs in the head and neck, making this the most common site. The locations most often encountered in the head and neck are the scalp (Figure 3), followed by the face and then the neck, often with a multifocal presentation (Figure 3). These tumours—also known as Wilson–Jones angiosarcoma, senile angiosarcoma or malignant angioendothelioma—are primarily found in elderly persons.22 Patients often present with skin lesions such as an enlarging bruise, a discoloured nodule or persistent ulceration. In the early stages, these lesions can be misdiagnosed for benign entities caused by cellulitis, infection or skin injuries. The differential diagnosis includes haemangioblastoma, Kaposi sarcoma, metastatic cancer from an unknown primary site, sinonasal squamous-cell cancer and Merkel cell carcinoma.23
Figure 3.

A 60-year-old male with cutaneous angiosarcoma of bilateral forehead. Axial (a) and sagittal (b) T2 weighted MR images show multifocal high signal intensity lesions arising from the skin of the forehead bilaterally (arrows). The lesions have exophytic components with associated osseous destruction of the outer table of the skull. No intracranial masses or extension.
MRI features include intermediate T1 signal intensity with possible areas of hyperintensity indicating the presence of haemorrhage and high T2 signal intensity (Figure 3). The most diagnostic finding is the presence of high-flow serpentine vessels (low signal intensity on both T1 and T2 weighted images) in an otherwise solid non-specific soft-tissue mass. Low flow vessels, however, may show hyperintensity on T2 weighted images. Angiosarcoma demonstrates enhancement after intravenous contrast administration and may show non-enhancing areas reflecting tumour necrosis.4 Although these imaging features are relatively non-specific, they often indicate malignancy and should prompt biopsy for further characterization.
Primary brain angiosarcoma is exceptionally rare, with only isolated cases reported in the literature. The imaging appearance is non-specific and differential diagnosis includes glioma, cavernoma and haemorrhagic metastasis.24 Similar to previously reported cases,25 we present one case of a primary brain angiosarcoma that exhibits inhomogeneous signal intensity on T1 weighted images with a focus of enhancement after contrast administration (Figure 4). Mixed signal intensity on T1 and T2 weighted images is thought to represent degraded blood products at different stages.24
Figure 4.

A 61-year-old male with biopsy-proven primary brain angiosarcoma. Axial T1 weighted images before (a) and after (b) intravenous contrast demonstrate a roughly well-defined lesion in the right occipital lobe. The lesion has heterogeneous signal intensity with hyperintense components due to the presence of blood degradation products. A small nodule of enhancement seen posteriorly (arrow).
Soft-tissue angiosarcoma (extremities, peritoneum, retroperitoneum and body wall)
The term “soft tissue” is a broad one that implies lack of solid organ origin and includes the deeper subcutaneous tissues, soft tissues of the upper and lower extremities, the abdominal and chest wall, peritoneum, retroperitoneum and mediastinum.9 Clinically, angiosarcoma of the extremities (Figures 5 and 6), and chest or abdominal wall (Figure 2), usually present as rapidly growing palpable masses, whereas deeper masses such as peritoneal (Figure 7), retroperitoneal and mediastinal tend to present more with pain or discomfort related to mass effect.3 These tumours can occur at any age; however, they are more prevalent in older patients between the ages of 60 and 70 years.23
Figure 5.

Two different patients with soft-tissue (a) and osseous angiosarcomas (b, c). (a) Axial T2 weighted MR image in a 63-year-old male with right thigh soft-tissue angiosarcoma. It demonstrates a large, well-defined, lobulated, solid, heterogeneous mass involving multiple muscles in the anterior compartment of the right thigh (white arrows). No underlying bone abnormality. (b, c) Plain radiograph and axial T2 weighted MR image in a 29-year-old male with angiosarcoma of the left femur. The plain radiograph (b) of the left femur demonstrates a large, expansile lytic lesion centred in the left femoral neck with associated pathological fracture and soft-tissue swelling. The axial T2 weighted MR image of the hip (c) shows a large, heterogeneous expansile aggressive tumour of the left proximal femur with extensive intramedullary involvement and pathological fracture as well as extension into the surrounding soft tissues (black arrows).
Figure 6.

A 69-year-old male with chronic lymphoedema-induced angiosarcoma of the right forearm. The patient has a history of radical mastectomy and chronic lymphoedema. (a) Ultrasound image with Doppler of the right forearm shows diffuse soft-tissue mass with heterogeneous echotexture and increased vascularity (white arrows). Axial T1 weighted MR images (b) without and (c) with intravenous contrast demonstrate diffuse soft-tissue oedema with diffuse, extensive, infiltrative cutaneous and subcutaneous multinodular soft-tissue tumour along the volar tissues of the right forearm with involvement of underlying fascia and musculature (black arrows).
Figure 7.

A 67-year-old male patient with primary peritoneal angiosarcoma presenting with haemoperitoneum. Contrast-enhanced CT images at presentation (a–c) show a diffuse peritoneal-based process with multifocal serosal implants on the liver (a, b) and large-volume high-attenuation peritoneal fluid consistent with haemorrhage. CT follow-up 1 month later (d) shows rapid progression of the heterogeneous peritoneal-based masses.
If left untreated, the tumour can enlarge to 20 cm or more. As the tumour size increases, ulceration, haemorrhage and oedema can develop.3 Even with radical surgical resection, the presence of multifocal disease and tissue infiltration often result in positive margins and local recurrence.26
On contrast-enhanced CT, soft-tissue angiosarcoma may manifest as an irregular, enhancing soft-tissue mass. In more advanced cases, underlying bone or adjacent solid organ invasion may be present (Figures 2 and 5).23 Soft-tissue calcifications can also be seen.
On MR, the signal characteristics include bright signal intensity on T2 weighted and intermediate signal intensity on T1 weighted images, with invasion of the surrounding tissues (Figure 5). Foci of high signal intensity indicating adjacent haemorrhage can also be seen on T1 weighted images.23 Vessels within the tumour may demonstrate high flow (low signal intensity on all pulse sequences) or low flow (increased signal intensity on T2 weighted images). There is tumour enhancement after administration of intravenous gadolinium-based contrast, potentially with central areas of necrosis (Figures 2 and 6).2 High soft-tissue vascularity and perfusion result in an increased rate of enhancement.4 On diffusion weighted imaging, there is a very low apparent diffusion coefficient, which is characteristic of soft-tissue malignancies.23 Angiosarcoma may appear as a plaque-like subcutaneous mass in the background of an enlarged extremity with oedema which represents underlying lymphoedema (Figure 6).4
On positron emission tomography (PET) imaging, avid fluorine-18 fludeoxyglucose (18F-FDG) uptake is usually seen.27
Angiosarcoma of the breast
Primary angiosarcoma of the breast is not common and is of unknown aetiology. It refers to those tumours that arise within the breast parenchyma as opposed to lesions from the overlying skin. It represents only 0.04% of all breast tumours and about 8% of breast sarcomas.18 Primary breast angiosarcoma is highly aggressive and tends to affect younger females, usually presenting as a palpable mass.28 Secondary breast angiosarcoma most often occurs following local radiation therapy with a latent period between 5 and 6 years.29,30 In addition, chronic lymphoedema of the breast is a risk factor for secondary breast angiosarcoma.14 Primary breast angiosarcoma has an unfavourable prognosis with a 5-year survival between 8% and 50%.18 Secondary angiosarcoma tend to have a poor prognosis. Five-year overall survival rate for radiation-associated angiosarcoma (RAS) ranges from 10–54% where local recurrence is seen in 45–64% of patients and results in a 25–30% mortality rate. Distant metastases are diagnosed in 27–42% of patients and are associated with a 75–89% mortality rate. Some of the prognostic factors affecting the outcome include completeness of surgical resection, patient's age, and tumour size. Local recurrence is an adverse prognostic indicator and is often associated with distant metastases.29,30,31 Surgical resection (mastectomy or lumpectomy), chemotherapy and irradiation are among treatment options for breast angiosarcoma.18
The imaging findings for breast angiosarcoma are varied and sometimes absent altogether, making diagnosis challenging. Mammography may show a solitary ill-defined uncalcified mass ranging in size from 3 to 6 cm in primary angiosarcoma while ultrasound shows predominantly hypoechoic solitary or multiple masses.32 However, hyperechoic masses have also been associated with angiosarcoma.33 In secondary angiosarcoma, mammographic findings are often non-specific, with skin thickening sometimes as the only relevant finding. It may be difficult to differentiate dermal lesions from skin thickening secondary to radiation.29,32
Since mammographic and sonographic findings can be non-specific, MR may be more helpful in characterizing these lesions.28 In primary angiosarcoma, MRI features include a heterogeneous mass of low signal intensity on T1 weighted and high signal on T2 weighted images. On dynamic post-contrast imaging, the tumours show rapid enhancement (Figure 1) with varying washout kinetics according to the tumour grade. High-grade tumours commonly show rapid washout, whereas lower grade tumours usually exhibit persistent or plateau enhancement curves.32,34 High signal haemorrhagic foci on T1 weighted images can be seen with high-grade tumours.29
Figure 1.

A 43-year-old female with unilateral breast angiosarcoma. Axial T1 weighted images (a) before contrast and (b) after intravenous contrast administration showing a large irregular hypointense heterogeneously enhancing mass that near completely replaces the right breast. The mass extends to appear to involve the nipple/areolar complex (arrows).
Chikarmane et al35 studied MRI characteristics of radiation-induced breast angiosarcoma. Diffuse T2 high-signal skin thickening was noted in all of the 16 patients studied. Low T2 signal intensity lesions were seen in six patients. All lesions showed rapid enhancement on contrast-enhanced T1 weighted images. Four patients showed breast parenchymal masses on mammography and MRI.
18F-FDG PET/CT shows intense metabolic activity in primary and secondary breast angiosarcoma, valuable both for initial evaluation and restaging.36
Cardiac and pulmonary artery angiosarcoma
Angiosarcoma is the most-common differentiated malignant neoplasm of the heart and accounts for 10–15% of primary cardiac malignancies.37 Approximately 80% of cardiac angiosarcoma cases originate in the right atrium near the atrioventricular groove (Figure 8). Patients often present with dyspnoea and chest pain as well as hypotension, tachycardia and syncope.37 because there is asymmetric involvement of the right heart, symptoms can be secondary to right-sided diastolic dysfunction and cardiac tamponade.38 Superior vena cava syndrome is a reported complication of cardiac angiosarcoma, and when recalcitrant to treatment approaches, palliative superior vena cava stenting may be considered.39
The differential diagnosis is broad and includes metastases, myxoma, thrombus, lymphoma, myxosarcoma and rhabdomyosarcoma.40,41 The prognosis is very poor with a mean survival of 3 months to 4 years. Because cardiac angiosarcoma is refractory to treatment, local recurrence and metastases (Figure 8) are seen usually within 1 year of attempted treatment. Treatment options include chemotherapy and palliative radiation. Palliative surgery may also prolong survival.5
Macroscopically, cardiac angiosarcoma has irregular borders and often invades the adjacent pericardium.42 There are two patterns of growth of cardiac angiosarcoma: (1) a well-defined mass with haemorrhagic and necrotic components that extend from the pericardium into the cardiac chamber and (2) a diffusely infiltrating mass with extension from the pericardium,37 with or without epicardial extension.43
On chest radiography, the most common finding is cardiomegaly. Other potential abnormalities include widened mediastinum, right-sided heart enlargement and hilar adenopathy.44 CT demonstrates a predominantly, highly vascular right atrial mass (Figure 8) that involves the cardiac chambers and may be nodular or irregular.44,45
Cardiac angiosarcomas are usually isointense on T1 weighted and hyperintense on T2 weighted MR images (Figure 8) and may demonstrate an irregular pattern of enhancement.46 Typically, they exhibit a tubular morphology and have internal flow voids, and they may have a characteristic lobulated “cauliflower” configuration (Figure 8).43 On gradient echo T1 weighted sequences, the hypointense layer with phase shift is thought to represent susceptibility effect secondary to haemorrhage within the tumour due to its highly vascular nature.40,46 In addition, a characteristic “sunray” appearance of pericardial involvement on contrast-enhanced MRI has been described.47 On steady-state free precession images, angiosarcomas appear hyperintense relative to the myocardium. Haemorrhage and necrosis are seen as foci of high and low signal intensities within the tumour on steady-state free precession images, respectively.38
Pulmonary artery angiosarcoma (Figure 9) is rare, and its identification is often delayed due to misdiagnosis as bland pulmonary embolism with subsequent anticoagulation.8,48 Pulmonary artery sarcomas have a poor prognosis, with more than half of the patients surviving less than 1 year.49 Pulmonary artery angiosarcoma is typically detected during contrast-enhanced CT chest imaging. CT imaging demonstrates a filling defect within the main pulmonary arteries that appears similar to pulmonary thromboembolism (Figure 9). Both entities have similar clinical and diagnostic features. However, CT features that are diagnostic of sarcoma over pulmonary embolism include an irregular low-attenuation filling defect with enhancement and extravascular extension (Figure 9).48,50
Figure 9.
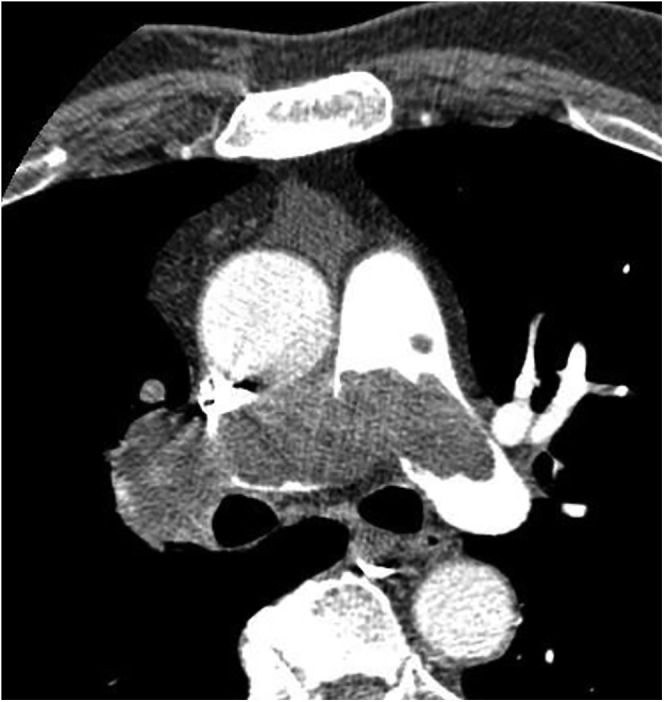
A 74-year-old male with angiosarcoma of the pulmonary artery. Axial CT angiography demonstrates massive filling defects in the main pulmonary artery, right and proximal left pulmonary arteries, simulating large pulmonary thromboembolism. Nodular borders, infiltration and enhancement help in distinguishing the mass from a bland thrombus.
Hepatic angiosarcoma
Hepatic angiosarcoma is uncommon and accounts for only 2% of all primary hepatic malignancies, but it is the most common malignant mesenchymal tumour of the liver.8,51 It predominantly affects patients aged 50–70 years, with a slight male predilection.52 In the majority of cases of primary hepatic angiosarcoma, no obvious risk factor can be identified. However, it is reported that the tumour has an association with environmental, iatrogenic and occupational exposure to certain carcinogens. These include thorium dioxide, vinyl chloride, arsenic and radiation. Haemochromatosis and von Recklinghausen disease are also known to be linked to angiosarcoma.3
As with other aggressive hepatic tumours, the most common presentation is abdominal pain, weakness, fatigue and weight loss with jaundice, hepatomegaly and ascites.19 These tumours have a very poor prognosis, with a median survival reported to be just 6 months.53 Metastases are commonly observed at the time of presentation, with the lungs and spleen being the most frequent sites.54 Portal vein thrombosis, Budd–Chiari syndrome and arteriovenous shunting-related high-output heart failure are some of the complications of hepatic angiosarcoma.55 Moreover, spontaneous rupture of tumours with massive intra-abdominal bleeding has been seen in multiple cases.56 Hepatic angiosarcomas are known to increase risk for haemorrhage after percutaneous liver biopsy.
The gross pathology and imaging findings of hepatic angiosarcoma can show four different patterns: multiple nodules (Figure 8), a large dominant mass (Figure 10), a combination of dominant mass and smaller nodules (Figure 11), and rarely, a diffusely infiltrating micronodular tumour.57–61 The two predominant patterns are large solitary mass and multifocal lesions.62
Figure 10.

A 54-year-old female with primary hepatic angiosarcoma. T1 weighted post-contrast MR images during arterial (a), portal venous (b) and delayed (c) phases show a dominant mass at the medial liver dome demonstrating progressive nodular enhancement that is similar to blood pool and resembles a cavernous haemangioma. However, multiple additional lesions are present, with a somewhat more irregular pattern of progressive enhancement. Similar to other vascular tumours, the masses are predominately hyperechoic at ultrasound (d). Contrast-enhanced CT images several months later in portal venous (e) and delayed (f) phases show marked interval tumour growth but with haemangioma-like enhancement pattern retained.
Figure 11.

A 64-year-old female presenting with hepatic angiosarcoma, over 50 years after exposure to thorium dioxide (Thorotrast®). Abdominal radiograph (a) shows dense, shrunken spleen and dense abdominal lymph nodes compatible with prior Thorotrast exposure. Contrast-enhanced CT in the arterial phase (b) shows the same splenic and nodal findings plus multifocal hypervascular liver lesions, demonstrating both nodular and ring-like enhancement patterns.
The CT appearance of angiosarcoma of the liver is consistent with that of an aggressive vascular tumour (Figures 10 and 11).54 Unenhanced CT demonstrates a predominantly hypoattenuating mass when compared with the surrounding liver parenchyma, with or without hyperattenuating foci, known to reflect haemorrhage.1,52,63 Hepatic angiosarcoma resulting from thorium dioxide exposure produces distinct imaging findings of a hypoattenuating mass on unenhanced CT with hyperattenuating linear meshwork appearance characteristic of residual Thorotrast, as well as a dense spleen and lymph nodes resulting from the agent (Figure 11).52,62
On contrast-enhanced CT, large dominant masses exhibit heterogeneous enhancement, which indicates central necrosis and fibrotic change (Figure 10). Small nodular lesions usually appear as hypoattenuating areas with some enhancing foci (Figure 10). Irregular or ring enhancement can also be seen (Figure 11). The enhancement is usually less than that of the aorta in the early phases with delayed progressive enhancement.64,65
On MRI, angiosarcoma has a heterogeneous appearance that reflects areas of haemorrhage, fibrosis and necrosis (Figure 10).64 If a dominant mass is present, the internal architecture is hyperintense on T2 with internal septae that are hypointense. T1 weighted images show low-intensity lesions with focal areas of increased signal due to haemorrhage. Angiosarcoma demonstrates a wide range of apparent diffusion coefficient (ADC) values, which is due to their heterogeneous composition. Lower ADC values may be attributed to haematoma, intermediate to progressively enhancing foci that may represent vascular channels and high values secondary to diffusion within cystic necrosis. There can be significant variability in the ADC map of different lesions, including those of the same patient.64
Several studies evaluated the dynamic enhancement of hepatic angiosarcoma on multiphasic CT and MRI.52,54,64,66 The largest case series published to date was conducted by Pickhardt et al64 and included 35 patients with biopsy-proven hepatic angiosarcoma. The findings of this study described the tumour enhancement in different phases of the dynamic post-contrast study. The lesions showed small foci of heterogeneous hyperenhacement on the first post-contrast phase that progressively expand on the subsequent phases, and typically follow blood pool with regard to CT attenuation and MR signal intensity (Figure 10). Other common findings were large hypovascular regions within the dominant lesions in addition to smaller hypovascular nodules (Figures 10 and 11). Smaller studies by Bruegel et al66 and Koyama et al52 showed progressive heterogeneous enhancement in most instances with multiphasic CT and/or MR.
The characteristic tumour progressive enhancement that follows the blood pool can sometimes resemble cavernous haemangiomas (Figure 10).1,67–69 However, the typical enhancement of hepatic angiosarcoma is of less attenuation than the aorta and hepatic artery; this is in contrast to haemangiomas that have regions of enhancement similar to the aorta during all phases of imaging.68,69 Additionally, enhancement of angiosarcoma is more irregular and disordered, even when there is progressive fill in from the periphery of the tumour (Figure 10). In their study, Pickhardt et al64 described centrifugal enhancement or a “reverse haemangioma” pattern and a complex mixture of progressive centripetal and centrifugal enhancement in cases of hepatic angiosarcoma. With precise review of the enhancement pattern, distinction from benign haemangiomas is typically possible.
On 18F-FDG PET CT, angiosarcoma is 18F-FDG avid and PET is generally used for detection of distant metastases, which are usually present at the time of presentation.11,70
In summary, cross-sectional imaging findings of hepatic angiosarcoma reflect the highly variable histopathological composition of angiosarcomas. Typical imaging features include multifocal, heterogeneous internal architecture often containing haemorrhage within large lesions; disordered, patchy arterial phase enhancement pattern is progressive during the later phases.64
Splenic angiosarcoma
Primary angiosarcoma of the spleen is very rare and is slightly more common in males (Figure 12). Patients may present with constitutional symptoms, abdominal pain from splenomegaly, anaemia and thrombocytopaenia.71,72 Splenic angiosarcoma has a 6-month survival rate of 20%.73 In cases of splenic rupture, the mean survival is only 4–5 months.74,75 Splenic angiosarcoma is usually treated with splenectomy because of the high risk of splenic rupture. However, treatment for splenic angiosarcoma is rarely curative due to the aggressive nature of the tumour.71 Splenectomy may expand the mean survival time to 14 months, emphasizing the value of prompt diagnosis.76
Figure 12.

A 62-year-old male with splenic angiosarcoma. (a) Unenhanced and (b) contrast-enhanced CT images show marked splenomegaly with irregular peripheral enhancement (b). Multiple liver lesions are also present, presumably from metastatic spread.
Many imaging modalities can be used for evaluating splenic angiosarcoma. Ultrasonography is often the initial imaging modality.71,77 The reported ultrasonography features include variable presence of splenomegaly with splenic masses of mixed echogenicity and increased internal Doppler flow.71,78,79
On CT, splenic angiosarcoma masses enhance heterogeneously with areas of necrosis (Figure 12); hyperdense haemorrhage may be seen within the tumour on non-contrast images or as extrasplenic in the event of rupture. On MR, areas of interspersed increased and decreased signal intensity reflect foci of solid tumour with areas of necrosis.71 Early spread to the liver may make determination of primary site difficult (Figure 12).
Osseous angiosarcoma
Angiosarcoma of the bone is rare and accounts for <1% of primary bone tumours.80 Approximately 6% of angiosarcomas originate in the bones.81 The peak incidence is in the third to fifth decades of life, and two-thirds of affected patients are males.58 The most common presenting symptom is swelling at the affected site with associated pain. Pathological fractures are common.59 Osseous angiosarcoma can occur in any bone type but the majority (60%) occurs in long bones, with the tibia being the most common site, followed by the femur, humerus and pelvis.82 The majority present as solitary tumours, but approximately one-third are multifocal.59
Osseous angiosarcoma is managed by surgical resection when possible, often combined with chemoradiation therapy.83 Risk factors for osseous angiosarcoma include history of prior radiation and/or lymphoedema at the tumour site, surgical implants near the tumour site, a previous bone infarction and Paget's disease.59,84,85 The tumour has a poor prognosis with one study reporting 66% of cases having distant metastases at the time of diagnosis and 5-year survival rate of 60%.81 Factors that negatively impact survival include intermediate- or high-grade tumours and tumours arising in an irradiated field.58
Typically, osseous angiosarcoma is initially detected on conventional radiography. The radiographic appearance is variable, but most exhibit a destructive, lytic pattern on radiographs without a sclerotic margin, which may be an indicator of aggressiveness (Figure 5).86 If the tumour is low grade, the bone may be expanded. High-grade tumours typically exhibit extension through the cortex and involvement of the regional soft tissues (Figure 5).86 Multicentric lesions may show a soap-bubble appearance with involvement of multiple adjacent vertebrae in the spine and extension up and down the cortex of a long bone.59
CT appearance is variable, demonstrating similar patterns overall to that seen on radiography.86 MRI appearance is compatible with an aggressive tumour associated with T1 hypointensity, heterogeneous T2 hyperintensity and irregular contrast enhancement (Figure 2 and 5). The lesions may exhibit peripheral vascularity.82 In addition, fluid–fluid levels may be present due to necrosis and haemorrhage.87 Cross-sectional imaging features are not specific enough to distinguish angiosarcoma from other similar primary osseous malignancies such as haemangiopericytoma and haemangioendothelioma.82 Therefore, tissue sampling is crucial for accurate diagnosis. Skeletal scintigraphy (99mTc-methylene diphosphonate) and 18F-FDG PET can be used to assess for multifocality, where a high degree of uptake is seen.86
Rare locations
Since angiosarcoma originates from vascular endothelium, it can arise anywhere in the body.
Angiosarcoma of the gastrointestinal tract is very rare and can present as a primary lesion or metastatic disease.88 Case reports in the literature discuss the pathological characteristics of these tumours but not their radiological characteristics.
Angiosarcoma of the urinary bladder is also an extremely rare and poorly characterized tumour. Patients usually present with pelvic pain and gross haematuria. CT and MRI demonstrate a large highly vascular mass with irregular margins. The tumour can protrude into the bladder lumen as well as extend into the extravesical region. Hydronephrosis, hydroureter and distant metastases are known associations.89
Angiosarcoma of the base of the penis is a rare entity that has been mentioned in only a few isolated case reports (Figure 13).90–92 Total penile amputation may be needed due to high propensity for local recurrence, even for a superficial lesion.92
Figure 13.

59- and 63-year-old males with penile and spermatic cord angiosarcomas. Contrast-enhanced axial CT images in two different patients with penile (a) and left spermatic cord angiosarcomas. There are heterogeneous diffusely infiltrative masses that involve the penis and encase the urethra in the first patient (a) as well as the left spermatic cord in the second patient (b) (arrows).
Primary testicular angiosarcoma is a rare tumour in males seen at all ages. It can be classified into two subgroups: one group comprised of young people in association with teratoma and the other occurring in older individuals without prior tumours. The latter may occur in association with chronic hydrocele.93 No imaging features have been reported for these tumours. Angiosarcoma arising in the spermatic cord can also be rarely seen (Figure 13).
ANGIOSARCOMA METASTASES
Regardless of its site of origin, angiosarcoma has a high tendency for metastatic multifocal disease.9,94 The dissemination of this tumour is predominantly haematogenous, early and aggressive. The lung is the most frequent site for metastatic angiosarcoma. Other frequent sites include the liver, bone and lymph nodes.9,95
As for the radiological appearance of metastatic angiosarcoma of the lungs, chest radiography shows bilateral peripheral solid nodules that are difficult to distinguish from those caused by other metastatic tumours. The most common CT manifestations are multiple solid nodular lesions, followed by multiple thin-walled cysts. Most of the solid lesions show inhomogeneous enhancement. In addition, mixed pattern tumours have been described, where both solid nodular and cystic lesions are seen concomitantly. Hilar adenopathy, punctate calcification, pneumothorax and pleural effusions have also been described in some patients with angiosarcoma lung metastases.94,95
Either of these presentations, solid or cystic, can often be observed with haemorrhagic change, which is considered a characteristic finding in metastatic angiosarcoma. Haemorrhagic changes may be due to the fragility of the neovascular tissue, which predisposes vessels to thrombosis and rupture. Radiologically, these haemorrhagic complications may present as air–fluid levels in thin-walled cysts, diffuse pulmonary infiltrates, haemothorax or areas of ground-glass attenuation.95
When discussing metastatic angiosarcoma of the liver, the common CT findings include multiple hypoattenuating lesions relative to the adjacent liver parenchyma. This is often associated with nodular enhancement and cystic attenuation with haemorrhagic change (e.g. fluid levels). On T1 weighted MR images, these tumours appeared heterogeneous and hypointense relative to adjacent liver parenchyma. Most show minimal peripheral enhancement after administration of intravenous gadolinium contrast.96
Few studies are available that have described the radiological presentation of metastatic angiosarcoma in the bone. Studies carried out on large cohorts or that have documented skeletal findings or metastatic pattern in the bone are not available.97 These reports are limited to case studies and several were secondary to primary angiosarcoma of the spleen despite being an uncommon site for primary tumours.97–99
In one study of a patient with primary splenic angiosarcoma, where pelvic MRI was followed by consecutive axial CT imaging at corresponding sacral levels, the radiographical findings presented as well-circumscribed osteosclerosis and, to a much lesser extent, osteolytic lesions.97 Furthermore, cortical breaching was minimal. These authors conclude that MRI did not provide significant additional information in the discrimination of the disease and found MR T2 weighted images to be insensitive for many of the osteoblastic lesions, whereas T1 weighted images depicted most lesions but not all lesions already clearly visible by CT. Thus, they favour its application in follow-up imaging for the evaluation of treatment response.97
Another study of a patient diagnosed with primary breast angiosarcoma revealed multiple well-defined bone lesions in the pelvis and lower spine, with high signal intensity on T2 weighted images and low signal intensity on T1 weighted images. These lesions showed persistent contrast enhancement on more delayed phases, suggestive of metastases, and were confirmed by CT-guided biopsy.100 According to this study, MRI was a more useful tool in identifying osteoblastic bone metastases that were not detected on CT scan or PET/CT.100
ANGIOSARCOMA MANAGEMENT
By and large, angiosarcomas have a poor prognosis with reported 5-year survival rates ranging from 12% to 35%.101 Treatment options include surgery, radiotherapy and chemotherapy, but outcomes vary widely and are impacted by site, size, resectability and tumour type (i.e. de novo vs radiation induced).101 Angiosarcomas of the scalp, compared with other sites, have a particularly poor prognosis with significant reduction in overall survival. Although angiosarcomas secondary to prior radiation have improved outcomes in some studies, other research contradict this finding.101
The mainstay of treatment has traditionally been wide-margin surgical resection. However, this approach alone is problematic in cases that are non-resectable (as is often the case in lesions of the scalp).102 Furthermore, it has generally resulted in poor outcomes.101 Thus, some studies have advocated for a more aggressive local multimodality approach that entails both surgical resection and radiotherapy whenever possible, citing improved survival rates.101 In one series, the median overall survival is about 36 months after adjuvant radiotherapy compared with 9 months without adjuvant radiotherapy, whereas in another, the 5-year overall survival is about 45% with adjuvant radiotherapy and about 20% with surgical resection alone.103 It is important to note that angiosarcoma of certain sites require specific considerations. For instance, lesions of the liver may benefit from embolization.102
As for adjuvant chemotherapy in the treatment of angiosarcoma, there remains some controversy with respect to its therapeutic role, as well as little agreement on the choice of agents.104,105 The most common chemotherapeutic drugs (targeting cytokines and their receptors) are Adriamycin®, ifosfamide, cyclophosphamide, vincristine, and dacarbazine, and paclitaxel is also typically administered weekly.105 Radiation-induced angiosarcomas seemed to fare as well to therapy as those that arose spontaneously.102 For recurrent cases of angiosarcoma, taxanes and pegylated liposomal doxorubicin have shown significant response rates. New and emerging therapeutic options are still under study, the most notable of which are the role of angiogenetic agents. Out of these numerous pro-angiogenetic factors, vascular permeability factor could be of major importance in the future of angiosarcoma management.102,103
CONCLUSIONS
Angiosarcoma is a rare, highly aggressive tumour that can affect any organ in the body but appears most commonly in its cutaneous form, which accounts for nearly 50% of all tumours reported. It has a rather poor prognosis, particularly when the patient is diagnosed with metastases at initial presentation, as is often the case. The aetiology is typically unknown but there are definite risk factors that have been identified for angiosarcoma; the two most important are the presence of chronic lymphoedema and prior radiation therapy. Other risk factors include environmental carcinogens (vinyl chloride, thorium dioxide and arsenic), foreign bodies such as synthetic grafts and certain familial syndromes. Multimodality imaging is necessary for proper staging and management. However, accurate radiological diagnosis of angiosarcoma is as much of a challenge as its treatment. What is certain, however, is that the radiological presentation is quite variable. Early diagnosis can be improved by correlation for risk factors, particularly in cases where the radiological findings suggest the presence of a vascular tumour with aggressive features.
Contributor Information
Ayman H Gaballah, Email: kmelsayes@mdanderson.org.
Corey T Jensen, Email: kmelsayes@mdanderson.org.
Sarah Palmquist, Email: kmelsayes@mdanderson.org.
Perry J Pickhardt, Email: ppickhardt2@uwhealth.org.
Alper Duran, Email: kmelsayes@mdanderson.org.
Gregory Broering, Email: kmelsayes@mdanderson.org.
Khaled M Elsayes, Email: kmelsayes@mdanderson.org.
REFERENCES
- 1.Peterson MS, Baron RL, Rankin SC. Hepatic angiosarcoma: findings on multiphasic contrast-enhanced helical CT do not mimic hepatic hemangioma. AJR Am J Roentgenol 2000; 175: 165–70. doi: https://doi.org/10.2214/ajr.175.1.1750165 [DOI] [PubMed] [Google Scholar]
- 2.Walker EA, Song AJ, Murphey MD. Magnetic resonance imaging of soft-tissue masses. Semin Roentgenol 2010; 45: 277–97. doi: https://doi.org/10.1053/j.ro.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 3.Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol 2010; 11: 983–91. doi: https://doi.org/10.1016/s1470-2045(10)70023-1 [DOI] [PubMed] [Google Scholar]
- 4.Walker EA, Salesky JS, Fenton ME, Murphey MD. Magnetic resonance imaging of malignant soft tissue neoplasms in the adult. Radiol Clin North Am 2011; 49: 1219–34, vi. doi: https://doi.org/10.1016/j.rcl.2011.07.006 [DOI] [PubMed] [Google Scholar]
- 5.Murinello A, Mendonca P, Abreu A, Santos AL, Roquete J, Pinto E, et al. Cardiac angiosarcoma—a review. Rev Port Cardiol 2007; 26: 577–84. [PubMed] [Google Scholar]
- 6.Lahat G, Dhuka AR, Hallevi H, Xiao L, Zou C, Smith KD, et al. Angiosarcoma: clinical and molecular insights. Ann Surg 2010; 251: 1098–106. doi: https://doi.org/10.1097/sla.0b013e3181dbb75a [DOI] [PubMed] [Google Scholar]
- 7.Rouhani P, Fletcher CD, Devesa SS, Toro JR. Cutaneous soft tissue sarcoma incidence patterns in the U.S. : an analysis of 12,114 cases. Cancer 2008; 113: 616–27. doi: https://doi.org/10.1002/cncr.23571 [DOI] [PubMed] [Google Scholar]
- 8.Harris E, Barry M, Kell MR. Meta-analysis to determine if surgical resection of the primary tumour in the setting of stage IV breast cancer impacts on survival. Ann Surg Oncol 2013; 20: 2828–34. doi: https://doi.org/10.1245/s10434-013-2998-2 [DOI] [PubMed] [Google Scholar]
- 9.Meis-Kindblom JM, Kindblom LG. Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998; 22: 683–97. [DOI] [PubMed] [Google Scholar]
- 10.Chang JH, Kim JH, Hong SH, Song ME, Ryu YJ, Lee JH, et al. Angiosarcoma presenting with spontaneous hydropneumothorax: report of a case and review of the literature. Open Respir Med J 2014; 8: 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almogy G, Lieberman S, Gips M, Pappo O, Edden Y, Jurim O, et al. Clinical outcomes of surgical resections for primary liver sarcoma in adults: results from a single centre. Eur J Surg Oncol 2004; 30: 421–7. doi: https://doi.org/10.1016/j.ejso.2004.01.004 [DOI] [PubMed] [Google Scholar]
- 12.Sturgis EM, Potter BO. Sarcomas of the head and neck region. Curr Opin Oncol 2003; 15: 239–52. doi: https://doi.org/10.1097/00001622-200305000-00011 [DOI] [PubMed] [Google Scholar]
- 13.Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer 1948; 1: 64–81. doi: https://doi.org/10.1002/1097-0142(194805)1:1<64::aid-cncr2820010105>3.0.co;2-w [DOI] [PubMed] [Google Scholar]
- 14.Huang J, Mackillop WJ. Increased risk of soft tissue sarcoma after radiotherapy in women with breast carcinoma. Cancer 2001; 92: 172–80. doi: https://doi.org/10.1002/1097-0142(20010701)92:1<172::aid-cncr1306>3.0.co;2-k [DOI] [PubMed] [Google Scholar]
- 15.Virtanen A, Pukkala E, Auvinen A. Angiosarcoma after radiotherapy: a cohort study of 332,163 Finnish cancer patients. Br J Cancer 2007; 97: 115–7. doi: https://doi.org/10.1038/sj.bjc.6603805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldblum JR, Weiss SW, Folpe AL. Enzinger and Weiss’s soft tissue tumors. ch. 23. Malignant Vascular Tumors: Mosby; 2014. pp. 703–32.
- 17.Huang NC, Wann SR, Chang HT, Lin SL, Wang JS, Guo HR. Arsenic, vinyl chloride, viral hepatitis, and hepatic angiosarcoma: a hospital-based study and review of literature in Taiwan. BMC Gastroenterol 2011; 11: 142. doi: https://doi.org/10.1186/1471-230X-11-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohan VS, Hanji AM, Patel JJ, Tankshali RA. Primary angiosarcoma of the breast in a postmenopausal patient. J Cancer Res Ther 2010; 6: 120–2. doi: https://doi.org/10.4103/0973-1482.63543 [DOI] [PubMed] [Google Scholar]
- 19.Bhatia K, Shiels MS, Berg A, Engels EA. Sarcomas other than Kaposi sarcoma occurring in immunodeficiency: interpretations from a systematic literature review. Curr Opin Oncol 2012; 24: 537–46. doi: https://doi.org/10.1097/cco.0b013e328355e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed I, Hamacher KL. Angiosarcoma in a chronically immunosuppressed renal transplant recipient: report of a case and review of the literature. Am J Dermatopathol 2002; 24: 330–5. doi: https://doi.org/10.1097/00000372-200208000-00009 [DOI] [PubMed] [Google Scholar]
- 21.Goedert JJ, Cote TR, Virgo P, Scoppa SM, Kingma DW, Gail MH, et al. Spectrum of AIDS-associated malignant disorders. Lancet 1998; 351: 1833–9. doi: https://doi.org/10.1016/s0140-6736(97)09028-4 [DOI] [PubMed] [Google Scholar]
- 22.Dhanasekar P, Karthikeyan VS, Rajkumar N, Chandra Sistla S, Manwar Ali S, Basu D, et al. Cutaneous angiosarcoma of the scalp masquerading as a squamous cell carcinoma: case report and literature review. J Cutan Med Surg 2012; 16: 187–90. doi: https://doi.org/10.1177/120347541201600309 [DOI] [PubMed] [Google Scholar]
- 23.Razek AA, Huang BY. Soft tissue tumors of the head and neck: imaging-based review of the WHO classification. Radiographics 2011; 31: 1923–54. doi: https://doi.org/10.1148/rg.317115095 [DOI] [PubMed] [Google Scholar]
- 24.La Corte E, Acerbi F, Schiariti M, Broggi M, Maderna E, Pollo B, et al. Primary central nervous system angiosarcoma: a case report and literature review. Neuropathology 2015; 35: 184–91. doi: https://doi.org/10.1111/neup.12178 [DOI] [PubMed] [Google Scholar]
- 25.Charman HP, Lowenstein DH, Cho KG, DeArmond SJ, Wilson CB. Primary cerebral angiosarcoma. Case report. J Neurosurg 1988; 68: 806–10. doi: https://doi.org/10.3171/jns.1988.68.5.0806 [DOI] [PubMed] [Google Scholar]
- 26.Pawlik TM, Paulino AF, McGinn CJ, Baker LH, Cohen DS, Morris JS, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer 2003; 98: 1716–26. doi: https://doi.org/10.1002/cncr.11667 [DOI] [PubMed] [Google Scholar]
- 27.Tateishi U, Yamaguchi U, Seki K, Terauchi T, Arai Y, Kim EE. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007; 245: 839–47. doi: https://doi.org/10.1148/radiol.2453061538 [DOI] [PubMed] [Google Scholar]
- 28.O'Neill AC, D'Arcy C, McDermott E, O'Doherty A, Quinn C, McNally S. Magnetic resonance imaging appearances in primary and secondary angiosarcoma of the breast. J Med Imaging Radiat Oncol 2014; 58: 208–12. doi: https://doi.org/10.1111/1754-9485.12100 [DOI] [PubMed] [Google Scholar]
- 29.Glazebrook KN, Magut MJ, Reynolds C. Angiosarcoma of the breast. AJR Am J Roentgenol 2008; 190: 533–8. doi: https://doi.org/10.2214/ajr.07.2909 [DOI] [PubMed] [Google Scholar]
- 30.Brenn T, Fletcher CD. Postradiation vascular proliferations: an increasing problem. Histopathology 2006; 48: 106–14. doi: https://doi.org/10.1111/j.1365-2559.2005.02293.x [DOI] [PubMed] [Google Scholar]
- 31.Cheseboro AL, Chikarmane SA, Gombos EC, Giardino AA. Radiation associated angiosarcoma of the breast: what the radiologist needs to know. AJR Am J Roentgenol 2016; 207: 217–225. [DOI] [PubMed] [Google Scholar]
- 32.Liberman L, Dershaw DD, Kaufman RJ, Rosen PP. Angiosarcoma of the breast. Radiology 1992; 183: 649–54. doi: https://doi.org/10.1148/radiology.183.3.1584913 [DOI] [PubMed] [Google Scholar]
- 33.Yang WT, Hennessy BT, Dryden MJ, Valero V, Hunt KK, Krishnamurthy S. Mammary angiosarcomas: imaging findings in 24 patients. Radiology 2007; 242: 725–34. doi: https://doi.org/10.1148/radiol.2423060163 [DOI] [PubMed] [Google Scholar]
- 34.Kikawa Y, Konishi Y, Nakamoto Y, Harada T, Takeo M, Ogata M, et al. Angiosarcoma of the breast—specific findings of MRI. Breast Cancer 2006; 13: 369–73. doi: https://doi.org/10.2325/jbcs.13.369 [DOI] [PubMed] [Google Scholar]
- 35.Chikarmane SA, Gombos EC, Jagadeesan J, Raut C, Jagannathan JP. MRI findings of radiation-associated angiosarcoma of the breast (RAS). J Magn Reson Imaging 2015; 42: 763–70. doi: https://doi.org/10.1002/jmri.24822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng W, Styblo TM, Li S, Sepulveda JN, Schuster DM. Breast angiosarcoma: FDG PET findings. Clin Nucl Med 2009; 34: 443–5. doi: https://doi.org/10.1097/rlu.0b013e3181a7d0eb [DOI] [PubMed] [Google Scholar]
- 37.Bendel EC, Maleszewski JJ, Araoz PA. Imaging sarcomas of the great vessels and heart. Semin Ultrasound CT MR 2011; 32: 377–404. doi: https://doi.org/10.1053/j.sult.2011.06.001 [DOI] [PubMed] [Google Scholar]
- 38.Sparrow PJ, Kurian JB, Jones TR, Sivananthan MU. MR imaging of cardiac tumors. Radiographics 2005; 25: 1255–76. doi: https://doi.org/10.1148/rg.255045721 [DOI] [PubMed] [Google Scholar]
- 39.Uchita S, Hata T, Tsushima Y, Matsumoto M, Hina K, Moritani T. Primary cardiac angiosarcoma with superior vena caval syndrome: review of surgical resection and interventional management of venous inflow obstruction. Can J Cardiol 1998; 14: 1283–5. [PubMed] [Google Scholar]
- 40.Bruna J, Lockwood M. Primary heart angiosarcoma detected by computed tomography and magnetic resonance imaging. Eur Radiol 1998; 8: 66–8. doi: https://doi.org/10.1007/s003300050341 [DOI] [PubMed] [Google Scholar]
- 41.Herrmann MA, Shankerman RA, Edwards WD, Shub C, Schaff HV. Primary cardiac angiosarcoma: a clinicopathologic study of six cases. J Thorac Cardiovasc Surg 1992; 103: 655–64. [PubMed] [Google Scholar]
- 42.Burke A. Primary malignant cardiac tumors. Semin Diagn Pathol 2008; 25: 39–46. doi: https://doi.org/10.1053/j.semdp.2007.10.006 [DOI] [PubMed] [Google Scholar]
- 43.Buckley O, Madan R, Kwong R, Rybicki FJ, Hunsaker A. Cardiac masses, part 2: key imaging features for diagnosis and surgical planning. AJR Am J Roentgenol 2011; 197: W842–51. doi: https://doi.org/10.2214/ajr.11.6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Best AK, Dobson RL, Ahmad AR. Best cases from the AFIP: cardiac angiosarcoma. Radiographics 2003; 23: S141–5. [DOI] [PubMed] [Google Scholar]
- 45.Marafioti T, Castorino F, Gula G. Cardiac angiosarcoma. Histological, immunohistochemical and ultrastructural study. Pathologica 1993; 85: 103–11. [PubMed] [Google Scholar]
- 46.Kaminaga T, Takeshita T, Kimura I. Role of magnetic resonance imaging for evaluation of tumors in the cardiac region. Eur Radiol 2003; 13(Suppl. 6): L1–10. [DOI] [PubMed] [Google Scholar]
- 47.Yahata S, Endo T, Honma H, Ino T, Hayakawa H, Ogawa M, et al. Sunray appearance on enhanced magnetic resonance image of cardiac angiosarcoma with pericardial obliteration. Am Heart J 1994; 127: 468–71. doi: https://doi.org/10.1016/0002-8703(94)90149-x [DOI] [PubMed] [Google Scholar]
- 48.Kim MJ, Kim MS, Park JH, Park KI, Lee CS, Na MH, et al. Pulmonary artery angiosarcoma confused with acute pulmonary thromboembolism: focusing on clinical and echocardiographic features in the differentiation of two categories. J Cardiovasc Ultrasound 2015; 23: 44–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cox JE, Chiles C, Aquino SL, Savage P, Oaks T. Pulmonary artery sarcomas: a review of clinical and radiologic features. J Comput Assist Tomogr 1997; 21: 750–5. doi: https://doi.org/10.1097/00004728-199709000-00018 [DOI] [PubMed] [Google Scholar]
- 50.Yi CA, Lee KS, Choe YH, Han D, Kwon OJ, Kim S. Computed tomography in pulmonary artery sarcoma: distinguishing features from pulmonary embolic disease. J Comput Assist Tomogr 2004; 28: 34–9. doi: https://doi.org/10.1097/00004728-200401000-00005 [DOI] [PubMed] [Google Scholar]
- 51.Chaudhary P, Bhadana U, Singh RA, Ahuja A. Primary hepatic angiosarcoma. Eur J Surg Oncol 2015; 41: 1137–43. doi: https://doi.org/10.1016/j.ejso.2015.04.022 [DOI] [PubMed] [Google Scholar]
- 52.Koyama T, Fletcher JG, Johnson CD, Kuo MS, Notohara K, Burgart LJ. Primary hepatic angiosarcoma: findings at CT and MR imaging. Radiology 2002; 222: 667–73. doi: https://doi.org/10.1148/radiol.2223010877 [DOI] [PubMed] [Google Scholar]
- 53.Buetow PC, Buck JL, Ros PR, Goodman ZD. Malignant vascular tumors of the liver: radiologic-pathologic correlation. Radiographics 1994; 14: 153–66; quiz 167–8. [DOI] [PubMed] [Google Scholar]
- 54.Qiu LL, Yu RS, Chen Y, Zhang Q. Sarcomas of abdominal organs: computed tomography and magnetic resonance imaging findings. Semin Ultrasound CT MR 2011; 32: 405–21. doi: https://doi.org/10.1053/j.sult.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 55.Harrison JR, Faust TW, Blackstone MO. Hepatic angiosarcoma: an unusual cause of congestive heart failure. South Med J 2001; 94: 336–8. doi: https://doi.org/10.1097/00007611-200103000-00012 [PubMed] [Google Scholar]
- 56.Locker GY, Doroshow JH, Zwelling LA, Chabner BA. The clinical features of hepatic angiosarcoma: a report of four cases and a review of the English literature. Medicine (Baltimore) 1979; 58: 48–64. doi: https://doi.org/10.1097/00005792-197901000-00003 [DOI] [PubMed] [Google Scholar]
- 57.Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med 2011; 135: 268–72. doi: https://doi.org/10.1043/1543-2165-135.2.268 [DOI] [PubMed] [Google Scholar]
- 58.Abraham JA, Hornicek FJ, Kaufman AM, Harmon DC, Springfield DS, Raskin KA, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol 2007; 14: 1953–67. doi: https://doi.org/10.1245/s10434-006-9335-y [DOI] [PubMed] [Google Scholar]
- 59.Abdelwahab IF, Klein MJ, Hermann G, Springfield D. Angiosarcomas associated with bone infarcts. Skeletal Radiol 1998; 27: 546–51. doi: https://doi.org/10.1007/s002560050435 [DOI] [PubMed] [Google Scholar]
- 60.Weitz J, Klimstra DS, Cymes K, Jarnagin WR, D'Angelica M, La Quaglia MP, et al. Management of primary liver sarcomas. Cancer 2007; 109: 1391–6. doi: https://doi.org/10.1002/cncr.22530 [DOI] [PubMed] [Google Scholar]
- 61.Kojiro M, Nakashima T, Ito Y, Ikezaki H, Mori T, Kido C. Thorium dioxide-related angiosarcoma of the liver. Pathomorphologic study of 29 autopsy cases. Arch Pathol Lab Med 1985; 109: 853–7. [PubMed] [Google Scholar]
- 62.White PG, Adams H, Smith PM. The computed tomographic appearances of angiosarcoma of the liver. Clin Radiol 1993; 48: 321–5. doi: https://doi.org/10.1016/s0009-9260(05)81240-1 [DOI] [PubMed] [Google Scholar]
- 63.Park YS, Kim JH, Kim KW, Lee IS, Yoon HK, Ko GY, et al. Primary hepatic angiosarcoma: imaging findings and palliative treatment with transcatheter arterial chemoembolization or embolization. Clin Radiol 2009; 64: 779–85. doi: https://doi.org/10.1016/j.crad.2009.02.019 [DOI] [PubMed] [Google Scholar]
- 64.Pickhardt PJ, Kitchin D, Lubner MG, Ganeshan DM, Bhalla S, Covey AM. Primary hepatic angiosarcoma: multi-institutional comprehensive cancer centre review of multiphasic CT and MR imaging in 35 patients. Eur Radiol 2015; 25: 315–22. doi: https://doi.org/10.1007/s00330-014-3442-0 [DOI] [PubMed] [Google Scholar]
- 65.Yu RS, Chen Y, Jiang B, Wang LH, Xu XF. Primary hepatic sarcomas: CT findings. Eur Radiol 2008; 18: 2196–205. doi: https://doi.org/10.1007/s00330-008-0997-7 [DOI] [PubMed] [Google Scholar]
- 66.Bruegel M, Muenzel D, Waldt S, Specht K, Rummeny EJ. Hepatic angiosarcoma: cross-sectional imaging findings in seven patients with emphasis on dynamic contrast-enhanced and diffusion-weighted MRI. Abdom Imaging 2013; 38: 745–54. doi: https://doi.org/10.1007/s00261-012-9967-2 [DOI] [PubMed] [Google Scholar]
- 67.Ginsberg F, Slavin JD, Jr, Spencer RP. Hepatic angiosarcoma: mimicking of angioma on three-phase technetium-99m red blood cell scintigraphy. J Nucl Med 1986; 27: 1861–3. [PubMed] [Google Scholar]
- 68.Okano A, Sonoyama H, Masano Y, Taniguchi T, Ohana M, Kusumi F, et al. The natural history of a hepatic angiosarcoma that was difficult to differentiate from cavernous hemangioma. Intern Med 2012; 51: 2899–904. doi: https://doi.org/10.2169/internalmedicine.51.7994 [DOI] [PubMed] [Google Scholar]
- 69.Itai Y, Teraoka T. Angiosarcoma of the liver mimicking cavernous hemangioma on dynamic CT. J Comput Assist Tomogr 1989; 13: 910–2. doi: https://doi.org/10.1097/00004728-198909000-00032 [DOI] [PubMed] [Google Scholar]
- 70.Maeda T, Tateishi U, Hasegawa T, Ojima H, Arai Y, Sugimura K. Primary hepatic angiosarcoma on coregistered FDG PET and CT images. AJR Am J Roentgenol 2007; 188: 1615–7. doi: https://doi.org/10.2214/ajr.05.0830 [DOI] [PubMed] [Google Scholar]
- 71.Vrachliotis TG, Bennett WF, Vaswani KK, Niemann TH, Bova JG. Primary angiosarcoma of the spleen—CT, MR, and sonographic characteristics: report of two cases. Abdom Imaging 2000; 25: 283–5. doi: https://doi.org/10.1007/s002610000034 [DOI] [PubMed] [Google Scholar]
- 72.Koutelidakis IM, Tsiaousis PZ, Papaziogas BT, Patsas AG, Atmatzidis SK, Atmatzidis KS. Spleen rupture due to primary angiosarcoma: a case report. J Gastrointest Cancer 2007; 38: 74–7. doi: https://doi.org/10.1007/s12029-008-9034-y [DOI] [PubMed] [Google Scholar]
- 73.Aranha GV, Gold J, Grage TB. Hemangiosarcoma of the spleen: report of a case and review of previously reported cases. J Surg Oncol 1976; 8: 481–7. doi: https://doi.org/10.1002/jso.2930080607 [DOI] [PubMed] [Google Scholar]
- 74.Smith VC, Eisenberg BL, McDonald EC. Primary splenic angiosarcoma. Case report and literature review. Cancer 1985; 55: 1625–7. doi: https://doi.org/10.1002/1097-0142(19850401)55:7<1625::aid-cncr2820550736>3.0.co;2-w [DOI] [PubMed] [Google Scholar]
- 75.Karakas HM, Demir M, Ozyilmaz F, Cakir B. Primary angiosarcoma of the spleen: in vivo and in vitro MRI findings. Clin Imaging 2001; 25: 192–6. doi: https://doi.org/10.1016/s0899-7071(01)00286-8 [DOI] [PubMed] [Google Scholar]
- 76.Montemayor P, Caggiano V. Primary hemangiosarcoma of the spleen associated with leukocytosis and abnormal spleen scan. Int Surg 1980; 65: 369–73. [PubMed] [Google Scholar]
- 77.Qi R, Yu JQ, Xu H, Zhou XP, Li XM. Primary angiosarcoma of the spleen as depicted on computed tomography. Clin Imaging 2012; 36: 619–22. doi: https://doi.org/10.1016/j.clinimag.2011.12.016 [DOI] [PubMed] [Google Scholar]
- 78.Nahman B, Cunningham JJ. Sonography of splenic angiosarcoma. J Clin Ultrasound 1985; 13: 354–6. doi: https://doi.org/10.1002/jcu.1870130513 [DOI] [PubMed] [Google Scholar]
- 79.Wafula JM. Ultrasound and CT demonstration of primary angiosarcoma of the spleen. Br J Radiol 1985; 58: 903–5. doi: https://doi.org/10.1259/0007-1285-58-693-903 [DOI] [PubMed] [Google Scholar]
- 80.Evans HL, Raymond AK, Ayala AG. Vascular tumors of bone: a study of 17 cases other than ordinary hemangioma, with an evaluation of the relationship of hemangioendothelioma of bone to epithelioid hemangioma, epithelioid hemangioendothelioma, and high-grade angiosarcoma. Hum Pathol 2003; 34: 680–9. doi: https://doi.org/10.1016/s0046-8177(03)00249-1 [DOI] [PubMed] [Google Scholar]
- 81.Yamashita H, Endo K, Teshima R. Angiosarcoma of the proximal humerus: a case report and review of the literature. J Med Case Rep 2012; 6: 347. doi: https://doi.org/10.1186/1752-1947-6-347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murphey MD, Fairbairn KJ, Parman LM, Baxter KG, Parsa MB, Smith WS. From the archives of the AFIP. Musculoskeletal angiomatous lesions: radiologic-pathologic correlation. Radiographics 1995; 15: 893–917. doi: https://doi.org/10.1148/radiographics.15.4.7569134 [DOI] [PubMed] [Google Scholar]
- 83.Asmane I, Litique V, Heymann S, Marcellin L, Metivier AC, Duclos B, et al. Adriamycin, cisplatin, ifosfamide and paclitaxel combination as front-line chemotherapy for locally advanced and metastatic angiosarcoma. Analysis of three case reports and review of the literature. Anticancer Res 2008; 28: 3041–5. [PubMed] [Google Scholar]
- 84.McDonald DJ, Enneking WF, Sundaram M. Metal-associated angiosarcoma of bone: report of two cases and review of the literature. Clin Orthop Relat Res 2002; 396: 206–14. doi: https://doi.org/10.1097/00003086-200203000-00031 [DOI] [PubMed] [Google Scholar]
- 85.Boulanger V, Chauveaux D, Kantor G, Loyer-Lecestre MJ, Rivel J, Coindre JM, et al. Primary angiosarcoma of bone in Paget's disease. Eur J Surg Oncol 1998; 24: 611–3. doi: https://doi.org/10.1016/s0748-7983(98)94112-9 [DOI] [PubMed] [Google Scholar]
- 86.Lomasney LM, Martinez S, Demos TC, Harrelson JM. Multifocal vascular lesions of bone: imaging characteristics. Skeletal Radiol 1996; 25: 255–61. doi: https://doi.org/10.1007/s002560050075 [DOI] [PubMed] [Google Scholar]
- 87.Griffith B, Yadam S, Mayer T, Mott M, van Holsbeeck M. Angiosarcoma of the humerus presenting with fluid-fluid levels on MRI: a unique imaging presentation. Skeletal Radiol 2013; 42: 1611–6. doi: https://doi.org/10.1007/s00256-013-1656-x [DOI] [PubMed] [Google Scholar]
- 88.Allison KH, Yoder BJ, Bronner MP, Goldblum JR, Rubin BP. Angiosarcoma involving the gastrointestinal tract: a series of primary and metastatic cases. Am J Surg Pathol 2004; 28: 298–307. doi: https://doi.org/10.1097/00000478-200403000-00002 [DOI] [PubMed] [Google Scholar]
- 89.Beyazal M, Pirincci N, Yavuz A, Ozkacmaz S, Bulut G. Computed tomography and magnetic resonance imaging findings of primary bladder angiosarcoma: a case report. Clin Imaging 2014; 38: 212–4. doi: https://doi.org/10.1016/j.clinimag.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 90.Webber RJ, Alsaffar N, Bissett D, Langlois NE. Angiosarcoma of the penis. Urology 1998; 51: 130–1. doi: https://doi.org/10.1016/s0090-4295(97)00471-8 [DOI] [PubMed] [Google Scholar]
- 91.Dehner LP, Smith BH. Soft tissue tumors of the penis. A clinicopathologic study of 46 cases. Cancer. 1970;25(6):1431–47.doi: https://doi.org/10.1002/1097-0142(197006)25:6<1431::aid-cncr2820250624>3.0.co;2-b [DOI] [PubMed] [Google Scholar]
- 92.Schellhammer P, Jordan G, Schelenbergh S. Tumours of the penis. In: Walsh P, Retik A, Stamey T, Vaughan E, eds. Campbell’s Urology. 6th edn; 1992. [Google Scholar]
- 93.Armah HB, Rao UN, Parwani AV. Primary angiosarcoma of the testis: report of a rare entity and review of the literature. Diagn Pathology 2007; 2: 23. doi: https://doi.org/10.1186/1746-1596-2-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bocklage T, Leslie K, Yousem S, Colby T. Extracutaneous angiosarcomas metastatic to the lungs: clinical and pathologic features of twenty-one cases. Mod Pathol 2001; 14: 1216–25. doi: https://doi.org/10.1038/modpathol.3880463 [DOI] [PubMed] [Google Scholar]
- 95.Tateishi U, Hasegawa T, Kusumoto M, Yamazaki N, Iinuma G, Muramatsu Y, et al. Metastatic angiosarcoma of the lung: spectrum of CT findings. AJR Am J Roentgenol 2003; 180: 1671–4. doi: https://doi.org/10.2214/ajr.180.6.1801671 [DOI] [PubMed] [Google Scholar]
- 96.Tateishi U, Hasegawa T, Muramatsu Y, Moriyama N. Hepatic metastases of soft tissue angiosarcoma: CT and MR imaging findings. Abdom Imaging 2003; 28: 660–4. doi: https://doi.org/10.1007/s00261-003-0008-z [DOI] [PubMed] [Google Scholar]
- 97.Jobke B, Werner M, Jundt G, Ostertag H, Freyschmidt J. Protracted disseminated skeletal metastases from angiosarcoma of the spleen. Clin Exp Metastasis 2010; 27: 117–22. doi: https://doi.org/10.1007/s10585-010-9308-1 [DOI] [PubMed] [Google Scholar]
- 98.Wang C, Rabah R, Blackstein M, Riddell RH. Bone marrow metastasis of angiosarcoma. Pathol Res Pract 2004; 200: 551–5. doi: https://doi.org/10.1016/j.prp.2004.05.003 [DOI] [PubMed] [Google Scholar]
- 99.Anoun S, Marouane S, Quessar A, Benchekroun S. Primary splenic angiosarcoma revealed by bone marrow metastasis. Turk J Haematol 2014; 4: 408–10. doi: https://doi.org/10.4274/tjh.2013.0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cucci E, Ciuffreda M, Tambaro R, Aquilani L, Barrassi M, Sallustio G. MRI findings of large low-grade angiosarcoma of the breast with subsequent bone metastases: a case report. J Breast Cancer 2012; 15: 255–7. doi: https://doi.org/10.4048/jbc.2012.15.2.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scott MT, Portnow LH, Morris CG, Marcus RB, Jr, Mendenhall NP, Mendenhall WM, et al. Radiation therapy for angiosarcoma: the 35-year University of Florida experience. Am J Clin Oncol 2013; 36: 174–80. doi: https://doi.org/10.1097/COC.0b013e3182436ea3 [DOI] [PubMed] [Google Scholar]
- 102.Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 2013; 27: 975–88. doi: https://doi.org/10.1016/j.hoc.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 103.Penel N, Marréaud S, Robin YM, Hohenberger P. Angiosarcoma: state of the art and perspectives. Crit Rev Oncol Hematol 2011; 80: 257–63. doi: https://doi.org/10.1016/j.critrevonc.2010.10.007 [DOI] [PubMed] [Google Scholar]
- 104.Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008; 113: 573–81. doi: https://doi.org/10.1002/cncr.23592 [DOI] [PubMed] [Google Scholar]
- 105.Bellitti R, Buonocore M, De Rosa N, Covino FE, Casale B, Santè P. Primary cardiac angiosarcoma in a 25-year-old man: excision, adjuvant chemotherapy, and multikinase inhibitor therapy. Tex Heart Inst J 2013; 40: 186–8. [PMC free article] [PubMed] [Google Scholar]