

Abstract
TMEM165 deficiencies lead to one of the Congenital Disorders of Glycosylation (CDG), a group of inherited diseases where the glycosylation process is altered. We recently demonstrated that the Golgi glycosylation defect due to TMEM165 deficiency resulted from Golgi manganese homeostasis defect and that Mn2+ supplementation was sufficient to rescue a normal glycosylation. In this paper we highlight TMEM165 as a novel Golgi protein sensitive to manganese. When cells were exposed to high Mn2+ concentrations, TMEM165 was degraded into lysosomes. The Mn2+ induced lysosomal targeting of TMEM165 occurred via a Rab7 and Rab5 independent pathways, suggesting a direct trafficking from the Golgi to lysosomes in response to Mn2+. Remarkably, the variant p.E108G recently identified in a novel TMEM165-CDG patient, was found insensitive to Mn2+ supplementation. Moreover, this mutation abolished the function of TMEM165, suggesting that a transport function may be necessary for its regulation.
Altogether our results identified the Golgi protein TMEM165 as a novel cytosolic Mn2+ sensor in mammalian cells and pointed to the crucial importance of the cytosolical ELGDK motif in both Mn2+ sensitivity and function.
Introduction
Manganese is a trace element essential for life. It is involved in the catalytic domain of many enzymes such as Golgi glycosyltransferases, mitochondrial enzymes, DNA and RNA polymerases. Regulation of its homeostasis is therefore particularly important. Manganese overexposure has been shown to induce neurological symptoms that can result in a Parkinson like disorder called manganism (1–3). On the contrary, a decrease in cellular Mn2+ has recently been shown to cause Congenital Disorders of Glycosylation (CDG). Mutations in SLC39A8, a putative plasma membrane manganese transporter, lead to severe glycosylation defects (4). We recently reported that TMEM165 deficiency was also linked with Golgi Mn2+ homeostasis (5).
Although progress has been made in identifying cellular Mn2+ transporters in mammals, the mechanisms of Mn2+ homeostasis are still unclear. Several different transporters have been involved in manganese transport mechanisms including the divalent metal transporter 1 (DMT1/NRAMP2/SLC11A2) (1, 6), NRAMP1 (7), the transferrin receptor, the transporters SLC30A10/ZNT8 (8), SLC39A8/ZIP8 (4) and SLC30A14/ZIP14 (9, 10). At the cellular level, most of these transporters are localized to the plasma membrane and/or endosomes. The secretory pathway, consisting of the ER, Golgi and associated vesicles is also crucial in regulating cellular Mn2+ homeostasis. In addition, the secretory pathway requires luminal Mn2+ concentration for quality control, proper targeting and processing of proteins. Current knowledge supports that this supply is realized via the action of SPCA1 (secretory pathway Ca-ATPase 1: ATP2C1) and SPCA2 (ATP2C2). SPCA1 is ubiquitously expressed and mediates the import of Ca2+/Mn2+ into the Golgi lumen (11, 12). The tissue expression of SPCA2 is more restricted. However the importance of the dual transport function in cellular processes is not yet completely deciphered (12). Overexpression of SPCA1 has been shown to facilitate Mn2+ accumulation into the Golgi (13) and it was thus proposed that SPCA1 was a way to detoxify cytosolic Mn2+ accumulation by sequestering it into the secretory pathway.
In 2012, we identified TMEM165 as a novel Golgi transmembrane protein causing CDG (14). It belongs to an uncharacterized family of membrane proteins named UPF0016 (Uncharacterized Protein Family 0016; Pfam PF01169). We recently demonstrated that the observed Golgi glycosylation defect resulted from Golgi Mn2+ homeostasis impairment (5). Based on these results, we hypothesized that TMEM165 could be a novel Golgi Mn2+ transporter. As studies of Mn2+ homeostasis in yeasts have indicated that most of the proteins involved in regulating intracellular Mn2+ concentrations are differentially targeted and/or degraded in response to Mn2+, the role of TMEM165 was tested.
The aim of this study was to decipher the impact of high extracellular Mn2+ concentrations on the subcellular localization and stability of TMEM165. This study demonstrates that low concentrations of extracellular Mn2+ lead to a rapid lysosomal degradation of TMEM165. We identified the highly conserved motif ELGDK, oriented toward the cytosol, as being the cytosolic Mn2+ sensor.
RESULTS
TMEM165 is rapidly and specifically degraded in response to Mn2+
Our previous work highlighted a link between TMEM165 and Golgi Mn2+ homeostasis (5). As many proteins involved in regulating intracellular Mn2+ homeostasis are directly impacted in their localization/stability by cellular Mn2+ homeostasis changes, the effect of Mn2+ on TMEM165 was tested. For this, a concentration of 500μM of MnCl2 was first used for different times and the stability of TMEM165 was assessed both by western blot and immunofluorescence experiments. We observed that in response to Mn2+, TMEM165 levels were significantly reduced (figure 1, panels A and B). As GPP130 has also been shown to be sensitive to high Mn2+ concentrations, we compared its time course degradation with TMEM165 (figure 1, panels A and B). Quantification indicated that TMEM165 loss exceeded 95% after 8h of Mn2+ treatment while only a 40% decrease was seen for GPP130. To further tackle the minimal Mn2+ concentration able to induce a loss of TMEM165, we analyzed the stability of TMEM165 with low MnCl2 concentrations (1 to 50 μM). While 100μM MnCl2 was sufficient to induce GPP130 degradation (15), our results showed that 1–25μM of Mn was already sufficient to induce a destabilization of TMEM165 (figure 1B). Altogether these results indicate that TMEM165, compared to GPP130, is more sensitive to manganese and likely suggests the existence of different degradation mechanisms in response to Mn2+. The impact of Mn2+ on TMEM165 was also seen by immunofluorescence where a decrease in TMEM165 fluorescence associated with Golgi was seen (supplementary figure 1A and 1B). We previously demonstrated that TMEM165 could be found at the plasma membrane (14). In order to assess the impact of Mn2+ on the plasma membrane targeted form of TMEM165, surface protein biotinylation was performed in absence and in presence of 500μM of MnCl2. Interestingly biotin-labelled cell surface TMEM165 displayed the same sensitivity to Mn2+ as the cellular TMEM165 suggesting that the Mn2+ induced degradation mechanism is not Golgi dependent (figure 2A). Previous studies have also demonstrated that in yeast, high environmental Ca2+ concentrations in gdt1Δ led to strong N-glycosylation deficiencies (16). The impact of Ca2+ on Mn2+-induced degradation of TMEM165 was assessed by western blot and immunofluorescence (figure 2B and 2C). Ca2+ alone had no significant effect on the stability of TMEM165. However, its combined presence with Mn2+ clearly decreased the Mn2+-induced degradation of TMEM165 (80% decrease for the Mn2+ treatment alone compared to 40% decrease for both Ca2+ and Mn2+) (Figure 2B). This was confirmed by confocal microscopy (figure 2C).
Figure 1. TMEM is rapidly degraded in response to Mn.
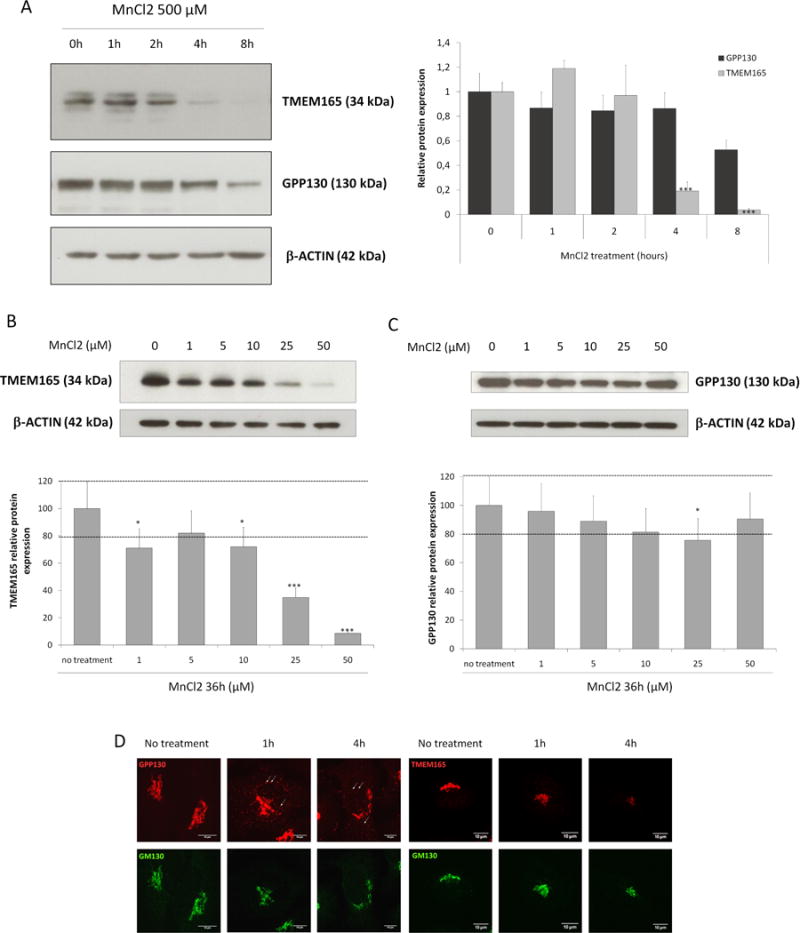
(A) Steady state cellular level of TMEM165 and GPP130. HeLa cells were treated with MnCl2 500 μM for 0 to 8h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Right panel shows the quantification of TMEM165 and GPP130 protein levels after normalization with actin (Number of experiments (N) = 2; *** = P value < 0,001). (B) Steady state cellular level of TMEM165. HEK293 cells were treated with MnCl2 from 0 to 50 μM for 36h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Lower panel shows the quantification of TMEM165 protein levels after normalization with actin (N = 2; *** = P value < 0,001). (C) Steady state cellular level of GPP130 in the same experimental conditions as described in (B). (D) HeLa cells were incubated with MnCl2 500 μM for 1 and 4h, fixed and labeled with antibodies against TMEM165, GPP130 and GM130 before confocal microscopy visualization. White arrows point to some GPP130 positive vesicles.
Figure 2. Plasma membrane TMEM165 is also degraded by Mn2+ and Ca2+ compete with Mn2+ for TMEM165 degradation.
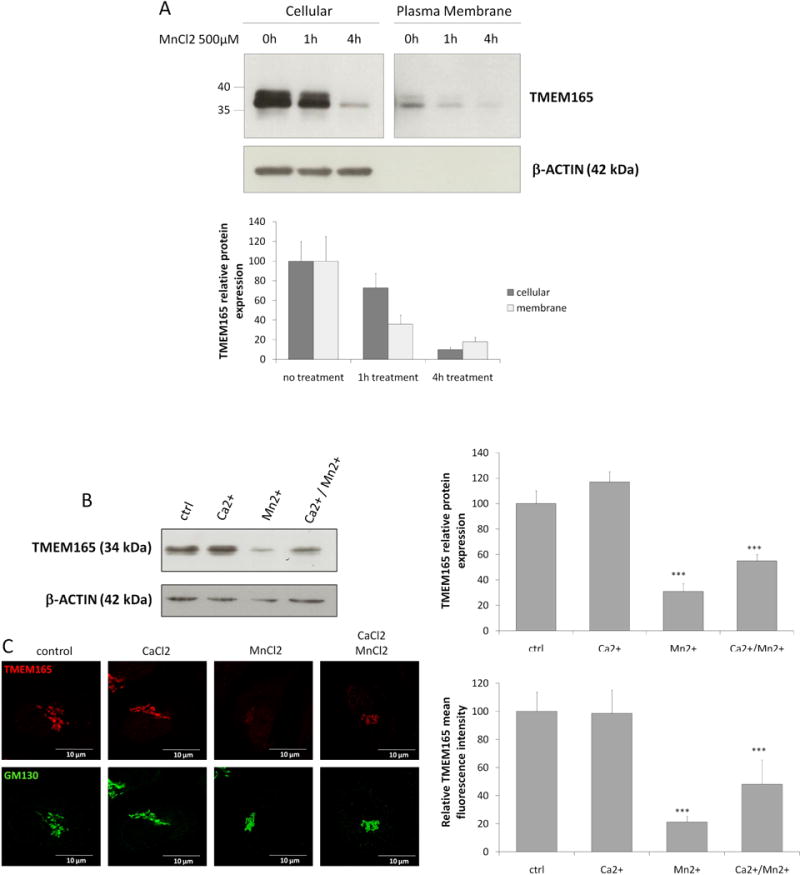
(A) Cell surface biotinylation was performed in absence and presence of MnCl2 500 μM. Samples were prepared as described in materials and methods and subjected to SDS-PAGE and Western blot with the indicated antibodies. Lower panel shows the quantification of TMEM165 protein levels. The plasma membrane panel is separated from the cellular panel as we had to longer expose films to reveal the bands. (B) HEK293 cells were incubated with MnCl2 500 μM and/or CaCl2 2mM for 4h, then subjected to SDS-PAGE and Western blot with the indicated antibodies. Right panel shows the quantification of TMEM165 protein levels after normalization with actin (N = 2; *** = P value < 0,001). (C) HEK293 cells were incubated with MnCl2 500 μM and/or CaCl2 2mM for 4h then fixed and labeled with antibodies against TMEM165 and GM130 before confocal microscopy visualization (N = 2; *** = P value < 0,001). Right panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 2; n = 50; *** = P value < 0,001).
Lysosomal degradation of TMEM165
As shown by Mukhopadhyay and collaborators, 500μM MnCl2 treatment induces rapid redistribution of GPP130 in vesicles before their lysosomal degradation via a Rab7-dependent mechanism (15). At the opposite of GPP130, no redistribution from the Golgi to peripheral punctate structures was observed for TMEM165 in response to high Mn2+ concentration (figure 1D). This absence of vesicles can either be explained by a decreased synthesis rate or an extremely fast degradation. To test the second hypothesis, the stability of TMEM165 in response to Mn2+ was studied by immunofluorescence in presence of chloroquine, a lysosomal inhibitor (figure 3A). Although cells treated with Mn2+ alone showed a dramatic loss of TMEM165, those treated in presence of chloroquine exhibited an accumulation of TMEM165 in punctate structures (figure 3A). Colocalization experiments with LAMP2, a lysosomal marker, confirmed the lysosomal accumulation of TMEM165 in cells treated with Mn2+ and chloroquine. To confirm the lysosomal Mn2+-induced degradation of TMEM165, an immunoblotting experiment was also performed. As shown in figure 3B, the Mn-induced degradation of TMEM165 was completely blocked by chloroquine. In order to figure out any putative contribution of the proteasomal degradation, similar experiments have been also done in the presence of MG132, a proteasome inhibitor. Such treatment did not block the observed Mn-induced degradation of GPP130 (data not shown). Given the similarity between the Mn induced degradation of GPP130 and TMEM165, the rab5 and rab7-dependent pathways were examined by using the dominant negative Rab7 (Rab7 T22N) that inhibits the trafficking from multi vesicular bodies (MVBs) to lysosomes (16, 17) and the constitutively active form of Rab5 (Q79L) that produces giant MVBs by causing fusion of early endosomes (EEs) and MVBs (18). To assess the involvement of these two different pathways, we examined the Mn2+ induced TMEM165 degradation in cells either expressing Rab7 T22N or Rab5 Q79L forms (figure 4A and 4B). Interestingly, in cells expressing either Rab7 T22N-GFP or Rab5Q79L-GFP, the Mn2+-induced degradation was not prevented and similar to the one observed in non-transfected cells (figure 4A and 4B). Overall, our results demonstrate that extracellular Mn2+ induces the lysosomal degradation of TMEM165 via a Rab7 and Rab5 independent pathways, suggesting a direct targeting of TMEM165 from the Golgi to lysosomes.
Figure 3. TMEM165 is targeted to lysosomal degradation after MnCl2 exposure.
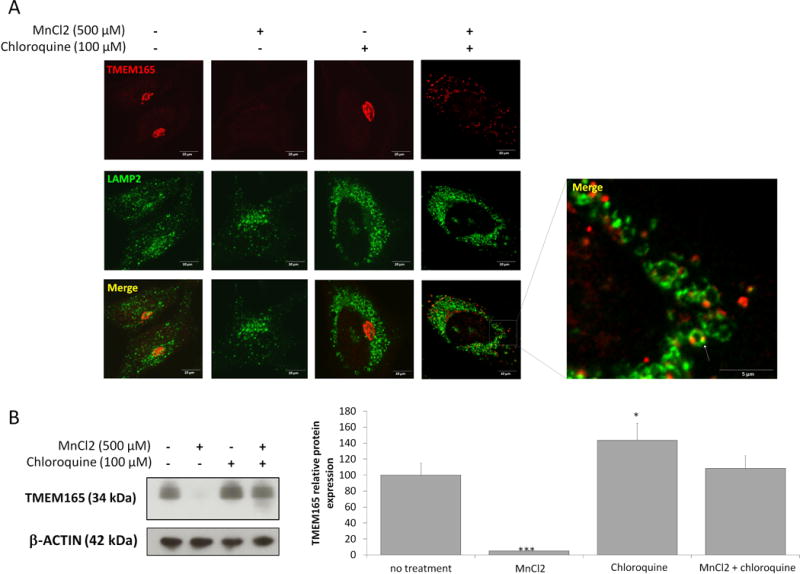
(A) HeLa cells were treated for 8h with MnCl2 500 μM and/or chloroquine 100 μM, fixed and labeled with antibodies against TMEM165 (upper panels) and LAMP2 (middle panels) before confocal microscopy visualization. (B) Western blot analysis of the same experiment described in (A). Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Right panel shows the quantification of TMEM165 protein level after normalization with actin (N = 2; *** = P value < 0,001).
Figure 4. TMEM165 lysosomal degradation is Rab7 and Rab5 independent.
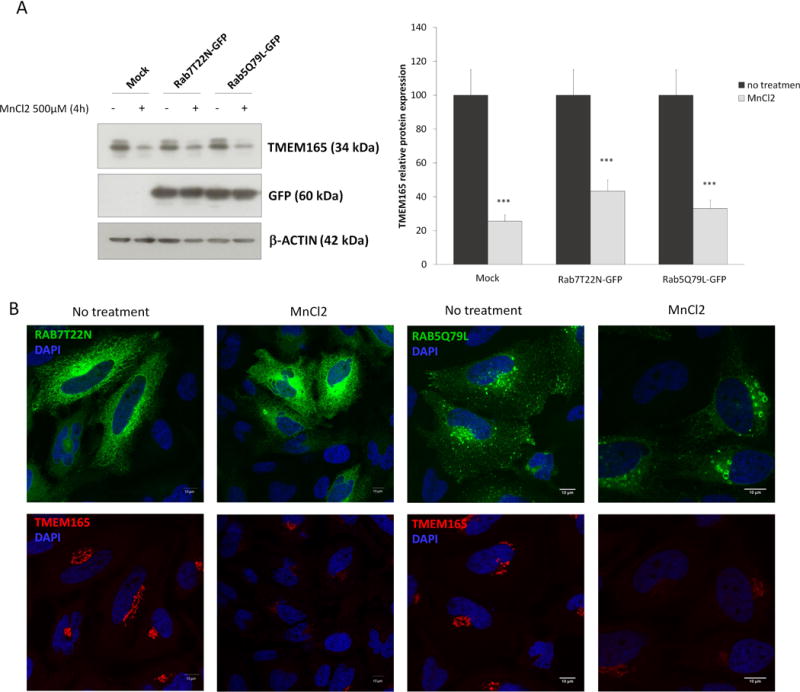
HeLa cells were transfected with empy vector (mock), Rab7T22N-GFP or Rab5Q79L-GFP. 36H after transfection, cells were treated or not for 4h with MnCl2 500 μM. (A) Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Right panel shows the quantification of TMEM165 protein levels after normalization with actin (N = 2; *** = P value < 0,001). (B) Immunofluorescence analysis by confocal microscopy of the same experiments as described in (A) (N = 2 ; n= 50).
The TMEM165 (69–172) domain confers Mn2+ sensitivity
In order to identiy the domain of TMEM165 sensitive to Mn2+, we took advantage of the existence of TMEM165 splice-transcripts isoforms leading to 129 aa (Short Form) and 259 aa (Long Form) protein isoforms, respectively (article in preparation) (Figure 5A). The resulting constructs were expressed in HeLa cells and subjected to Mn2+ addition. The stability of the construct as well as of endogenous TMEM165 was analyzed and compared by western blot experiments (figure 5B). While the endogenous form of TMEM165 was clearly sensitive to MnCl2 exposure, none of the two different TMEM165 constructs responded to Mn2+. This result strongly suggests that the Mn2+ sensitive domain of TMEM165 is comprised between residues 69 and 172.
Figure 5. The TMEM165 (69–172) domain confers Mn sensitivity.
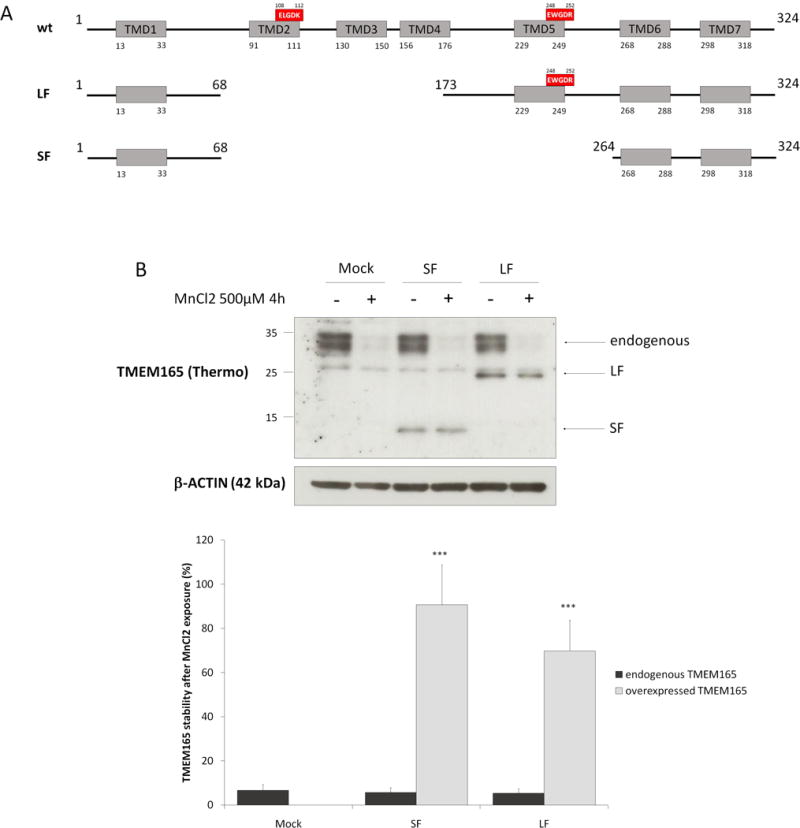
(A) TMEM165 full length, short isoform and long isoform protein organization. Grey boxes represent transmembrane domains and the red boxes are the well conserved acidic motifs (B) HeLa cells were transfected with empty vector, SF or LF for 36h then treated with MnCl2 500 μM for 4h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Lower panel shows the percentage of TMEM165 stability after normalization with actin (N = 2; * = P value < 0,05; *** = P value < 0,001).
The ELGDK motif is involved in Mn2+ sensitivity
Intriguingly the highly conserved ELGDK motif belongs to this domain (figure 5A). We then wondered whether this conserved motif could confer the Mn2+ sensitivity to TMEM165. To tackle this important aspect, we took advantage of a newly reported missense mutation c.323 A>G (p.E108G), in the two newly TMEM165 deficient siblings (19). To test the putative role of this motif in Mn2+ responsiveness, immunofluorescence and western blot experiments were performed in absence and in presence of MnCl2 (figure 6A and 6B). As observed for wt-TMEM165, the mutant form is Golgi localized in fibroblasts for the two siblings. This indicates that the mutation does not disturb the subcellular localization of the mutated form of TMEM165 (figure 6B). The impact of Mn2+ treatment was then investigated during an 8h time course by western blot and immunofluorescence experiments (figure 6A and 6B). As expected, the wt form of TMEM165 was very sensitive to Mn2+ exposure (figure 6A). However, the mutated form of TMEM165 (E108G) remained very stable (figure 6A and 6B). No changes neither in localization, nor stability was observed by immunofluorescence. Quantification of the western blot results indicated that wt-TMEM165 loss exceeded 95% at the 6h time point while only 20% loss was observed for the mutated form p.E108G.
Figure 6. The ELDGK motif is crucial for Mn2+ sensitivity.
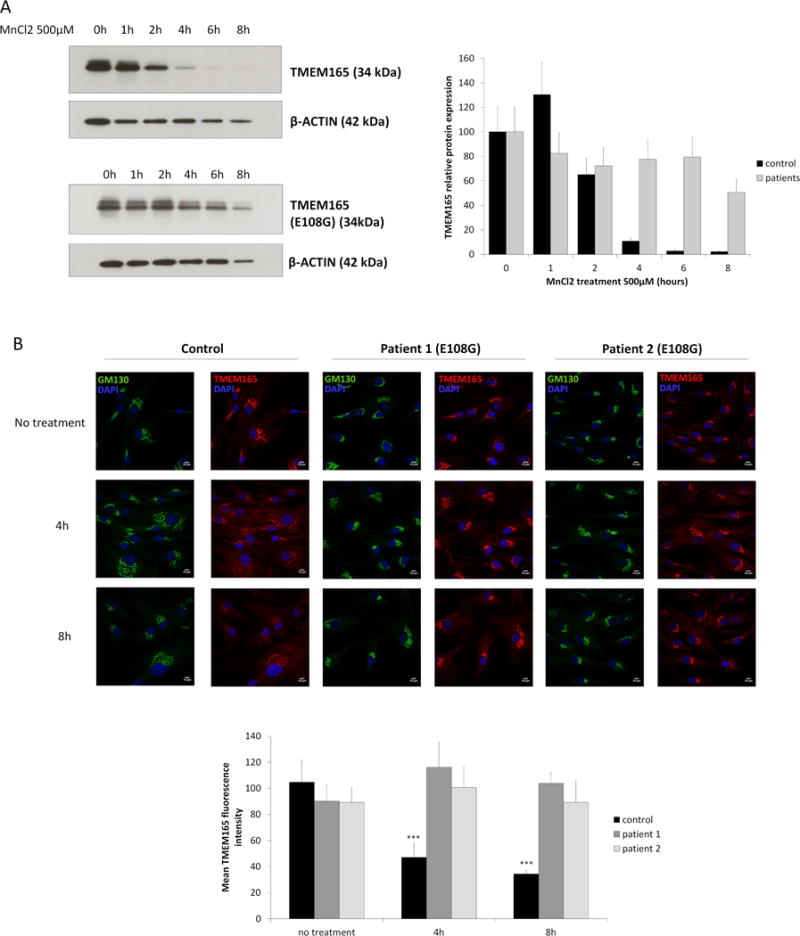
(A) Healthy skin fibroblasts (upper left) and patients skin fibroblasts (lower left) carrying E108G mutation were treated with MnCl2 500μM for 0 to 8h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. Right panel shows the quantification of TMEM165 protein levels after normalization with actin (N = 2; *** = P value < 0,001). (B) Healthy skin fibroblasts and patients skin fibroblasts carrying E108G mutation were treated with MnCl2 500μM for 0, 4 and 8h. Cells were then fixed and labeled with antibodies against TMEM165 and GM130 before confocal microscopy visualization (N = 2; *** = P value < 0,001). Lower panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 2; n = 30; *** = P value < 0,001).
To avoid any bias resulting from the different genetic background between control and patient cells, the TMEM165 (E108G)-GFP construct (named mutac1) was used to test for Mn2+ sensitivity. HEK293 cells were transfected and exposed to Mn2+ for 4h (figure 7A). The stability of the construct as well as of the endogenous TMEM165 was evaluated. Remarkably, Mn2+ treatment did not alter the stability of the construct while the endogenous TMEM165 was strongly decreased. In summary, our results support the evidence that the glutamic acid (E) of the highly conserved ELGDK motif is crucial in mediating the lysosomal degradation of TMEM165 in response to Mn2+. To determine whether the E108G mutation could also affect TMEM165 activity, the Golgi glycosylation of LAMP2 was assessed in TMEM165 KO HEK293 cells generated by CRISPR-Cas9. While the expression of the wt-TMEM165 clearly complemented the observed glycosylation defect of LAMP2, the E108G TMEM165 completely failed to restore the observed glycosylation defect. This strongly supports the evidence that the TMEM165 E108G mutant protein is not functional.
Figure 7. Mutations in the ELGDK motif are not functional and insensitive to Mn2+.
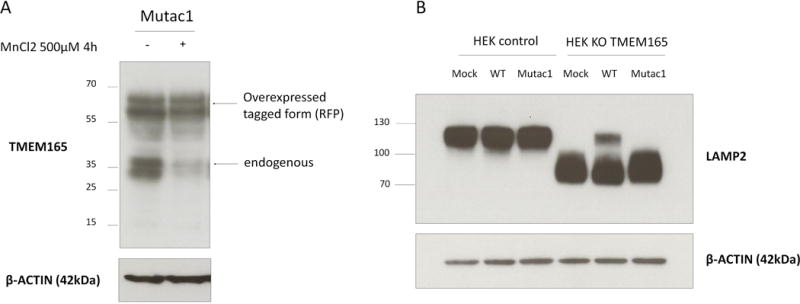
(A) HEK293 cells were transfected with mutac1 -RFP for 36h then treated with MnCl2 500 μM for 4h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies. (B) HEK293 control cells and HEK293 KO TMEM165 cells were transfected with empty-vector, wild-type or mutac1 plasmid for 36h. Total cell lysates were prepared, subjected to SDS-PAGE and Western blot with the indicated antibodies.
Altogether, this demonstrates the crucial importance of the ELGDK domain in both Mn2+ responsiveness and functionality. This also suggests that the metal transport function may be necessary for its regulation.
Topology of TMEM165
Human TMEM165 encodes a 7-transmembrane (7-TM) spanning protein of 324 amino acids. To analyse the topology of TMEM165 and thus the orientation of the ELGDK motif, we used the two available commercial antibodies against TMEM165 each recognizing two differentially oriented epitopes; the Sigma antibodies recognizing the immunogen sequence (aa176–aa229) and the antibodies provided by Thermofischer directed against the immunogen sequence (aa17–aa45) (figure 8A). The topology was determined by selective membrane permeabilization and immunofluorescence analysis (figure 8B). Under conditions that allowed antibody access to all cellular compartments, both epitopes were detectable and showed co-localization with the Golgi marker GM130 (figure 8B). Selective permeabilization of the plasma membrane with low concentrations of digitonin allowed visualization of the cytosolic epitope only recognized by the Sigma antibody. On the basis of these results, we can propose a model where the loop encompassing the aa 176 to 229 is cytosolic and where the ELGDK motif is facing the cytosol (figure 8A).
Figure 8. TMEM165 topology.
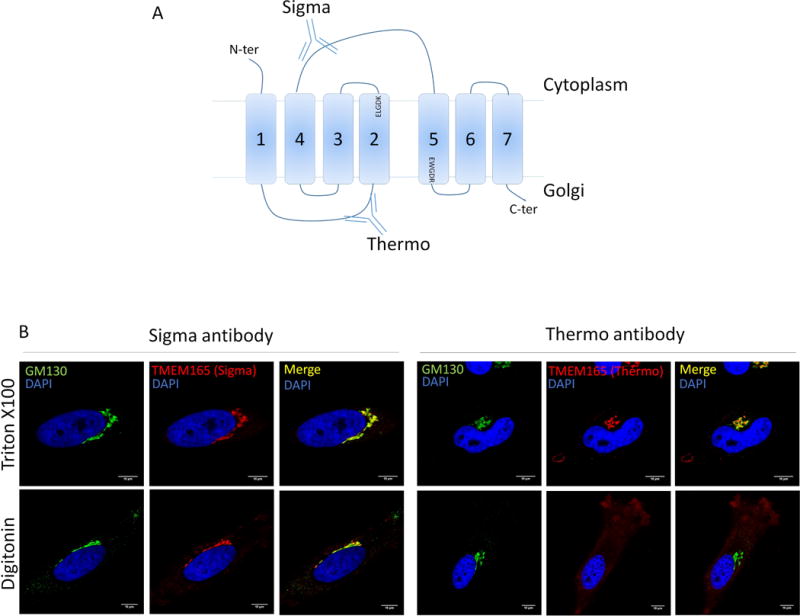
(A) Representation of TMEM165 predicted topology. The two anti-TMEM165 antibodies (Sigma-Aldrich and Thermo Fisher Scientific) depicted here recognize two different parts of the protein. The Sigma one recognize the cytoplasmic loop between the fourth and fifth transmembrane domains. The Thermo one recognize the short luminal loop between the first and the second transmembrane domain. (B) Cells were fixed with paraformaldehyde 4% and treated as described in materials and methods. Selective permeabilization was done by using triton ×100 or digitonin. Cells were labeled with antibodies against TMEM165 and GM130 before confocal microscopy visualization.
Discussion
Our previous work has shown that the observed Golgi glycosylation defect due to a lack of Gdt1p/TMEM165 resulted from Golgi Mn2+ homeostasis defect then leading to strong Golgi glycosylation abnormalities. Interestingly, we demonstrated that such defects could totally be suppressed by manganese supplementation, strongly suggesting that TMEM165 could somehow be involved in the Golgi transport of Mn2+. It has been shown in yeast that Smf1p and Smf2p, members of the Nramp family of metal transporters, are tightly regulated by different intracellular Mn2+ concentrations (20, 21). When cells are exposed to toxic Mn2+ concentrations, Smf1p and Smf2p are targeted to the vacuole for degradation thus stopping the Mn2+ cellular entry. To test whether TMEM165 falls under the same regulation, TMEM165 stability for Mn2+ was tested. Our results showed that TMEM165 was highly sensitive to Mn2+ as manganese supplementation targets TMEM165 under the way of lysosomal degradation (figure 9). Although intriguing, the molecular mechanisms by which TMEM165 is degraded followed Mn2+ exposure is currently not known. Another mammalian Golgi protein GPP130 has been reported to be sensitive to Mn2+ (15). While the obtained results are very similar to the one observed for GPP130, several lines of evidences tend to prove that the molecular mechanisms are different. First and in contrast to GPP130, we demonstrated that the manganese induced degradation of TMEM165 is Rab7 and Rab5 independent. Second, the manganese sensitivity is also different as 25μM of manganese is sufficient to engage TMEM165 in the lysosomal degradation pathway while 100μM is required for GPP130. Third, the manganese induced degradation rate of TMEM165 is faster than of GPP130 as TMEM165 accumulation was never seen in punctate structures under Mn2+ supplementation. Because GPP130 and TMEM165 present high sensitivity to manganese, we can’t exclude that these two proteins somehow interact. This can either be at the functional level if TMEM165 is a Mn2+ transporter or at the physical level by stabilizing GPP130.
Figure 9. Model for TMEM165 degradation in excess of Mn.
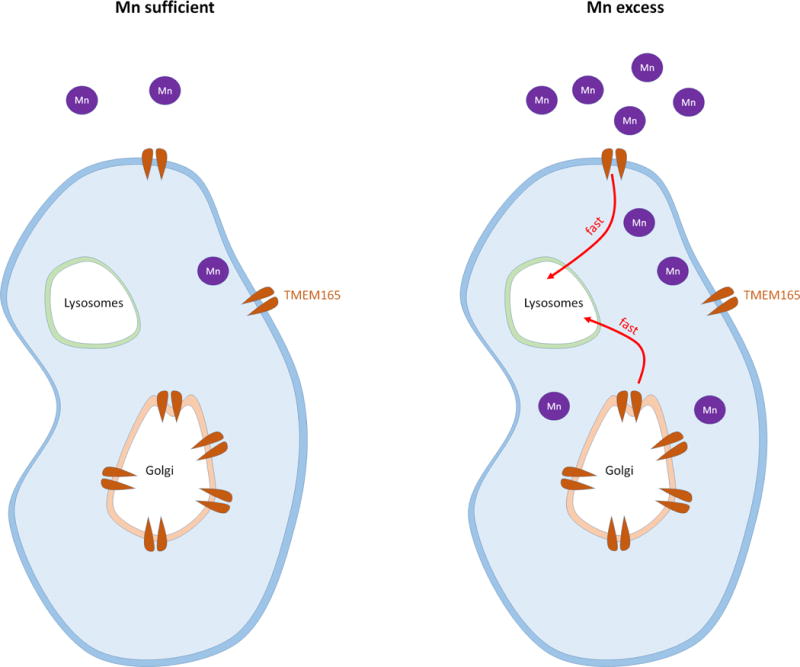
The left panel present the steady state TMEM165 levels in physiological conditions. The right panel shows TMEM165 lysosomal degradation in excess of Mn.
Interestingly, we mapped the Mn2+ responsiveness of TMEM165 to its first highly conserved ELGDK motif as E108G TMEM165 is insensitive to Mn2+. According to the prediction of TMEM165 membrane topology, this motif, belonging to a larger conserved domain (ELGDKTFFIAAIMAMR), is cytosolically oriented and located between the second and the third transmembrane domain of TMEM165. Our data also show that the E108G TMEM165 mutant form fails to rescue the glycosylation defect. This highlights both the crucial importance of the ELGDK motif in Mn2+ sensitivity and functionality of TMEM165 and suggests that the transport function may be necessary for its regulation.
The other important question is why TMEM165 is degraded by high cytosolic Mn2+ concentration? While we currently don’t have the answer, our data raise several hypotheses. As a slight fraction of TMEM165 can be found at the plasma membrane, the degradation could be a mechanism to prevent the plasma membrane Mn2+ entry. As mammalian cells can however transport the metal by other plasma membrane transporters, this hypothesis is not likely. When cells are exposed to high manganese concentrations, the plasma membrane transporters import the dangerous metal in the cytosol where it accumulates and impairs many fundamental cellular processes. It is critical for the cell to detoxify the cytosol. In such situation, SPCA1, the Golgi P-type ATPase essential to import cytosolic Ca2+ but also Mn2+ inside the Golgi lumen, appears to be the major way for eliminating the surplus of cytosolic Mn2+ from the cell. As TMEM165 is degraded when the manganese level becomes toxic, we hypothesize that this mechanism participates in detoxification. If TMEM165 is a Mn2+/Ca2+ transporter that imports Mn2+ into the Golgi lumen by using the electrochemical gradient of Golgi Ca2+, we imagine that TMEM165 is degraded to 1_increase the cytosolic Mn2+ concentration and thus specifically engage SPCA1 in the way of detoxification and 2_prevent Golgi Ca2+ exit. We also suggest the possibility that, in the presence of high Mn2+ concentration in the Golgi, TMEM165 would work in the opposite direction, importing Ca2+ into the Golgi lumen by using the electrochemical gradient of Golgi Mn2+ generated by SPCA1. In that case, the specific degradation of TMEM165 in response to Mn2+ would prevent the Golgi Mn2+ recapture back into the cytosol, a mechanism that would definitely annihilate the efforts made by SPCA1. Although the way of functioning is still unclear, we hypothesize that TMEM165 may operate in both forward and reverse directions simultaneously, depending on the combined effects of Mn2+ and Ca2+ gradients.
Overall, our studies highlight TMEM165 as a novel Golgi Mn2+ cytosolic sensor of mammalian cells. This discovery sheds light on a novel actor involved in the regulation of intracellular Mn2+ homeostasis and the pathophysiological mechanisms in TMEM165-CDG patients.
Material and Methods
Antibodies and other reagents
Anti-TMEM165 and anti-β Actin antibodies were from Sigma–Aldrich (St Louis, MO, USA). The other anti-TMEM165 antibody was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-GM130 antibody was from BD Biosciences (Franklin lakes, NJ, USA). Anti-GPP130 antibody was purchased from Covance (Princeton, NJ, USA). Goat anti-rabbit or goat anti-mouse immunoglobulins HRP conjugated were purchased from Dako (Glostrup, Denmark). Polyclonal goat anti-rabbit or goat anti-mouse conjugated with Alexa Fluor were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Manganese (II) chloride tetrahydrate was from Riedel-de-Haën (Seelze, Germany). All other chemicals were from Sigma-Aldrich unless otherwise specified.
Constructs, vector engineering and mutagenesis
TMEM165-RFP plasmid is the same as described by Rosnoblet et al (Rosnoblet et al., 2013). Rab7T22N-GFP and Rab5Q79L-GFP are kind gifts from Dr M.Zerial (Max Planck Institute, Dresden, Germany). For TMEM165-wt, SF and LF, first strand cDNA synthesis were achieved using 1 μg of total RNA from cultured cell lines (Hela cells and primary skin fibroblasts). After synthesis of the first strand cDNA, PCR were performed with exon1/6-specific primers (5′-GGTGCTGACTGCTCCCTAAG-3 and 5′-CCACACTTAGAAGTCAGGTTTCTTT-3′) or exon3′/6-specific primers (5′-ATTATCCGATGTTAGGCCAGTG-3′ and 5′-GATGAACAAACAGCGTTAAAAA-3′). PCR amplifications were performed with the Taq polymerase from Taq PCR Core Kit (Qiagen GmbH, Germany) according to provider’s instructions. PCR products obtained were size separated by agarose gel electrophoresis, subcloned into pCR2.1 TOPO TA cloning (Invitrogen, Thermo Fisher Scientific, Waltham, MA USA) and sequenced by GATC Biotech AG (Constance, Germany).
Cell culture and transfections
All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland), at 37°C in humidity-saturated 5% CO2 atmosphere. Transfections were performed using Lipofectamine 2000® (Thermo Scientific) according to the manufacturer’s guidelines. For drug treatments, incubations were done as described in each figures.
Immunofluorescence staining
Cells were seeded on coverslips for 12 to 24h, washed once in Dulbecco’s Phosphate Buffer Saline (DPBS, Lonza) and fixed either with 4% paraformaldehyde (PAF) in PBS pH 7.3, for 30 min at room temperature or with ice-cold methanol for 10 minutes at room temperature. Coverslips were then washed three times with PBS. Only if the fixation had been done with PAF, cells were permeabilized with 0.5% Triton X-100 in PBS for 15 min then washed three times with PBS. Coverslips were then put in saturation for 1h in blocking buffer [0.2% gelatin, 2% Bovin Serum Albumin (BSA), 2% Fetal Bovine Serum (FBS) (Lonza) in PBS], followed by the incubation for 1h with primary antibody diluted at 1:100 in blocking buffer. After washing with PBS, cells were incubated for 1h with Alexa 488-, Alexa 568- or Alexa 700-conjugated secondary antibody (Life Technologies) diluted at 1:600 in blocking buffer. After three washing with PBS, coverslips were mounted on glass slides with Mowiol. Fluorescence was detected through an inverted Zeiss LSM780 confocal microscope. Acquisition were done using the ZEN pro 2.1 software (Zeiss, Oberkochen, Germany). For selective membrane permeabilization, we have used digitonine. Stock solution was prepared at 5mg/mL in absolute ethanol, 0,3M sucrose, 0,1M KCl, 2,5 mM MgCl2, 1 mM EDTA, 10 mM HEPES, pH 6,9. Permeabilization was done at 4°C during 15min.
Image Analyses
Immunofluorescence images were analyzed using TisGolgi, an homemade imageJ (Schneider et al., 2012) (http://imagej.nih.gov/ij) plugin developed by TISBio and available upon request. Basically, the program automatically detects and discriminates Golgi and vesicles, based on morphological parameters such as size and sphericity. Then, the program calculates for each image the number of detected objects, their size and mean fluorescence intensity.
Western Blotting
Cells were scraped in DPBS and then centrifuged at 4500 rpm for 3 min. Supernatant was discarded and cells were then resuspended in RIPA buffer [Tris/HCl 50mM pH 7.9, NaCl 120mM, NP40 0.5%, EDTA 1mM, Na3VO4 1mM, NaF 5mM] supplemented with a protease cocktail inhibitor (Roche Diagnostics, Penzberg, Germany). Cell lysis was done by passing the cells several times through a syringe with a 26G needle. Cells were centrifuged for 30 min at 20 000g. The supernatant containing protein was estimated with the micro BCA Protein Assay Kit (Thermo Scientific). 20 μg of total protein lysate were put in NuPAGE LDS sample buffer (Invitrogen) pH 8.4 supplemented with 4% β-mercaptoethanol (Fluka). Samples were heated 10 min at 95°C and then separated on 4%–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membrane Hybond ECL (GE Healthcare, Little Chalfont, UK). The membrane were blocked in blocking buffer (5% milk powder in TBS-T [1× TBS with 0.05% Tween20]) for 1 hr at room temperature, then incubated overnight with the primary antibodies in blocking buffer, and washed three times for 5 min in TBS-T. The membranes were then incubated with the peroxidase-conjugated secondary goat anti-rabbit or goat anti-mouse antibodies (Dako; used at a dilution of 1:10,000) in blocking buffer for 1 hr at room temperature and later washed three times for 5 min in TBS-T. Signal was detected with chemiluminescence reagent (ECL 2 Western Blotting Susbtrate, Thermo Scientific) on imaging film (GE Healthcare, Little Chalfont, UK).
Cell surface biotinylation
Cells were plated to reach 70–80% confluency on the day of the experiment. Cells were kept on ice all time. Cells were washed 4 times with PBS +/+ (containing Ca and Mg) pH8. 1,5 mL PBS +/+ pH 8 with 7,5μL biotin was added per dish (Biotin : EZ Link Sulfo-NHS-SS-Biotin (Life Technologies, Carlsbad, CA, USA) final concentration 0,5mg/ml in DMSO). Cells were then incubated 30min in cold room on slow rocking then washed 3 times with PBS+/+ pH8. Cells were quenched 15min with 1,5ml PBS+/+ glycine 100mM, BSA 0,5 % in cold room on slow rocking then washed 3 times with PBS+/+ glycine. Cells were scraped in 200μl lysis buffer (50mM Hepes pH 7,2, 100mM NaCl, 1% Triton X-100, protease inhibitors) then incubated 20min on ice. Centrifuge 15 min at 20000g at 4°C. Supernatant was kept and the protein concentration was measured. For the pulldown, put the maximum amount of protein, ideally 500μg in 1mL final (lysis buffer) + 30 μL streptavidin beads. Incubate 4h at 4°C on a wheel then centrifuge at 4000g for 1min at 4°C. Wash 3 times with 1mL lysis buffer (not supplemented with protease inhibitors), mix well by inverting the tubes 30 times. Centrifuge at 4000g for 1min at 4°C and remove supernatant. Centrifuge at 4000g for 1min at 4°C and remove supernatant with a flat end tip. Add NuPAGE LDS sample buffer (Invitrogen) pH 8.4 supplemented with 4% β-mercaptoethanol (Fluka). Samples are boiled at 70°C for 10min (do not boil if you want to reveal TMEM165 on western blot after), then centrifuged at 1000g for 1min and the supernatant was collected. Samples were frozen at −20°C. Samples are ready to load on gel.
Statistical Analysis
Comparisons between groups were performed using Student t-test for 2 variables with equal or different variances, depending on the result of the F-test.
Supplementary Material
Supplementary Figure 1. Confirmation of TMEM165 degradation by immunofluorescence experiments (A) HeLa cells were incubated with MnCl2 500 μM for 0 to 8h, fixed and labeled with antibodies against TMEM165 before confocal microscopy visualization. Right panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 3; number of cells (n) = 50; *** = P value < 0,001). (B) HeLa cells were treated with MnCl2 for 8h at different concentrations, then fixed and labeled with antibodies against TMEM165 and GM130 before confocal microscopy visualization. Lower panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 2; n = 50; *** = P value < 0,001).
Acknowledgments
We are indebted to Dr Dominique Legrand for the Research Federation FRABio (Univ. Lille, CNRS, FR 3688, FRABio, Biochimie Structurale et Fonctionnelle des Assemblages Biomoléculaires) for providing the scientific and technical environment conducive to achieving this work.
Funding
‘This work was supported by the French National Research Agency [SOLV-CDG to F.F]; and the Mizutani Foundation for Glycoscience [to F.F].’
Footnotes
Conflict of interests
None.
References
- 1.Au C, Benedetto A, Aschner M. Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. 2008;29:569–576. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Squitti R, Gorgone G, Panetta V, Lucchini R, Bucossi S, Albini E, Alessio L, Alberici A, Melgari JM, Benussi L, et al. Implications of metal exposure and liver function in Parkinsonian patients resident in the vicinities of ferroalloy plants. J Neural Transm Vienna Austria 1996. 2009;116:1281–1287. doi: 10.1007/s00702-009-0283-0. [DOI] [PubMed] [Google Scholar]
- 3.Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol. 2007;66:675–682. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- 4.Park JH, Hogrebe M, Grüneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am J Hum Genet. 2015;97:894–903. doi: 10.1016/j.ajhg.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potelle S, Morelle W, Dulary E, Duvet S, Vicogne D, Spriet C, Krzewinski-Recchi MA, Morsomme P, Jaeken J, Matthijs G, et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum Mol Genet. 2016;25:1489–1500. doi: 10.1093/hmg/ddw026. [DOI] [PubMed] [Google Scholar]
- 6.Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, Moore EG, Hainsworth LN, Umbreit JN, Conrad ME, et al. DMT1: a mammalian transporter for multiple metals. Biometals Int J Role Met Ions Biol Biochem Med. 2003;16:41–54. doi: 10.1023/a:1020702213099. [DOI] [PubMed] [Google Scholar]
- 7.Forbes JR, Gros P. Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood. 2003;102:1884–1892. doi: 10.1182/blood-2003-02-0425. [DOI] [PubMed] [Google Scholar]
- 8.Chen P, Bowman AB, Mukhopadhyay S, Aschner M. SLC30A10: A novel manganese transporter. Worm. 2015;4:e1042648. doi: 10.1080/21624054.2015.1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, Wang B, Dalton TP, Nebert DW. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol. 2008;73:1413–1423. doi: 10.1124/mol.107.043588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen P, Chakraborty S, Mukhopadhyay S, Lee E, Paoliello MMB, Bowman AB, Aschner M. Manganese homeostasis in the nervous system. J Neurochem. 2015;134:601–610. doi: 10.1111/jnc.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dode L, Andersen JP, Vanoevelen J, Raeymaekers L, Missiaen L, Vilsen B, Wuytack F. Dissection of the functional differences between human secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and 2 isoenzymes by steady-state and transient kinetic analyses. J Biol Chem. 2006;281:3182–3189. doi: 10.1074/jbc.M511547200. [DOI] [PubMed] [Google Scholar]
- 12.He W, Hu Z. The role of the Golgi-resident SPCA Ca2+/Mn2+ pump in ionic homeostasis and neural function. Neurochem Res. 2012;37:455–468. doi: 10.1007/s11064-011-0644-6. [DOI] [PubMed] [Google Scholar]
- 13.Mukhopadhyay S, Linstedt AD. Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc Natl Acad Sci U S A. 2011;108:858–863. doi: 10.1073/pnas.1013642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foulquier F, Amyere M, Jaeken J, Zeevaert R, Schollen E, Race V, Bammens R, Morelle W, Rosnoblet C, Legrand D, et al. TMEM165 Deficiency Causes a Congenital Disorder of Glycosylation. Am J Hum Genet. 2012;91:15–26. doi: 10.1016/j.ajhg.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol Biol Cell. 2010;21:1282–1292. doi: 10.1091/mbc.E09-11-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: A Key to Lysosome Biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Progida C, Cogli L, Piro F, Luca AD, Bakke O, Bucci C. Rab7b controls trafficking from endosomes to the TGN. J Cell Sci. 2010;123:1480–1491. doi: 10.1242/jcs.051474. [DOI] [PubMed] [Google Scholar]
- 18.Stenmark H, Parton RG, Steele-Mortimer O, Lütcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Althoff SS, Grüneberg M, Reunert J, Park JH, Rust S, Mühlhausen C, Wada Y, Santer R, Marquardt T. TMEM165 Deficiency: Postnatal Changes in Glycosylation. JIMD Rep. 2015 doi: 10.1007/8904_2015_455._455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Confirmation of TMEM165 degradation by immunofluorescence experiments (A) HeLa cells were incubated with MnCl2 500 μM for 0 to 8h, fixed and labeled with antibodies against TMEM165 before confocal microscopy visualization. Right panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 3; number of cells (n) = 50; *** = P value < 0,001). (B) HeLa cells were treated with MnCl2 for 8h at different concentrations, then fixed and labeled with antibodies against TMEM165 and GM130 before confocal microscopy visualization. Lower panel shows the quantification of the associated TMEM165 fluorescence intensity (N = 2; n = 50; *** = P value < 0,001).