

Abstract
Malbrancheamide is a dichlorinated fungal indole alkaloid isolated from both Malbranchea aurantiaca and Malbranchea graminicola that belongs to a family of natural products containing a characteristic bicyclo[2.2.2]diazaoctane core. The introduction of chlorine atoms on the indole ring of malbrancheamide differentiates it from other members of this family and contributes significantly to its biological activity. In this study, we characterized the two flavin-dependent halogenases involved in the late-stage halogenation of malbrancheamide in two different fungal strains. MalA and MalA’ catalyze the iterative dichlorination and monobromination of the free substrate premalbrancheamide as the final steps in the malbrancheamide biosynthetic pathway. Two unnatural bromo-chloro-malbrancheamide analogs were generated through MalA-mediated chemoenzymatic synthesis. Structural analysis and computational studies of MalA’ in complex with three substrates revealed that the enzyme represents a new class of zinc-binding flavin-dependent halogenases, and provides new insights into a potentially unique reaction mechanism.
Keywords: halogenase, flavin, fungal, malbrancheamide, Malbranchea aurantiaca, Malbranchea graminicola, MalA, natural products, biocatalysis
Graphical Abstract
Introduction
The prevalence of halogenated natural products has led to significant advances in understanding various classes of halogenases involved in secondary metabolism. Most halogenases characterized thus far can be placed into three classes: haloperoxidases (heme-containing and vanadium-containing), non-heme Fe(II)α-ketoglutarate-dependent, and flavin-dependent enzymes. Haloperoxidases are generally nonselective and perform halogenation through a mechanism utilizing freely diffusing hypohalous acid. By contrast, Fe(II)α-ketoglutarate-dependent halogenases proceed through a radical mechanism, typically halogenating aliphatic, unactivated carbons.1 Flavin-dependent halogenases (FDHs) also proceed through a hypohalous acid intermediate, with the reactive reagent captured by a lysine residue that appears to control the regioselectivity of halogenation on aromatic substrates.2,3 The FDH-derived hypohalous acid is generated through a reaction between the flavin C4a-peroxide adduct and the bound chloride ion. FDHs are thought to proceed through an electrophilic aromatic substitution where the catalytic lysine residue provides the chloramine halogenating agent and a catalytic glutamate facilitates the reaction by deprotonating the positively charged intermediate generated during catalysis.2
The majority of previously characterized FDHs are of bacterial origin, with relatively few reported from eukaryotes,4–12 and fewer still characterized biochemically.4–6,10–12 The bacterial FDHs have been found to catalyze reactions on both free,2,13–15 and carrier-protein bound substrates,16,17 including precursor amino acids in natural product biosynthesis. The well-characterized eukaryotic FDHs Rdc24 and ChlA6 catalyze late-stage C–H functionalization reactions in the biosynthesis of halogenated metabolites. However, structural data for these two enzymes have not been reported, and it has remained unclear how they control site-selective halogenation on large, structurally complex substrates.
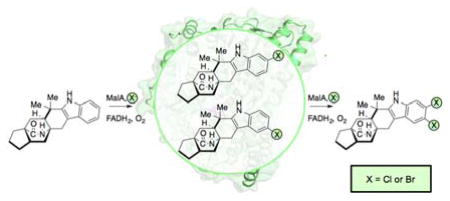
Malbrancheamide (1) is a complex halogenated indole alkaloid produced by the terrestrial fungus Malbranchea aurantiaca RRC181318 and the marine-derived fungus Malbranchea graminicola (086937A).19 The discovery of malbrancheamide was enabled by a search for calmodulin antagonists, and several studies have characterized its significant vasorelaxant effect.20,21 Along with malbrancheamide, a close structural relative, spiromalbramide, was isolated from M. graminicola.19 The two strains are highly related, with 99% sequence identity overall, and their biosynthetic pathways for malbrancheamide are proposed to be identical (Scheme 1). Malbrancheamide (1) belongs to a family of prenylated indole alkaloids formed through peptide coupling by a nonribosomal peptide synthetase (NRPS), addition of an isoprene unit by a prenyltransferase, and a proposed [4+2] Diels-Alder cycloaddition to form the characteristic bicyclo[2.2.2]diazaoctane ring of premalbrancheamide (2) (Scheme 1).22–26 Premalbrancheamide is then proposed to be dichlorinated through an iterative mechanism, but whether this halogenation is performed by one or two halogenases remained to be determined.25 Since the chlorination of the indole ring differentiates this molecule from the rest of its class and significantly contributes to its biological activity,21 we were motivated to probe the mechanism of its iterative late-stage halogenation at two adjacent positions on the indole ring system.
Scheme 1.

Malbrancheamide biosynthetic pathway in M. aurantiaca and M. graminicola, where spiromalbramide is produced only in the latter.
In earlier efforts to elucidate the malbrancheamide biosynthetic pathway, precursor incorporation studies were performed in M. aurantiaca. This led to the conclusion that premalbrancheamide (2) is indeed incorporated into the monochlorinated malbrancheamide B (3) and that both compounds are natural metabolites of M. aurantiaca.25 We had previously proposed that there is a site-selective chlorination of the C9 position prior to functionalization of C8 for production of malbrancheamide (1).25 However, the isolation of both C8 (isomalbrancheamide B (4)) and C9 (malbrancheamide B (3)) monohalogenated metabolites from M. aurantiaca20 and from M. graminicola19 conflicted with the proposed C9 selectivity hypothesis, providing further motivation to investigate the malbrancheamide halogenation process.
Previous genome sequencing and bioinformatic analyses of M. aurantiaca and M. graminicola led to the identification of MalA and MalA’, respectively. These two FDHs are 99% identical, differing by only two amino acids, and are proposed to catalyze dihalogenation as the last step in the malbrancheamide biosynthetic pathways of each organism (Figure 1).26 The late-stage halogenation of free substrate by a flavin-dependent halogenase from an NRPS-containing gene cluster is unusual. Halogenation typically occurs as the first step prior to activation of an amino acid in bacterial non-ribosomal peptide biosynthesis.1 Of the previously characterized flavin-dependent halogenases, most act on subunit substrates such as single amino acids,2,13–15,27 or carrier protein-tethered small molecules.16,17,28,29 In terms of late-stage activity on a complex polycyclic substrate, the closest comparison to MalA is the cyanobacterial-derived WelO5 non-heme Fe(II)α-ketoglutarate-dependent halogenase, which acts on fischerindole and hapalindole alkaloids.30 In addition to substrate scope analyses, halogen selectivity has also been explored in flavin-dependent halogenases, and the majority were found to catalyze both chlorination and bromination reactions. In precursor incorporation studies using high bromide salt concentrations in the marine fungal strain M. graminicola, bromination of premalbrancheamide (2) led to the production of malbrancheamide C (5) and isomalbrancheamide C (6) (Figure 2).19
Figure 1.
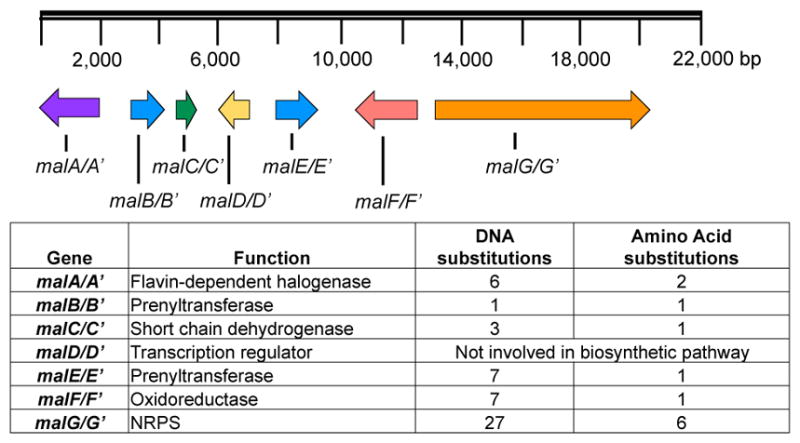
Malbrancheamide biosynthetic gene clusters in M. aurantiaca and M. graminicola.
Figure 2.
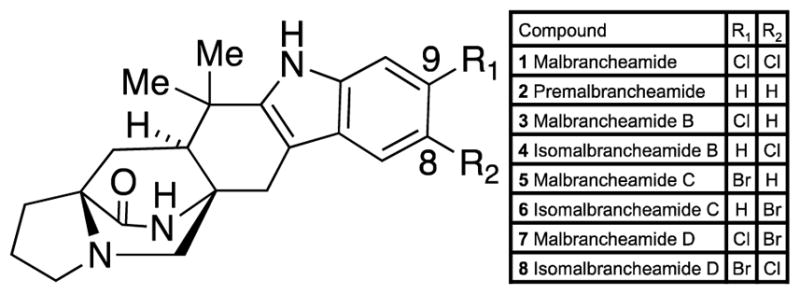
Malbrancheamide and related metabolites. M. aurantiaca isolates included 1, 2, 3, and 4, while M. graminicola also produced 5 and 6. Compounds 7 and 8 had not been previously described from either organism.
The ability to selectively halogenate C–H bonds in highly complex molecules through synthetic methods has posed a formidable challenge due to the abundance of chemically equivalent C–H bonds, and the inability to overcome inherent steric or electronic bias for reactivity.31,32 The large number of biologically active natural products that undergo late-stage functionalization by tailoring enzymes provides a unique opportunity to leverage the power of halogenating enzymes to perform difficult chemical transformations. Early efforts to modulate the selectivity of these enzymes achieved shifts in site-selectivity and slight modifications to substrate scope,33,34 while recent efforts have succeeded in engineering FDHs for a broad range of site-selectivities and substrates.35–39 Through this work, we sought to identify and characterize the versatile halogenase involved in malbrancheamide biosynthesis and demonstrate its potential as a biocatalyst for late-stage halogenation of the structurally complex substrate, premalbrancheamide (2).
Results
Isolation of malbrancheamides from M. aurantiaca. From a 1.5 L growth of M. aurantiaca, we obtained the following yields of the naturally occurring malbrancheamides: 1.6 mg/L premalbrancheamide (2), 2.6 mg/L isomalbrancheamide B (4), 4.4 mg/L malbrancheamide B (3), and 5.8 mg/L malbrancheamide (1). These materials enabled subsequent biochemical and structural studies of MalA and MalA’.
Biochemical activity of MalA
Purification of MalA by Ni-affinity chromatography and gel filtration provided pure protein for in vitro assays, and MalA was found to catalyze the iterative chlorination and monobromination of the natural precursor premalbrancheamide (2). In reactions with the monochlorinated 3 and 4, MalA was also able to chlorinate and brominate these compounds to generate 1, 7, and 8, of which the latter two are novel indole alkaloids. The chlorination reaction of MalA was confirmed by co-elution with standards isolated from M. aurantiaca (Figure 3). 1H-NMR analysis confirmed the halogenation site on the indole ring and the high-resolution mass spectrometry data for all compounds were consistent with the expected masses and isotope peak patterns for the halogenated products (SI).
Figure 3.
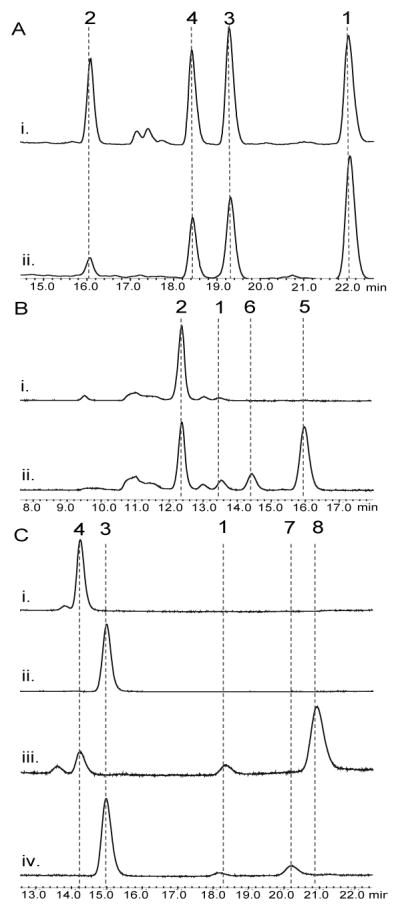
HPLC traces (240 nm) for the MalA in vitro reactions (A) chlorination of 2 (ii) compared to standards from the fungal extract (i), (B) bromination of 2 (ii) compared to no enzyme control (NEC) (i), and (C) bromination of 4 (iii) compared to NEC (i) and bromination of 3 (iv) compared to NEC (ii).
The percent conversions of the halogenation reactions were determined by isolated yields. The chlorination of 2 to produce 3, 4, and 1 in a 5 mg in vitro reaction showed 34%, 26%, and 24% conversion, respectively. The bromination of 2 in a 4 mg reaction generated 23% 5 and 18% 6 by isolated yield, but the calculated conversions based on standard curves displayed a C9 selectivity with an 8:1 ratio of 5 to 6. The methodology for separation of these monohalogenated intermediates by HPLC is well resolved compared to previous reports, thus the NMR data for the individual molecules significantly adds to the literature of these brominated indole alkaloids. MalA was also used as a biocatalyst for the generation of two novel malbrancheamide analogs 7 and 8 (Figures 3 and 4). The bromination of 3 in a 2 mg reaction produced 24% 7 and 15% 1 as a side product. The bromination of 4 in a 2.3 mg reaction produced 78% 8. The structural assignments of 7 and 8 were confirmed through extensive 1D and 2D NMR analyses. The structures were confirmed using key gHMBCAD correlations (Figure 4) where a significant downfield shift was observed for the chlorinated carbon as opposed to the brominated carbon. The positions of the halogens on the indole ring were confirmed by the two singlet peaks observed in the 1H-NMR aromatic region of each spectrum. Moreover, 5 and 6 can also be chlorinated to produce 8 and 7, respectively (see Figure 4 in SI).
Figure 4.
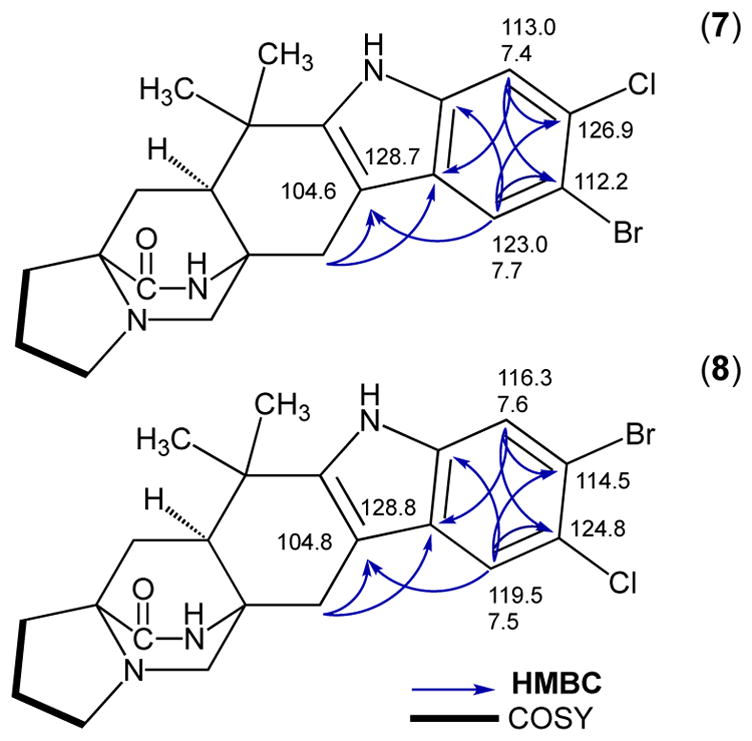
gHMBCAD and gCOSY NMR correlations for the indole region used to determine the sites of halogenation of new malbrancheamide analogs.
Kinetic characterization of MalA
Michaelis-Menten kinetic parameters were determined for the natural chlorination reactions of MalA. They revealed that the enzyme has similar kcat and Km values for both the initial and second chlorination reactions. The kcat from 2 to 3, 2 to 4, 3 to 1, and 4 to 1 were 0.08, 0.09, 0.12, and 0.12 min−1, respectively, which are comparable values to those of FDH PrnA (0.10 min−1).2 The Km values for 2 to 3, 2 to 4, 3 to 1, and 4 to 1 were 7.0, 7.5, 4.4, and 4.0 μM. The catalytic efficiencies were calculated for each of the four reactions resulting in the kcat/Km values of 11.5, 12.0, 27.3, and 29.7 min−1mM−1, respectively (see Table 2 in SI). These catalytic efficiencies are fairly high compared to those of the eukaryotic FDH Rdc2 which are 2.93 min−1mM−1 for the initial chlorination reaction and 0.11 min−1mM−1 for the second chlorination reaction.4
Structural characterization of the substrate complexes of MalA’
To further elucidate the unique functionality of MalA/MalA’ reactivity at two sites, the co-crystal structures of MalA’ in complex with premalbrancheamide (2), malbrancheamide B (3), and isomalbrancheamide B (4) were determined. MalA and MalA’ are 99% identical, differing at only two amino acid positions (Leu276 and Arg428 in MalA; Pro276 and Pro428 in MalA’), and have comparable catalytic activities, but only MalA’ was amenable to crystallization. To verify that MalA’ was a viable substitute for MalA, the activities of the two were compared, and it was determined that MalA’ was essentially identical to MalA under the reaction conditions tested. The structures of the ternary complexes with FAD, chloride ion, and each of the three substrates (2, 3, and 4) were determined at 2.36 Å, 2.09 Å, and 2.04 Å, respectively. MalA’ has a similar overall structure to bacterial FDHs with the addition of a few unique motifs including a Zn2+-binding C-terminus and a large active site capable of accommodating the structurally complex substrates. The natural substrates 2, 3, and 4 have a similar binding mode in the MalA’ active site (Figure 5). Specific interactions include a hydrogen bond of the indole nitrogen to Glu494 and a Cl-π interaction of 3 and 4 with Phe489 (Figures 5 and 8). The roles of amino acids in the active site were analyzed through site-directed mutagenesis (Figures 6 and 7), and Lys108 was determined to be necessary for catalytic activity.
Figure 5.
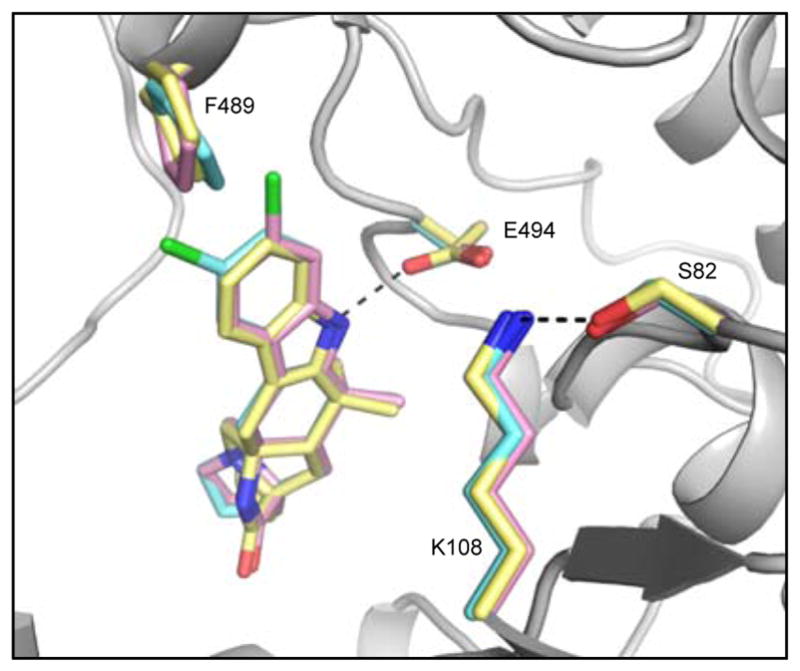
MalA’ active site overlay of complexes with substrates 2 (yellow), 3 (pink), and 4 (cyan).
Figure 8.

Interactions between MalA’ Phe489 (orange) and substrates (A) premalbrancheamide (2), (B) malbrancheamide B (3), and (C) isomalbrancheamide B (4).
Figure 6.
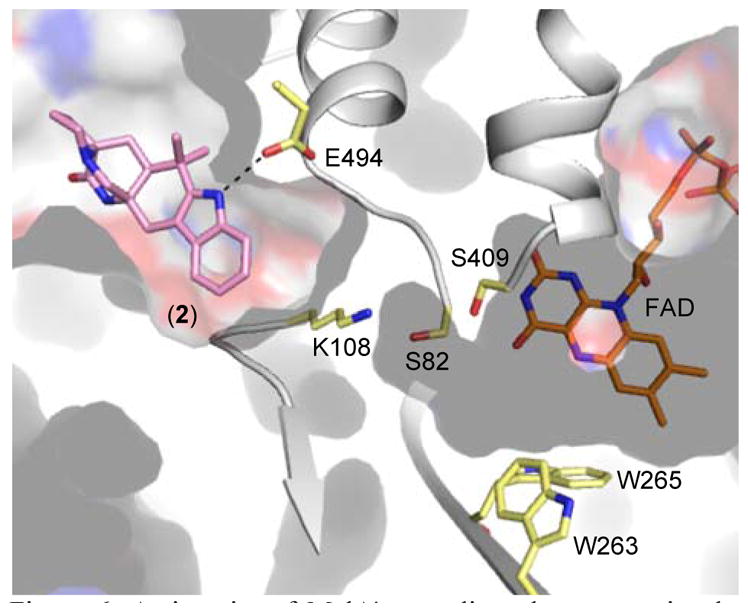
Active site of MalA’, revealing clear separation between the substrate and FAD binding pockets.
Figure 7.

Percent conversion with mutants versus wild-type MalA in reactions with 2 to produce 4 (green), 3 (blue), and 1 (red). MalA K108A and E494A/Q were inactive and MalA S82A was insoluble, precluding functional analysis.
Trp263 and Trp265 form a characteristic flavin-binding motif proposed to aid in cofactor binding (Figure 6). While MalA W265A showed a drastic decrease in activity, MalA W263A showed a more modest decrease in product formation (60% production of 4, 50% production of 3, and 5% production of 1) relative to wild-type MalA. A residue key to binding the substrate (Phe489) was substituted with histidine to ascertain its significance and was found to decrease MalA activity as well. Phe489 is analogous to the phenylalanine whose mutagenesis altered the site-selectivity in the FDH PrnA.33
In a preliminary effort to probe the mechanism of MalA, Glu494 was substituted with a variety of other residues including alanine, glutamine, and aspartate. While E494A and E494Q inactivated MalA, E494D maintained slight activity. Glu494 forms a hydrogen bond with the proton of the indole nitrogen, facilitating binding of the substrate. The substitution of aspartate at this position shifted the substrate away from the catalytic lysine, thus decreasing the activity (Figures 7 and 9).
Figure 9.
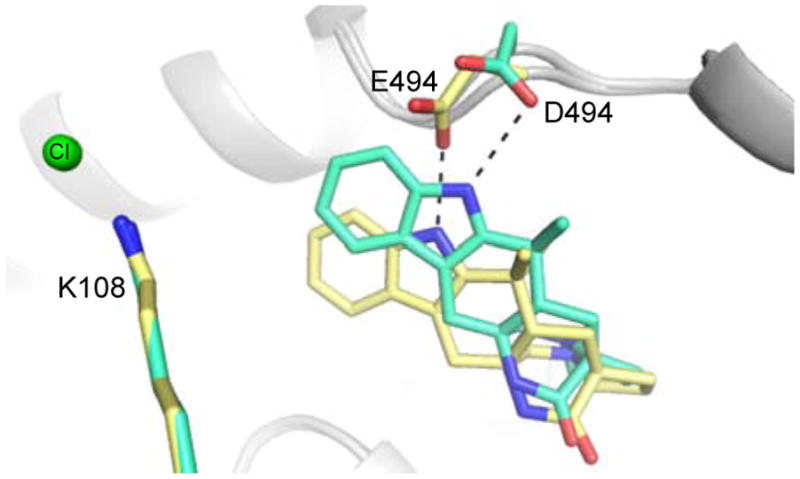
Comparison of wild-type MalA’ (yellow) and MalA’ E494D (cyan) co-crystallized with 2.
Initial efforts to probe the mechanism of site-selectivity in MalA included substitution of His253 with alanine, phenylalanine, and other amino acids. MalA H253A was selective for producing the C9-chlorinated 3, while MalA H253F displayed selectivity for producing the C8-chlorinated 4. Co-crystal structures of MalA’ H253A in complex with 2 and 3 revealed no evident changes in the protein that would lead to the observed site-selectivity. On the other hand, the co-crystal structure of MalA’ H253F in complex with 2 revealed a shift in S129, a residue near the indole ring of the substrate (Figure 10). When S129 was substituted with alanine, the C8 selectivity of MalA H253F was abolished, leading to the conclusion that Ser129 is involved in the selectivity induced by the Phe substitution at position 253 (Figure 7). Interestingly, the MalA H253A and MalA H253F mutants did not display the same site-selectivity profile for the bromination reaction. Instead we observed the same product profile as with the wild-type MalA (see Figure 5 in SI)
Figure 10.
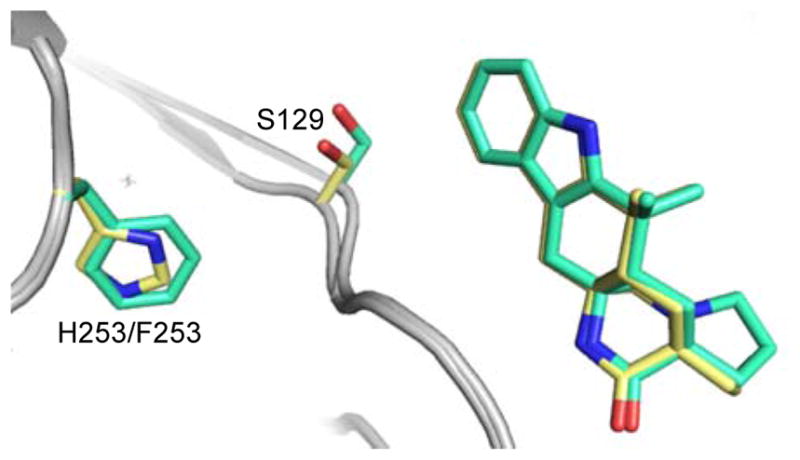
Comparison of wild-type MalA’ (yellow) and MalA’ H253F (cyan) co-crystallized with 2.
The structures of MalA’ also revealed a unique zinc site with coordination by four cysteine residues (Cys597, Cys600, Cys613, Cys616) near the C-terminus. The Zn2+ ion was identified using anomalous scattering experiments with diffraction data recorded at X-ray energies bracketing the zinc K-edge (9.6586 keV). Anomalous difference density was present only in the map using data from the energy above the edge (see Figure 11 in SI). A double mutant MalA C613S/C616S was prepared and the protein was insoluble, thus no biochemical activity assays were performed in the absence of Zn2+.
Molecular dynamics simulations and QM models
We conducted molecular dynamics (MD) simulations to gain further insight into the structure and activity of the protein, starting from the MalA’ crystal structures in their apo and substrate bound forms. In the latter case, the Lys108 chloramine intermediate has been considered integral to the mechanism discussed below.
The analysis of the MD trajectories revealed the structural role played by the Zn2+ counterion in the protein structure. Residues within the Zn2+ binding region (597–616) exhibited a low root-mean-square fluctuation (RMSF) compared to the very flexible adjacent region (621–646). These simulations indicate that the flexible region acts as a substrate channel lid, having two main open/closed conformations that were explored during the 500 ns MD simulations in both the apo and substrate-bound states (see Figure 13 and Figure 14 in SI).
From the apo state trajectories, the pKa of the Lys108 and Glu494 side chains was estimated (see Figure 23 in SI). Glu494 has a relatively high pKa (≈6.0–7.5), while Lys108 has an estimated pKa of 7.2–8.3 (see Figure 23 in SI).
The analysis of possible polar interactions between the substrate and the enzyme active site showed that, although the backbone carbonyls of Gly131 and Ala132 could potentially interact with the substrate amide nitrogen, these interactions are not as important as the Glu494 – H(N-indole) hydrogen bond. The latter corresponds to the main and stronger interaction between the substrate and protein active site residues, and it is observed with all bound substrates (2, 3 and 4) during the entirety of the MD trajectory simulations (see Figure 16 in SI). The basicity of Glu494 can thus enhance the hydrogen bonding between the carboxyl side chain and the indole ring of the substrate, positioning it to effectively interact with the catalytic Lys108 residue.
MD simulations including the proposed active chloramine Cl-Lys108 species showed that when substrate 2 is bound into the active site and the channel lid is closed, the Cl atom is placed very close to the C8/C9 positions of the substrate, due in part to the positioning of the substrate by the Glu494 residue (see Figure 11a). When the lid is in its open conformation, the substrate binding is slightly displaced although still H-bonding with Glu494, but then the Cl-Lys108 side chain conformation changes to place the Cl atom closer to the FAD cofactor (see Figure 15 in SI). This indicates that the protein conformational change between the substrate bound open/closed states also controls the positioning of Cl-Lys108 active species, which can explore two main conformations to bring the Cl atom from the flavin cofactor to the substrate (see Figure 15 in SI). This observation supports that Cl-Lys108 is the actual chlorinating species. When Cl-Lys108 is in this catalytically competent pose, the distances (Cl-C) and angles (Cl-C-H) measured for both C8 and C9 positions are very similar, indicating that Cl-Lys108 is preorganized to chlorinate both positions (see Figure 21 in SI).
Figure 11.

a) Representative snapshot (at 40 ns) taken from the 500 ns MD simulation of MalA’ and substrate 2 bound complex, including the chloramine adduct at Lys108. b) Computed DFT reaction pathways for the three models: 1a-indole and methyl chloramine; 1b-indole, methyl chloramine and a water molecule coordinating at the C8 or C9 positions, respectively; 1c-indole, methyl chloramine, and MeOH coordinating to H-C8. c) DFT optimized geometries of some representative structures (see SI for additional material). Gibbs energies are given in kcal/mol, and distances in Å.
MD simulations also revealed a key interaction between Cl-Lys108 and the backbone carbonyl of the neighboring Asp109 residue, effectively positioning Cl-Lys108 towards a catalytically competent arrangement (Figure 11a). The Cl atom from Lys108 chloramine active species can only be placed close to C8/C9 positions when this H-bond is present (Figure 17a and 17b in SI). The essential role of Asp109 was further explored by mutagenesis experiments, and the D109A mutant has very low activity (Figure 7). MD simulations on this particular mutant showed that the Ala109 backbone carbonyl is pointing towards a different position than in the original Asp109, thus eliminating a key H-bond interaction. This is caused by the different conformation of the amino acid side chain, which is pointing outside the protein cavity and exposed to the solvent in Asp109, while in Ala109 it is displaced (see Figure 20 in SI). Indeed, in our 500 ns simulations for the D109A mutant, Cl-Lys108 never explores any conformation in which the Cl atom approaches the C8/C9 positions to affect chlorination (see Figure 20 in SI).
MD simulations on MalA’ Cl-Lys108 with bound substrate 2 highlighted the arrangement of the Ser129 side chain with respect to H-C8 in 2. The distance between Ser129 Og and H-C8 is particularly short (between 2.5–3.5 Å) when the substrate is in an appropriate orientation for the electrophilic aromatic substitution (see Figure 17 in SI). This interaction was not observed along the MD trajectory of MalA’ H253A, but was prominent in the MalA’ H253F trajectory (see Figures 18 and 19 in SI). This interaction is quite important since Ser129 is one of the few polar residues in a very hy drophobic region of the active site pocket. Along the MD trajectories, the solvation shell estimated for the C8 and C9 positions of 2 showed a more apolar environment for wild-type MalA’ and MalA’ H253A (i.e. fewer surrounding water molecules) than for MalA’ H253F (see Figure 22 in SI).
Based on the experimental evidence and computational modeling of the enzyme active site, we propose a mechanism for the MalA halogenase (Figure 11, Scheme 2) involving the formation of a Lys108-chloramine intermediate active species, which then interacts with C8 or C9 to carry out an electrophilic aromatic substitution (EAS), with generation of a Wheland intermediate (W) before a final deprotonation step (Figure 11b and Scheme 2). This deprotonation can be effected by a water molecule acting as a base, or in the case of C8 by the well-positioned Ser129. To gain insight on the proposed reaction mechanism, density functional theory (DFT) calculations were conducted, using three different computational models (see SI Materials and Methods for computational details). The first model considers only an indole ring and a protonated methyl chloramine as the active species (1a); the second adds a water molecule close to H-C8 or H-C9 positions (1b); and the third model includes a methanol molecule near H-C8 position to mimic Ser129 (1c). Our calculations show that the intrinsic rate-limiting step of the reaction is the initial chlorination, while the deprotonation step occurs slightly faster. A water molecule or methanol interacting with the C8/C9 protons accelerates the chlorination steps because hydrogen bonding enhances the nucleophilicity of these carbons. The computed reaction barrier for C8-chlorination (TS1a-C8) was decreased from 6.4 to 5.5 kcal/mol by a coordinating H2O (TS1b-C8), and further decreased to 3.5 kcal/mol when MeOH coordinates to the H-C8 (TS1c-C8). On the other hand, the computed barrier for C9 chlorination (TS1a-C9) decreased from 7.0 to 5.2 kcal/mol by a coordinating H2O molecule at H-C9 (TS1b-C9), as shown in Figure 11, and to 4.9 kcal/mol when MeOH interacted with H-C8 (TS1c-C9), as shown in Figure 22 in the SI. This highlights the role of Ser129 in directing the selectivity towards the formation of the C8 chlorinated product.
Scheme 2.
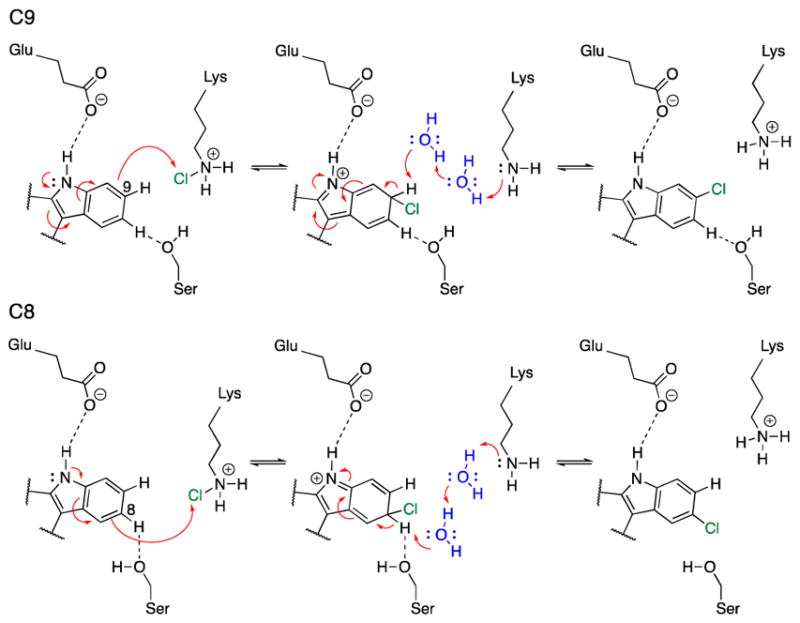
Proposed mechanism for MalA catalysis.
The Wa-C9 Wheland intermediate is 1.3 kcal/mol more stable than Wa-C8, but they become almost isoenergetic when coordinating water molecules are considered (Wb-C8 and Wb-C9). An apolar environment favors the formation of the C9 chlorinated product 3. Finally, once the Wheland intermediates are formed, re-aromatization by deprotonation occurs rapidly. The computed deprotonation barriers for the two positions are 4.1 and 3.5 kcal/mol for C8 (TS2b-C8) and C9 (TS2b-C9) respectively when a water molecule acts as the base, and 0.6 kcal/mol for C8 when MeOH acts as a base (TS2c-C8).
The DFT optimized structures for the reactant complexes and transition states are highly similar. The catalytically competent arrangement of Cl-Lys108 near C8 and C9 was found in our MD simulations (represented in Figures 11a, 11c). Taking together our QM models, MD simulations, and the pre-organization of the Cl-Lys108 versus the substrate previously described, we conclude that the proposed reaction mechanism involving a Cl-Lys108 intermediate is the most plausible for the MalA flavin-dependent halogenase.
Discussion
These results provide an example of a unique subclass of flavin-dependent halogenases that performs iterative late-stage halogenation of complex substrates independent of a carrier protein. MalA is encoded in a gene cluster containing an NRPS, but the evidence provided herein demonstrates that this protein catalyzes effective late-stage functionalization on free substrates. We propose a new mechanism, involving Ser129, for deprotonation of the positively charged Wheland intermediate in MalA/MalA’ halogenation (Scheme 2). The hydrogen bond between Glu494 and the indole nitrogen is proposed to increase the nucleophilicity of the aromatic ring. This facilitates the EAS reaction, producing the positively charged intermediate. A water molecule or serine side chain can then deprotonate the Wheland intermediate, leading to re-aromatization of the indole ring system.
Two critical residues in the active site significantly contribute to substrate binding, Glu494 and Phe489. Glu494 hydrogen bonds with the indole nitrogen and Phe489 facilitates a favorable hydrophobic interaction with the aromatic ring of the indole. The activity of MalA F489H was significantly decreased relative to the wild-type, especially for the second chlorination reaction. We hypothesize that the phenylalanine residue in the back of the active site aids in substrate binding, and maintains interactions with the monochlorinated products to facilitate a second halogenation reaction.
The Michaelis-Menten model kinetics displayed equal rates of monochlorination at both the C9 and C8 sites of 2, and equal rates for chlorination of 3 and 4, leading to the conclusion that MalA is equally selective for both sites of the indole ring. Interestingly, the catalytic efficiency (kcat/Km) values for the second chlorination were twice those observed for the first chlorination, thus the initial chlorination is proposed to prime the substrate for the second halogenation. This effect can be correlated with the structural data where the chlorine atom on the indole substrates interacts favorably with Phe489, potentially facilitating a better mode of binding. Additionally, the dichlorinated malbrancheamide (1) did not bind in crystals, suggesting a faster dissociation rate for the dichlorinated than either of the monochlorinated products, which bound readily in MalA’ crystals.
In efforts elucidate the mechanism of selectivity in MalA, a histidine residue near the catalytic lysine was used to probe the active site region. MalA H253A displayed selectivity for chlorination at the C9 position of 2, while MalA H253F was selective for the C8 position of 2. MD simulations and DFT calculations demonstrated how key interactions between polar amino acid side chains (Ser129) or water molecules in the active site with the different C–H positions of the indole can control the selectivity of the chlorination reaction. This is accomplished by enhancing the nucleophilicity of the carbon atom during the EAS, but also by pre-organizing a base to carry out the final deprotonation step. An alanine substitution at His253 prevented the interaction of Ser129 and H-C8, leading to an overall apolar active site environment, favoring chlorination at C9. On the other hand, the C8 selectivity observed in MalA H253F can be explained by a more effective Ser129 interaction with H-C8. When investigating these mutants in the the site-selectivity of the bromination reaction, the wild-type product profile was observed. Compared to HOCl, HOBr is a milder halogenating reagent, thus favoring the inherently more reactive C9 site of 2. These findings demonstrate that even small modulations to the active site region can lead to significant changes in the site-selectivity of the halogenation reaction.
The designation of MalA into a new class of flavin-dependent halogenases is exemplified not only by its reactivity but also by its unique structural motifs: a Zn2+-binding C-terminus and an expansive active site, able to accommodate complex substrates. The discovery of this Zn2+-binding motif provides a fingerprint for use in mining sequence data for MalA homologs in pursuit of biocatalysts for late-stage halogenation (Figure 12). Investigation of MalA/MalA’ has provided new insights into the biocatalytic mechanism for iterative late-stage halogenation of complex substrates. With a starting model that we have shown is equally reactive toward C8 and C9 of premalbrancheamide, future efforts will focus on rational engineering of MalA to mediate its regioselectivity, substrate scope, and iterative function.
Figure 12.

Phylogenetic tree of characterized and uncharacterized (labeled with producing organism) FDHs. Putative fungal FDHs with high amino acid identity (>50%) to MalA cluster in the blue region and are proposed to contain the Zn2+-binding motif based on sequence alignments. The green region contains previously characterized bacterial FDHs (the exception is the eukaryotic ChlA) and the yellow region denotes a new subclass of putative FDHs from fungi. Phylogenetic tree prepared using Lasergene MegAlignPro (DNASTAR).40
Supplementary Material
Acknowledgments
Funding Sources
The authors thank the National Institutes of Health for financial support (R01 CA070375 to RMW and DHS, NIH/NCI R01 CA047135 to PC, and NIGMS R01 086374 to RS) as well as the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1205646), the Hans W. Vahlteich Professorship (to DHS), and the Margaret J. Hunter Professorship (to JLS).
M.G.-B. thanks the Ramón Areces Foundation for a post-doctoral fellowship. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575).
We thank Dr. Rachel Mata and Dr. Abraham Madariaga-Mazón for their helpful discussions on culturing Malbranchea aurantiaca for the isolation of malbrancheamide compounds.
We thank Dr. David Ballou for generously providing the phaC flavin reductase plasmid from Pseudomonas aeruginosa.
GM/CA@APS is supported by the National Institutes of Health, National Institute of General Medical Sciences (AGM-12006) and National Cancer Institute (ACB-12002). The Advanced Photon Source is a U.S. Department of Energy (DOE) Office of Science User Facility operated by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Accession codes:
Atomic structure factors and coordinates have been deposited in the Protein Data Bank: 5WGR, 5WGS, 5WGT, 5WGU, 5WGV, 5WGW, 5WGX, 5WGY, 5WGZ
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Full experimental details and NMR spectra are available free of charge via the Internet at http://pubs.acs.org
Methods, Tables, and Figures (PDF)
References
- 1.Weichold V, Milbredt D, van Pée KH. Angew Chem Int Ed. 2016;55:2–18. doi: 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- 2.Dong C, Flecks S, Unversucht S, Haupt C, van-Pée K-H, Naismith JH. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Biochemistry. 2007;46(5):1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 4.Zeng J, Zhan J. ChemBioChem. 2010;11:2119–2123. doi: 10.1002/cbic.201000439. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara M, Perrone G, Gambacorta L, Epifani F, Solfrizzo M, Gallo A. Appl Environ Microbiol. 2016;82(18):5631–5641. doi: 10.1128/AEM.01209-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann CS, Walsh CT, Kay RR. Proc Natl Acad Sci U S A. 2010;107(13):5798–803. doi: 10.1073/pnas.1001681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen MT, Nielsen JB, Anyaogu DC, Holm DK, Nielsen KF, Larsen TO, Mortensen UH. PLoS One. 2013;8(8):1–10. doi: 10.1371/journal.pone.0072871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chankhamjon P, Boettger-Schmidt D, Scherlach K, Urbansky B, Lackner G, Kalb D, Dahse HM, Hoffmeister D, Hertweck C. Angew Chem Int Ed. 2014;53:13409–13413. doi: 10.1002/anie.201407624. [DOI] [PubMed] [Google Scholar]
- 9.Sato M, Winter JM, Kishimoto S, Noguchi H, Tang Y, Watanabe K. Org Lett. 2016;18:1446–1449. doi: 10.1021/acs.orglett.6b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chankhamjon P, Tsunematsu Y, Ishida-Ito M, Sasa Y, Meyer F, Boettger-Schmidt D, Urbansky B, Menzel KD, Scherlach K, Watanabe K, Hertweck C. Angew Chem Int Ed. 2016;55:11955–11959. doi: 10.1002/anie.201604516. [DOI] [PubMed] [Google Scholar]
- 11.Cacho RA, Chooi YH, Zhou H, Tang Y. ACS Chem Biol. 2013;8:2322–2330. doi: 10.1021/cb400541z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menon BRK, Brandenburger E, Sharif HH, Klemstein U, Shepherd SA, Greaney MF, Micklefield J. Angew Chem Int Ed. 2017 doi: 10.1002/anie.201706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh E, Garneau S, Walsh CT. Proc Natl Acad Sci U S A. 2005;102(11):3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seibold C, Schnerr H, Rumpf J, Kunzendorf A, Hatscher C, Wage T, Ernyei AJ, Dong C, Naismith JH, Van Pée KH. Biocatal Biotransform. 2006;24(6):401–408. [Google Scholar]
- 15.Zehner S, Kotzsch A, Bister B, Süssmuth RD, Méndez C, Salas JA, van Pée KH. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc Natl Acad Sci U S A. 2005;102(39):13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Gamal A, Agarwal V, Diethelm S, Rahman I, Schorn MA, Sneed JM, Louie GV, Whalen KE, Mincer TJ, Noel JP, Paul VJ, Moore BS. Proc Natl Acad Sci U S A. 2016;113(14):3797–3802. doi: 10.1073/pnas.1519695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martínez-Luis S, Rodríguez R, Acevedo L, González MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]
- 19.Watts KR, Loveridge ST, Tenney K, Media J, Valeriote FA, Crews P. J Org Chem. 2011;76(15):6201–6208. doi: 10.1021/jo2009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figueroa M, González-Andrade M, Sosa-Peinado A, Madariaga-Mazón A, Del Río-Portilla F, Del Carmen González M, Mata R. J Enzyme Inhib, Med Chem. 2011;26(3):378–385. doi: 10.3109/14756366.2010.518964. [DOI] [PubMed] [Google Scholar]
- 21.Madariaga-Mazón A, Hernández-Abreu O, Estrada-Soto S, Mata R. J Pharm Pharmacol. 2015;67(4):551–558. doi: 10.1111/jphp.12346. [DOI] [PubMed] [Google Scholar]
- 22.Klas K, Tsukamoto S, Sherman DH, Williams RM. J Org Chem. 2015;80:11672–11685. doi: 10.1021/acs.joc.5b01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stocking EM, Williams RM. Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]
- 24.Finefield JM, Frisvad JC, Sherman DH, Williams RM. J Nat Prod. 2012;75:812–833. doi: 10.1021/np200954v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y, Greshock TJ, Miller KA, Sherman DH, Williams RM. Org Lett. 2008;10(21):4863–4866. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S, Krithika S, Tran H, Yu F, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. MedChemComm. 2012;3:987–996. doi: 10.1039/C2MD20029E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng J, Zhan J. Biotechnol Lett. 2011;33:1607–1613. doi: 10.1007/s10529-011-0595-7. [DOI] [PubMed] [Google Scholar]
- 28.Buedenbender S, Rachid S, Müller R, Schulz GE. J Mol Biol. 2009;385(2):520–530. doi: 10.1016/j.jmb.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 29.Podzelinska K, Latimer R, Bhattacharya A, Vining LC, Zechel DL, Jia Z. J Mol Biol. 2010;397:316–331. doi: 10.1016/j.jmb.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 30.Hillwig ML, Liu X. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 31.Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 32.Chung W, Vanderwal CD. Angew Chem Int Ed. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang A, Polnick S, Nicke T, William P, Patallo EP, Naismith JH, van Pée KH. Angew Chem Int Ed. 2011;50:2951–2953. doi: 10.1002/anie.201007896. [DOI] [PubMed] [Google Scholar]
- 34.Glenn WS, Nims E, O’Connor SE. J Am Chem Soc. 2011;133:19346–19349. doi: 10.1021/ja2089348. [DOI] [PubMed] [Google Scholar]
- 35.Shepherd SA, Karthikeyan C, Latham J, Struck AW, Thompson ML, Menon BRK, Styles MQ, Levy C, Leys D, Micklefield J. Chem Sci. 2015;6:3454–3460. doi: 10.1039/c5sc00913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shepherd SA, Menon BRK, Fisk H, Struck AW, Levy C, Leys D, Micklefield J. ChemBioChem. 2016;17:821–824. doi: 10.1002/cbic.201600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Payne JT, Andorfer MC, Lewis JC. Angew Chem Int Ed. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Payne JT, Poor CB, Lewis JC. Angew Chem Int Ed. 2015;54:4226–4230. doi: 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andorfer MC, Park HJ, Vergara-Coll J, Lewis JC. Chem Sci. 2016;7:3720. doi: 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MegAlign Pro ®. Version 12.0. DNASTAR; Madison, WI: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.