

Abstract
A number of consensuses regarding cancer immunology have recently emerged from both preclinical immunotherapy models and analysis of cancer patients. First and foremost, the natural state of endogenous tumor reactive T cells is characterized by general hyporesponsiveness or anergy. This is likely due to a number of mechanisms that tumors use to induce tolerance as they develop. While many of the newer generation vaccines can effectively transfer antigen to and activate dendritic cells, T-cell tolerance remains a major barrier that is difficult to overcome by vaccination alone. Preclinical models demonstrate that for poorly immunogenic tumors, once tolerance has been established, therapeutic vaccines alone are ineffective at curing animals with a significant established tumor burden. However, combination strategies of vaccination together with inhibitors of immunologic checkpoints and agonists for co-stimulatory pathways are proving capable of overcoming tolerance and generating significant anti-tumor responses even in cases of established metastatic cancer.
Historically, interest in cancer immunology stemmed from the perceived potential activity of the immune system as a weapon against cancer cells. In fact, the term “magic bullet”, commonly used to describe many visions of cancer therapy, was coined by Paul Erlich in the late 1800s in reference to antibodies targeting both microbes and tumors. Central to the concept of successful cancer immunotherapy are the dual tenets that tumor cells express an antigenic profile distinct from their normal cellular counterparts and that the immune system is capable of recognizing these antigenic differences. Support for this notion originally came from animal models of carcinogen induced cancer in which it was demonstrated that a significant number of experimentally induced tumors could be rejected upon transplantation into syngeneic immunocompetent animals.1 Extensive studies by Prehn on the phenomenon of tumor rejection suggested that the most potent tumor rejection antigens were unique to the individual tumor.2
As cancer genetics and genomics has exploded over the past decade, it is now quite clear that altered genetic and epigenetic features of tumor cells indeed result in a distinct tumor antigen profile. Overexpression of “oncogenic” growth factor receptor tyrosine kinases such as HER2/Neu and epidermal growth factor receptor (EGFR) via epigenetic mechanisms has provided clinically relevant targets for one arm of the immune system—antibodies.3,4 In general, we have learned that tumors employ mechanisms of tolerance induction to turn off T cells specific for tumor-associated antigens. Oncogenic pathways in tumors result in the elaboration of factors that organize the tumor microenvironment in ways that are quite hostile to anti-tumor immune responses. This review will outline the major features of tumor–immune system interactions and set the stage for molecularly based approaches to manipulate immune responses for successful cancer therapy.
HOW DO TUMORS DIFFER FROM SELF TISSUES?
Tumors differ fundamentally from their normal tissue counterparts in both antigenic composition and biologic behavior. Genetic instability, a basic hallmark of cancer, is a primary generator of true tumor-specific neo-antigens. The most common genetic alteration in cancer—mutations—arise from defects in DNA damage repair systems of the tumor cell.5 Recent estimates from genome-wide sequencing efforts suggest that many tumor types contain hundreds to thousands of mutations in coding regions.6 The major histocompatibility complex (MHC) presentation system for T-cell recognition makes peptides derived from all cellular proteins available on the cell surface as peptide MHC complexes capable of being recognized by T cells. There are a few recent examples of T-cell responses to mutation-derived neo-antigens. Most are unique to the individual tumor and have no obvious oncogenic relevance; they are likely “passenger mutations”.7,8 However, there are a growing number of examples of tumor-specific mutations that are shared. As with non-shared mutations, these common tumor-specific mutations all occur in intracellular proteins, and therefore require T-cell recognition of MHC-presented peptides for immune recognition. Indeed, both the Kras codon 12 G→A and the BrafV600E mutations result in “neopeptides” capable of being recognized by human leukocyte antigen (HLA) class I– and class II–restricted T cells.9
The other major difference between tumor cells and their normal counterparts derives from epigenetics.10 Global alterations in DNA methylation as well as chromatin structure in tumor cells results in dramatic shifts in gene expression. All tumors overexpress hundreds of genes relative to their normal counterparts, and in many cases, turn on genes that are normally completely silent in their normal cellular counterparts. Overexpressed genes in tumor cells represent the most commonly targeted tumor antigens by both antibodies and cellular immonotherapies. The most dramatic examples of tumor-selective expression of epigenetically altered gene are the so-called “cancer-testis antigens”.11 These genes appear to be highly restricted in their expression in the adult. Many are expressed selectively in the testis of males and are not expressed at all in females. Their expression in tumors appears to be purely the consequence of epigenetic instability rather than functional selection, and antigen-negative variants are easily selected out in the face of immunotherapeutic targeting.
The most commonly generated melanoma-reactive T cells from melanoma patients recognize melanocyte antigens.12 While one cannot formally call tissue-specific antigens tumor-specific, they are nonetheless potentially viable targets for therapeutic T-cell responses when the tissue is dispensable (ie, prostate cancer or melanoma).
From the standpoint of T-cell targeting, tumor antigens upregulated as a consequence of epigenetic alterations represent “self antigens” and are therefore likely to induce some level of immune tolerance. This notion has led to the popular recent view that mutation-dependent tumor neo-antigens are more immunogenic because the immune system is less tolerant of them. However, at present, no careful comparison has been made in humans of the potential repertoires and state of natural T-cell responsiveness to genetically versus epigenetically generated tumor antigens.
In contrast to normal tissues, cancers are constantly confronted with inflammatory responses as they invade tissues and metastasize. In some circumstances these inflammatory and immune responses can potentially eliminate a tumor—so-called “immune surveillance”. However, oncogenic pathways in the tumor appear to organize the immunologic component of the microenvironment in a fashion that not only protects itself from anti-tumor immune responses but that can qualitatively shift immune responses to those that actually support and promote tumor growth. Thus, tumors can entice the immune system to the “dark side”. It is these elements of the cancer–immune system interaction that will be the central targets of future immunotherapeutic strategies.
EVIDENCE PRO AND CON FOR IMMUNE SURVEILLANCE OF CANCER
The fundamental tenet of the immune surveillance hypothesis, first conceived nearly a half century ago,13 is that a major role of the immune system is to survey the body for tumors as it does for infection with pathogens, recognizing and eliminating them based on their expression of tumor-associated antigens. In animal models, carcinogen-induced tumors can be divided into those that grow progressively (termed progresser tumors) and those that are rejected after an initial period of growth (termed regresser tumors).1,2 A fundamental prediction of the immune surveillance hypothesis is that immunodeficient individuals would display a dramatic increase in tumor incidence. After an extensive analysis of spontaneous tumor formation in immunodeficient nude mice, which have significantly reduced numbers of T cells and T-cell–dependent immune responses, no increased incidence of tumors was observed.14 These studies were taken as a major blow to the immune surveillance hypothesis. However, a caveat to the interpretation of these results is that nude mice still produce diminished numbers of T cells via thymus-independent pathways and therefore can mediate some degree of T-cell–dependent immunity. In addition, nude mice frequently display compensatory increases in innate immunity that may provide an alternative to adaptive immunity and could contribute to immune surveillance of cancer.
In humans, the most common cancers that arise in immunodeficient individuals are virus-induced and thus are not considered to be tumor-specific immune surveillance as it was originally conceived. These include Epstein-Barr virus (EBV)-associated lymphomas, Kaposi’s sarcoma-associated herpesvirus (KSHV), and human papilloma virus (HPV)-associated cervical and anal cancers.15–17 However, non-virus–associated tumors, particularly skin cancers, have been associated with pharmacologic immunosuppression in the context of organ transplantation.18
A number of recent studies re-evaluating tumor immune surveillance in genetically manipulated mice has revealed clear-cut evidence that various components of the immune system can at least modify, if not eliminate, both carcinogen-induced and spontaneously arising cancers. In a series of studies by Schreiber and colleagues re-examining cancer incidence in mice rendered immunodeficient via genetic knockout of either the RAG2 gene (deficient in both B and T cells), the γ-interferon receptor gene, STAT 1 gene, or the type 1 interferon receptor gene.19 Even animals not crossed onto a cancer-prone genetic background or treated with carcinogens developed an increased incidence of invasive adenocarcinomas when observed over their entire lifespan.20 Furthermore, γ-interferon, RAG2 double knockout mice developed a broader spectrum of tumors than RAG2 knockout mice. All of the tumors that arise in these genetically manipulated immunodeficient animals behave as regresser tumors when transplanted into immunocompetent animals. Regression in immunocompetent animals has, in some cases, been linked to T-cell responses against a single mutation-derived antigen. Thus, the presence of a competent immune system “sculpts” the tumor through a processes that has been termed “immunoediting”. While the classic concepts of immune surveillance of cancer remain unsupported by experimental evidence, studies on tumorigenesis in genetically manipulated immunodeficient mice indeed suggest that developing tumors must actively adapt themselves to their immune microenvironment in order to exist within the context of a competent immune system.
IMMUNE TOLERANCE AND IMMUNE EVASION—THE HALLMARK OF A SUCCESSFUL TUMOR
While natural immune surveillance plays a much smaller role than originally envisioned by Thomas and Burnet, developing tumors need to adapt to their immunologic milieu in a manner that either turns off potentially harmful (to the tumor) immune responses or creates a local microenvironment inhibitory to the tumoricidal activity of immune cells that could inadvertently become activated. These processes—tolerance induction and immune evasion—have become a central focus of cancer immunology efforts and will undoubtedly provide the critical information necessary for development of successful immunotherapies that break tolerance to tumor antigens and breakdown the resistance mechanisms operative within the tumor microenvironment (Figure 1).
Figure 1.
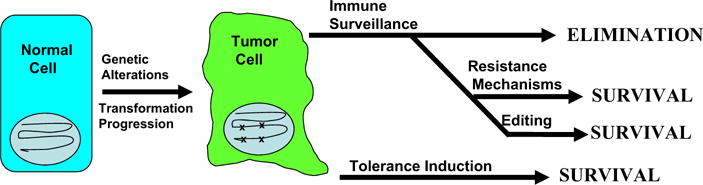
The balance between immune surveillance, resistance and tolerance. Transformation of normal cells to cancer cells involves the creation of true neoantigens due to mutation, as well as up-regulation of self-antigens due to epigenetic forces. Successful immune surveillance of tumors based on recognition of these tumor-specific antigens would lead to tumor elimination at early stages. Clinically relevant tumor survival and progression requires that tumors develop resistance mechanisms that inhibit tumor-specific immune responses to kill tumor cells. Alternatively, if the tumor develops mechanisms to induce immune tolerance to its antigens, anti-tumor effecter responses do not develop. Evidence is accumulating that tumors actively develop immune resistance mechanisms and immune tolerance mechanisms to survive despite displaying antigens capable of recognition by the immune system.
Evidence from both murine tumor systems and human tumors strongly demonstrates the capacity of tumors to induce tolerance to their antigens. This capacity to induce immune tolerance may very well be the single most important strategy that tumors use to protect themselves from elimination by the host’s immune system. Tolerance to tumors appears to operate predominately at the level of T cells. Numerous adoptive transfer studies have demonstrated the potent capacity of T cells to kill growing tumors, either directly through cytotoxic T lymphocyte (CTL) activity, or indirectly through multiple CD4-dependent effector mechanisms.21–23 It is thus likely that induction of antigen-specific tolerance among T cells is of paramount importance for tumor survival.
The first direct evidence for induction of T-cell tolerance by tumors was provided by Bogen and colleagues who examined the response of T-cell receptor (TCR) transgenic T cells specific for the idiotypic immunoglobulin expressed by a murine myeloma tumor.24 They first demonstrated induction of central tolerance to the myeloma protein followed by peripheral tolerance. Using influenza hemaglutinin (HA) as a model tumor antigen, Levitsky and colleagues demonstrated that adoptively transferred HA-specific TCR transgenic T cells were rapidly rendered anergic by HA-expressing lymphomas and HA-expressing renal carcinomas.25 In a model of prostate tumorigenesis, Drake and colleagues evaluated CD4 responses to HA and double transgenic animals expressing HA and SV40 T antigen under control of the prostate-specific probasin promoter.26 Development and progression of prostate tumors did not result in enhanced activation of adoptively transferred HA-specific T cells. Tolerance to HA as a normal prostate antigen occurred largely through ignorance since there was no evidence for antigen recognition by HA-specific T cells. However, increased recognition was observed upon either androgen ablation (which causes massive apoptosis within the prostate) or development of prostate cancer. Blankenstein and colleagues evaluated T-cell responses and rejection in a model of sporadic induction of tumors associated with expression of a tumor-specific antigen only at the time of transformation.27 They found that pre-immunization of mice against the tumor-associated antigen prevented the development of tumors. However, non-immunized mice developed spontaneous tumors without any significant evidence of natural immune surveillance in the absence of pre-immunization. They further demonstrated that an initial antigen-dependent activation of tumor-specific T cells could be observed at the time of spontaneous tumor induction but that this recognition ultimately resulted in an anergic form of T-cell tolerance similar to that observed by Drake and colleagues in the prostate system.
The capacity of spontaneously arising tumors to tolerize T cells has not been uniformly observed. A contrasting result by Ohashi and colleagues was observed when lymphocytic choriomeningitis virus (LCMV) GP33-specific TCR transgenic CD8 T cells were adoptively transferred into double transgenic mice expressing both SV40 T antigen and LCMV GP33 under control of the rat insulin promoter.28 These animals develop pancreatic islet cell tumors that express GP33. The investigators found that as tumors progressed in the mice, enhanced T-cell activation occurred. CD8 T-cell activation was demonstrated through bone marrow chimera experiments to occur exclusively via cross presentation in the draining lymph nodes. Despite the activation of tumor specific T cells, the tumors grew progressively, indicating that the degree of immune activation induced by tumor growth was insufficient to ultimately eliminate the tumors. These results suggest that developing tumors can induce immune responses but may titrate their level of immune activation to one that ultimately does not “keep up” with tumor progression. This mechanism in humans may explain, in part, the success of checkpoint blockade (see below), which requires a potential endogenous anti-tumor repertoire of T cells whose activity against tumors can be enhanced enough to yield clinically relevant tumor regression or disease stabilization.
It has been more difficult to obtain definitive evidence that human cancers tolerize tumor-specific T cells since humans cannot be manipulated the way mice are. However, the T cells that are grown out of patients with cancer tend to be either of low affinity for their cognate antigen or recognize antigens that bind poorly to their presenting HLA (human MHC) molecule, resulting in inefficient recognition by T cells.29
REGULATORY T CELLS AND CANCER
Over the past 10 years, regulatory T cells (Tregs), defined by expression of the canonical transcription factor, Foxp3, have emerged as a central player in maintenance of the tolerant state, as well as general downregulation of immune responses to pathogens.30 Not surprisingly, they appear to play a role in tolerance to tumor antigens, as well as the resistance of tumors to immune-mediated elimination.31,32 Mechanisms of immune suppression by regulatory T cells vary and include production of inhibitory cytokines such as interleukin (IL)-10 and transforming growth factor (TGF)-β. In keeping with the emerging appreciation that tumors are by nature highly tolerogenic, numerous murine studies have demonstrated that Tregs expand within tumors and significantly limit the potency of anti-tumor immune responses—either natural or vaccine-induced.33,34 As new cell membrane molecules that define Tregs are identified, the capacity to block Treg activity with antibodies to these molecules presents new opportunities for immunotherapeutic strategies to break tolerance to tumor antigens.
ONCOGENIC PATHWAYS ACTIVELY MEDIATE TUMOR–IMMUNE SYSTEM INTERACTIONS
The previous sections outlined the experimental evidence that the immune system is in general tolerant to tumors and their antigens under circumstances in which a tumor has established and is expanding within the host. There is much evidence that specific oncogenic pathways drive the “toleragenic immune microenvironment” of cancer.
The best studied oncogenic pathway to play a role in tumor immune evasion is the STAT3 pathway. STAT3 is one of two STATs (the other being STAT5A) to be constitutively activated in many diverse tumor types.35 Activation of STAT3 involves tyrosine phosphorylation resulting in homodimerization in the cytosol that leads to nuclear transport where it participates in transcriptional activation (and in some cases repression) of diverse genes. Although synthetic mutations in STAT3 can confer upon it oncogenic activity, constitutive activation of STAT3 in tumors is not a consequence of mutation. Instead, STAT3 is downstream of a number of important oncogenic tyrosine kinases. A number of receptor tyrosine kinases, including those associated with epidermal growth factor receptor (EGFR), HER2/Neu, and cMet, signal in part through STAT3. In addition, src and potentially other src family tyrosine kinases can activate STAT3.36 In fact, the original association of STAT3 with oncogenisis came from the demonstration that src-dependent transformation required STAT3.
Beyond its cell-intrinsic role in promoting tumor growth, STAT3 activation in tumors has been shown to repress the production of pro-inflammatory cytokines and chemokines that could enhance antitumor immune responses.37,38 These include pro-inflammatory cytokines such as type I interferons and tumor necrosis factor (TNF), as well as pro-inflammatory chemokines such as RANTES and IP-10. Thus, blockade of STAT3 signaling in tumor cells results in the release of multiple pro-inflammatory mediators and consequent infiltration with cells of both the innate and adaptive immune system that ultimately inhibit tumor growth. Beyond simply repressing the production and release of molecules that could promote anti-tumor immune responses, STAT3 signaling also induces the release of factors that inhibit activation of multiple immune cell types in the tumor microenvironment. These include dendritic cells, natural killer (NK) cells and granulocytes, which, though present in significant numbers within tumors, are generally found in an un-activated state. Some of the STAT3-regulated factors that induce this “quiescent microenvironment” include IL-10, VEGF, IL-6, and IL-23.37–40
As will be described below, some of these cytokines promote distinct forms of immune responses that promote rather than inhibit tumor growth. The receptors for each of these factors are expressed on cells of the hematopoietic system and themselves signal through STAT3. Thus, infiltrating hematopoietic cells within the tumor microenvironment are found to also express constitutively activated STAT3. Blockade of STAT3 in the hematopoietic system (for example via hematopoietic specific STAT3 knockout) results in dramatically enhanced activation of dendritic cells and cells in the innate immune system (such as NK cells and granulocytes) and leads to anti-tumor immune responses. In fact, even aggressive tumors fail to grow when transplanted into animals with hematopoietic STAT3 knockout.39 Thus, STAT3 appears to be an important global signaling pathway that restrains antitumor immunity. In part, STAT3 signaling skews T-cell responses to Th17 and away from Th1. Th17 responses are characterized by production of IL-17, while Th1 responses are characterized by production of interferon (IFN)-γ and promotion of cytotoxic T lymphocyte (CTL) responses. While Th1 responses generally produce anti-tumor immunity and are associated with better prognoses, Th17 responses are pro-carcinogenic and have been associated with worse prognosis when dominating in established tumors.41,42
The example of STAT3 described above demonstrates that oncogenic pathways in tumors play important roles in orchestrating the interaction between the tumor cell and its immune microenvironment such that immune responses induced by the invasion and metastasis process do not eliminate the tumor cell itself. While most of the focus on the function of oncogenic and tumor-suppressor pathways has been on cell autonomous functions within the tumor such as growth regulation, there is growing appreciation that these pathways additionally affect the immune cells of the tumor microenvironment.
IMMUNOLOGIC CHARACTERISTICS OF THE TUMOR MICROENVIRONMENT AND OPPORTUNITIES TO MANIPULATE THEM
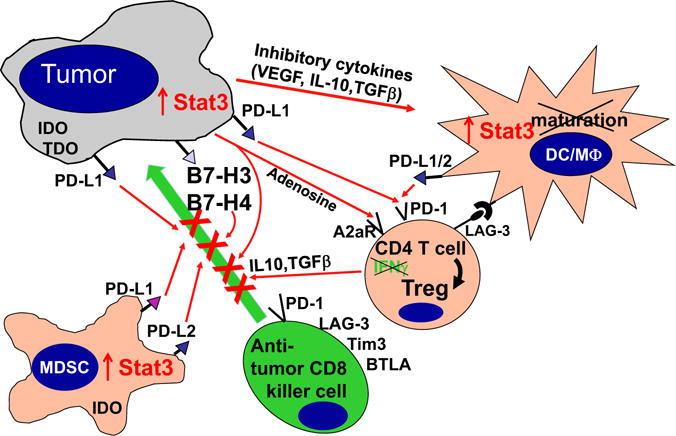
Ultimate understanding of the relationship between the tumor and the host immune system requires elucidation of local crosstalk at the level of the tumor microenvironment. As mentioned at the outset, the hematopoietic/immune system is a major component of the tumor microenvironment. The systemic tolerance to tumor antigens begins with events that occur in this microenvironment. Beyond mechanisms that skew tumor-specific T cells toward immune tolerance, the tumor microenvironment is replete with mechanisms that dampen antitumor immune responses locally. These inhibitory signals, commonly referred to as checkpoints, have emerged as leading targets for immunotherapy as will be covered in subsequent reviews in this issue of Seminars. They represent an important barrier to successful immunotherapy even when activated effector responses can be generated with vaccines (Figure 2). As the specific cells and molecules within the tumor microenvironment that mediate this hostile immune environment are elucidated, inhibitors are being developed and tested to use as adjuncts to vaccination that will allow activated immune cells to function more effectively within the tumor microenvironment.
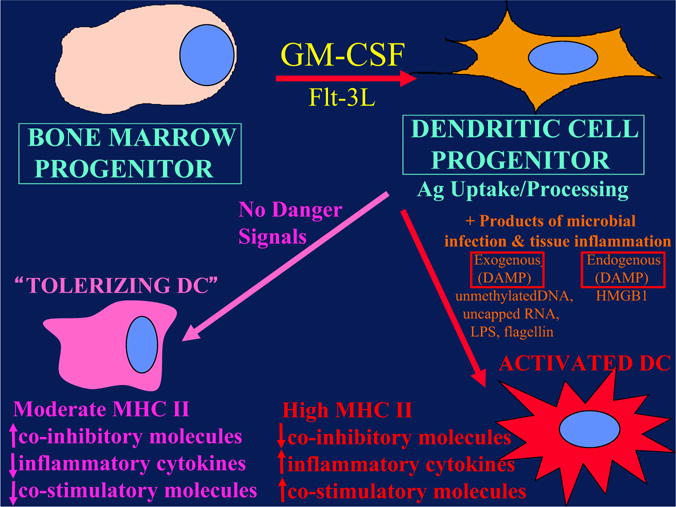
Figure 2.
Dendritic cells (DC) can either activate adaptive immunity or tolerize T cells depending on their state of maturation. DC progenitors develop from hematopoietic (bone marrow–derived) progenitors under the influence of various cytokines, particularly granulocyte-macrophage colony-stimulating factor (GM-CSF). Under circumstances of microbial infection, specific pathogen-associated molecular patterns (termed PAMPs) engage pattern recognition receptors (PAMPs), leading to release of pro-inflammatory danger signals that induce DC maturation. DC maturation leads to up-regulation of co-stimulatory molecules, MHC and chemokines that result in activation of T cells to effecter cells (right side of figure). In the absence of these “danger signals”, DCs follow a default pathway (left side of figure) in which they become “tolerizing DCs” that present antigen to T cells in the absence of co-stimulatory signals. This represents a steady-state pathway for continuous presentation of self-antigens. The consequence is that these T cells are turned off (anergy), inducing tolerance.
The previous section described how an oncogenic pathway in the tumor cell directly affected the immune microenvironment of the tumor. In addition to its role in inhibiting the activation and effector function of dendritic cells, granulocytes, and NK cells in the tumor microenvironment, STAT3 signaling has also been reported to play a role in guiding immature myeloid cells (iMC) in the tumor microenvironment to differentiate into myeloid suppressor cells (MSC) rather than dendritic cells with antigen-presenting cell (APC) activity. iMC43,44 and MSC45–48 represent a cadre of myeloid cell types, including tumor-associated macrophages (TAM), that share the common feature of inhibiting both the priming and effector function of tumor-reactive T cells. It is still not clear whether these myeloid cell types represent distinct lineages or different states of the same general immune inhibitory cell subset. In mice, iMC and MSC are characterized by co-expression of CD11b (considered a macrophage marker) and Gr1 (considered a granulocyte marker) while expressing low or no MHC class II or the CD86 co-stimulatory molecule. In humans, they are defined as CD33+ but lacking markers of mature macrophages, dendritic cells, or granulocytes and are HLA-DR-negative. A number of factors produced by tumors tend to drive iMC/MSC accumulation. These include IL-6, conlony-stimulating factor (CSF)-1, IL-10, and gangliosides. IL-6 and IL-10 are potent inducers of STAT3 signaling.
A number of mechanisms have been proposed to explain how iMC/MSC inhibit T-cell responses within the tumor microenvironment.49,50 Most include the production of reactive oxygen species (ROS) and or reactive nitrogen species (RNS). Nitric oxide (NO) production by iMC/MSC as a result of arginase activity, which is high in these cells, has been well documented and inhibition of this pathway with a number of drugs can mitigate the inhibitory effects of iMC/MSC. ROS, including H2O2, have been reported to block T-cell function associated with the downmodulation of the z chain of the TCR signaling complex,51 a phenomenon well recognized in T cells from cancer patients and associated with generalized T-cell unresponsiveness.
Another mediator of T-cell unresponsiveness associated with cancer is the production of indolamine-2,3 dioxygenase (IDO).52 IDO appears to be produced by dendritic cells either within tumors or in tumor-draining lymph nodes. Interestingly, IDO in dendritic cells has been reported to be induced via backward signaling by B7-1/2 upon ligation with CTLA-4.53,54 Apparently, the major IDO producing dendritic cell subset is either a plasmacytoid dendritic cell (PDC) or a PDC-related cell that is B220+.55 IDO appears to inhibit T-cell responses through catabolism of tryptophan. Activated T cells are highly dependent on tryptophan and are therefore sensitive to tryptophan depletion. Thus, Munn and Mellor have proposed a bystander mechanism, whereby dendritic cells in the local environment deplete tryptophan via IDO upregulation, thereby inducing metabolic apoptosis in locally activated T cells.
A major inhibitory cytokine produced by many cell types that has been implicated in blunting anti-tumor immune responses is TGF-β, which is produced by a variety of cell types, including tumor cells, Tregs, and iMC, and which has pleotropic physiological effects. From an immunological perspective, TGF-β possesses broadly immunosuppressive properties and TGF-β knockout mice develop widespread inflammatory pathology and corresponding accelerated mortality.56,57 Interestingly, a majority of these effects seem to be T-cell–mediated, as targeted disruption of T-cell TGF-β signaling also results a similar autoimmune phenotype.58 Experiments by Chen et al rather convincingly demonstrated a role for TGF-β in Treg-mediated suppression of CD8 T-cell anti-tumor responses.59 In these experiments, adoptive transfer of CD4+ CD25+ Tregs inhibited an anti-tumor CD8 T-cell effector response, and this inhibition was ameliorated when the CD8 T cells came from animals with a dominant negative TGF-β1 receptor that rendered them TGF-β–resistant.
PATTERN RECOGNITION RECEPTORS AND DENDRITIC CELL FUNCTION—IMPLICATIONS FOR TUMOR TOLERANCE AND ANTI-TUMOR IMMUNITY
An emerging concept is that immature or not fully matured dendritic cells are critical in presenting self-antigens to induce T-cell tolerance in the absence of danger signals associated with infection.60 The receptors in the immune system that generate these signals are termed pattern recognition receptors (PRRs). The best studied class of PRRs is the toll-like receptor (TLR) group that reside on the cell surface or in endosomal compartments. There are 10–12 TLRs, each of which recognizes specific pathogen-associated molecular patterns (PAMP) that are biochemical classes of molecules produced by bacteria, fungi, and viruses but not by eucaryotes.61 The best characterized PAMP is lipopolysaccharide (LPS), which is made by most Gramnegative bacteria and is recognized by the PRR, TLR4.62 Interestingly, tumor cell killing by certain chemotherapy agents induces a phenomenon called “immunogenic cell death” that mechanistically involves the release of a self-protein, HMGB1, which binds to and activates TLR4.63 Normally, HMBG1 is a nuclear protein in intact cells but is released into the extracellular milieu under certain types of cell death, normally associated with infection. These damage-associated molecular patterns (DAMPs) represent a putative adjunct mechanism to stimulate innate immunity (and dendritic cells) via the PRRs normally used to sense PAMPs. The importance of DAMPs relative to the better characterized PAMPs in activation of innate immunity and dendritic cells is not yet clear. More recently, a number of cytosolic PRRs have been identified that recognize RNA structures unique to viruses (RIG-I and MDA5).64 STING is a cytosolic PRR that recognizes double-stranded DNA from certain viruses and intracellular bacteria, as well as cyclic dinucleotides (CDN), produced by many bacteria.65,66 STING has been postulated to represent a potential sensor of tumor DNA.67 PRRs are expressed by many cells in the innate immune system and also by non-hematopoietic cells. Their engagement is a major inducer of inflammation, largely via induction of type 1 IFN production via IRF-3 activation and production of additional inflammatory cytokines regulated by nuclear factor (NF)-κB signaling. These same PRRs induce the maturation of dendritic cells into activated APCs with high levels of MHC and co-stimulatory molecules. Only activated dendritic cells express the necessary levels of co-stimulatory molecules, such as CD80, CD86, ICOSL, CD137, OX40, and T-cell stimulatory cytokines, such as IL-1, IFN-α/β, IL-12, and TNF-α, to appropriately activate CD4 T-cell to Th cells and CD8 cells to CTL function. In fact, a major mechanism for induction of T-cell tolerance is presentation of antigens by immature (unactivated) dendritic cells, whose PRRs have not been engaged (Figure 3). The majority of “older generation” cancer vaccines failed to include agonists for PRRs in their formulation and it is not surprising that they were not particularly effective in breaking established tolerance among tumor-specific T cells. More recent vaccine formulations are indeed incorporating natural and synthetic TLR agonists, as well as agonists for STING.68,69 Alternatively, recombinant viral and bacterial vaccines take advantage of the PAMPs that are naturally provided by the vector. These newer vaccines are certainly more potent in activating immune responses but their ability to break established tolerance to tumors remains to be determined.
Figure 3.
The hostile immune microenvironment of the tumor. Activation of oncogenic pathways and inactivation of tumor-suppressor pathways in the tumor lead to a cascade of molecular and cellular processes in the tumor microenviroment that block the killing function of innate immune effectors such as NK cells and granulocytes and block DC maturation. In addition, multiple cell membrane molecules such as IL-10, TGF-β, B7-H1, and B7-H4 are up-regulated. These molecules bind to receptors that inhibit T-cell effector function. Immature myeloid cells (iMC) produce nitric oxide (NO) that inhibits T cells and immature plasmacytoid DC (iPDC) produce indoleamine dioxygenase (IDO), which depletes tryptophan. Regulatory T cells also accumulate in the tumor microenvironment, further blunting anti-tumor T-cell responses.
Unquestionably, dendritic cells found within the tumor microenvironment, where there are no PAMPs, have a relatively immature, unactivated phenotype characterized by low levels of pro-inflammatory cytokine production, CD86, and surface MHC class II expression. These immature “activation-inhibited” dendritic cells clearly represent a prime candidate for the induction of tumor specific T cell tolerance. As such, recent interest has grown in the intra-tumoral injection of oncolytic viruses and other PRR agonists in an attempt to activate dendritic cells within the tumor so that tumor antigens are presented in a fashion that activates, rather than tolerizes, tumor-specific T cells.66 This maneuver, if successful, will represent a key approach to “auto-vaccinate” individuals against the unique non-shared mutation-dependent neoantigens expressed by their tumor. Likewise, the implications of the release of DAMPs in “immunogenic cell death” induced by certain chemotherapy agents and also radiation therapy is that endogenous tumors can potentially release their antigens upon chemotherapy or radiotherapy induced death in a fashion that can potentially prime the immune response, particularly when given together with a checkpoint inhibitor.
CO-INHIBITORY LIGANDS AND RECEPTORS THAT DOWN-MODULATE TUMOR IMMUNITY
Without question, the major molecules to be successfully targeted in clinical cancer immunotherapy are the growing class of ligand–receptor pairs, commonly referred to as immune checkpoints. In considering the mechanism(s) of action of inhibitors of various checkpoints, it is critical to appreciate the diversity of immune functions that they regulate. For example, the two immune checkpoint receptors that have been most actively studied in the context of clinical cancer immunotherapy, CTLA-4 (CD152) and PD-1 (CD279), regulate immune responses at very different levels and by very different mechanisms. The clinical activity of blocking antibodies for each of these receptors implies that anti-tumor immunity can be enhanced at multiple levels and that combinatorial strategies can be intelligently designed, guided by mechanistic considerations and preclinical models. This section will focus particular attention upon the CTLA-4 and PD-1 pathways since they were the two checkpoints whose inhibition has revolutionized clinical cancer immunotherapy. However, it is important to emphasize that multiple additional checkpoints represent promising targets for therapeutic blockade based on preclinical experiments and inhibitors of many of these are under active development.
The CTLA-4 Checkpoint—A Global Regulator of T-Cell Activation
CTLA-4, the first immune checkpoint receptor to be clinically targeted, is expressed exclusively on T cells where it primarily regulates the amplitude of the early stages of T-cell activation. CTLA-4 knockout mice die within 3 weeks from immune destruction of multiple organs, which attests to its critical role as an inhibitory regulator of T-cell–dependent immune responses. Primarily, CTLA-4 counteracts the activity of the T cell costimulatory receptor CD28.70 CD28 does not affect T-cell activation unless the TCR is first engaged by cognate antigen (Box 1). Once antigen recognition occurs, CD28 signaling strongly amplifies the TCR signal to activate T cells. CD28 and CTLA-4 share identical ligands: CD80 (B7.1) and CD86 (B7.2).71–74 Because CTLA-4 has a much higher overall affinity for both ligands, its expression on the surface of T cells dampens the activation of T cells by both out-competing CD28 in binding CD80 and CD86, as well as by actively delivering inhibitory signals to the T cell.75 The specific signaling pathways by which CTLA-4 blocks T-cell activation are still under investigation, although a number of studies suggest that CTLA-4 signaling disrupts kinase signals induced by TCR and CD28.76 However, CTLA-4 also confers “signaling-independent” T-cell inhibition through sequestration of CD80 and CD86 from CD28 engagement, as well as active removal of these molecules from the APC surface.77 The central role of CTLA-4 in maintaining T-cell activation in check is dramatically demonstrated by the systemic immune hyperactivation phenotype of CTLA-4 knockout mice.78,79
Even though CTLA-4 is expressed by activated CD8 killer T cells, the major physiologic role of CTLA-4 appears to be through distinct effects on the two major subsets of CD4 T cells—down-modulation of helper T-cell activity and enhancement of regulatory T-cell suppressive activity. CTLA-4 blockade results in a broad enhancement of immune responses dependent on helper T cells and conversely, CTLA-4 engagement on Treg enhances their suppressive function. CTLA-4 is a target gene of the transcription factor Foxp3,80 the expression of which determines the Treg lineage, and Tregs therefore express CTLA-4 constitutively. While the mechanism by which CTLA-4 enhances the inhibitory function of Tregs is not known, Treg-specific CTLA-4 knockout or blockade significantly inhibits their ability to regulate both autoimmunity and anti-tumor immunity.80 Thus, in considering the mechanism of action for CTLA-4 blockade, both enhancement of effector CD4 T cell activity and inhibition or elimination of Tregs by antibodies can be important factors. As will be discussed in subsequent reviews in this issue of Seminars, blockade of CTLA-4 with antibodies, first shown to have an anti-tumor effect in murine tumor models, was the first checkpoint blocking antibody to be approved for clinical use, based on the demonstration that, despite inducing a high degree of autoimmune side effects, it prolonged survival in patients with advanced melanoma.81
Biology of the PD-1 Checkpoint—A Pathway That Functions Within the Tumor Microenvironment
Another immune checkpoint receptor, PD-1, is emerging as a promising target, emphasizing the diversity of potential molecularly defined immune manipulations capable of inducing anti-tumor responses by the patient’s own immune system.
In contrast to CTLA-4, the major role of PD-1 is to limit the activity of T cells in the peripheral tissues at the time of an inflammatory response to infection and to limit autoimmunity. This translates to a major immune resistance mechanism within the tumor microenvironment.82,83 PD-1 expression is induced when T cells become activated. When engaged by one of its ligands, PD-1 inhibits kinases involved in T-cell activation via the phosphatase SHP2, although additional signaling pathways are also likely induced and because PD-1 engagement inhibits the TCR stop signal, this pathway could modify the duration of T cell/APC or T cell/target cell contact.84 Similar to CTLA-4, PD-1 is highly expressed on Tregs, where it may enhance their proliferation in the presence of ligand.85 Because many tumors are highly infiltrated with Tregs that likely further suppress effector responses, PD-1 pathway blockade may also enhance anti-tumor responses by diminishing the number and/or suppressive activity of intra-tumoral Tregs.
The two ligands for PD-1 are PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273).86–88 These B7 family members share 37% sequence homology and arose via gene duplication, positioning them within 100 kB of each other in the genome.88 Recently, an unexpected molecular interaction between PD-L1 and CD80 was discovered,89 whereby CD80 expressed on T cells (and possibly APC) can potentially behave as a receptor rather than a ligand, delivering inhibitory signals when engaged by PD-L1; the relevance of this interaction in tumor immune resistance has not yet been determined.
PD-1 is more broadly expressed than CTLA-4; it is induced on other activated non-T lymphocyte subsets, including B cells and NK cells, limiting their lytic activity. Thus, while PD-1 blockade is typically viewed as enhancing the activity of effector T cells in tissues and in the tumor microenvironment, it likely also enhances NK activity in tumors and tissues and may also enhance antibody production either indirectly or through direct affects on PD-1+ B cells.
In addition, chronic antigen exposure, such as occurs with chronic viral infection and cancer, can lead to high levels of persistent PD-1 expression, which induces a state of exhaustion or anergy among cognate antigen-specific T cells. This state, which has been demonstrated in multiple murine and human chronic viral infections, appears to be partially reversible by PD-1 pathway blockade.90 Finally, while the PD-1 pathway plays its major role in limiting immune effector responses in tissues (and tumors), it can also shift the balance from T-cell activation to tolerance at early stages in T-cell responses to antigen within secondary lymphoid tissues (ie, at a similar point as CTLA-4). Taken together, these findings imply a complex set of mechanisms of action for PD-1 pathway blockade.
PD-1 is expressed on a large proportion of tumor-infiltrating lymphocytes (TILs) from many different tumor types. Some of the enhanced PD-1 expression among CD4 TILs reflects a generally high level of PD-1 on Tregs, which, as noted above, can represent a large fraction of intra-tumoral CD4 T cells. Increased PD-1 expression on CD8 TILs may reflect an anergic/exhaused state, as has been suggested by decreased cytokine production by PD-1+ versus PD-1− TILs from melanomas.91
Just as PD-1 is highly expressed on TILs from many cancers, the PD-1 ligands are commonly up-regulated on many different human tumors.82,92 On solid tumors, the major PD-1 ligand to be expressed is PD-L1. Forced expression of PD-L1 on murine tumors inhibits local anti-tumor T cell responses. Indeed, this combination of findings provides the basis for PD-1 pathway blockade to enhance anti-tumor effector function in the tumor microenvironment. As immunohistochemistry techniques and flow cytometry analysis of surface expression has been employed, it has become clear that the selective up-regulation of PD-1 ligands in various human tumor types is heterogeneous at a number of levels.93 Expression patterns of PD-1 ligands may very well be critical in choosing suitability for therapeutic blockade of this pathway since its primary role in cancer is thought to be immune inhibition within the tumor microenvironment and PD-1 only inhibits lymphocyte function when it is engaged by cognate ligand.
Initially, the majority of melanoma, ovarian and lung cancer samples were reported to have high expression of PD-L1 and subsequently, many other human cancers were reported to up-regulate PD-L1. In addition to tumor cells, PD-L1 is commonly expressed on myeloid cells in the tumor microenvironment. An initial report in renal cancer demonstrated that expression of PD-L1 on either tumor cells or infiltrating leukocytes in primary tumors predicted a worse prognosis, ie, decreased overall survival relative to PD-L1− tumors.94 In other tumor types, such as melanoma, PD-L1 status correlates with better prognosis.83 Variability in IHC technique, cancer type, stage of cancer analyzed (most analyses are of primary, not metastatic lesions) and treatment history in the analyzed cohort all likely contribute to the wide range of reported outcomes.
While most of the analyses of PD-1 ligand expression has focused on PD-L1, PD-L2 has also been reported to be upregulated on a number of tumors. It is highly upregulated on certain B-cell lymphomas such as primary mediastinal, follicular cell B-cell lymphoma and Hodgkin disease. Up-regulation in these lymphomas is commonly associated with gene amplification or rearrangement to the CIITA locus, which is highly transcriptionally active in B-cell lymphomas.95
Given the heterogeneity of expression and potential relevance as a biomarker for blockade of the PD-1 pathway, it is important to understand the signals that induce expression of PD-1 ligands on tumor cells and also hematopoietic cells within the tumor microenvironment. Two general mechanisms for regulation of PD-L1 have emerged: innate and adaptive. For some tumors such as glioblastoma, it has been demonstrated that PD-L1 is driven by constitutive oncogenic signaling pathways in the tumor cell. Expression on glioblastomas is enhanced upon deletion or silencing of PTEN, implicating the PI3K-AKT pathway.96 Similarly, constitutive ALK signaling, observed in certain lymphomas and occasionally in lung cancer, has been reported to drive PD-L1 expression via STAT3 signaling.97
The alternative mechanism for PD-L1 up-regulation on tumors that has emerged from both clinical and preclinical studies reflects their adaptation to endogenous tumor-specific immune responses, a process termed adaptive resistance.76 In adaptive resistance, the tumor utilizes the natural physiology of the PD-1 ligand induction for tissue protection in the face of an immune response to infection in order to protect itself from an anti-tumor response. Expression of PD-L1 as an adaptive response to endogenous anti-tumor immunity can occur because it is induced on most cancers in response to interferons—pre-dominantly γ-IFN, similarly to epithelial and stromal cells in normal tissues. This mechanism represents an alternative to the conventional drug resistance mechanisms that involve mutation of drug targets. It also contrasts with mechanisms of viral immune escape that involve mutation of immunodominant epitopes. The mechanism of adaptive resistance intrinsically implies that immune surveillance does exist even in some advanced cancers but the tumor ultimately resists immune elimination by up-regulating ligands for inhibitory receptors on tumor-specific lymphocytes that turn off anti-tumor responses within the tumor microenvironment.
A number of preclinical and clinical studies support the adaptive resistance hypothesis. Gajewski and colleagues have demonstrated that melanomas can be roughly divided into “inflammatory” and “non-inflammatory” catagories defined by expression of multiple inflammatory genes, including those involved in the interferon pathway.98 A recent study in melanoma demonstrated a very high correlation between cell surface PD-L1 expression on tumor cells and both lymphocytic infiltration and intra-tumoral ?-IFN expression. This correlation was not only seen among tumors but within individual PD-L1+ tumors at the regional level, in which regions of lymphocyte infiltration were exactly the regions where PD-L1 was expressed on both tumor cells and infiltrating leukocytes.83
As will be discussed in other reviews in this issue of Seminars, multiple clinical antibodies to PD-1 and PD-L1, all of which block their interaction, have shown impressive durable remissions originally in patients with either melanoma, renal cancer, or lung cancer99–101 and subsequently in those with other histologies, including head and neck cancer, gastro-esophageal cancer, ovarian cancer, bladder cancer, and Hodgkin lymphoma.
ADDITIONAL CHECKPOINTS PARTICIPATE IN TUMOR IMMUNE RESISTANCE AND TOLERANCE
Successful clinical outcomes of CTLA-4 and PD-1 pathway targeting have garnered great interest in a number of additional immune checkpoints. Basic immunologic studies have demonstrated that a number of checkpoint receptors are expressed coordinately under circumstances of tolerance to self-antigens and chronic infections, as well as in inflammatory settings. In addition to defined lymphocyte inhibitory receptors, a number of B7-family inhibitory ligands—in particular B7-H3 (CD276) and B7-H4—do not yet have defined receptors but murine knockout experiments support an inhibitory role for both of these molecules.102 In addition, they are up-regulated on tumor cells or tumor-infiltrating cells.103 B7-H3 appears to be up-regulated on endothelial cells of the tumor vasculature and B7-H4 has been reported to be expressed on tumor-associated macrophages.104 Preclinical tumor models have been used to demonstrate that blockade of many of these individual immune checkpoint ligands or receptors can enhance anti-tumor immunity and dual blockade of coordinately expressed receptors can produce additive or synergistic anti-tumor activity. Inhibitors for a number of these immune checkpoint targets are either entering the clinic or are under active development. Those described below are targets with currently available blocking antibodies or small molecule inhibitors but do not represent a comprehensive list.
LAG-3 (CD223), 2B4 (CD244), BTLA (CD272), Tim-3, A2aR, and the family of killer inhibitory receptors have each been associated with inhibition of lymphocyte activity and in some cases induction of lymphocyte anergy. Antibody targeting of these receptors, either alone or in combination with a second immune checkpoint blocker has been shown to enhance anti-tumor immunity in animal models of cancer. Because many tumors express multiple inhibitory ligands and TIL express multiple inhibitory receptors, there are many opportunities to enhance anti-tumor immunity via dual or triple blockade of immune checkpoints. While human blocking antibodies specific for a number of these “second-generation” inhibitory receptors are under development, none have entered the clinic at this time. Most of these receptors are induced upon T-cell activation, in keeping with the biologic theme that they play roles in feedback inhibition of T-cell responses when their cognate ligands are present. In addition to providing inhibitory signals to activated effector T cells, some of these receptors such as LAG-3 are highly expressed on Tregs, where they are important to amplify their inhibitory activity.104 This implies that as with CTLA-4 and PD-1, these receptors play a dual role in ultimately inhibiting effector immune responses and enhancing Treg suppressive function. Blocking antibodies therefore have multiple potential mechanisms of action.
LAG-3 was cloned over 20 years ago as a CD4 homologue but its function in the immune checkpoint was only defined in 2005 when it was shown to play a role in enhancing Treg function.104 LAG-3 also inhibits CD8 effector function independently of its role on Tregs. The only known ligand for LAG-3 is MHCII, which is up-regulated on some epithelial cancers (generally in response to IFN-??) but is also expressed on tumor infiltrating macrophages and dendritic cells. The role of the LAG-3/MHCII interaction in LAG-3–mediated inhibition of T-cell responses is unclear since anti–LAG-3 antibodies that do not block the LAG-3/MHCII interaction nonetheless enhance T-cell proliferation and effector function in vitro and in vivo.104 The MHCII interaction of LAG-3 may be most important for its role in enhancing Treg function. LAG-3 is one of a number of immune checkpoint receptors coordinately up-regulated on both Tregs and anergic T cells and simultaneous blockade can result in enhanced reversal of this anergic state relative to blockade of either receptor alone. In particular, PD-1 and LAG-3 are commonly co-expressed on anergic or exhausted T cells.105,106 Dual blockade of LAG-3 and PD-1 provide synergy in reversing anergy among tumor specific CD8 T cells as well as virus specific CD8 T cells in the setting of chronic infection. Dramatic evidence of the affects of coordinate T-cell inhibition by PD-1 and LAG-3 comes from PD-1/LAG-3 double knockout mice, which completely reject even poorly immunogenic tumors in a T-cell–dependent fashion but also develop autoimmune syndromes much more quickly than PD-1 or LAG-3 single knockouts that are ultimately fatal (though not as quickly as CTLA-4 knockouts).107 These findings emphasize the balance between anti-tumor effects and autoimmune side effects that must be taken into consideration in all of the immune checkpoint blockade strategies.
Tim-3, the ligand of which is galectin-9 (a galectin reported to be up-regulated in a number of cancer types such as breast cancer), inhibits Th1 responses108 and anti–Tim-3 antibodies enhance anti-tumor immunity.109 Tim3 has also been reported to be co-expressed with PD-1 on tumor specific CD8 T cells and dual blockade of both molecules significantly enhances the in vitro proliferation and cytokine production of human T cells when stimulated by the cancer-testes antigen NY-ESO-1.110 In animal models, coordinate blockade of PD-1 and Tim3 was reported to enhance antitumor responses and tumor rejection under circumstances where only modest effects from blockade of each individual molecule were observed.111
The A2a receptor for adenosine inhibits T-cell responses, in part by driving CD4 T cells to express Foxp3 and develop into Tregs.112 Knockout of this receptor results in enhanced and sometimes pathologic inflammatory responses to infection. This receptor is particularly relevant in tumor immunity because the rate of cell death in tumors from cell turnover is high and dying cells release adenosine. In addition, Tregs express high levels of the exoenzymes CD39, which converts extracellular ATP to AMP, and CD73, which converts AMP to adenosine.113 Given that A2a receptor engagement by adenosine drives T cells to become Tregs, this can produce a self-amplifying loop within the tumor. Indeed, tumors grow more slowly in A2aR knockout mice and tumor vaccines are much more effective against established tumors in these mice.114 A2aR can be inhibited either by antibodies that block adenosine binding or by adenosine analogues, some of which are fairly specific for A2aR. While these drugs have been used in clinical trials for Parkinson’s disease, they have not yet been tested clinically in cancer patients.
CLINICAL IMPLICATIONS FOR MANIPULATION OF THE IMMUNE RESPONSE TO TUMOR CELLS
Fundamentally, we now have clear-cut evidence that antibodies and T cells can selectively recognize and kill cancer cells in patients. Cancer genetics, epigenetics, and genomics have provided us with a far better understanding of the nature and specificity or selectivity of tumor antigens, providing new opportunities for targeted antigen specific immunotherapy. We know much more about the details of antigen recognition by T cells allowing for the opportunity to modify antigens at critical residues to provide for enhanced immune stimulatory capacity. Finally, we are learning much about the ligands, receptors, and signaling pathways that regulate immune responses and how they are expressed within the tumor microenvironment. Elucidation of these regulatory pathways has demonstrated that the outcome of antigen recognition is in large part determined by the balance between co-stimulatory signals and inhibitory signals. These relatively recent insights into the molecular basis of immune regulation are demonstrating profound significance for the development of more potent combinatorial immunotherapy approaches to cancer.
Footnotes
Conflicts of interest: none declared.
References
- 1.Baldwin RW. Immunity to methylcholanthrene-induced tumours in inbred rats following atrophy and regression of the implanted tumours. Br J Cancer. 1955;9:652–7. doi: 10.1038/bjc.1955.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prehn RT, Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957;18:769–78. [PubMed] [Google Scholar]
- 3.Ciardiello F, Bianco R, Damiano V, et al. Antitumor activity of sequential treatment with topotecan and anti-epidermal growth factor receptor monoclonal antibody C225. Clin Cancer Res. 1999;5:909–16. [PubMed] [Google Scholar]
- 4.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 6.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 7.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kvistborg P, Philips D, Kelderman S, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6:254. doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 9.Abrams SI, Khleif SN, Bergmann-Leitner ES, et al. Generation of stable CD4+ and CD8+ T cell lines from patients immunized with ras oncogene-derived peptides reflecting codon 12 mutations. Cell Immunol. 1997;182:137–51. doi: 10.1006/cimm.1997.1224. [DOI] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Der Bruggen P, Zhang Y, Chaux P, et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64. doi: 10.1034/j.1600-065x.2002.18806.x. [DOI] [PubMed] [Google Scholar]
- 12.Kawakami Y, Robbins PF, Wang RF, et al. The use of melanosomal proteins in the immunotherapy of melanoma. J Immunother. 1998;21:237–46. doi: 10.1097/00002371-199807000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 14.Rygaard J, Povlsen CO. The nude mouse vs. the hypothesis of immunological surveillance. Transplant Rev. 1976;28:43–61. doi: 10.1111/j.1600-065x.1976.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 15.Gaidano G, Dalla-Favera R. Biologic aspects of human immunodeficiency virus-related lymphoma. Curr Opin Oncol. 1992;4:900–6. doi: 10.1097/00001622-199210000-00013. [DOI] [PubMed] [Google Scholar]
- 16.Mesri EA, Cesarman E, Arvanitakis L, et al. Human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J Exp Med. 1996;183:2385–90. doi: 10.1084/jem.183.5.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boshart M, Gissmann L, Ikenberg H, et al. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984;3:1151–7. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Euvrard S, Kanitakis J, Pouteil-Noble C, et al. Skin cancers in organ transplant recipients. Ann Transplant. 1997;2:28–32. [PubMed] [Google Scholar]
- 19.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 20.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 21.Staveley-O’Carroll K, Sotomayor E, Montgomery J, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95:1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Getnet D, Maris CH, Hipkiss EL, et al. Tumor recognition and self-recognition induce distinct transcriptional profiles in antigen-specific CD4 T cells. J Immunol. 2009;182:4675–85. doi: 10.4049/jimmunol.0803400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 24.Bogen B. Peripheral T cell tolerance as a tumor escape mechanism: deletion of CD4+ T cells specific for a monoclonal immunoglobulin idiotype secreted by a plasmacytoma. Eur J Immunol. 1996;26:2671–9. doi: 10.1002/eji.1830261119. [DOI] [PubMed] [Google Scholar]
- 25.Staveley-O’Carroll K, Sotomayor E, Montgomery J, et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95:1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drake CG, Doody AD, Mihalyo MA, et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell. 2005;7:239–49. doi: 10.1016/j.ccr.2005.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437(7055):141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen LT, Elford AR, Murakami K, et al. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J Exp Med. 2002;195:423–35. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox AL, Skipper J, Chen Y, et al. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264(5159):716–9. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 30.Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 31.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 32.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–61. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 33.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–32. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ercolini AM, Ladle BH, Manning EA, et al. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu CL, Meyer DJ, Campbell GS, Larner AC, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269(5220):81–3. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 37.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 38.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10(1):48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 39.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 40.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15(2):114–23. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tosolini M, Kirilovsky A, Mlecnik B, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71(4):1263–71. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- 43.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young MR, Petruzzelli GJ, Kolesiak K, et al. Human squamous cell carcinomas of the head and neck chemoattract immune suppressive CD34(+) progenitor cells. Hum Immunol. 2001;62:332–41. doi: 10.1016/s0198-8859(01)00222-1. [DOI] [PubMed] [Google Scholar]
- 45.Bronte V, Apolloni E, Cabrelle A, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–46. [PMC free article] [PubMed] [Google Scholar]
- 46.Bronte V, Serafini P, De Santo C, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–8. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 47.Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 48.Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 49.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;4:348–53. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 50.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60. [PubMed] [Google Scholar]
- 52.Munn DH, Sharma MD, Lee JR, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 53.Baban B, Hansen AM, Chandler PR, et al. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–19. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 54.Mellor AL, Chandler P, Baban B, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 55.Munn DH, Sharma MD, Hou D, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–90. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 57.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 58.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–81. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 59.Chen ML, Pittet MJ, Gorelik L, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102:419–24. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227(1):221–33. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 61.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 62.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;82(5396):2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 63.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 64.Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. 2014;5:342. doi: 10.3389/fimmu.2014.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328(5986):1703–5. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14(1):19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 67.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–88. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 68.Fu J, Malm IJ, Kadayakkara DK, Levitsky H, et al. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74:4042–52. doi: 10.1158/0008-5472.CAN-13-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 71.Azuma M, Ito D, Yagita H, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76–9. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 72.Freeman GJ, Gribben JG, Boussiotis VA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993;262:909–11. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 73.Hathcock KS, Laszlo G, Dickler HB, et al. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. 1993;262:905–7. doi: 10.1126/science.7694361. [DOI] [PubMed] [Google Scholar]
- 74.Linsley PS, Brady W, Urnes M, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–9. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–5. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 76.Schneider H, Smith X, Liu H, Bismuth G, Rudd CE. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur J Immunol. 2008;38:40–7. doi: 10.1002/eji.200737423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qureshi OS, Zheng Y, Nakamura K, et al. Transendocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–3. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 79.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 80.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 81.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 83.Taube JM, A R, Xu H, Sharma R, et al. B7-H1 expression co-localizes with inflammatory infiltrates in benign and malignant melanocytic lesions: Implications for immunotherapy. Sci Translat Med. 28(4):127ra37. [Google Scholar]
- 84.Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–92. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong H, Zhu G, Tamada K, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 87.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 88.Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–46. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 91.Gros A, Robbins PF, Yao X, et al. PD-1 identifies the patient-specific CD8⁺ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–59. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Topalian Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thompson RH, Gillett MD, Cheville JC, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101:17174–9. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–81. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 97.Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1) Proc Natl Acad Sci U S A. 2008;105:20852–7. doi: 10.1073/pnas.0810958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. 2010;16:399–403. doi: 10.1097/PPO.0b013e3181eacbd8. [DOI] [PubMed] [Google Scholar]
- 99.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yi KH, Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev. 2009;229:145–51. doi: 10.1111/j.1600-065X.2009.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.He C, Qiao H, Jiang H, et al. The inhibitory role of b7-h4 in antitumor immunity: association with cancer progression and survival. Clin Dev Immunol. 2011:695834. doi: 10.1155/2011/695834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grosso JF, Kelleher CC, Harris TJ, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117:3383–92. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grosso JF, Goldberg MV, Getnet D, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182:6659–69. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 109.Ngiow SF, von Scheidt B, Akiba H, et al. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated anti-tumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 110.Fourcade J, Sun Z, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8⁺ T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045–55. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sakuishi K, Apetoh L, Sullivan JM, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zarek PE, Huang CT, Lutz ER, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–9. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Waickman AT, Alme A, Senaldi L, et al. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. 2012;61:917–26. doi: 10.1007/s00262-011-1155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]