

Abstract
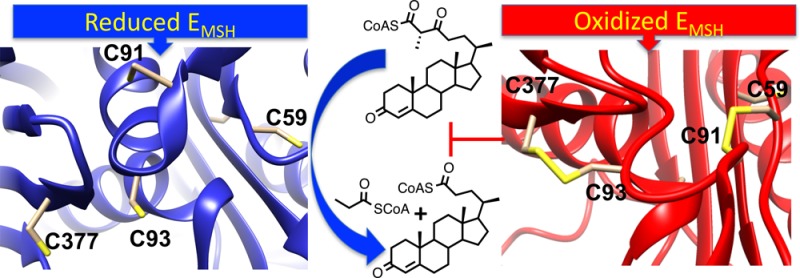
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is a highly successful human pathogen and has infected approximately one-third of the world’s population. Multiple drug resistant (MDR) and extensively drug resistant (XDR) TB strains and coinfection with HIV have increased the challenges of successfully treating this disease pandemic. The metabolism of host cholesterol by Mtb is an important factor for both its virulence and pathogenesis. In Mtb, the cholesterol side chain is degraded through multiple cycles of β-oxidation and FadA5 (Rv3546) catalyzes side chain thiolysis in the first two cycles. Moreover, FadA5 is important during the chronic stage of infection in a mouse model of Mtb infection. Here, we report the redox control of FadA5 catalytic activity that results from reversible disulfide bond formation between Cys59-Cys91 and Cys93-Cys377. Cys93 is the thiolytic nucleophile, and Cys377 is the general acid catalyst for cleavage of the β-keto-acyl-CoA substrate. The disulfide bond formed between the two catalytic residues Cys93 and Cys377 blocks catalysis. The formation of the disulfide bonds is accompanied by a large domain swap at the FadA5 dimer interface that serves to bring Cys93 and Cys377 in close proximity for disulfide bond formation. The catalytic activity of FadA5 has a midpoint potential of −220 mV, which is close to the Mtb mycothiol potential in the activated macrophage. The redox profile of FadA5 suggests that FadA5 is fully active when Mtb resides in the unactivated macrophage to maximize flux into cholesterol catabolism. Upon activation of the macrophage, the oxidative shift in the mycothiol potential will decrease the thiolytic activity by 50%. Thus, the FadA5 midpoint potential is poised to rapidly restrict cholesterol side chain degradation in response to oxidative stress from the host macrophage environment.
Keywords: regulation, disulfide, metabolism, structure, mycothiol, domain swap
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is a highly successful human pathogen and has infected approximately one-third of the world’s population.1 TB infection in most industrialized countries was once controlled by highly successful antibiotics. However, multiple drug resistant (MDR) and extensively drug resistant (XDR) TB strains have emerged as a pandemic, and in combination with HIV coinfection, the consequences for global health are severe.2 Despite the worldwide efforts to discover new anti-TB therapeutics, only one new compound, bedaquiline,3 has been added to the anti-TB arsenal since 1970. Therefore, new drugs targeting novel pathways are urgently needed to combat TB, one of the most neglected diseases in the 21st century.
The Mtb cholesterol metabolism pathway presents opportunities for new drug discovery.4,5 An 82-gene cluster in Mtb encodes the majority of the cholesterol catabolism enzymatic pathway.5 Mutant Mtb strains lacking the ability to uptake and metabolize cholesterol fail to establish an effective infection in mouse models.6−8 Most cholesterol catabolic genes are up-regulated during infection of human macrophages by Mtb.9 Recently, a phenotypic macrophage screen carried out by VanderVen and co-workers10 identified a large set of inhibitors targeting cholesterol metabolism in Mtb, emphasizing the importance of the cholesterol metabolic pathway as a potential drug target.
FadA5 is encoded in the 82-gene cholesterol catabolism cluster and catalyzes the thiolytic cleavage of β-keto-acyl-CoA thioesters.4,11 Structural and biochemical characterization established that FadA5 is a degradative thiolase, and we identified the thiolytic cleavage of cholesterol β-keto-acyl-CoA side chain intermediates as the physiological function of this enzyme.11
Sequence and structural alignments of FadA5 with other thiolases reveal that the catalytic residues12 are conserved in FadA5 (Figure 1). Specifically, Cys93 is the nucleophile that attacks the β-keto carbonyl moiety of the steroid acyl-CoA; Cys377 and His347 function as general acid/base residues. In addition to the catalytic cysteines in FadA5, there is a third cysteine in the active site, Cys91. Cys91 is located near the nucleophilic Cys93, but the thiol of Cys91 is oriented away from Cys93. Interestingly, Cys91 and Cys59 are conserved only among FadA5 orthologs and are not present in other thiolases (Figure 1B). The orthologs are found primarily in the actinomycetales genuses of mycobacteria, rhodococci, gordonia, and pseudonocardia.
Figure 1.

Cysteine conservation in thiolases. (A) Catalytic mechanism of FadA5 thiolase. (B) Sequence alignments of thiolases from different species. The conserved catalytic residues (Cys93, His347, and Cys377) are highlighted in yellow and labeled on the basis of the amino acid numbering in FadA5. Cys 59 and Cys 91 are only present in FadA5 orthologues and are highlighted in cyan. Disulfide bonds are present in Arabidopsis thaliana (blue), Clostridium acetobutylicum (green), and Mycobacterium tuberculosis (red). (Sequence alignments were obtained using Clustal Omega. Next to the protein sequences, PDB codes, if structures are available, or NCBI Reference Sequence Project accession numbers were added.)
A peroxisomal thiolase from Arabidopsis thaliana forms a disulfide bond between the nucleophile Cys138 and Cys192, a noncatalytic cysteine (Figure 1B).13 The formation of the disulfide bond necessarily eliminates the catalytic activity of the thiolase. The reduction midpoint of the Cys138-Cys192 disulfide bond is −283 mV, suggesting tight control of the thiolase activity by the redox environment in the plant peroxisome.14 Similarly, the biosynthetic Clostridium acetobutylicum thiolase forms a reversible disulfide bond between its two catalytic cysteine residues, the nucleophile Cys88 and the general base/acid Cys378 (Figure 1B).15 The disulfide bond has a reduction potential of −270 mV and acts as a redox switch to control the production of butanol versus acetic acid in Clostridium acetobutylicum as a function of the cellular metabolic state.15 These studies suggest disulfide bond formation in thiolases as a physiologic control mechanism. However, different structural motifs are utilized depending on the physiological context.
During infection, Mtb experiences a wide range of host-generated stresses accompanied by changes in intracellular redox potential.16 The precedent of redox control through disulfides in plants and bacteria inspired us to explore whether FadA5 disulfide bond formation can act as a switch to control the cholesterol degrading capacity. Here, we report the redox control of FadA5 catalytic activity that results from reversible disulfide bond formation between Cys59-Cys91 and Cys93-Cys377. The midpoint potential of FadA5 is the same as the intracellular mycothiol potential of Mtb in the activated macrophage. This equivalence suggests disulfide control as a motif for rapid Mtb response to environmental challenges from the host macrophage to control propionyl-CoA production.
Results and Discussion
The Thiolytic Activity of WT FadA5 Is Sensitive to Oxidation
We tested whether FadA5 catalytic activity is modulated by disulfide formation. The catalysis of thiolysis by FadA5 was monitored by MALDI-TOF analysis.11 We found that incubation of WT FadA5 in oxidizing buffer (10 mM cystamine, CSSC, 25 °C) for 10 min abolished the catalytic activity of FadA5 (Figure S1). Ultrafiltration and resuspension of CSSC-treated WT FadA5 in reducing buffer (20 mM cysteamine, CSH, 25 °C) for 10 min fully restored the thiolytic activity as determined by specific activity measurements and product analysis (Figure S1). Thus, we conclude that the catalytic activity of WT FadA5 is sensitive to oxidizing conditions and that oxidation is reversible.
We performed the same oxidation–reduction experiment with the C91A mutant of FadA5. Oxidation did not abolish the catalytic activity of the C91A mutant (Figure S1). Mutation of Cys91 to Ala thus prevents the oxidation of the essential catalytic site residues C93 and C377. The C93A and C377A mutants are catalytically inactive, and therefore, analogous oxidation–reduction experiments could not be performed with these mutants.
Protein Secondary Structure and Stability
Upon observing that FadA5s catalytic activity is sensitive to the oxidizing potential of the assay buffer, we analyzed whether FadA5 undergoes significant conformational changes.
The CD spectra of the wild-type protein as well as the spectra of the C93A and the C91A mutants were normalized to the same concentration based on molar absorptivity. The spectra clearly show that the WT and mutant proteins do not differ in their overall secondary structure elements (Figure 2). Furthermore, a comparison of the CD spectra of the wild-type protein in the fully oxidized and reduced states shows that there are no significant changes in secondary structure upon oxidation (Figure 2). Regardless of their oxidation state, the WT, C91A, and C93A proteins display the same circular dichroism minima17,18 at 208 and 222 nm (Figure 2).
Figure 2.

WT and mutant proteins do not differ significantly in their overall secondary fold whether oxidized or reduced. Oxidation or reduction of WT, C91A, or C93A FadA5 does not change the overall secondary structure. (A) Superposition of the buffer corrected and normalized CD spectra for the following oxidized samples: WT (green), C93A (orange), and C91A (blue). The native WT spectrum is shown for comparison (black). (B) Superposition of the buffer corrected and normalized CD spectra for the following reduced samples: WT (green), C93A (orange), and C91A (blue). The native WT spectrum is shown for comparison (black).
The melting temperature of each protein was assessed under both reducing and oxidizing conditions using a thermofluor assay. Two different buffer types were used to ensure that the protein’s stability was not influenced by the respective buffer systems used for crystallization and CD measurements. The melting temperature of the WT protein and the two mutants differed. Whereas, the WT protein displays the highest melting temperature (Tm) at ∼65 °C regardless of the buffer system (20 mM bicine–NaOH, pH 8.5, 250 mM NaCl, or 10 mM potassium/sodium phosphate, pH 7.5), the C91A mutant has a slightly lower melting temperature compared to the WT protein with a Tm of 63 °C. In contrast, the C93A mutant has a melting temperature which is reduced by almost 10 °C with a Tm of 55 °C. Interestingly, the addition of reducing or oxidizing agents such as cystamine, cysteamine, hydrogen peroxide (data not shown), or potassium hexacyanoferrate (II) (data not shown) did not further influence the melting temperature of the WT protein or the mutants.
The Formation of Two Disulfide Bonds Is the Cause of FadA5 Oxidative Inactivation
We obtained crystals of apo FadA5 in the hexagonal space group, P6322, with cell dimensions of a = b = 120.3 Å and c = 206.0 Å at 2.2 Å resolution (Table 1). Previously, we described the structure of FadA5 in its apo form which crystallized in the tetragonal P41212 space group (PDB entry 4UBW) as well as in the presence of different ligands (PDB entries 4UBV, 4UBU, and 4UBT).11 The published apo FadA5 structure is referred to as apo I and the new apo structure reported here will be referred to as apo II in the following discussion.
Table 1.
| FadA5 WT apo structure II (oxidized) | FadA5 WT apo II, anomalous data | |
|---|---|---|
| Relevant Buffers | ||
| protein buffer | 60 mM bicine–NaOH, pH 8.5, 250 mM NaCl | |
| crystallization condition | 0.1 M citrate, pH 5.5, 0.7 M (NH4)2HPO4, 0.3 M NaCl | |
| cryoprotectant solution | 0.1 M citrate, pH 5.5, 1.0 M (NH4)2HPO4, 0.2 M NaCl, 25% glycerol | |
| Data Collection | ||
| wavelength (Å) | 0.8726 | 1.8000 |
| temperature (K) | 100 | 100 |
| space group | P6322 | P6322 |
| unit cell parameters | ||
| a/b/c (Å) | 120.3/120.3/206.0 | 119.8/119.8/205.6 |
| α/β/γ | 90°/90°/120° | 90°/90°/120° |
| resolution (Å) | 46.17–2.2 | 46.30–2.65 |
| number of FadA5 molecules | 2 | 2 |
| total reflections | 451 732 (37 977) | 626 808 (5990) |
| unique reflections | 46 018 (3897) | 21 619 (1016) |
| completeness (%) | 100.0 (100.0) | 83.0 (31) |
| redundancy | 9.8 (9.7) | 29 (5.9) |
| ⟨I/σ(I)⟩ | 15.2 (2.6) | 32.2 (1.2) |
| Rmergeb (%) | 10.0 (90.9) | 8.40 (100.1) |
| Wilson B factor (Å2)c | 39.0 | 54.3 |
| Refinement | ||
| total number of atoms | 5606 | |
| Rwork/Rfree (%) | 18.4/22.5 | |
| RMSD | ||
| bond angle (deg) | 1.6 | |
| bond length (Å) | 0.015 | |
| average B factor (Å2)d | 72.9 | |
| chain A | 49.1 | |
| chain B | 102.2 | |
| most favored (%) | 98.0 | |
| allowed (%) | 2.0 | |
| disallowed (%) | 0.0 | |
| PDB code | 5ONC | |
Apo I FadA5 (PDB entry 4UBW) assumes a typical thiolase fold13 with three subdomains. The N- (I) and the C-terminal (III) subdomains are very similar as they both comprise a central β-sheet, consisting of β-strands β1, β3, β4, β5, and β8 in the N-terminal subdomain and β9, β10, β11, and β12 in the C-terminal subdomain. The β-sheet is surrounded by α helices α1, α2, and α3 in subdomain I and α8, α9, and α11 in subdomain III. In contrast to these two subdomains, the third subdomain, II or lid subdomain, displays a more extended structure with two shorter β-strands, β6 and β7, and three α-helices, α4, α5, and α6. It is located on top of subdomains I and III and occludes the catalytic triad, C93, H347, and C377, from the solvent. This overall fold was observed for all previously analyzed FadA5 structures with or without ligand bound in the catalytic site.
The apo II structure (PDB code 5ONC) contains two FadA5 molecules per asymmetric unit with 366 residues in chain A and 359 residues in chain B. Like the apo I structure, a large disordered region, comprising residues V126 to H147 (chain A) and S124 to P149 (chain B) is not resolved. The data were refined to Rwork and Rfree values of 18.4% and 22.5%, respectively. Validation of the apo II structure proved to be difficult as significant problems in geometry and clashing were observed. The overall electron density of monomer B was not as well-defined compared to monomer A, so that many side chains lack electron density and the monomer has a total of three gaps in the polypeptide chain. This undefined and partially noncontinuous electron density probably reflects enzyme regions with higher atomic disorder, which is also indicated by extraordinarily high atomic B factors, especially for chain B (Table 1). The differences in electron density between molecule A and B are also reflected by the differences in crystal contacts. Whereas the crystal contacts of molecule A amount to a surface area of 1388 Å2, the contacts for molecule B only amount to 991 Å2, indicating less ordered packing for molecule B supporting higher flexibility.
A comparison of the two FadA5 apo structures with each other by a superposition of monomers A of each dimer clearly shows that the individual subunits are quite similar; however, the position of the B monomer relative to the A monomer is shifted significantly between these structures (Figure 3A,B). This shifted orientation of the two monomers relative to each other is especially evident in a comparison of the respective B chains (Figure 3B). The apo II structure employs a slightly altered order of the secondary structure motifs compared to the apo I structure (Figure 3C,D). A smaller β-strand is not formed; some 310 helices are missing, as for example the catalytic C93 harboring η3, and helix α3 is shortened by four amino acids.
Figure 3.

Comparison of the FadA5 apo structures and illustration of the domain architecture of the oxidized enzyme. (A) Superposition of the two apo structure dimers, apo I (PDB entry 4UBW) with chain A, green, chain B, magenta; apo II (this publication) with chain A, blue, chain B, yellow in cartoon presentation. The monomers of apo I are related to each other by the indicated 2-fold axis. A conversion between the monomers of the apo II structure by the same axis is not possible. The N-termini of the monomers are indicated except for the N-terminus of the B chain within the apo II structure since it is not visible in this orientation. (B) Position of the two B chains resulting from the superposition of the A chains. The magenta (apo I) and the yellow (apo II) monomer are not congruently superimposed when A chains are aligned. They are in a different orientation with respect to monomer A, and thereby, the proteins form two different dimer interfaces. (C) The subdomain structure of the oxidized FadA5 apo II monomer is shown as a cartoon, consisting of the N-terminal subdomain (I, yellow), the lid subdomain (II, green), and the C-terminal subdomain (III, blue). The brackets highlight the swapped domain consisting of the α2-turn-ß3 motif (η = 310helix). (D) The subdomain structure of the apo I monomer is shown as a cartoon, consisting of the N-terminal subdomain (I, orange), the lid subdomain (II, blue), and the C-terminal subdomain (III, green). The monomer orientation is chosen according to panel C. Due to this orientation, η4, η6, and ß10 are not visible. The swapped domains here comprise the α2-turn-ß4 motif (η = 310helix).
Intriguingly, a domain swap, of the η1-α2-β4 (α2-β3 in the new structure) motif with respect to subdomain I is observed (Figures 4 and S2A,B). This domain swap leads to the formation of an extra protein interface between the two monomers but results in the absence of η1 and η3 as well as the shortening of α3 (Figure 3C,D). The domain swap is confirmed by continuous electron density ranging from residues D68 to C91 (Figure S2B). Residues at the beginning of the swapped domain are not resolved (Q62 to S67 in chain A and F63 to Q66 in chain B), but residues T88 to C91, which adopt a different fold compared to the apo I structure, are clearly defined (Figure S2B).
Figure 4.

The oxidized structure shows a swapped domain caused by the formation of a disulfide bond. (A) Superposition of the apo structures’ A chains (apo I, green; apo II; blue) is shown in transparent cartoon presentation. According to our activity data, the green monomer is active, and the blue monomer is inactive. The nontransparent secondary structure motifs, η1-α2-turn-ß4 (green) and α2-turn-ß3 (blue), are the regions of FadA5 which are swapped when comparing the apo I (active) with the apo II (inactive) protein structure. Arrows 1 indicate the starting and the end points of the reoriented domain (blue). The green domain is folding back to chain A, whereas the blue domain is pointing away from its monomer and forms an additional interface with chain B of the here described new apo II structure. (B) The apo II dimer is shown in cartoon presentation with the swapped domains being highlighted in black. The cysteines, which are forming the disulfide bonds (C59–C91 and C93–C377) in each monomer, are shown in stick presentation. Chain A is shown as transparent blue and chain B as transparent yellow cartoon. (C) Zoom of (B) to the residues that are forming the disulfide bonds in each monomer. (D) Superposition of the catalytic residues and the disulfide bond forming cysteines of the apo structures I (active, green) and II (inactive, blue) from chain A in stick presentation in the respective color code. The swapped domains of the apo II (inactive, blue) and apo I (active, green) structure of chain A are shown.
The buried surface area, resulting from dimer formation of each apo structure, was calculated with the PDBePISA server.22 The dimer interface of the apo I structure buries 1969 Å2, which is comparable to the 2087 Å2 buried in the apo II structure. The similarities in the buried surface area are consistent with the equivalent Tm’s of the reduced and oxidized WT FadA5s. If the domain swap is omitted, a hypothetical dimer interface would only comprise a buried surface area of approximately 700 Å2. This area is comparably low, with respect to the dimer interface in the apo I structure, but also following the definition of protein interfaces by Kim et al.23 This clearly reduced buried surface area would most likely not provide sufficient stabilization for the dimer, and it is therefore likely that the domain swap is required to maintain the dimer. The formation of two disulfide bonds, one between C59 and C91 and the second between C93 and C377, accompanies this domain reorientation (Figure 4B,C) which involves a repositioning of the catalytic cysteines relative to the cysteine positions in the apo I structure (Figure 4D). In contrast, the location of H347 is not affected. We confirmed the location of the disulfide bonds by generating and analyzing an anomalous electron density map, for which a data set was collected at a wavelength of 1.80 Å (Figure S2C,D).
Interestingly, a difference in dimer formation was also observed in the oxidized and reduced structures of an Arabidopsis thaliana 3-ketoacyl-CoA thiolase.13,14 While the reduced form of the A. thaliana thiolase dimer is very similar to the reduced form of the FadA5 dimer, the oxidized form of the A. thaliana thiolase dimer displays a significantly different orientation of the two monomers relative to its reduced state and also differs with respect to the oxidized FadA5 structure. In addition, the oxidized A. thaliana dimer does not display a domain swap as observed in the oxidized FadA5 structure and only harbors one disulfide bond which can be expected since C59 and the C91 × 92C93 sequence is only conserved in FadA5 orthologs (Figure 1).
The Reduction Potential of WT FadA5 Is Poised at −223 mV
We undertook the determination of the midpoint reduction potential of WT FadA5 by measuring the catalytic activity at different solution redox potentials at pH 7.0. The initial rate of thiolysis using 3,22-dioxo-chol-4-ene-24-oyl CoA and CoA as substrates was measured as a function of the redox potential.
The midpoint reduction potential of WT FadA5 is −223 ± 3 mV (Figure 5A). This result was consistent with the product analysis by MALDI-TOF spectrometry (Figure S1) in which the catalytic activity is lost under oxidizing conditions and can be restored upon subsequent reduction.
Figure 5.
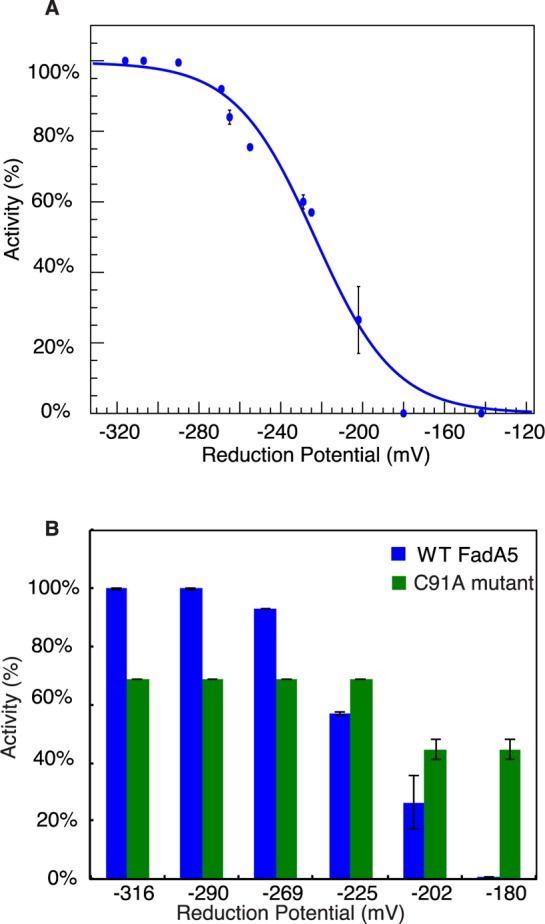
Redox control of WT and C91A FadA5’s catalytic activity. (A) Percentage of reduced WT FadA5 as a function of its solution reduction potential at pH 7.0. The midpoint reduction potential is determined to be at −223 ± 3 mV. Error bars are standard error of measurement. (B) Relative catalytic activity of WT and C91A FadA5 at different solution reduction potentials, pH 7.0. Catalytic activities are normalized to WT FadA5 activity at −316 mV.
Poising FadA5 Midpoint Potential at −223 mV Requires Two Disulfides
We undertook an assessment of the C93–C377 active site disulfide midpoint potential in the absence of the C59–C91 disulfide utilizing a C91A mutant to block the C59–C91 disulfide formation and monitored the catalytic activity as a function of the solution redox potential.
The activity of the C91A mutant did not change when the reduction potential was raised to −225 from −320 mV. The C91A FadA5 catalytic activity decreased moderately (∼30%) when the reduction potential was raised above −202 mV. On the basis of the catalytic activity, the active site C93–C377 disulfide of the C91A mutant was not fully formed even at −180 mV, the most oxidizing potential tested (Figure 5B). This result was consistent with the earlier product analysis by MALDI-TOF spectrometry (Figure S1). Therefore, mutation of Cys91 to Ala prevents not only the formation of the C91–C59 disulfide but also oxidation of the essential active site residues C93 and C377. We conclude that the formation of the C91–C59 disulfide is required for the oxidation of the catalytic cysteines C93 and C377.
The C93A mutant is catalytically inactive, and formation of the C59–C91 disulfide could not be monitored by activity measurements. We attempted to use intrinsic tryptophan fluorescence to monitor environmental changes associated with disulfide formation. However, due to the combination of multiple oxidation intermediates and domain swapping, spectral shifts could not be correlated with disulfide bond formation. Therefore, we were unable to assign a midpoint potential for the isolated C59–C91 disulfide.
Previously identified thiolases with a redox switch control mechanism only comprise one intramolecular disulfide bond with a midpoint potential at a more reducing level: −270 to −283 mV.14,15 The combined motifs of two intramolecular disulfide bonds and a domain interface swap appear to shift the FadA5 activity midpoint to a more oxidizing potential (−223 mV). The elevation of the midpoint potential in the C91A mutant implies that formation of the C59–C91 disulfide bond is a prerequisite for the rearrangement of C93 and C377 into conformations compatible with disulfide bond formation as observed in the crystal structure. The absence of this two disulfide motif and α2-turn-β3 motif in thiolases, other than FadA5s, indicates a unique adaptation. In the case of Mtb, the essentiality of fadA5 for growth on cholesterol4 suggests cholesterol utilization is adjusted for intracellular survival in the cholesterol-rich host environment.
The Reduction Potential of FadA5 Suggests in Vivo Catalytic Activity Can Be Sensitively Adjusted in Response to Environmental Changes
Mtb is an intracellular pathogen that is phagocytosed by the host macrophage. Mtb resides and replicates in the host phagoendosome, and lysosomal fusion is arrested. In the macrophage, Mtb encounters numerous stresses imposed by host defense mechanisms, such as hypoxia, starvation, reactive oxygen intermediates (ROI), reactive nitrogen intermediates (RNI), and acidic pH.24,25 The mycothiol reduction potentials26 (EMSH) of mycobacteria residing inside macrophages range from reducing (−300 ± 6 mV) to basal (−275 ± 5 mV) or oxidizing (−240 ± 3 mV) levels. The fraction of the mycobacterial population at a specific potential varies with the stage of infection and state of the macrophage.
In resting macrophages, the drop in phagosomal pH extracellular to the mycobacterium signals a shift to generate more mycothiol (MSH), thereby reducing the internal mycobacterial reduction potential. Generation of additional MSH is accomplished through the biosynthesis of MSH and/or the reduction of mycothiol disulfide (MSSM) to MSH by mycothiol disulfide reductase.16 The shift to reductive EMSH is accompanied by induction of lipid anabolism gene expression in the mycobacterium. With a midpoint reduction potential at −223 mV, FadA5 is expected to be fully active when the mycobacterium shifts to a reducing EMSH. This reductive potential would ensure full FadA5 catalytic activity, thereby maximizing Mtb β-oxidation of the cholesterol side chain to provide sufficient lipid precursors, e.g., acetyl-CoA and propionyl-CoA, and reductants, NADH/NADPH, for lipid anabolism, as well as ATP generation in the mycobacterium.
In the activated macrophage, Mtb is in an acidic vacuole with a pH of ∼4.5 and the synergistic effect of acidic pH with ROI and RNI generation by the activated macrophage results in a shift to a larger population of mycobacteria with an oxidizing EMSH.26 When the mycobacterium EMSH is oxidizing (−240 mV), only 50% of FadA5 is expected to be active. Thus, the generation of acetyl-CoA and propionyl CoA, products of Mtb cholesterol β-oxidation, would be reduced, but not eliminated, in activated macrophages where oxidative stress is dominant. Redox control of lipid anabolism in Mtb is primarily controlled by WhiB3.16whiB3 is induced by cholesterol4 highlighting the tight linkage between generation of lipid precursors and lipid biosynthesis as a function of the mycobacterial microenvironment which in turn is determined by the state of the infected macrophage.
Conclusion
FadA5’s catalytic activity is controllable by a thiol redox switch. Importantly, formation of the catalytically inactive FadA5 requires the formation of two disulfide bonds. One disulfide bond is in the active site and directly blocks FadA5 catalytic activity. The second disulfide bond resides at the dimer interface adjacent to a strand-loop-helix motif that undergoes a domain swap upon oxidation. This motif is unique within the FadA5 subfamily.
Formation of the second disulfide is required for poising the FadA5 midpoint potential at −223 mV, which is close to the potential of intracellular mycobacteria exposed to low pH and oxidative stress. The shifted midpoint potential and unique motif compared to other redox-controlled thiolases suggests a role for adjusting cholesterol metabolite flux between precursor generation for ATP and lipid generation and alternative pathways. We propose that cholesterol metabolites may be used to combat the environmental stresses encountered in activated macrophages. In support of this concept, the fadA5 mutant does not accumulate apparent upstream metabolites when grown on cholesterol,4 suggesting they may be utilized in alternate pathways when FadA5 activity is curtailed. Further elucidation of potential alternate pathways is currently underway.
Experimental Procedures
Materials and Methods
The C91A and C93A mutants were prepared as described for C93S FadA5 by quick-change mutagenesis.11 The protein was purified as described in Schaefer et al.11 with the exception that the protein was eluted from the size exclusion column with a buffer containing 60 mM bicine–NaOH, pH 8.5, and 250 mM NaCl. 3,22-dioxo-chol-4-ene-24-oyl-CoA was prepared as described.11
Redox-Controlled FadA5 Activity Measurements
FadA5 and the C91A mutant were assayed for thiolytic activity with 3,22-dioxo-chol-4-ene-24-oyl-CoA and CoA as substrates under different redox conditions. The thiolytic activity was followed at 303 nm using a UV/vis spectrophotometer (Shimadzu Scientific Instruments) at 30 °C by monitoring the disappearance of the Mg2+-3,22-dioxo-chol-4-ene-24-oyl-CoA complex.4,27 The assay was carried out with 28 nM WT FadA5 or C91A in 100 mM HEPES, pH 7.0, containing 25 mM MgCl2, 10 μM 3,22-dioxo-chol-4-ene-24-oyl-CoA, and 10 μM CoA. FadA5 and C91A were preincubated in redox buffers (pH = 7.0) with different ratios of CSSC and CSH. The total sulfur concentration of CSSC and CSH buffers was kept constant at 20 mM. The reaction was also analyzed by MALDI-TOF spectrometry (Bruker Autoflex II TOF/TOF) to confirm the formation or absence of thiolytic products. The midpoint reduction potential of WT FadA5 was determined by fitting the percentage of reduced WT FadA5 against the buffer reduction potential E with eq 1 where L is the curve’s maximum value, k is the steepness of the curve, and E1/2 is the midpoint reduction potential.
| 1 |
Circular Dichroism Experiments
Measurements were performed with FadA5 WT as well as the C93A and C91A mutants at 20 °C using 4.4 to 6.3 μM protein. Prior to the experiment, the proteins, FadA5 WT, C93A, and C91A, were dialyzed into a buffer containing 10 mM Na/K phosphate, pH 7.5. To compare the protein folding in the oxidized, reduced, and native state, a third of the protein sample was preincubated with a 1000-fold molar excess of cystamine or a 2000-fold molar excess of cysteamine, for 1 h at 25 °C each. To reduce the background signal by the reagents, they were removed by extensive buffer exchange. The samples were then centrifuged for 30 min at 16 100 × g to remove dust particles or precipitated protein. Measurements were performed in 1 mm cuvettes employing a Jasco J-810 spectrometer at 20 °C using 4.4–6.3 μM protein. The spectrum was measured at wavelengths from 260 to 185 nm with a data pitch of 0.5 nm and a scan speed of 50 nm/min. The bandwidth was 1 nm; the response was 1 s, and the data were registered with standard sensitivity. Ten spectra were accumulated to optimize the signal-to-noise ratio. Finally, the spectra were corrected by the buffer only control and normalized.
Thermofluor Analysis
To analyze the protein’s stability,28 FadA5 WT, C93A, and C91A were preincubated with a 1000-fold molar excess of CSSC or a 2000-fold molar excess of CSH in different buffers for 1 h at 25 °C. The final concentration of the protein in every setup was 2.5 mg/mL. After incubation, the protein was analyzed in the presence and absence of the respective agent and compared to the unmodified, native protein samples. All protein samples were mixed with 0.1% (v/v) Sypro Orange and the respective protein buffer (10 mM Na/K phosphate, pH 7.5, or 20 mM bicine–NaOH, pH 8.5, 250 mM NaCl) for analysis. Protein unfolding was performed and monitored with the Stratagene Mx3005P qPCR System from Agilent Technologies. The experiments were executed utilizing a temperature gradient ranging from 25 to 95 °C with steps of 1 °C per minute and subsequently analyzed with the MxPro QPCR Software.
FadA5 Crystallization
The detailed crystallization and cryoprotectant conditions are listed in Table 1. FadA5 WT apo (12 mg/mL) was mixed in a one to one ratio with the crystallization buffer and was set up for crystallization at 20 °C. The crystals grew after approximately 5 months. Prior to flash-freezing, the crystals were briefly transferred into a cryoprotectant solution. A native data set was collected at a wavelength of 0.873 Å at the ESRF beamline ID23-2 with a 225 mm MarMOSAIC detector, and an anomalous data set was collected at the ESRF ID29 beamline at a wavelength of 1.800 Å with a Pilatus 6 M detector.
Structure Determination
The native data set was integrated with iMosflm29 and scaled with Aimless.30 Phasing was performed by molecular replacement with Phaser31 using chain A from the FadA5 C93S mutant structure with bound steroid and CoA but excluding ligands and solvent molecules (PDB entry 4UBT) as a search model. Manual model building was performed with Coot,32 and the structure was further refined with Refmac5.33 MolProbity21 analysis was performed to validate the structure. Figures of the structure were generated using PyMOL34 after secondary structure assignment by DSSP.35,36 The anomalous data set was integrated with XDS37 and scaled with Aimless.30 This data set was used to generate anomalous maps with the help of Cad and FFT (CCP4 package20) to display the location of the sulfur atoms.
Acknowledgments
This work was supported by NIH AI134054, AI092455, and NIH RR021008 (N.S.S.), NSF BIO1039771 (N.M.N.), and the Deutsche Forschungsgemeinschaft SFB630 and Forschungszentrum FZ82 (C.K.). We thank the staff of beamlines ID23-2 and ID29 at the ESRF, Grenoble, for technical support and Shruthi Krishnan for support to collect the CD spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00072.
FadA5-catalyzed reaction and MALDI-TOF spectrometric analysis; additional comparisons and electron density maps for the oxidized FadA5 protein (PDF)
Accession Codes
Atomic coordinates and structure factors for WT apo II FadA5 have been deposited in the Protein Data Bank with accession code 5ONC.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. (2016) Global tuberculosis report, http://www.who.int/tb/publications/global_report/en/.
- WHO. (2016) Multidrug-resistant tuberculosis (MDR-TB), http://www.who.int/tb/challenges/mdr/mdr_tb_factsheet.pdf.
- Koul A.; Dendouga N.; Vergauwen K.; Molenberghs B.; Vranckx L.; Willebrords R.; Ristic Z.; Lill H.; Dorange I.; Guillemont J.; Bald D.; Andries K. (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3 (6), 323–324. 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- Nesbitt N. M.; Yang X.; Fontan P.; Kolesnikova I.; Smith I.; Sampson N. S.; Dubnau E. (2010) A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect. Immun. 78 (1), 275–282. 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Geize R.; Yam K.; Heuser T.; Wilbrink M. H.; Hara H.; Anderton M. C.; Sim E.; Dijkhuizen L.; Davies J. E.; Mohn W. W.; Eltis L. D. (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 104 (6), 1947–1952. 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. C.; Harik N. S.; Liao R. P.; Sherman D. R. (2007) Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J. Infect. Dis. 196 (5), 788–795. 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- Chang J. C.; Miner M. D.; Pandey A. K.; Gill W. P.; Harik N. S.; Sassetti C. M.; Sherman D. R. (2009) igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191 (16), 5232–5239. 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. K.; Sassetti C. M. (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A. 105 (11), 4376–4380. 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D.; Ehrt S.; Voskuil M. I.; Liu Y.; Mangan J. A.; Monahan I. M.; Dolganov G.; Efron B.; Butcher P. D.; Nathan C.; Schoolnik G. K. (2003) Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 198 (5), 693–704. 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVen B. C.; Fahey R. J.; Lee W.; Liu Y.; Abramovitch R. B.; Memmott C.; Crowe A. M.; Eltis L. D.; Perola E.; Deininger D. D.; Wang T.; Locher C. P.; Russell D. G. (2015) Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 11 (2), e1004679. 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer C. M.; Lu R.; Nesbitt N. M.; Schiebel J.; Sampson N. S.; Kisker C. (2015) FadA5 a thiolase from Mycobacterium tuberculosis: a steroid-binding pocket reveals the potential for drug development against tuberculosis. Structure 23 (1), 21–33. 10.1016/j.str.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapalainen A. M.; Merilainen G.; Wierenga R. K. (2006) The thiolase superfamily: condensing enzymes with diverse reaction specificities. Trends Biochem. Sci. 31 (1), 64–71. 10.1016/j.tibs.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy R.; Micossi E.; Alphey M. S.; Germain V.; Bryce J. H.; Smith S. M.; Leonard G. A.; Hunter W. N. (2006) The crystal structure of a plant 3-ketoacyl-CoA thiolase reveals the potential for redox control of peroxisomal fatty acid beta-oxidation. J. Mol. Biol. 359 (2), 347–357. 10.1016/j.jmb.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Pye V. E.; Christensen C. E.; Dyer J. H.; Arent S.; Henriksen A. (2010) Peroxisomal plant 3-ketoacyl-CoA thiolase structure and activity are regulated by a sensitive redox switch. J. Biol. Chem. 285 (31), 24078–24088. 10.1074/jbc.M110.106013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Jang Y. S.; Ha S. C.; Ahn J. W.; Kim E. J.; Lim J. H.; Cho C.; Ryu Y. S.; Lee S. K.; Lee S. Y.; Kim K. J. (2015) Redox-switch regulatory mechanism of thiolase from Clostridium acetobutylicum. Nat. Commun. 6, 8410. 10.1038/ncomms9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta M.; Rajmani R. S.; Singh A. (2016) Mycobacterium tuberculosis WhiB3 Responds to Vacuolar pH-induced Changes in Mycothiol Redox Potential to Modulate Phagosomal Maturation and Virulence. J. Biol. Chem. 291 (6), 2888–2903. 10.1074/jbc.M115.684597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodero V. I.; Quirolo Z. B.; Sequeira M. A. (2011) Biomolecular studies by circular dichroism. Front. Biosci., Landmark Ed. 16, 61–73. 10.2741/3676. [DOI] [PubMed] [Google Scholar]
- Kelly S. M.; Jess T. J.; Price N. C. (2005) How to study proteins by circular dichroism. Biochim. Biophys. Acta, Proteins Proteomics 1751 (2), 119–139. 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- French S.; Wilson K. (1978) On the treatment of negative intensity observations. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 34 (4), 517–525. 10.1107/S0567739478001114. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (1), 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372 (3), 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kim W. K.; Henschel A.; Winter C.; Schroeder M. (2006) The many faces of protein-protein interactions: A compendium of interface geometry. PLoS Comput. Biol. 2 (9), e124. 10.1371/journal.pcbi.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.; Tanaka K.; Carroll D.; Flynn J.; Bloom B. R. (1995) Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect. Immun. 63 (2), 736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K. H.; Abramovitch R. B.; Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2 (5), 352–364. 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Bhaskar A.; Chawla M.; Mehta M.; Parikh P.; Chandra P.; Bhave D.; Kumar D.; Carroll K. S.; Singh A. (2014) Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 10 (1), e1003902. 10.1371/journal.ppat.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton B. (1974) The kinetic mechanism and properties of the cytoplasmic acetoacetyl-coenzyme A thiolase from rat liver. Biochem. J. 139 (1), 109–121. 10.1042/bj1390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson U. B.; Hallberg B. M.; DeTitta G. T.; Dekker N.; Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357 (2), 289–298. 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40 (4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2006) The PyMOL Molecular Graphics System, http://www.pymol.org/.
- Joosten R. P.; te Beek T. A.; Krieger E.; Hekkelman M. L.; Hooft R. W.; Schneider R.; Sander C.; Vriend G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res. 39 (Database issue), D411–D419. 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 (12), 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.