

Abstract
G protein coupled receptors have historically been one of the most druggable classes of cellular proteins. The members of this large receptor gene family couple to primary effectors, G proteins, that have built in mechanisms for regeneration and amplification of signaling with each engagement of receptor and ligand, a kinetic event in itself. In recent years GPCRs, have been found to interact with arrestin proteins to initiate signal propagation in the absence of G protein interactions. This pinnacle observation has changed a previously held notion of the linear spectrum of GPCR efficacy and uncovered a new paradigm in GPCR research and drug discovery that relies on multidimensionality of GPCR signaling. Ligands were found that selectively confer activity in one pathway over another, and this phenomenon has been referred to as ‘biased agonism’ or ‘functional selectivity’. While great strides in the understanding of this phenomenon have been made in recent years, two critical questions still dominate the field: How can we rationally design biased GPCR ligands, and ultimately, which physiological responses are due to G protein versus arrestin interactions? This review will discuss the current understanding of some of the key aspects of biased signaling that are related to these questions, including mechanistic insights in the nature of biased signaling and methods for measuring ligand bias, as well as relevant examples of drug discovery applications and medicinal chemistry strategies that highlight the challenges and opportunities in this rapidly evolving field.
Keywords: Biased ligand, Functional selectivity, GPCR, Drug discovery
G protein-coupled receptors (GPCRs) are seven-transmembrane domain proteins that function to transmit signals into the intracellular space. It is estimated that nearly 36% of all FDA-approved drugs target at least one member of the GPCR gene family.1,2 Classically, the categorization of GPCR-ligands has been done based on their efficacies for activation of G proteins, as full agonists, partial agonists, antagonists, or inverse agonists, depending on their abilities to elicit a receptor-mediated response. However, it has become evident that GPCRs can mediate multiple signaling outcomes. For instance, βarrestins, which have been long associated with receptor desensitization, can also lead to signaling events.3
Arrestins are cytosolic proteins originally discovered in the visual system, which include arrestin-1 (also referred to as visual arrestin), arrestin-2 (βarrestin1), arrestin-3 (βarrestin2) and arrestin-4 (termed a cone arrestin).4,5 While arrestins-1 and -4 are localized in retinal rods and cones, molecular cloning revealed two ubiquitously expressed isoforms termed βarrestin1 and βarrestin2 due to their high homology with the visual arrestin and their potent functional regulation of the β2 adrenergic receptor (β2AR).6–8 More recently, two additional families of arrestin-related proteins have been found to be broadly expressed in eukaryotes, namely, a group of vacuolar protein sort 26 (Vps26)-related proteins and αarrestins. Their tertiary structure is similar to that of the visual and βarrestins, although their physiological role and potential involvement in regulating GPCR signaling are still not fully understood.9,10
While arrestins were named based on their initially discovered ability to arrest (turn off) GPCR signaling, it is now evident that βarrestins regulate GPCR trafficking as well as G protein-independent signaling.11–13 A general scheme for the βarrestin-mediated regulation of GPCR function is depicted in Figure 1. Agonist binding to GPCR stabilizes an active conformation(s) of the receptor that promotes its interaction with heterotrimeric G proteins, Gαβγ (Fig. 1a). This is followed by the GDP to GTP exchange at the Gα subunit, and the subsequent dissociation of G proteins (Fig. 1b). The dissociated G proteins interact with and modulate the activity of downstream effectors (e.g., adenylyl cyclase and phospholipases) that produce second messengers such as cAMP and inositol phosphate (G protein-dependent signaling).14
Figure 1.

Paradigms of GPCR-mediated signaling and multiple roles of βarrestins: binding of an agonist (a) results in activation of signaling pathways by G proteins (b), as well as βarrestins (f), in addition to desensitization and internalization by βarrestins (d and e).
Since one activated receptor can sequentially couple to multiple G proteins with signal amplification occurring through to the enzymatic activity of receptor second messengers (e.g., cyclases and phospholipases), desensitization mechanisms have evolved to turn off the potentially deleterious effects of sustained signaling.15 Desensitization is considered a two-step process, which starts with phosphorylation of the agonist-occupied GPCR by the second messenger-dependent protein kinases, such as protein kinase A or C (PKA or PKC), or by a corresponding GPCR Kinase (GRK, Fig. 1c).4 This triggers the second desensitization step, which involves the recruitment of βarrestin to active phosphorylated receptors and the consequent steric hindrance of further G protein activation (Fig. 1d).16 The initial discovery of the critical involvement of βarrestins in GPCR desensitization was subsequently followed up by studies that indicated their important role in receptor internalization. It has been found that the receptor-bound βarrestins interact with several key components of the receptor endocytotic process (Fig. 1e), including the adaptor-protein2 (AP2) complex,17 the clathrin heavy chain,18 and the E3 ubiquitin ligase Mdm2.19 The complex formed upon the binding of clathrin/AP2 to the receptor-bound arrestin translocates to the clathrin-coated pit, which is then sequestered off the plasma membrane and hindered from subsequent stimuli.20
While the receptor–G protein interaction that takes place upon receptor activation is unstable and brief, the receptor–arrestin complex can be relatively stable and exist on a time scale of minutes to hours.21,22 Analysis of the agonist-mediated arrestin translocation to multiple GPCRs identified two major classes of receptor–arrestin complexes, based on their strength and longevity. Class A complexes are transient and receptor–βarrestin interactions rapidly dissociate upon receptor internalization, as exemplified by the β2 adrenergic, μ-opioid, endothelin type A, dopamine D1, and α1 adrenergic receptors. Receptors in this class are rapidly recycled back to the plasma membrane. In contrast, Class B complexes have more stable receptor–βarrestin interactions that persist even as the receptor undergoes endosomal sorting. Receptors in this class are sequestered in endosomes and recycled slowly or undergo degradation, for example, the angiotensin II type 1A, neurotensin 1, vasopressin V2, thyrotropin-releasing hormone, and substance P receptors.23
Recent βarrestin crystallographic,24 mutation,25 and biophysical studies26 suggest that βarrestins undergo extensive conformational changes upon binding to the phosphorylated GPCR. In their basal cytosolic receptor-unbound state, arrestins are elongated molecules, which consist of two (N- and C-) domains (Fig. 2a) and the C-terminus anchored in a polar core between them, unavailable for interaction with partner proteins (Fig. 2b).27
Figure 2.

βarrestin1 crystal structures. (a) Superimposed structures of βarrestin1 in a basal state (inactive state: blue; PDB 1G4 M) and in complex with V2Rpp (Vasopressin Receptor 2, partial protein, active state: green; PDB 4JQI) reveal marked conformational differences, including the release of the C-terminus which contains binding sites for partner proteins such as clathrin; (b) inactive state: the polar core consisting of five interacting residues keeping the C-terminus locked in the place is thought to be a critical stabilizer of the βarrestin1 inactive state (Arg393–Asp290 salt bridge highlighted by the broken black circle); (c) active state: upon V2Rpp binding (omitted for clarity) the C-terminal strand residue Arg393 is displaced, and its interacting partner Asp297 undergoes a large movement together with the rest of the lariat loop, resulting in significant conformational rearrangement in the polar core of the activated βarrestin1.
The recently reported crystal structure of βarrestin1 in complex with a fully phosphorylated 29-amino-acid carboxy-terminal (derived from the vasopressin receptor-2 (V2Rpp) and shown to functionally and conformationally activate βarrestin1) revealed extensive conformational changes in the C-terminus, which is released and becomes available for interactions with clathrin and AP2 (Fig. 2c).24 The V2Rpp–βarrestin1 crystal structure also revealed a 20° twist between the N- and C-domains. A similar 20° rotation has been observed in the crystal structure of the mouse visual arrestin bound to a constitutively active form of human rhodopsin.28 It has been suggested that the twisting movement of the two domains is part of the general mechanism by which arrestins, upon activation, may expose an additional interface for interacting with their numerous binding partners.
Over the past decades, a growing list of binding partners has implicated βarrestins in a number of important cellular functions in addition to GPCR desensitization and trafficking. In particular, arrestins have been found to link activated GPCRs to signaling effectors such as the Src family tyrosine kinases,29 components of the extracellular signal-regulated kinase 1/2 (ERK 1/2) and the c-Jun N-terminal kinase 3 (JNK3),30 mitogen-activated protein kinase (MAPK) cascades,31 Akt,32,33 and effectively convey the G protein-independent signaling (Fig. 1f). Because arrestin binding can uncouple G protein from the activated receptor, the G protein-dependent and arrestin-dependent signaling can be segregated. Furthermore, the two signaling pathways, proved to be spatially and temporally distinct events, are also pharmacologically separable.34 In recent years, there has been a movement to develop ligands that will preferentially segregate GPCR to one pathway or another. These functionally selective ligands, also referred to as the ‘functionally selective’ or ‘biased’ agonists, have demonstrated the potential to harness GPCR signaling and have collectively opened new possibilities in GPCR drug discovery.35
Although the exact molecular mechanism of biased signaling is not well understood, the studies reported to date suggest that the GPCR conformation, stabilized by a G protein-biased agonist, is distinct from the conformation stabilized by a βarrestin-biased agonist. For example, the fluorescence-based study on the arginine vasopressin type 2 receptor (V2R) activation using biased and unbiased agonists provided experimental evidence suggesting that the transmembrane helix 6 (TM6) and third intracellular loop (icl3) movements are associated with selective G protein signaling, whereas the movements between its TM7 and helix 8 (H8) region are required for selective βarrestin recruitment.36 Similar conclusions resulted from the 19F NMR study of β2AR in complex with a range of ligands with different functional profiles.37 The NMR signal changes of the site-specific 19F-labels located in the cytoplasmic region revealed that binding of an unbiased agonist (e.g., formoterol) shifts the receptor conformational equilibrium toward the specific ‘G protein active’ state of TM6, while the βarrestin-biased ligands (e.g., carvedilol) impact primarily the conformational state of the TM7 helix. These findings are consistent with the crystallographic38 and electron microscopy39 studies of the β2AR–Gαβγ heterotrimer complex that showed that the Gα subunit contacts primarily the intracellular regions of TM5 and TM6.
A study of the patterns of β2AR phosphorylation by individual GRKs, a prerequisite for β-arrestin binding, showed that GRKs target primarily the short H8 helix adjacent to the TM7 region.40 In addition, a detailed mapping of the β2AR phosphorylation in this study demonstrated that the βarrestin-biased ligand (carvedilol) induced a phosphorylation pattern that was distinct from that of isoproterenol, an unbiased full agonist.40 This suggests that by inducing distinct receptor conformations, the biased ligands are able to recruit distinct GRKs and, consequently, produce a distinct phosphorylation pattern. In fact, the experimental evidence accumulated over the recent years indicates that the biased ligands can stabilize both the receptor and βarrestin in conformations that are distinct from those associated with unbiased ligands. For example, by employing an intramolecular bioluminescence resonance energy transfer (BRET)-based biosensor of βarrestin2 and a combination of biased ligands and/or biased mutants of three different GPCRs, Shukla and colleagues at Duke University provided evidence suggesting that βarrestins can adopt multiple ‘active’ conformations. 41 One can speculate that these multiple βarrestin conformers with distinct surface topologies complex with different binding partners, and thereby engage in different downstream signaling pathways.
Another important advance toward unraveling the molecular basis of biased signaling was the recently reported crystal structure of two 5-hydroxytryptamine serotonin receptor subtypes, namely 5-HT1B and 5-HT2B, both bound to ergotamine (ERG, Fig. 3a).42,43 ERG is an alkaloid produced by the ergot fungus, which has been shown to exhibits a strong βarrestin-biased agonism at 5-HT2B, and a relatively weak bias at the 5-HT1B receptor.
Figure 3.

(a) Superimposed crystal structures of 5-HT1B (blue; PDB 4IAR) and 5HT2B (gold; PDB 4IB4) in complex with ergotamine (ERG); (b) prominent conformational difference of ERG bound to 5-HT1B (gray) and 5HT2B (green) is highlighted by the broken black circle. Contacts with additional residues observed in ERG-5HT2b structure are also highlighted (gold); (c) conserved DRY motif exist in an ‘active’ state (the Asp152–Arg153 salt bridge broken) in 5HT1B structure (blue), and ‘inactive’ state (the Asp146–Arg147 salt bridge intact) in 5HT2B structure (gold).
A close inspection of the two data sets revealed a small shrinkage of the ligand binding pocket in the ERG-5-HT2B structure, with ERG in slightly different binding conformation and hydrophobic contacts with several additional residues when compared with the 5-HT1B structure (Fig. 3b). Intriguingly, a similar network of additional interactions has been found in the structure of carvedilol, a βarrestin-biased agonist bound to the β1AR.44 The analysis of the two crystal structures also revealed that the ERG-bound 5HT1B complex exists in a classical full agonist-induced active-like state, whereas the ERG-bound 5HT2B structure displays conformational characteristics that are attributed to both the active and inactive states. For example, both structures showed the conserved NPxxY motif (TM7) in an active-like conformation with the intracellular TM7 helix moved toward the receptor core. However, only the ERG-5HT1B structure displayed another highly conserved GPCR feature, the DRY motif in TM3, in an active state with the salt bridge between the Asp146–Arg147 residues being broken. In the ERG-5HT2B structure, the DRY motif salt bridge between Asp152 and Arg153 was intact, which is consistent with all inactive-state GPCR structures known to date (Fig. 3c). Consequently, it is tempting to attribute this ‘intermediate’ state of the 5HT2B receptor structure to the βarrestin-biased signaling conformation characterized by a strong βarrestin and an inefficient G protein coupling. However, it is important to note that the observed difference between the two structures may also reflect distinct signal transduction mechanisms between the two receptor subtypes used in this study. Additionally, it is critical to take into account the fact that crystal structures represent only a static snapshot of a potentially complex ensemble of conformations. It is now widely recognized that GPCRs are highly dynamic systems that assume many conformations associated with different signaling states, which are stabilized by different ligands.45,46 Further research efforts, most likely involving crystallography in conjunction with other biophysical and computational47 methods to study GPCRs as dynamic systems, will be required to reliably delineate the biased signaling mechanism at the molecular level.
Functional selectivity (biased signaling): definition, assays, quantification
The definition and identification of biased ligands has been challenging. It appears that initially, differences in relative efficacy or potency were used as the determinants of ligand bias.48–51 The realization that more efficient approaches for determining bias were needed, led to the development of different methods for identifying biased ligands.52 However, not always a consensus was reached about the most appropriate way of measuring functional selectivity.53,54 Both qualitative and quantitative methods have been described.52,55,56 These methods compare test ligands with reference agonists in order to determine if a compound is biased for one signaling pathway relative to another.52 In the case of the quantitative methods, a ‘bias factor’ defining the degree of bias of the test ligand is generated.52
One of the most simplistic methods, developed by Ehlert and colleagues,57,58 utilizes data from standard four-parameter Hill equations that display slopes close to unity. This method uses ratios of relative efficacies (Emax) by potencies (EC50) (relative activities) to quantify ligand bias, as shown in the equation bellow:
Additional methods use the Black and Leff operational model to quantify functional selectivity. As shown in the equation bellow, the Black and Leff model defines the terms τ (coupling efficiency) and KA (conditional affinity) as the ligands’ (A) intrinsic efficacy and dissociation constant of agonist–receptor–signal transducer complex, respectively (n = transducer slope).59
The Black and Leff operational model is as a mathematical rearrangement of two distinct Hill equations (concentration of agonist by receptor occupancy and receptor occupancy by effect) with the introduction of τ (τ = receptor density/receptor occupancy needed for producing 50% of the system’s maximal effect).59 By plotting ligand’s functional data into the Black and Leff equation, ratios of τ/KA quantify pharmacological responses regardless of signal amplification and receptor reserve.52,60 This feature is very desirable for ligand bias analyses and has been developed by Kenakin and colleagues as ‘transduction coefficients’ according to the following equation:60
The transduction coefficients and the relative activity ratios become proportional (and, thus, result in the same bias factors) when slopes of 1 are obtained from the dose–response curves.52,55 The transduction coefficients have been recently modified in order to more accurately accommodate data from weak partial agonists. 61 This new method entitled ‘competitive model’ utilizes ligand curves in agonist and antagonist mode to more precisely define τ and KA values for weak partial agonists.61
The utility of the methods of measuring ligand bias has been studied. For instance, the transduction coefficients have been successfully incorporated into SAR studies to aid in the development of biased ligands for the dopamine D2 and the κ-opioid receptors. 62–64 It also appears that these analyses can provide mechanistic insight on the activation of more complex signaling pathways that are downstream of immediate GPCR signal transducers.55 Moreover, bias analyses have been successfully implemented in studies to investigate ligand-specific functional consequences of different point mutations in the muscarinic M1 receptor.65 It is important to note that bias is a result of how a ligand interacts with the receptor (molecular bias, which translates across assays and expression systems) plus how the receptor response is observed (an experimental artifact that is system- and assay-dependent and may not translate to other expression systems or assays).52 As shown in the methods above, the solution found to correct for this observational issue has been to use a reference ligand for comparisons of relative activities within specific assay systems.52 Therefore, bias is always a measure relative to the reference ligand and assay systems. A reference agonist is typically chosen based on its ability to robustly activate the receptor in the assays that are to be compared. In some cases, the endogenous ligand of a receptor is used as the reference compound. However, nature may also exploit biased signaling and, thus, endogenous ligands may also present functional selectivity.66–71 This allows for a test ligand being biased compared to an endogenous agonist, but that does not preclude the endogenous ligand from displaying bias compared to another reference ligand.52 Therefore, it is important to emphasize that bias is not an absolute property, but is highly dependent on context of the assay and performance of the reference compound.
Another type of bias that needs to be considered in functional selectivity studies is the case of ‘perfect bias’. Ligands that display perfect bias are agonists for one signaling pathway and do not display any detectable activity for another pathway.52 Caution should be taken in the classification of those ligands, since assay systems that display low amplification or have low receptor reserve might not result in any detectable responses for partial agonists. Therefore, it has been suggested that those ligands should be defined as ‘extremely biased’.72 Different assay systems with higher signal amplification and receptor reserve should be employed for the analyses of ligands that display this type of bias.
The cases of perfect or extreme bias also expose a deficiency in the current quantitative methods for measuring ligand bias. That is, these methods cannot accommodate data from compounds that display antagonism or inverse agonism for one of the signaling pathways analyzed.55 Extremely biased compounds have been described.49,73,74 However, a framework for quantitatively ranking the degree of bias of these compounds has only just begun to emerge.61,75 As these quantitative, mechanistic methods become more accepted and elaborative there is certain to be a broader interest in both the discovery and interpretation of the molecular and physiological implications of extremely biased ligands. Again, it is important to emphasize that bias is context dependent and bias observed in one condition (i.e., cell-based signaling assay) may not translate to the receptor expressed in an endogenous setting (i.e., synapse).
Drug discovery and medicinal chemistry highlights
The realization that βarrestins mediate cellular signaling, which is independent and pharmacologically separable from G protein signaling, has changed a previously held notion of the linear spectrum of GPCR efficacy and uncovered a new paradigm in drug discovery that relies on the multidimensionality of GPCR signaling. Over the past couple of decades a number of GPCR ligands displaying biased profiles were reported in the literature, most discovered retrospectively. In this section we discuss the hypothesis-driven drug discovery and medicinal chemistry efforts that were intentionally focused from the outset on the design and development of biased GPCR ligands toward safer and/or more efficacious medicines. For a more general coverage outside of the scope of this section readers are referred to recently published reviews of this topic.76,77
One of the earliest identified opportunities for the biased GPCR agonist approach in drug discovery steamed from the studies reported in 1999, 2000 and 2005 by Bohn and colleagues. These studies showed that morphine administered to βarrestin2 KO mice produce enhanced and prolonged analgesia with reduced tolerance and fewer adverse events, when compared to wild-type mice.78–80 More recently, a similar profile of improved morphine potency with reduced side effects has been observed in mice and rats with βarrestin ablated by siRNA.81,82 Opioids produce powerful analgesia, by activating μ-opioid receptors (MOR), in addition to many efficacy-limiting adverse effects, which include tolerance, nausea, vomiting, sedation, constipation, and respiratory depression.83–85 Numerous MOR agonists have been developed over the years as part of a global effort to improve the opioid safety and tolerability profile. However, all of these structurally diverse opioids display morphine-like adverse effects, suggesting that the analgesic efficacy, as well as the side effects, is directly related to MOR activation. Intriguingly, experiments in the βarrestin2 KO mice imply not only that βarrestins reduce morphine’s analgesic potency, but it may also be associated with producing morphine-induced side effects.
This hypothesis suggests that it would be beneficial to develop agonists that could selectively activate the G protein signaling pathway without engaging βarrestins as a means to preserve analgesic efficacy while avoiding side effects. The first ligand identified that displayed this separation between signaling pathways was a neoclerodane diterpene derived from the kappa opioid receptor natural product agonist, salvinorin A.86 This compound, herkinorin, is structurally intriguing as it has no basic nitrogen, a highly conserved property of GPCR ligands.87,88 In antinociception studies assessing formalin-induced pain responses, herkinorin proved efficacious without tolerance; supporting the general hypothesis that the antinociceptive properties could be dissected from tolerance by driving G protein signaling over βarrestin2 recruitment.89 However, herkinorin was not shown to be acting at the same sites as morphine and therefore, it is difficult to conclude whether its bias properties produced the differences in physiological responses.
Further motivated by this hypothesis, Chen and colleagues at Trevena Inc. set out to identify G protein-biased MOR agonists as potential novel analgesics with improved efficacy and side effect profile.90 They initiated a hit finding effort by screening the internal compound collection in the cAMP accumulation assay (G protein signaling) and the βarrestin2-based β-galactosidase fragment compartmentalization assay (βarrestin signaling). Both assays were developed in the HEK-293 cell line expressing human MOR.
This effort led to the discovery of tetrahydropyran 1 (Table 1) with a submicromolar G protein MOR potency and a low MOR βarrestin recruitment activity (32% vs 100% in comparison to morphine), which was deemed a suitable starting point for an optimization program. The optimization efforts around compound 1, which primarily focused on improving the MOR G protein potency while diminishing βarrestin recruitment, resulted in the discovery of thiophenyl analogue 2. It is worth noting that some closely related analogues displayed an unbiased full agonist profile, such as compound 3, which showed high efficacy in both the cAMP and βarrestin assays (104% and 197%). Displaying the desired in vitro profile, compound 2 was progressed to the in vivo evaluation and benchmarking against morphine’s efficacy and potential for adverse effects. These studies showed that at equianalgesic doses, established in the mouse hot-plate assay, morphine (6 mg/kg) exhibited a more severe constipation effect than compound 2 (3 mg/kg) in a mouse glass bead retention model of colonic motility (158 min vs 65 min retention times, respectively). However, despite the favorable in vivo pharmacology profile, the potent hERG channel inhibition (IC50 = 2.3 μM) and the cardiac abnormalities observed in the rabbit ventricular wedge assay prevented further progression of compound 2. Encouraged by the initial in vivo data that reaffirmed the original hypothesis, the authors continued their optimization efforts around 2, which led to the discovery of 3-methoxy thiophenyl derivative 4 with not only an improved biased MOR profile (Table 1), but also reduced cardiovascular liabilities with hERG IC50 >200-fold over MOR cAMP EC50 value of 8 nM. The authors calculated a bias factor of 3 based on the equiactive comparison method based on the intrinsic relative activity model outlined by Griffin et al.58 and Rajagopal et al.56,91
Table 1.
G protein-biased MOR agonists90
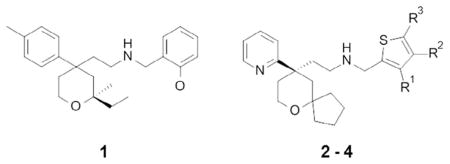
| |||||
|---|---|---|---|---|---|
| Compound | R1, R2, R3 | MOR cAMP pEC50 | MOR cAMP Emax (%) | MOR βarr2 pEC50 | MOR βarr2 Emax (%) |
| Morphine | NA | 7.4 | 100 | 6.3 | 100 |
| 1 | NA | 6.3 | 74 | 5.7 | 32 |
| 2 | H, H, H | 7.8 | 95 | 6.6 | 15 |
| 3 | H, Me, Me | 8.3 | 104 | 6.3 | 197 |
| 4 (TRV-130) | OMe, H, H | 8.1 | 84 | 7.3 | 15 |
On the basis of its in vitro profile, compound 4 (TRV-130) was progressed for in vivo evaluation, where it displayed a 5–10 times higher potency than morphine in the mouse (ED50 values 0.9 mg/kg vs 4.9 mg/kg) and rat (ED50 0.32 mg/kg vs 3.2 mg/kg) hot-plate assays, as well as an improved safety profile in the mouse glass bead colonic motility assay (Emax = 53% for TRV-130 compared to 100% for morphine at equianalgesic doses). Despite showing at higher doses an effect on blood gases, TRV-130 therapeutic index proved superior to morphine in respect of the analgesia versus respiratory suppression in the rat, with no effect on blood CO2 and O2 levels at doses of up to eight-fold over the equianalgesic dose.91 Interestingly, the in vitro MOR biased profile of compound TRV-130 was found to be species dependent. It showed higher βarrestin potency and efficacy at the mouse-(EC50 = 12.6 nM; efficacy = 74%) and dog-(EC50 = 9.4 nM; efficacy = 24%) receptors, compared to the rat MOR (inactive) and human MOR (EC50 = 40 nM; efficacy = 14%). This phenomenon, potentially resulting from differences in phosphorylation sites and/or receptor expression, is important to remember when designing or interpreting translational in vivo data for biased ligands.
Gratifyingly, the observed pre-clinical profile of TRV-130 proved translatable in the clinic, where in healthy volunteers it produced analgesia with less reduction in a respiratory drive and a less severe nausea than morphine.92 However, in a recently reported phase 2, randomized, placebo- and active-controlled study in acute pain following bunionectomy, intravenously administered TRV130 produced greater categorical pain relief compared with morphine (P <0.005), with tolerability similar to morphine and no serious adverse effects; the respiratory effects in this study did not provide a statistically significant benefit over morphine but it is hoped that the improved analgesic benefit will offset this difference. 93 These results provide an early clinical translation of ligand bias as an important new paradigm in GPCR-targeted pharmacotherapy. However, considering a profound effect seen in βarrestin2 KO mice, one may be disappointed with the relatively limited improvements brought about by TRV-130 in both the animal models and clinical trials. For example, in comparison to morphine, TRV-130 showed only approximately a two-fold improved therapeutic index (analgesia vs respiratory depression).92 This raises the question of whether this result is the maximum one could expect from this approach, or if a more biased G protein MOR agonist would show a safer profile with an even greater therapeutic index than TRV-130.
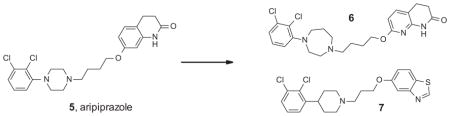
Similarly to the MOR for pain, the dopamine D2 receptor has been for a long time a focus of research and drug discovery for psychiatric disorders. With the exception of aripiprazole 5, reportedly a D2 partial agonist, all clinically used antipsychotics are unbiased antagonists of the D2 receptor.94 The D2 antagonism is thought to underlie the therapeutic benefit in the treatment of psychotic symptoms, as well as serious extrapyramidal side effects including catalepsy and other dyskinesias.95 Investigating a possibility of separating the D2 antipsychotic efficacy from the target-related side effects, Chen and colleagues were interested in developing a βarrestin signaling biased D2 receptor agonist.96 They selected the FDA approved antipsychotic aripiprazole as a starting point because of its unique D2 profile and hoping that its closely related analogues may share its favorable pharmacokinetic properties, including CNS exposure. A range of aripiprazole analogues were synthesized using diversity-oriented high throughput synthetic methods. The compounds were profiled in the cAMP accumulation (G protein signaling) and βarrestin recruitment assays, in which aripiprazole itself displayed an unbiased partial agonist profile (Table 2).
Table 2.
From aripiprazole to the βarrestin-biased D2 agonists96
| ||||
|---|---|---|---|---|
| Compound | D2 cAMP EC50 (nM) | D2 cAMP Emax (%) | D2 βarr2 EC50 (nM) | D2 βarr2 Emax (%) |
| 5 | 1.0 | 51 | 4.0 | 62 |
| 6 | Inactive | <20 | 1.6 | 47 |
| 7 | Inactive | <20 | 50 | 97 |
This effort resulted in the discovery of potent and βarrestin-biased D2 agonists 6 and 7 (Table 2). Compound 6 displayed a potent partial agonist profile in the D2 βarrestin assay, similar to the parent compound 5, while in the same assay 7 was found to be a slightly less potent but full agonist. Importantly, both compounds displayed a potent antipsychotic-like efficacy in the amphetamine-induced hyperlocomotion studies in mouse, without inducing motor side effects that are characteristic for unbiased D2 antagonists such as haloperidol. At the same doses both compounds produced a haloperidol-like catalepsy in βarrestin2 KO mice.97 Taken together, these results suggest that the D2 receptor βarrestin2 signaling blocking is not required for antipsychotic efficacy, while the βarrestin2 agonist activity may protect from the motor side effects triggered by D2-mediated G protein antagonism. It remains to be seen if these encouraging preclinical data for the βarrestin-biased D2 agonists can be translated to the clinic.
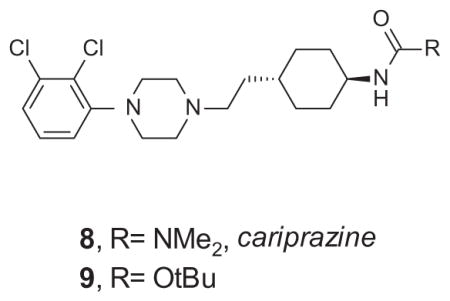
A similar literature-inspired approach in the design of βarrestin-biased D2 receptor ligands has been taken by Shonberg and colleagues at Monash University.62 The focus of their structure–functional selectivity exploration was on cariprazine 8, another D2 partial agonist currently in clinical development for schizophrenia.98 Again, to assess G protein-related signaling, the compounds were profiled in the D2 cAMP accumulation assay, whereas βarrestin signaling was evaluated by measuring phosphorylation of extracellular signal-regulated kinase 1/2 (ERK 1/2). A particularly interesting aspect of this work is the application of the transduction coefficients, as shown in Table 3.
Table 3.
From cariprazine to the G protein-biased D2 agonists62
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | cAMP
|
pERK1/2
|
cAMP–pERK1/2
|
||||||
| pKA | Logτ | Log(τ/KA) | Δlog(τ/KA) | pKA | Logτ | Log(τ/KA) | Δlog(τ/KA) | ΔΔlog(τ/KA) | |
| 8 | 8.71 | 0.85 | 9.59 | 1.04 | 7.61 | −0.37 | 7.25 | −1.3 | 2.34 |
| 9 | 7.11 | 0.76 | 7.92 | −0.63 | 7.51 | −0.31 | 7.21 | −1.34 | 0.71 |
In this testing paradigm, cariprazine 8 displayed a 230-fold bias toward the cAMP pathway compared with dopamine, whereas a closely related carbamate 9 showed a 42-fold bias decrease. Interestingly, while all cariprazine derivatives disclosed in this Letter showed a bias toward the G protein signaling pathway, subtle changes of the D2 unbiased partial agonist 10, a structurally related aryl piperazine reported by Tschammer et al.,99 led to analogues that displayed a strong bias toward βarrestin signaling, for example, 11; ΔΔlog(τ/KA) = −1.2 (Fig. 4).
Figure 4.

Aryl piperazine analogues of cariprazine and aripiprazole with different D2 receptor functional selectivity profiles.99
Cardiovascular disease is another therapeutic area where biased agonists are extensively investigated as an approach to improve the current standards of care. Angiotensin II is a powerful endogenous vasoconstrictor that exerts its physiological effects through activation of angiotensin II receptors (AT1R), a Gαq-coupled GPCR.100 AT1R antagonism proved to be an effective approach for the treatment of hypertension since the 1985 launch of losartan, the first of the AT1 receptor blockers (ARBs) currently used in the clinic.101 However, in addition to blocking adverse effects of angiotensin II, such as hypertension and cardiac remodeling, ARBs also prevent beneficial effects of angiotensin II, including the AT1R-mediated inotropy which supports cardiac function. Consequently, reduced cardiac output was long thought to be an inseparable side effect associated with AT1R antagonism and ARBs in general. This notion has been recently challenged by studies showing that, in a stark contrast to angiotensin II (a balanced AT1R agonist) a βarrestin-biased analogue Sar1Ile4Ile8-angiotensin (SII) robustly promotes contractility of isolated cardiac myocytes.102 This finding suggested that the AT1R G protein signaling pathway is the primary mediator of the angiotensin II vasoconstrictive effect, whereas βarrestin signaling is solely responsible for the ionotropic effect of AT1R stimulation. In line with this hypothesis, another biased AT1R ligand that blocks the G proteins but activates the βarrestin signaling pathway, Sar-Arg-Val-Tyr-Ile-His-Pro-D-Ala-OH (TRV027), has been shown to reduce blood pressure and improve cardiac performance in rats, whereas the unbiased ARB losartan decreased both blood pressure and cardiac performance.103 It has been shown that the in vivo effect of a selective βarrestin-biased AT1R agonist on cardiac contractility is lost in βarrestin2 KO animals, which further supports the hypothesis that positive ionotropic effects of AT1R stimulation are specific to βarrestin-mediated signaling.104 Since administration of a βarrestin-biased AT1R agonist does not lead to increased intracellular levels of secondary messengers such as calcium, IP1 or DAG, it is hypothesized that the observed βarrestin-dependent inotropic effect is likely related to enhanced sensitivity of the myofilament to calcium. In addition to the positive effect on cardiac contractility, βarrestin-biased AT1R agonists were also found to promote cell survival during cardiac injury. Together, these findings present intriguing opportunities for cardiac therapy in congestive heart failure in general and ischemic injury in particular, which is often characterized clinically by peripheral arterial vasoconstriction and diminished cardiac contractility. Indeed, the preclinical efficacy and safety profile of TRV027 has been translated into the clinic where it lowered blood pressure in healthy volunteers on a salt restricted diet, as well as in patients with acute heart failure (AHF), without significant adverse safety signals. On the basis of these data TRV027 has recently progressed to a larger Phase 2b study in AHF patients.105
Concluding remarks and future perspectives
The classical understanding of GPCR signaling106,107 has undergone major revisions in recent years with the introduction of the biased agonism paradigm. 108–111 The realization that βarrestin has a role in GPCR signaling and the discovery of ‘biased’ or ‘functionally selective’ ligands that can produce specific receptor activation profiles have stimulated significant interest in the physiological, pharmacological and biostructural mechanisms that underline this paradigm. 33,64,78–80,112–116 Based on experimental evidence suggesting that the physiological effects of GPCR activation are pathway dependent, functional selectivity has been proposed as a strategy for improving current drug therapies that target GPCRs.66
Emerging reports of drug discovery programs aimed to evaluate potential therapeutic benefits of the functional selectivity paradigm provide an insight into the current challenges and opportunities in this rapidly evolving field. For example, understanding SAR data is one of the commonly reported challenges for medicinal chemists working on biased ligand programs, since even small structural modifications are often found to result in large shifts in the functional selectivity. Interestingly, this phenomenon has been successfully leveraged as an effective hit generation strategy, where a systematic SAR exploration around a known ligand, regardless of its functional selectivity, led to identification of a suitable starting compound with a desired biased profile. However, these steep and often unpredictable ‘SAR cliffs’ present a significant challenge at the hit/lead optimization stage, when SAR-driven medicinal chemistry efforts are focused on preserving the desired biased profile while optimizing other properties that are required in a successful clinical candidate. Great strides have been made in unravelling the biostructural mechanisms of the biased signaling. 117 However, to better understand the SAR and enable a rational design of biased ligands, more atomic-level biostructural information about the nature of ligand–receptor–effector interactions will be required. The most useful information in this context will likely come from research that effectively combines the crystallographic and other biophysical and computational methods to study GPCRs as dynamic systems.
Availability of high quality biased ligands is a critical prerequisite to addressing the ultimate challenge of deconvoluting in vivo physiological effects that are associated with different GPCR signaling pathways. This is proving particularly difficult, as the manner in which a receptor signals is highly context dependent and the diversity of expression of GPCRs in vivo is contextually rich. When translating in vitro bias to in vivo pharmacology one should be aware of possible complications resulting from potential species differences, which may affect ligand potency or functional selectivity in vivo as indicated earlier. One should also consider potential effects of in vivo pharmacokinetics, which may result in suboptimal ligand exposure at the site of action, or formation of active metabolites with unbiased agonist profiles. For example buprenorphine, a clinically used opioid analgesic, is reportedly a partial and biased agonist of the MOR, whereas its main metabolite in humans, norbuprenorphine, is a potent, full and balanced MOR agonist.118 Interestingly, since norbuprenorphine is a P-gp substrate actively effluxed at the blood brain barrier, it does not affect buprenorphine’s centrally-mediated pharmacology.119
Moreover, many receptors were found to couple to different types of Gα subunits of G proteins,120,121 as well as the multiple isoforms and combinations of Gβγ subunits, which could result in distinct signaling events and further increase the overall complexity of GPCR signaling.122–124 The concept of biased signaling will inevitably broaden to encompass additional complexities and continue to evolve as an important approach toward safer and more effective medications. Indeed, the sheer extent of the current scientific literature and ongoing clinical developments for a range of indications assure increasingly prominent role of the biased agonism paradigm in GPCR drug discovery.
References and notes
- 1.Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Disc. 2006;5:993. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Rask-Andersen M, Almen MS, Schioth HB. Nat Rev Drug Disc. 2011;10:579. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz R, Shenoy S. Science. 2005;308:512. doi: 10.1126/stke.2005/308/cm10. [DOI] [PubMed] [Google Scholar]
- 4.Shukla AK, Xiao K, Lefkowitz RJ. Trends Biochem Sci. 2011;36:457. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefkowitz RJ. Prog Mol Biol Transl Sci. 2013;118:3. doi: 10.1016/B978-0-12-394440-5.00001-2. [DOI] [PubMed] [Google Scholar]
- 6.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Science. 1990;248:1547. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 7.Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, Benovic JL. J Biol Chem. 1993;268:15640. [PubMed] [Google Scholar]
- 8.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Proc Natl Acad Sci USA. 1987;84:8879. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aubry L, Klein G. Prog Mol Biol Transl Sci. 2013;118:21. doi: 10.1016/B978-0-12-394440-5.00002-4. [DOI] [PubMed] [Google Scholar]
- 10.Kang DS, Tian X, Benovic JL. Curr Opin Cell Biol. 2014;27:63. doi: 10.1016/j.ceb.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurevich VV, Gurevich EV, Cleghorn WM. Handb Exp Pharmacol. 2008:15. doi: 10.1007/978-3-540-72843-6_2. [DOI] [PubMed] [Google Scholar]
- 12.Lefkowitz RJ, Rajagopal K, Whalen EJ. Mol Cell. 2006;24:643. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Tian X, Kang DS, Benovic JL. Handb Exp Pharmacol. 2014;219:173. doi: 10.1007/978-3-642-41199-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birnbaumer L. Biochim Biophys Acta. 2007;1768:756. doi: 10.1016/j.bbamem.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Science. 1989;246:235. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Ferguson SS, Barak LS, Aber MJ, Giros B, Lefkowitz RJ, Caron MG. Receptors Channels. 1997;5:193. [PubMed] [Google Scholar]
- 17.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. Proc Natl Acad Sci USA. 1999;96:3712. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Nature. 1996;383:447. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 19.Shenoy SK, Barak LS, Xiao K, Ahn S, Berthouze M, Shukla AK, Luttrell LM, Lefkowitz RJ. J Biol Chem. 2007;282:29549. doi: 10.1074/jbc.M700852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Praefcke GJ, McMahon HT. Nat Rev Mol Cell Biol. 2004;5:133. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 21.Charest PG, Terrillon S, Bouvier M. EMBO Rep. 2005;6:334. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfleger KD, Dromey JR, Dalrymple MB, Lim EM, Thomas WG, Eidne KA. Cell Signal. 2006;18:1664. doi: 10.1016/j.cellsig.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Luttrell LM, Gesty-Palmer D. Pharmacol Rev. 2010;62:305. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ. Nature. 2013;497:137. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurevich VV, Gurevich EV. Pharmacol Ther. 2006;110:465. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nobles KN, Guan Z, Xiao K, Oas TG, Lefkowitz RJ. J Biol Chem. 2007;282:21370. doi: 10.1074/jbc.M611483200. [DOI] [PubMed] [Google Scholar]
- 27.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Structure. 2001;9:869. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 28.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE. Nature. 2015;523:561. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Science. 1999;283:655. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 30.Seo J, Tsakem EL, Breitman M, Gurevich VV. J Biol Chem. 2011;286:27894. doi: 10.1074/jbc.M111.260448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chavkin C, Schattauer SS, Levin JR. Handb Exp Pharmacol. 2014;219:281. doi: 10.1007/978-3-642-41199-1_14. [DOI] [PubMed] [Google Scholar]
- 32.Schmid CL, Bohn LM. J Neurosci. 2010;30:13513. doi: 10.1523/JNEUROSCI.1665-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. J Biol Chem. 2013;288:22387. doi: 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. J Biol Chem. 2004;279:35518. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopal S, Rajagopal K, Lefkowitz RJ. Nat Rev Drug Disc. 2010;9:373. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P, Baneres JL, Mouillac B, Granier S. Proc Natl Acad Sci USA. 2012;109:6733. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Science. 2012;335:1106. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westfield GH, Rasmussen SG, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, Chae PS, Liu T, Li S, Woods VL, Jr, Steyaert J, Kobilka BK, Sunahara RK, Skiniotis G. Proc Natl Acad Sci USA. 2011;108:16086. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ. Sci Signal. 2011;4:1. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Proc Natl Acad Sci USA. 2008;105:9988. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Science. 2013;340:615. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Science. 2013;340:610. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warne T, Edwards PC, Leslie AG, Tate CG. Structure. 2012;20:841. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ. Nat Chem Biol. 2011;7:692. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK. Cell. 2013;152:532. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costanzi S. Trends Pharmacol Sci. 2014;35:277. doi: 10.1016/j.tips.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Urban J, Clarke W, von Zastrow M, Nichols D, Kobilka B, Weinstein H, Javitch J, Roth B, Christopoulos A, Sexton P, Miller K, Spedding M, Mailman R. J Pharmacol Exp Ther. 2007;320:1. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 49.Gay EA, Urban JD, Nichols DE, Oxford GS, Mailman RB. Mol Pharmacol. 2004;66:97. doi: 10.1124/mol.66.1.97. [DOI] [PubMed] [Google Scholar]
- 50.Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, Thomas WG. Mol Pharmacol. 2002;61:768. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- 51.Sneddon WB, Magyar CE, Willick GE, Syme CA, Galbiati F, Bisello A, Friedman PA. Endocrinology. 2004;145:2815. doi: 10.1210/en.2003-1185. [DOI] [PubMed] [Google Scholar]
- 52.Kenakin T, Christopoulos A. Nat Rev Drug Disc. 2013;12:205. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 53.Kenakin T, Christopoulos A. Nat Rev Drug Disc. 2013;12:483. doi: 10.1038/nrd3954-c2. [DOI] [PubMed] [Google Scholar]
- 54.Rajagopal S. Nat Rev Drug Disc. 2013;12:483. doi: 10.1038/nrd3954-c1. [DOI] [PubMed] [Google Scholar]
- 55.Brust TF, Hayes MP, Roman DL, Burris KD, Watts VJ. J Pharmacol Exp Ther. 2015;352:480. doi: 10.1124/jpet.114.220293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajagopal S, Ahn S, Rominger D, Gowen-MacDonald W, Lam C, Dewire S, Violin J, Lefkowitz R. Mol Pharmacol. 2011;80:367. doi: 10.1124/mol.111.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ehlert FJ. Naunyn-Schmiedeberg’s Arch Pharmacol. 2008;377:549. doi: 10.1007/s00210-008-0260-4. [DOI] [PubMed] [Google Scholar]
- 58.Griffin MT, Figueroa KW, Liller S, Ehlert FJ. J Pharmacol Exp Ther. 2007;321:1193. doi: 10.1124/jpet.107.120857. [DOI] [PubMed] [Google Scholar]
- 59.Black JW, Leff P. Proc R Soc London, Ser B. 1983;220:141. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- 60.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. ACS Chem Neurosci. 2012;3:193. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stahl EL, Zhou L, Ehlert FJ, Bohn LM. Mol Pharmacol. 2015;87:866. doi: 10.1124/mol.114.096503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shonberg J, Klein Herenbrink C, Lopez Munoz L, Christopoulos A, Scammells PJ, Capuano B, Lane JR. J Med Chem. 2013 doi: 10.1021/jm401318w. [DOI] [PubMed] [Google Scholar]
- 63.Lovell KM, Frankowski KJ, Stahl EL, Slauson SR, Yoo E, Prisinzano TE, Aube J, Bohn LM. ACS Chem Neurosci. 2015;6:1411. doi: 10.1021/acschemneuro.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aube J, Bohn LM. J Biol Chem. 2013;288:36703. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keov P, Lopez L, Devine SM, Valant C, Lane JR, Scammells PJ, Sexton PM, Christopoulos A. J Biol Chem. 2014;289:23817. doi: 10.1074/jbc.M114.582874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luttrell LM, Maudsley S, Bohn LM. Mol Pharmacol. 2015;88:579. doi: 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmid CL, Raehal KM, Bohn LM. Proc Natl Acad Sci USA. 2008;1079:105. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Groer CE, Schmid CL, Jaeger AM, Bohn LM. J Biol Chem. 2011;286:31731. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. Proc Natl Acad Sci USA. 2010;107:628. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. J Biol Chem. 2004;279:23214. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 71.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Proc Natl Acad Sci USA. 2009;106:9649. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kenakin T. J Pharmacol Exp Ther. 2011;336:296. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 73.Brust TF, Hayes MP, Roman DL, Watts VJ. Biochem Pharmacol. 2015;93:85. doi: 10.1016/j.bcp.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Proc Natl Acad Sci USA. 2003;100:10782. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kenakin T. Mol Pharmacol. 2015 [Google Scholar]
- 76.Correll CC, McKittrick BA. J Med Chem. 2014;57:6887. doi: 10.1021/jm401677g. [DOI] [PubMed] [Google Scholar]
- 77.Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR. Med Res Rev. 2014;34:1286. doi: 10.1002/med.21318. [DOI] [PubMed] [Google Scholar]
- 78.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Nature. 2000;408:720. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 79.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Science. 1999;286:2495. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 80.Raehal KM, Walker JK, Bohn LM. J Pharmacol Exp Ther. 2005;314:1195. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 81.Li Y, Liu X, Liu C, Kang J, Yang J, Pei G, Wu C. Int J Mol Sci. 2009;10:954. doi: 10.3390/ijms10030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang CH, Huang HW, Chen KH, Chen YS, Sheen-Chen SM, Lin CR. Br J Anaesth. 2011;107:774. doi: 10.1093/bja/aer291. [DOI] [PubMed] [Google Scholar]
- 83.Pasternak GW. J Clin Oncol. 2014;32:1655. doi: 10.1200/JCO.2013.53.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Massotte D, Kieffer BL. Essays Biochem. 1998;33:65. doi: 10.1042/bse0330065. [DOI] [PubMed] [Google Scholar]
- 85.Gaveriaux-Ruff C, Kieffer BL. Neuropeptides. 2002;36:62. doi: 10.1054/npep.2002.0900. [DOI] [PubMed] [Google Scholar]
- 86.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci USA. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. Mol Pharmacol. 2007;71:549. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 89.Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Drug Alcohol Depend. 2012;121:181. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen XT, Pitis P, Liu G, Yuan C, Gotchev D, Cowan CL, Rominger DH, Koblish M, Dewire SM, Crombie AL, Violin JD, Yamashita DS. J Med Chem. 2013;56:8019. doi: 10.1021/jm4010829. [DOI] [PubMed] [Google Scholar]
- 91.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. J Pharmacol Exp Ther. 2013;344:708. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 92.Soergel DG, Subach RA, Sadler B, Connell J, Marion AS, Cowan CL, Violin JD, Lark MW. J Clin Pharmacol. 2014;54:351. doi: 10.1002/jcph.207. [DOI] [PubMed] [Google Scholar]
- 93.Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, Soergel DG, Subach RA, Cook E, Skobieranda F. Pain. 2015 doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 94.Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu J, Gainetdinov R, Caron M. Proc Natl Acad Sci USA. 2008;105:13656. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kapur S, Mamo D. Prog Neuropsychopharmacol Biol Psychiatry. 2003;1081:27. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 96.Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. J Med Chem. 2012;55:7141. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Allen J, Yost J, Setola V, Chen X, Sassano M, Chen M, Peterson S, Yadav P, Huang X, Feng B, Jensen N, Che X, Bai X, Frye S, Wetsel W, Caron M, Javitch J, Roth B, Jin J. Proc Natl Acad Sci USA. 2011;108:18488. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agai-Csongor E, Domany G, Nogradi K, Galambos J, Vago I, Keseru GM, Greiner I, Laszlovszky I, Gere A, Schmidt E, Kiss B, Vastag M, Tihanyi K, Saghy K, Laszy J, Gyertyan I, Zajer-Balazs M, Gemesi L, Kapas M, Szombathelyi Z. Bioorg Med Chem Lett. 2012;22:3437. doi: 10.1016/j.bmcl.2012.03.104. [DOI] [PubMed] [Google Scholar]
- 99.Tschammer N, Bollinger S, Kenakin T, Gmeiner P. Mol Pharmacol. 2011;79:575. doi: 10.1124/mol.110.068106. [DOI] [PubMed] [Google Scholar]
- 100.Skeggs LT, Dorer FE, Kahn JR, Lentz KE, Levine M. Am J Med. 1976;60:737. doi: 10.1016/0002-9343(76)90888-3. [DOI] [PubMed] [Google Scholar]
- 101.Burnier M. Circulation. 2001;103:904. doi: 10.1161/01.cir.103.6.904. [DOI] [PubMed] [Google Scholar]
- 102.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, Lefkowitz RJ. Proc Natl Acad Sci USA. 2006;103:16284. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, Lark MW. J Pharmacol Exp Ther. 2010;335:572. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 104.Kim KS, Abraham D, Williams B, Violin JD, Mao L, Rockman HA. Am J Physiol Heart Circ Physiol. 2012;303:H1001. doi: 10.1152/ajpheart.00475.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Violin JD, Crombie AL, Soergel DG, Lark MW. Trends Pharmacol Sci. 2014;35:308. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 106.Stephenson RP. Br J Pharmacol Chemother. 1956;11:379. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weiss JM, Morgan PH, Lutz MW, Kenakin TP. J Ther Biol. 1996;181:381. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- 108.Deupi X, Kobilka BK. Physiology (Bethesda) 2010;25:293. doi: 10.1152/physiol.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Katritch V, Cherezov V, Stevens RC. Annu Rev Pharmacol Toxicol. 2013;53:531. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kenakin T. BMC Pharmacol Toxicol. 2012;13:3. doi: 10.1186/2050-6511-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Nature. 2013;494:185. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 112.Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Mol Pharmacol. 2004;66:106. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- 113.Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. J Neurosci. 2007;27:11614. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bruchas MR, Macey TA, Lowe JD, Chavkin C. J Biol Chem. 2006;281:18081. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Raehal KM, Schmid CL, Groer CE, Bohn LM. Pharmacol Rev. 2011;1001:63. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Whalen E, Rajagopal S, Lefkowitz R. Trends Mol Med. 2011;17:126. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shukla AK, Singh G, Ghosh E. Trends Biochem Sci. 2014;39:594. doi: 10.1016/j.tibs.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 118.McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. Mol Pharmacol. 2010;78:756. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alhaddad H, Cisternino S, Decleves X, Tournier N, Schlatter J, Chiadmi F, Risede P, Smirnova M, Besengez C, Scherrmann JM, Baud FJ, Megarbane B. Crit Care Med. 2012;40:3215. doi: 10.1097/CCM.0b013e318265680a. [DOI] [PubMed] [Google Scholar]
- 120.Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, Schultz G. Proc Natl Acad Sci USA. 1996;93:116. doi: 10.1073/pnas.93.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Offermanns S, Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G, Jakobs KH. Mol Pharmacol. 1994;45:890. [PubMed] [Google Scholar]
- 122.Dupre DJ, Robitaille M, Rebois RV, Hebert TE. Annu Rev Pharmacol Toxicol. 2009;49:31. doi: 10.1146/annurev-pharmtox-061008-103038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbe JC, Miller GJ, Hebert TE. Pharmacol Rev. 2013;65:545. doi: 10.1124/pr.111.005603. [DOI] [PubMed] [Google Scholar]
- 124.Lin Y, Smrcka AV. Mol Pharmacol. 2011;80:551. doi: 10.1124/mol.111.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]