

Visual Abstract

Key Words: phagocytosis, CD47, macrophage
Abbreviations and Acronyms: IAP, integrin associated protein; MI, myocardial infarction; CM, cardiomyocyte; LV, left ventricular; Mϕ, macrophage; NO, nitric oxide
Highlights
-
•
New therapies are needed to enhance myocardial salvage after myocardial ischemia and reperfusion.
-
•
Human and murine hearts express CD47 and calreticulin that increased after ischemia and reperfusion.
-
•
Phagocytic efficiency of dying cardiac myocytes was enhanced after antibody-mediated blockade of either myocyte CD47 or macrophage CD47-ligand, SIRPα.
-
•
After ischemia and reperfusion, enhancement of dead myocyte clearance by macrophages, after CD47 blockade, improved inflammation resolution, reduced infarct size, and preserved cardiac systolic function.
Summary
Our data suggest that, after a myocardial infarction, integrin-associated protein CD47 on cardiac myocytes is elevated. In culture, increased CD47 on the surface of dying cardiomyocytes impairs phagocytic removal by immune cell macrophages. After myocardial ischemia and reperfusion, acute CD47 inhibition with blocking antibodies enhanced dead myocyte clearance by cardiac phagocytes and also improved the resolution of cardiac inflammation, reduced infarct size, and preserved cardiac contractile function. Early targeting of CD47 in the myocardium after reperfusion may be a new strategy to enhance wound repair in the ischemic heart.
Although clinical management of acute myocardial infarction (AMI) has significantly reduced morbidity and mortality, the consequence of these advances include an emerging incidence of post-MI heart failure (1). In turn, new approaches that enhance cardiac wound healing and are complementary to the current standards of care have the potential to improve on left ventricular (LV) systolic function (2).
A critical determinant of heart failure susceptibility after AMI is infarct size (3). Infarct size and loss of nonregenerative cardiomyocytes (CMs) by acute necrosis directly correlates with ventricular dysfunction and heart failure (4). Infarct necrosis can also expand during reperfusion injury or after maladaptive inflammation, leading to accelerated and adverse ventricular remodeling 5, 6.
During wound healing, clearance of dying cells must occur efficiently to prevent secondary necrosis and prolonged inflammation. Efficient phagocytic clearance of apoptotic cells by macrophages (Mϕs) via efferocytosis, actively programs cellular inflammation-resolution and tissue repair signaling pathways (7). This clearing is particularly important in the heart, where inefficient removal of necrotic CMs may lead to collateral myocyte loss and infarct expansion 5, 8. In a previous study, we experimentally linked the efficiency of phagocytic clearance to infarct size expansion and discovered that Mϕs defective for efferocytosis led to deteriorated cardiac function after MI (9). These studies begged additional questions, including the natural efficiency of CM clearance by phagocytic cells and whether strategies that target phagocytic enhancement might enhance heart healing. In this study, we focused on CM-intrinsic factors, namely CD47, that regulate phagocytic interactions with Mϕs (10).
Dying cell engulfment requires cell surface presentation of so-called “eat-me” signals, such as phosphatidylserine and calreticulin, which must occur in tandem with downregulation of antiphagocytic “don’t-eat-me” markers, including CD47 (10). Independent of programmed cell death per se, blockade of CD47 and concomitant overexpression of calreticulin can permit phagocyte ingestion of viable cells. In addition to experimental blockade (11), natural reduced expression of CD47 occurs in vivo, for example, in senescent erythrocytes; this is associated with red blood cell erythrophagocytosis and clearance from the circulation by resident tissue Mϕs (12). A recent study published in the journal Nature (13) highlights the potential of CD47 blocking strategies in atherosclerotic cardiovascular disease.
Although CD47 expression has been profiled in neonatal CMs (14), much less is known about its expression and function in adult CMs. CD47 deficiency is associated with enhanced cardiac performance after administration of vasoactive agents (15). In skeletal muscle, CD47 can regulate PGC-1α–dependent mitochondrial biogenesis through recognition of its thrombospondin ligand (16). CD47 has also been linked to signaling in smooth muscle cells (17). The significance of CD47 after AMI is unclear.
Our preliminary mechanistic studies suggested elevated CD47 expression in CMs relative to other cardiac cells, leading us to hypothesize that CMs may exhibit natural resistance to phagocytic removal after injury. To test in principle whether acute CD47 blockade during reperfusion of ischemic myocardium enhances cardiac repair, in association with CM phagocytosis, we used an experimental MI model in mice and injected anti–CD47-blocking antibodies during reperfusion. Importantly, CD47 targeting was limited to a single time and dose to focus actions during the height of CM phagocytosis and to avoid targeting alternative postacute CD47 signaling pathways.
Materials and Methods
Human samples
Human cardiac samples were obtained from the Northwestern University Department of Pathology and the office of the Medical Examiner of Chicago. Under the protocol approved by the institutional review board at Northwestern University (#STU00079445), archived formalin-fixed paraffin-embedded cardiac autopsy samples were obtained and analyzed at Northwestern University Feinberg School of Medicine, as previously described (18). Specifically, autopsy cases were evaluated by a combination of factors, including serum cardiac enzymes and gross evidence at time of autopsy. Gross evidence consisted of pale or yellow myocardial areas with or without a hyperemic border. Age of infarct ranged from ∼6 h to several days. Time from death to autopsy ranged from 18 h to 7 days. Autopsy tissue blocks of myocardium from acutely infarcted areas versus noninfarcted areas and noninfarcted individuals were fixed in 10% formalin and serial 6-μm sections were stained with hematoxylin and eosin. In this study, a total of 22 human cardiac autopsy samples were used, 11 from AMI cases and 11 from non-AMI cases.
Murine samples
Murine hearts were perfused and fixed with 10% phosphate-buffered formalin at physiological pressures. Hearts were cut transversely, parallel to the atrioventricular groove and coronary sulcus. Fixed hearts were embedded in paraffin at the Northwestern University Mouse Histology and Phenotyping Laboratory. Blocks were serially cut 6 μm apart. Alternating sections were stained with hematoxylin and eosin. For frozen sections for immunohistochemistry, samples were rinsed and incubated overnight in 7% sucrose and frozen in freezing medium unless otherwise indicated. Transverse cryosections were cut at a thickness of 10 μm on a Leica cryostat and placed on super frost plus-coated slides for analysis.
Mice
B6; D2-Tg(Myh6*-mCherry)2Mik/J mice were from Jackson Laboratory (stock No: 021577, Bar Harbor, Maine). The alphaMHC-mCherry transgene has a modified mouse alpha myosin heavy chain promoter sequence directing cardiac-specific mCherry expression in CMs. Mice were housed in temperature- and humidity-controlled environments and kept on a 12:12-h day–night cycle with access to standard mouse chow and water ad libitum. All studies were approved and reviewed by the Institutional Animal Care and Use Committee at Northwestern University (Chicago, Illinois). Cd47-/- mice were obtained from the Jackson Laboratory (stock #003173).
Materials and antibodies
Tissue culture dishes were from Corning (Corning, New York), and fetal bovine serum was from Gibco (Gaithersburg, Maryland). Chemical reagents were from Sigma Chemical Co. (St. Louis, Missouri), unless stated otherwise.
Antibodies
CD47 monoclonal and polyclonal antibodies were as previously described (19). Anti-CD68 was obtained from Abcam (ab125212, Cambridge, United Kingdom). Monoclonal mouse anti-Desmin (clone DE-U-10) from mouse ascites fluid was from Sigma (D1033). Rat antimouse Mϕ antibody Mac-2 was from Cedarlane (Burlington, Ontario, Canada). Rat antineutrophil antibody was from Serotec (Raleigh, North Carolina).
Primers for semiquantitative and real-time PCR in this study
Ccl2: F: CCT GGA TCG GAA CCA AAT GA, R: ACC TTA GGG CAG ATG CAG TTT TA. TNF-α: F: CGG AGT CCG GGC AGG T, R: GCT GGG TAG AGA ATG GAT GAA CA. IL-6: F: GAGGATACCACTCCCAACAGACC, R: AAG TGC ATC ATC GTT GTT CAT ACA, IL-10: F: GCC AAG CCT TAT CGG AAA TG, R: GGG AAT TCA AAT GCT CCT TGA T. Gapdh: F: GGT GGC AGA GGC CTT TG, R: TGC CCA TTT AGC ATC TCC TT. hCD47: F: 5’-AGC TCT AAA CAA GTC CAC TGT CCC-3’. hCD47: R: 5’-TCC TGT GTG TGA GAC AGC ATC ACT-3’. Control, hTBP: F 5’-TGA GTT GCT CAT ACC GTG CTG CTA-3′, hTBP: R: 5’-CCC TCA AAC CAA CTT GTC AAC AGC-3′.
Ischemia and reperfusion
Ischemia and reperfusion injury was performed in male and female B6 mice 8 to 12 weeks of age. Ischemia was produced by occluding the left coronary artery with a 7-0 silk suture on a tapered tube for 45 min as we have previously described (20) followed by reopening of the ligature to allow for reperfusion. Mice were anesthetized with tribromoethanol (Avertin) and received buprenorphine (0.1 mg/kg subcutaneously) before surgery and after survival surgery, every 12 h up to 48 h for pain management.
Laser capture microdissection
RNA from myocardial sections was captured by laser capture microdissection using a Zeiss P.A.L.M. laser microdissection system as previously described (21). Total RNA was isolated using the RNAqueous-Micro kit from Ambion (Waltham, Massachusetts) and reverse-transcribed into cDNA using SuperScript III First-Strand Synthesis Mix (Invitrogen, Carlsbad, California).
Semiquantitative and quantitative reverse transcriptase polymerase chain reaction
Hearts were snap-frozen for RNA. Total RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany). cDNA was synthesized from 4 μg of total RNA using oligo (dT) and Superscript II (Invitrogen). cDNA was subjected to quantitative reverse transcriptase polymerase chain reaction amplification using a SYBR Green PCR Master Mix (Applied Biosystems, Foster City, California).
Administration of anti-CD47 via intramyocardial delivery
Five minutes before the ligature was released, 100 μg anti-CD47 (mIAP301) monoclonal antibody was injected at 3 sites into the ischemic myocardium, just distal to the coronary ligation with 30 μl of antibody solution per site. At the end of 45 min of occlusion, the ligature was released, and the heart was reperfused.
Adenovirus administration
CM specific-CD47 expression was induced as previously described (22). Adeno-associated virus1-cTNTp-GFP-2A-mCD47-WPRE and adeno-associated virus1-cTNTp-GFP were from Vector Biolabs (Malvern, Pennsylvania). We administered 1 × 1012 adeno-associated virus into 6-week-old B6 mice and hearts were harvested for immunohistochemical staining to check for increased protein.
Flow cytometry preparation (cardiac)
Flow cytometric analysis of cells after MI
Mice were anesthetized with isoflurane after MI. Peripheral blood was drawn via retro-orbital bleed with citrate solution (100 mmol/l Na-citrate, 130 mmol/l glucose, pH 6.5), as an anticoagulant. Spleens were removed, triturated in Hank's Balanced Salt Solution (Mediatech, Inc., Manassas, Virginia) at 4°C with the end of a 3-ml syringe and filtered through a nylon mesh. Cell suspension was pelleted and red blood cells were lysed with ACK lysis buffer and resuspended in flow cytometry buffer.
For infarct tissue
Hearts were harvested, perfused with saline to remove peripheral cells, minced with fine scissors, and placed into a cocktail of collagenase and DNase (Sigma-Aldrich, and Worthington Biochemical Corp., Lakewood, New Jersey) and shaken at 37°C for 1 h. Cells were triturated through nylon mesh or 70 μm strainer and centrifuged at 15 min at 500×g and 4°C. Total cell numbers were determined by Trypan blue staining. The resulting single-cell suspensions were rinsed with Hank's Balanced Salt Solution supplemented with 0.2% (wt/vol) bovine serum albumin and 1% wt/vol fetal calf serum. Flow cytometry was performed as previously described (9) and with indicated antibodies in figure legends.
Cardiac infarct size measurement
At time of humane killing, mice were anesthetized with a ketamine hydrochloride and xylazine hydrochloride solution C-IIIN from Sigma (K113). The abdominal wall below the rib cage was opened and the diaphragm was cut after lifting the sternum with tweezers. The lower part of rib cage was removed to expose the heart for removal. Fluorescent latex beads were perfused retrograde to determine the area at risk. Subsequently, hearts were sliced into 1-mm transverse cross sections and then incubated with 1% 2,3,5-triphenyl-tetrazolium chloride solution (Sigma T8877) in saline for 15 min at 37°C. Hearts were then placed in 10% formalin. Viable tissue stained red and infarcted tissue white. Heart sections were weighed. Digital photomicrographs were taken and images of infarcts were blinded for analysis. Infarct size was determined as a percentage of the left ventricle. Infarct area and the total area of LV myocardium were traced manually in the digital images. Infarct size, expressed as a percentage of the area at risk, was calculated by dividing the sum of infarct areas from all sections by the sum of LV areas from all sections and multiplying by 100.
Gadolinium-enhanced magnetic resonance imaging
Digital photomicrographs were taken and images of infarcts were blinded for analysis. Infarct size was determined as a percentage of the left ventricle. Infarct area and the total area of LV myocardium were traced manually in the digital images. Infarct size, expressed as a percentage of area at risk, was calculated by dividing the sum of infarct areas from all sections by the sum of LV areas from all sections and multiplying by 100. Magnetic resonance imaging was performed on a 7-T Clinscan system (Bruker, Ettlingen, Germany) equipped with actively shielded gradients (BGA12) with a full strength of 440 mT/m and a slew rate of 3,440 mT/m/ms. Images were acquired using a receive only 4-channel phased array radiofrequency coil (using a body coil for radiofrequency transmission) and an magnetic resonance-compatible physiological monitoring and gating system for mice (SA Instruments, Inc., Stony Brook, New York). The magnetic resonance imaging was performed on day 1 after the MI. The magnetic resonance imaging protocol included multislice localizer imaging to select a short axis LV slice. Late gadolinium imaging was performed using a cardiorespiratory gated multislice inversion recovery sequence covering the entire LV from base to apex. Typical imaging parameters included TE/TR, 2.3/4.7 ms; slice thickness, 1 mm; number of slices, 7 to 8; inversion time, 550 ms; number of averages, 2; spatial resolution, 0.2 × 0.2 mm2; flip angle, 300; and scan time, 7 min. Gadolinium (0.5 mmol/kg body weight) was injected intraperitoneally and late gadolinium enhancement imaging was started 20 minutes after the injection. Image analysis was performed on a workstation using Segment software (Medviso, Lund, Sweden). Using the software, epicardial and endocardial contours were drawn and Otsu thresholding method was used to automatically delineate the infarct zone. Infarct size was calculated as a percentage of the total LV mass.
Cardiac efferocytosis assays
Transgenic Myh6-driven mCherrry mice underwent the surgery as described and flow cytometry of myocardial extract was performed after the MI to identify phagocyte, mCherry double positive cells, as previously described (18). Cells were trypsinized to dissociate cell–cell interactions and reveal only internalized mCherry signal. Cardiac phagocytosis was also measured in situ by counting the number of free and ingested apoptotic cells in individual sections. Apoptotic cells were considered “free” when they were not surrounded by, or in contact with, Mϕs. The analysis was performed in a blinded fashion by 2 independent observers.
Echocardiography
Two-dimensional transthoracic echocardiography was performed using a 25-MHz probe (Vevo 770, Visualsonics, Toronto, Canada) with mice in a supine position. The mouse chest was treated with a depilatory agent and mice were anesthetized with isoflurane (2% in O2) and heart rate was monitored. Left ventricle dimensions were assessed on Visualsonics software in a short axis and long axis views from the mid-left ventricle, just below the papillary muscles. In short axis, 2D (B-mode) images were taken every millimeter starting from apex to base; subsequently, an M-mode was taken 1 mm before, at, and after the papillaries. All measurements were made in 2 to 6 consecutive cardiac cycles and the averaged values used for analysis. The LV end-diastolic and end-systolic dimensions and the thickness of the interventricular septum and posterior wall were made from the M-mode tracings and fractional shortening (ratio between diameter of the LV in diastole or relaxed versus diameter when contracted) was also measured as an indicator of systolic function. Dimensions were measured between the anterior wall and posterior wall. Diastolic measurements were made at the point of minimal cavity dimension, using the leading edge method of the American Society of Echocardiography.
Cell types
For isolation of primary Mϕs, when indicated, bone marrow–derived Mϕs were isolated from murine femurs and cultured in L-cell–conditioned medium, containing macrophage colony-stimulating factor, as described elsewhere (9). Special care was taken to cultivate Mϕs in normoglycemic (5.5 mmol/l glucose) conditions in Dulbecco's modified Eagle's medium culture and to carefully monitor medium pH and lactate accumulation. For peritoneal Mϕs, thioglycolate-elicited Mϕs were prepared as described previously (23). The isolation of cardiomyocytes and cardiac fibroblasts from adult hearts was performed as previously described (24). In brief, 6- to 8-week-old B6 mice were injected intraperitoneally with 0.5 ml heparin solution before being anesthetized with tribromoethanol. The excised heart was immediately mounted to a modified Langendorff apparatus and perfused with perfusion buffer, digestion buffer, and stopping buffer in sequential order. After enzyme dissociation, CMs were enriched through a gradient of calcium reintroduction buffer, and were plated accordingly for downstream experimentation.
In vitro apoptosis
To compare relative efferocytosis efficiency across cell types, a common apoptotic stimulus was selected: staurosporine. CMs, cardiac fibroblasts, and Mϕs were incubated with 0.5 μmol/l staurosporine. The morphology of apoptotic blebbing was quantified and confirmed by annexin V staining. Staurosporine was used as a robust means to induce apoptosis across different cell types (25) and also owing to our empirical observations that CMs are relatively resistant to ultraviolet light–induced apoptosis. Fibroblasts, primary Mϕs, and CMs were induced to undergo apoptosis with 0.5 μmol/l of staurosporine treatment for 2 h followed by harvest of nonadherent floating cells (approximately 70% of the total cells). Apoptosis was confirmed by microscopic observation of cell size, morphology, and flow cytometric analysis of phosphadylserine exposure by binding of annexin V–FITC (catalog no. 556419, 1:50, BD Pharmingen, Franklin Lakes, New Jersey) according to the recommendations of the manufacturer (14).
In vitro efferocytosis assay
Peritoneal Mϕs were plated at 2 × 105 in 24-well plates 1 day before the experiment. Apoptotic cells were prelabeled with Calcein AM before apoptosis was induced. The Mϕ to CM ratio was 1:5. Cells were cocultured for 1 hour before 3 subsequent rinses to remove nonengulfed cells. The phagocytes were then fixed with 4% paraformaldehyde and analyzed by fluorescence microscopy.
Statistical analysis
Data are presented as mean ± SEM Data fit a normal distribution. Thus, the Student t test was used to determine statistical significance and p < 0.05 were considered significant. SPSS Statistics (SPSS, Inc., Chicago, Illinois) version 17 was used for statistical analyses.
Results
Evidence for CM-specific CD47 expression in humans and CD47-induction after experimental MI
A previous study characterized CD47 levels from total ventricular biopsy extracts (26); however, our understanding of CM-specific expression in humans is limited. We, therefore, first used immunohistochemistry of human hearts after autopsy to test for CM-specific CD47. As shown in Figure 1A, human (Supplemental Table 1, Supplemental Figures 1 and 2) myocardial CD47 expression was found to colocalize to CMs; reduced colocalization was identified on other human cardiac cells, such as endothelial cells (Supplemental Figure 3). In specimens from patients who had succumbed to MI, CD47 staining intensity was interestingly increased (Figure 1B). Some of this CD47 colocalized with prophagocytic ligand calreticulin (Supplemental Figure 4), leading us to hypothesize that increased CD47 might interfere with phagocytic efficiency (10). Indeed, increased human CM CD47 levels after acute ischemia is consistent with an impediment to dying myocyte phagocytic clearance and healing. Therefore, we set forth to measure CM CD47 under experimental ischemia and test if CD47 blockade could enhance myocyte phagocytosis and cardiac repair. Similar findings were seen in experimental mouse hearts, where the CD47 signal colocalized with CMs, in contrast with vimentin-positive cardiac fibroblasts (Figures 1C and 1D). To measure CM CD47 levels after MI, experimental mice were subjected to coronary occlusion, as described in the Methods. Interestingly, CD47 levels after MI were significantly increased in the ischemic area at risk versus the remote myocardium (Figure 1E). Ex vivo analysis corroborated these findings, revealing significant CD47 protein in isolated adult CMs. Interestingly, CD47 protein was further induced after treatment with stimuli that induced CM death (Supplemental Figure 5). Cell death-induced CD47 was confirmed on the cell surface, and in contrast with primary cardiac fibroblasts.
Figure 1.

CM CD47 Levels in Human Hearts and After Murine Experimental MI
(A) Human myocardial samples after autopsy were assessed for pathological evidence of myocardial infarction (MI) as described in Materials and Methods. MI and non-MI sections were stained with antibodies against CD47 (red) and CM-marker alpha-actinin (green). Representative images are shown. Scale bar is 50 μm. (B) Quantitation of human CD47 fluorescent signal intensity from (A) in non-MI versus MI specimens. N = 10 per group. p = 0.03 between groups. (C) Mouse myocardial samples were stained with antibodies against CD47 (red from Abcam ab175388), alpha-actinin (blue from Abcam ab9465), and fibroblast marker vimentin (green from Abcam ab11256). MERGE is of red CD47 and green vimentin (green). Scale bar is 100 μm. (D) Quantitation of mouse CD47 fluorescent signal intensity that colocalized with cardiac fibroblasts (vimentin) versus that which colocalized with cardiomyocytes (alpha-actinin). n = 10 per group. p = 0.009 between groups. (E) Relative gene expression of CD47 after laser capture microdissection of ischemic areas at risk (AAR) versus remote (outside the AAR) myocardium. The number of animals is 8 per group. p = 0.04. I/R= ischemia/reperfusion.
Ex vivo uptake of CM apoptotic bodies by Mϕs is regulated by the CD47–SIRPα axis
We previously reported that phagocytosis is a significant early regulator of cardiac repair after an MI (9), and furthermore that CMs are engulfed by Mϕs at a lower efficiency than other cell types that turnover after an MI (18). In that the Henson (10) and Weissman groups (11) have shown the potential of blocking CD47 to enhance phagocytosis, we reasoned that increased CD47 might render CMs particularly sensitive to CD47–phagocytic regulation. To test the potential of CD47 blockade for rescuing basal CM phagocytosis inefficiency, we added anti-CD47 blocking antibodies to cocultures of primary adult differentiated murine CMs and Mϕs. Figure 2A shows that CD47 blockade significantly heightened CM efferocytosis, over and above isotype control, and specifically for apoptotic CMs (Figure 2B, Supplemental Figure 6). Efferocytosis enhancements required CD47, because CMs prepared from Cd47-/- mice were resistant to the effect of anti-CD47 blocking and Cd47-/- CMs were more susceptible to phagocytic clearance at baseline (Figure 2C). These findings predicted similar responses by blocking CD47 ligand, Mϕ SIRP-1α. Indeed, anti–SIRP-1α monoclonal antibody, added to Mϕs, also increased phagocytosis (Figure 2D) of Cd47+/+ CMs. Similar findings were found when inducing induced pluripotent stem cell derived cardiomyocytes to apoptosis and cocultivating with human Mϕs (Supplemental Figure 7).
Figure 2.

Blockade of the CD47-SIRP-1α Pathway Specifically Enhances CM Phagocytosis by Mϕs in Culture
(A) Cocultivations of macrophages (Mϕs) and dying adult mouse ventricular CMs with or without anti-CD47 (a47) versus isotype (C). The CMs were labeled with red R18 dye. The Mϕs were labelled with calcein-AM green. (Left) Scale bar, 300 μm. (Right) Scale bar is 20 μm. (B) Quantification of percent phagocytosis in CMs versus cardiofibroblasts. Zoom is 600%. *p = 0.01 relative to CM “c”. aCD47, blocking antibody; c, isotype control. (C) Percent phagocytosis of Cd47+/+ versus Cd47-/- CMs by Mϕs. *p < 0.03. (D) Similar to in (A), except that the antibody treatment is anti–SIRP-1α versus isotype (Rat IgG2) versus anti-Fc control. Here, CMs were labelled green and Mϕs are not labelled. *p = 0.04 versus control. Scale bar is 50 μm. CF = cardiac fibroblast; other abbreviations as in Figure 1.
Having tested proof of principle ex vivo, we next set out to test physiological relevance. Because of the potential therapeutic implications of our approach, we decided on a reperfusion model of acute murine MI. Also, because of potential nonphagocytic pathways impacted by CD47 signaling, we further decided to limit the blocking of CD47 to acute treatment. Thus, germline deficiency was neither a favorable strategy for our questions nor a viable approach, owing to previously published studies that indicated significant compensatory responses in Cd47-/- mice (27).
In vivo testing
Our experimental outline is shown in Supplemental Figure 8. Experimental B6 mice were subjected to ligation of the left anterior descending artery, followed by reperfusion, with or without anti-CD47 treatment. After performing a titration of anti-CD47 concentrations to find the optimal dose for affecting infarct size, we settled on 100 μg. In consideration of potential CD47–thrombospondin interactions, no significant changes in markers of neovascularization were measured (Supplemental Figure 9B). Furthermore, given that global Cd47-/- knockout mice have been reported to exhibit an increase in arterial diastolic and systolic pressure and that CD47 has also been shown to suppress nitric oxide (NO) signaling in vascular cells after long-term thombospondin binding (28), we assessed hemodynamics. However, no differences in systolic blood pressure were found after CD47 blockade (Supplemental Figure 9A).
Acute CD47 blockade during reperfusion after MI enhances myocardial phagocytosis
To specifically measure effects on myocardial phagocytosis, we used mice transgenic for Mhc6-mCherry, the fluorescence of which is restricted to CMs (18). In line with our hypothesis, mice administered anti-CD47 blocking antibody during reperfusion (Figure 3A), exhibited an increased profile of myocardial CD11b+ phagocytes with associated mCherry fluorescence (Figure 3A and 3B), an indicator of CM phagocytosis. Strikingly, mice administered adeno-associated virus CD47, which increased CD47 levels, exhibited reduced markers of myocardial phagocytosis (Figure 3C). Overexpression of CD47 did not affect infarct size, potentially owing to phagocytosis-independent signaling (Supplemental Figure 10). Importantly, anti-CD47 blocking strategies also led to other predicted consequences of enhanced clearance, namely, faster kinetics of cardiac innate immune cell resolution (Figure 3D), such as reduced Ly6cHI monocytes and heightened Ly6cLO monocytes cells (29). Because no differences in neutrophil turnover were found, this suggested specificity for CM uptake. Importantly, administration of antibodies did not affect initial levels or recruitment of blood-borne monocytes (Supplemental Figure 11).
Figure 3.

In Vivo CD47 Blockade Versus CD47 Induction Regulates CM Phagocytosis Efficiency and Cardiac Inflammation Resolution
(A) Flow cytometric measure of Mϕ engulfment of transgenic mCherry CMs. Gating strategy is shown gating CD11b myeloid cells that are mCherry+ and Ly6g low (non-neutrophil phagocytes or Mϕs). Mice were subjected to 45 minutes of ischemia followed by injection of isotype IgG control (ctrl) versus CD47 monoclonal antibodies in phosphate-buffered saline, just before unoccluding the left anterior descending coronary artery. Right image depicts sorted Mϕs with internalized CM mCherry. (B) Dot plots, histogram, and bar graph of flow cytometry analysis of CM mCherry-association with myocardial CD11b+ phagocytes after ischemia and reperfusion of transgenic Myh6-mCherrry mice. (C) Adeno-associated virus CD47 (myocyte Cre) was injected intravenously before surgery and fluorescent intensity (scale bar, 100 μm) is a marker of adeno-associated virus expression and quantified as a function of viral titer. To the right is cardiac phagocytosis enumerated as in (B and C). (D) Inflammation resolution kinetics at indicated day (D) time points. Flow cytometric analysis of (CD45-hi CD11b-hi Ly6g-lo) Ly6c and CD11c cells in the myocardium. (Left) Percentage of CD45-hi CD11b-hi Ly6g-hi neutrophils and polymorphonuclear neutrophils. (Right) (CD45-hi CD11b-hi Ly6g-lo) Ly6C-hi monocytes and Ly6c-lo Mo/Mϕs. *p ≤ 0.05 relative to ctrls. ns, not significant. Abbreviations as in Figures 1 and 2.
The effects of acute anti-CD47 on infarct size, systolic function, and cardiac scarring
Impressively, singular administration of anti-CD47 suppressed infarct size and cardiac troponin release (Figures 4A to 4C). Blockade of CD47 also enhanced systolic cardiac function as indicated by increased LV ejection fraction (Figures 5A to 5C). Anti-CD47 monoclonal antibody treatment limited ventricular remodeling, because treated mice exhibited reduced scarring, demonstrated by a 2-fold reduction in collagen fraction after Masson’s Trichrome staining (Figure 5D). Taken together, our data suggest a significant potential for incorporating CD47 blocking strategies toward the amelioration of cardiac injury. This approach is unique in its association and targeting of phagocytosis-associated repair.
Figure 4.
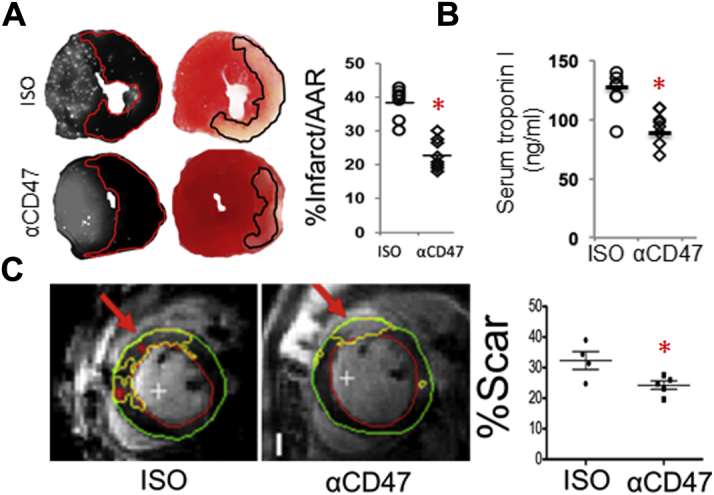
Intramyocardial Injection of Anti-CD47 Reduces Acute Infarct Size
(A) Fluorescent microspheres were used to measure area at risk (left) at 24 h after reperfusion. Staining with 2,3,5-triphenyl-tetrazolium chloride was performed to measure infarct size (right). Shown here are representative images of myocardial short axis slices and quantification. There were 10 animals in each group (p = 0.0001). (B) Serum troponin levels after anti-CD47 treatment at 24 h (p = 0.01). (C) CD47 monoclonal antibody injection reduced infarct size as measured by gadolinium enhanced magnetic resonance imaging at 24 h after reperfusion. Example segmentation of the late gadolinium enhancement images. The infarct zones are indicated by the red arrows. The green contours represent the epicardial border, red contours represent the endocardial border, and the yellow contours depict the infarct zones as delineated by automatic thresholding. To the right is quantification of infarct size; n = 7 vs 7. p < 0.05 versus control.
Figure 5.

Intramyocardial Injection of Anti-CD47 Improves Systolic Function and Reduces Fibrosis
(A) Assessment of heart function after ischemia-reperfusion in CD47-antagonized mice at indicated days (d) after I/R. Images are M-mode tracings. Control average heart rate = 474 and anti-CD47 heart rate = 507. (B) Echocardiography analysis of percent ejection fraction. Day 7, p = 0.03; day 21, p = 0.03; day 28, p = 0.03 (comparisons are between ISO and anti-CD47 at each indicated day). (C) Left ventricular (LV) systolic and diastolic dimensions. The LV anterior wall diastolic thickness (LVAWDd) (mm) on day 28 from IsoIgG and α-CD47 antibody-injected mice (*p = 0.04). (D) Hearts harvested 21 to 28 days after I/R for staining with Masson’s trichrome blue. ISO, isotype control. Scale bar, 1.5 mm. *p = 0.04 relative to control. The number of animals in each group = 10 versus 10. Abbreviations as in Figure 1.
Discussion
Although cardiac reperfusion therapies have reduced patient mortality after AMI, improved survival has also led to increases in the frequency of heart failure. A contributing factor to both the benefit and detriment of cardiac repair after ischemia and reperfusion is the innate immune response. Immune cells directly interact with the injured tissue and this includes through binding to the molecular ligand CD47 on parenchymal cells. Herein, we target CD47 with blocking antibodies to enhance phagocytic clearance of dying CMs and in turn, augment cardiac wound healing after MI. Although the gold standard to test molecular requirements is the genetic knockout, Cd47 germline deficiency tolerizes mice to the CD47–Sirp1α–phagocytosis pathway, rendering phagocytosis CD47 independent (27). In this context, CD47-blocking strategies have most notably been pioneered by the Weissman group and to promote tumor cell uptake (11). During wound healing, CD47 inhibition accelerates wound closure after dermal thermal injury in mice, independent of CD47 ligand thrombospondin-1 (30). Herein, a similar approach is tested in the heart, but with distinct mechanistic and clinical implications. Although multiple cell types turnover after MI 31, 32, 33, distinct myocardial patterns of expression during ischemia (Figure 1, Supplemental Figure 4) and CM-specific mechanisms may render CMs uniquely sensitive to the effects of CD47-blockade. Also, dying cells must both downregulate antiphagocytic signals such as CD47 and concomitantly induce prophagocytic signals. Thus, strategies that target CD47 for phagocytosis should be specific to dying cells, which further require induction of prophagocytic signals.
Previous studies suggest that phagocytosis of adult CMs by Mϕs is naturally inefficient (18). Indeed, diseases of aging such as MI do not exert evolutionary pressure to optimize CM clearance after severe MI. During aging, phagocytosis is compromised (34), including in the heart (35). In the case of CMs, we speculate that low phagocytic efficiency is linked to the low regenerative potential of adult myocytes and could require enhanced mechanisms to prevent phagocytic removal. For example, our data indicate that CD47 surface levels are uniquely heightened after apoptosis (Supplemental Figure 5) and our unpublished experiments suggest novel interactions with membrane scaffolding proteins, which may in turn regulate CD47 cell surface half-life and interactions with other prophagocytic molecules, including calreticulin (Supplemental Figure 4). In the CM, the function, spatial organization, and interactome of CD47 remains understudied.
Phagocytosis-targeting approaches might be enhanced by the administration of opsonins. For example, natural defects in efferocytosis in cardiovascular disease have been hypothesized to be the result of deficient Gas6 expression (36), a critical bridging molecule that facilitates Mϕ-mediated engulfment. Similarly, a polymorphism associated with coronary artery disease leads to reduced calreticulin expression on vascular smooth muscle cells and decreased efferocytsosis (37). On the Mϕ side, blocking SIRP-1α may also enhance phagocytic efficiency. This approach is attractive because of the potential to target peripheral blood phagocytes, destined for the heart. Recent advances include the engineering of high-affinity SIRPα variants, which are capable of enhancing CD47 blockade (38). However, other studies indicate SIRP-1α is critical to platelet activation (39) and neutrophil and Mϕ migration (40), potentially precluding their efficacy in this regard. Furthermore, cardiac SIRP-1α protects against myocardial hypertrophy through disruption of TLR4 signaling (41).
Study limitations
This study has several limitations, including the restricted extent to which murine studies can be extrapolated to humans. Further studies are necessary to test CD47 blockade in large animal models and examine conservation and reproducibility across species. Also, a closer look at our echocardiography data reveals that the slope of the reduction in the ejection fraction in the anti-CD47 cohort was greater than that in the control group (Figure 5). Therefore, additional studies should test the chronic benefit versus the transient nature of this treatment. It is important to consider phagocytosis-independent effects of CD47 blockade. For instance, CD47 targeting during tissue ischemia and reperfusion injury is associated with angiogenesis (42) and efferocytosis in heart can trigger vascular endothelial growth factor A (43). However, we did not detect significant increases in vascular density (Supplemental Figure 9). CD47 also has an inhibitory effect on NO signaling. Hypoxia can induce NO synthases and increased NO signaling can improve cell survival under ischemia, in Cd47-null mice. In endothelial cells, NO production induces stimulation of vascular smooth muscle cells to promote vascular relaxation; however, induced CD47 ligand thrombosponidin-1 inhibits NO production and stimulates production of reactive oxygen species (44). In our own hands, CD47 blockade does not affect CM viability in vitro. Also, the effects on blood pressure were not found during CD47 blockade (Supplemental Figure 9). Finally, excessive phagocytic uptake has been shown to be detrimental in the central nervous system through phagocytosis-induced cell death (45). In this context, CD47 may in some instances act as a prophagocytic signal (46).
Conclusions
An increased understanding of the basic mechanisms of CD47 regulation in CMs holds the potential to uncouple nonphagocytic CD47 signaling to selectively enhance the clearance of dying cells after MI and improve tissue repair. The acute nature of infarct-associated CM death, paired with standards of percutaneous coronary intervention, offer a tractable opportunity for CD47-targeted approaches during clinical reperfusion.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: CD47, also known as integrin-associated protein, is known to regulate multiple distinct cellular processes, including the inhibition of phagocytosis of dying cells, or efferocytosis, by immune cell Mϕs. Our preclinical studies indicate that CD47-blocking antibodies enhance the phagocytic removal of dying cardiac myocytes, after reperfusion in MI. This intervention also improved the resolution of cardiac inflammation and reduced infarct size.
TRANSLATIONAL OUTLOOK: Further studies are needed to enhance the pharmacological route of administration for anti-CD47 inhibitors and to test the efficacy of humanized anti-CD47 antibodies.
Footnotes
Dr. Zhang is supported by an American Heart Association predoctoral grant. Dr. Thorp is supported by 1R01HL122309-01 and funding from the Chicago Biomedical Consortium. Publication of this research was supported by the Sidney & Bess Eisenberg Memorial Fund. Dr. Frazier is a consultant for Vasculox. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Robert Roberts, MD, served as Guest Editor for this paper.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Velagaleti R.S., Pencina M.J., Murabito J.M. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jhund P.S., McMurray J.J. Heart failure after acute myocardial infarction: a lost battle in the war on heart failure? Circulation. 2008;118:2019–2021. doi: 10.1161/CIRCULATIONAHA.108.813493. [DOI] [PubMed] [Google Scholar]
- 3.Whelan R.S., Kaplinskiy V., Kitsis R.N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 4.Cesselli D., Jakoniuk I., Barlucchi L. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis N.G. The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol. 2014;63:185–195. doi: 10.1097/FJC.0000000000000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panizzi P., Swirski F.K., Figueiredo J.L. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–1638. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandivier R.W., Henson P.M., Douglas I.S. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 8.Lambert J.M., Lopez E.F., Lindsey M.L. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–158. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan E., Yeap X.Y., Dehn S. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. 2013;113:1004–1012. doi: 10.1161/CIRCRESAHA.113.301198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardai S.J., McPhillips K.A., Frasch S.C. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 11.Jaiswal S., Jamieson C.H., Pang W.W. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khandelwal S., van Rooijen N., Saxena R.K. Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion. 2007;47:1725–1732. doi: 10.1111/j.1537-2995.2007.01348.x. [DOI] [PubMed] [Google Scholar]
- 13.Kojima Y., Volkmer J.P., McKenna K. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briassouli P., Komissarova E.V., Clancy R.M., Buyon J.P. Role of the urokinase plasminogen activator receptor in mediating impaired efferocytosis of anti-SSA/Ro-bound apoptotic cardiocytes: implications in the pathogenesis of congenital heart block. Circ Res. 2010;107:374–387. doi: 10.1161/CIRCRESAHA.109.213629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isenberg J.S., Qin Y., Maxhimer J.B. Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress. Matrix Biol. 2009;28:110–119. doi: 10.1016/j.matbio.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frazier E.P., Isenberg J.S., Shiva S. Age-dependent regulation of skeletal muscle mitochondria by the thrombospondin-1 receptor CD47. Matrix Biol. 2011;30:154–161. doi: 10.1016/j.matbio.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X.Q., Frazier W.A. The thrombospondin receptor CD47 (IAP) modulates and associates with alpha2 beta1 integrin in vascular smooth muscle cells. Mol Biol Cell. 1998;9:865–874. doi: 10.1091/mbc.9.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S., Yeap X.Y., Grigoryeva L. Cardiomyocytes induce macrophage receptor shedding to suppress phagocytosis. J Mol Cell Cardiol. 2015;87:171–179. doi: 10.1016/j.yjmcc.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindberg F.P., Gresham H.D., Schwarz E., Brown E.J. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J Cell Biol. 1993;123:485–496. doi: 10.1083/jcb.123.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeap X.Y., Dehn S., Adelman J., Lipsitz J., Thorp E.B. Quantitation of acute necrosis after experimental myocardial infarction. Method Mol Biol. 2013;1004:115–133. doi: 10.1007/978-1-62703-383-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorp E.B. Methods and models for monitoring UPR-associated macrophage death during advanced atherosclerosis. Methods Enzymol. 2011;489:277–296. doi: 10.1016/B978-0-12-385116-1.00016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miao W., Luo Z., Kitsis R.N., Walsh K. Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J Mol Cell Cardiol. 2000;32:2397–2402. doi: 10.1006/jmcc.2000.1283. [DOI] [PubMed] [Google Scholar]
- 23.Li S., Sun Y., Liang C.P. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. 2009;105:1072–1082. doi: 10.1161/CIRCRESAHA.109.199570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connell T.D., Rodrigo M.C., Simpson P.C. Isolation and culture of adult mouse cardiac myocytes. Method Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 25.Murphy B.M., O'Neill A.J., Adrain C., Watson R.W., Martin S.J. The apoptosome pathway to caspase activation in primary human neutrophils exhibits dramatically reduced requirements for cytochrome C. J Exp Med. 2003;197(5):625–632. doi: 10.1084/jem.20021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharifi-Sanjani M., Shoushtari A.H., Quiroz M. Cardiac CD47 drives left ventricular heart failure through Ca2+-CaMKII-regulated induction of HDAC3. J Am Heart Assoc. 2014;3:e000670. doi: 10.1161/JAHA.113.000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Madariaga M.L., Wang S., Van Rooijen N., Oldenborg P.A., Yang Y.G. Lack of CD47 on nonhematopoietic cells induces split macrophage tolerance to CD47null cells. Proc Natl Acad Sci U S A. 2007;104:13744–13749. doi: 10.1073/pnas.0702881104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isenberg J.S., Ridnour L.A., Perruccio E.M., Espey M.G., Wink D.A., Roberts D.D. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nahrendorf M., Swirski F.K., Aikawa E. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soto-Pantoja D.R., Shih H.B., Maxhimer J.B. Thrombospondin-1 and CD47 signaling regulate healing of thermal injury in mice. Matrix Biol. 2014;17:00087–00090. doi: 10.1016/j.matbio.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narula J., Haider N., Virmani R. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 32.Vivar R., Humeres C., Ayala P. TGF-beta1 prevents simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis by activation of both canonical and non-canonical signaling pathways. Biochim Biophys Acta. 2013;1832:754–762. doi: 10.1016/j.bbadis.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 34.Aprahamian T., Takemura Y., Goukassian D., Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. 2008;152:448–455. doi: 10.1111/j.1365-2249.2008.03658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bujak M., Kweon H.J., Chatila K., Li N., Taffet G., Frangogiannis N.G. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurtado B., Munoz X., Recarte-Pelz P. Expression of the vitamin K-dependent proteins GAS6 and protein S and the TAM receptor tyrosine kinases in human atherosclerotic carotid plaques. Thromb Haemost. 2011;105:873–882. doi: 10.1160/TH10-10-0630. [DOI] [PubMed] [Google Scholar]
- 37.Kojima Y., Downing K., Kundu R. Cyclin-dependent kinase inhibitor 2B regulates efferocytosis and atherosclerosis. J Clin Invest. 2014;124:1083–1097. doi: 10.1172/JCI70391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiskopf K., Ring A.M., Ho C.C. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung J., Gao A.G., Frazier W.A. Thrombspondin acts via integrin-associated protein to activate the platelet integrin alphaIIbbeta3. J Biol Chem. 1997;272:14740–14746. doi: 10.1074/jbc.272.23.14740. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y., Buhring H.J., Zen K. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J Biol Chem. 2002;277:10028–10036. doi: 10.1074/jbc.M109720200. [DOI] [PubMed] [Google Scholar]
- 41.Jiang D.S., Zhang X.F., Gao L. Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension. 2014;63:96–104. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isenberg J.S., Shiva S., Gladwin M. Thrombospondin-1-CD47 blockade and exogenous nitrite enhance ischemic tissue survival, blood flow and angiogenesis via coupled NO-cGMP pathway activation. Nitric Oxide. 2009;21:52–62. doi: 10.1016/j.niox.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howangyin K.Y., Zlatanova I., Pinto C. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation. 2016;133:826–839. doi: 10.1161/CIRCULATIONAHA.115.020857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers N.M., Sharifi-Sanjani M., Csanyi G., Pagano P.J., Isenberg J.S. Thrombospondin-1 and CD47 regulation of cardiac, pulmonary and vascular responses in health and disease. Matrix Biol. 2014;37:92–101. doi: 10.1016/j.matbio.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neher J.J., Emmrich J.V., Fricker M., Mander P.K., Thery C., Brown G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A. 2013;110:E4098–E4107. doi: 10.1073/pnas.1308679110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burger P., Hilarius-Stokman P., de Korte D., van den Berg T.K., van Bruggen R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood. 2012;119:5512–5521. doi: 10.1182/blood-2011-10-386805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.