

Abstract
Midkine (MDK) is a cytokine and neurotrophic factor that is more highly expressed in the brains of human alcoholics and in mice predisposed to drink large amounts of ethanol, suggesting that MDK may regulate ethanol consumption. Here we measured ethanol consumption in male and female Mdk knockout (−/−) mice using the two-bottle choice and the drinking in the dark (DID) tests. We found that Mdk −/− mice consumed significantly more ethanol than wild type controls in both tests. To determine if MDK acts in the ventral tegmental area (VTA) to regulate ethanol consumption, we delivered lentivirus expressing a Mdk shRNA into the VTA of male C57BL/6J mice to locally knockdown Mdk and performed the DID test. Mice expressing a Mdk shRNA in the VTA consumed more ethanol than mice expressing a control non-targeting shRNA, demonstrating that the VTA is one site in the brain through which MDK acts to regulate ethanol consumption. Since MDK also controls the expression of inflammatory cytokines in other organs, we examined gene expression of interleukin-1 beta (Il1b), tumor necrosis factor alpha (Tnf) and the chemokine (C-C motif) ligand 2 (Ccl2) in the VTA of Mdk −/− mice and in mice expressing Mdk shRNA in the VTA. Expression of Ccl2 was elevated in the VTA of Mdk −/− mice and in mice expressing Mdk shRNA in the VTA. These results demonstrate that MDK functions in the VTA to limit ethanol consumption and levels of CCL2, a chemokine known to increase ethanol consumption.
Keywords: Alcohol, addiction, dopamine, chemokine, cytokine, innate immune, Mdk, Ccl2, Il1b, ventral tegmental area
Introduction
Alcohol addiction is a complex psychiatric disorder with genetic and environmental influences. Whole-genome approaches in humans and rodents have aided in the identification of novel genes and pathways that may predispose individuals to alcohol use disorder, with the dual goals of understanding the biological underpinnings of this behavioral disorder and finding potential new therapeutic targets. As an example, transcriptional profiling of mouse brain has yielded thousands of genes whose expression is significantly different between strains that consume high vs. low amounts of ethanol (Mulligan et al., 2006). The challenge remains to validate individual genes as potential predisposing factors for alcohol addiction. One of these genes, Mdk, is expressed at higher levels in the brains of high alcohol-preferring (HAP) mice compared with low-alcohol-preferring (LAP) mice, suggesting that Mdk may be involved in the genetic predisposition to alcohol addiction (Mulligan et al., 2006).
Mdk encodes a secreted growth factor and cytokine known as midkine (MDK). In addition to being more abundant in the brains of HAP mice, Mdk expression is induced in the brain and in cultured neuroblastoma cells by alcohol exposure (He et al., 2015, Mulligan et al., 2011). MDK protein levels are also higher in the prefrontal cortex of alcoholics, suggesting that expression of this gene is not only responsive to ethanol, but may also contribute to excessive alcohol drinking in humans (Flatscher-Bader et al., 2005, Flatscher-Bader & Wilce, 2006, Flatscher-Bader & Wilce, 2008). Recently, Vicente-Rodriguez et al. demonstrated that Mdk knockout (−/−) mice, in contrast to wild type mice, develop ethanol conditioned place preference at a low dose of ethanol (1 g/kg), indicating that MDK may act to limit the rewarding properties of ethanol (Vicente-Rodriguez et al., 2014). Mdk −/− mice also demonstrate a delayed recovery of ethanol-induced ataxia on the rotarod (Vicente-Rodriguez et al., 2014).
Although Mdk modulates these behavioral responses to ethanol, the effect of Mdk deletion on ethanol consumption has not been determined. Based on the behavioral phenotypes of Mdk −/− mice and the fact that Mdk expression is increased in HAP mice, we hypothesized that Mdk −/− mice would have altered ethanol consumption compared with wild type mice. Here, we tested ethanol consumption in Mdk −/− mice using two paradigms, the two-bottle choice ethanol consumption test, which measures moderate ethanol consumption, and the drinking in the dark (DID) test, which measures binge-like ethanol consumption.
We also performed viral-mediated RNA interference (RNAi) to locally knockdown Mdk in a specific neuroanatomical location in the adult mouse. We injected a lentiviral vector expressing a short-hairpin RNA (shRNA) targeting Mdk into the ventral tegmental area (VTA) and tested mice for binge-like ethanol consumption. The VTA was chosen because dopamine neurons in this brain region regulate ethanol consumption (Gonzales et al., 2004). Moreover, MDK is a survival factor for dopamine neurons in culture, and Mdk −/− mice exhibit decreased dopamine levels in the striatum (Kikuchi et al., 1993, Ohgake et al., 2009, Prediger et al., 2011).
In addition to its effects on the dopamine system, MDK is also an important modulator of inflammatory conditions such as rheumatoid arthritis, diabetic nephropathy, inflammatory bowel disease, and multiple sclerosis (Weckbach et al., 2011). Interestingly, the innate immune system in the brain is activated by chronic alcohol exposure, and deletion of cytokine and chemokine genes and their receptors in mice or treatment of rodents with pharmacological agents that act on neuroimmune targets alters ethanol consumption (Crews & Vetreno, 2015, Robinson et al., 2014). Thus, MDK might also regulate ethanol consumption through an effect on the innate immune response. To test this, we examined transcript levels of the pro-inflammatory cytokine and chemokine genes tumor necrosis factor (Tnf), interleukin 1 beta (Il1b), and chemokine (C-C motif) ligand 2 (Ccl2) in the VTA of mice with reduced levels of Mdk. We found that Mdk knockdown in the VTA increased expression of Ccl2 and Il1b and that Mdk −/− mice had higher expression of Ccl2.
This study establishes MDK as a factor that limits ethanol consumption in mice and suggests that it may also restrict the induction of neuroimmune genes in response to alcohol exposure. Manipulating MDK activity or signaling pathways may be a novel strategy to reduce excessive alcohol drinking and the brain immune response to chronic ethanol exposure.
Materials and methods
Subjects
Mdk −/− mice have been described previously (He et al., 2015, Nakamura et al., 1998). We re-derived Mdk −/− mice from frozen embryos obtained from the Riken BioResource Center (Ibaraki, Japan; Riken number RBRC02422, strain name B6.129S2-Mdk<tm1Tmu>) at the University of Illinois at Chicago Transgenic Production Service. Mdk −/− mice were maintained on a C57BL/6J background (previously backcrossed 2 generations). Heterozygous Mdk (+/−) mice were bred to obtain homozygous male and female knockout (−/−) and wild-type (+/+) littermates for behavioral testing. Mice were housed with same-sex littermates and kept in a temperature- (21°C) and humidity-controlled room on a 14 h light/dark cycle with lights on at 6 a.m. and off at 8 p.m., unless otherwise indicated. Male C57BL/6J mice (8 weeks of age) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were tested for behavior between the ages of 10–16 weeks. Food (Harlan Teklad Diet T.7912, Indianapolis, IN, USA) and water were available ad libitum. All procedures with mice were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Illinois at Chicago Animal Care Committee. All efforts were taken to minimize pain and discomfort.
Two-bottle choice experiments
Two-bottle choice experiments for ethanol, saccharin, and quinine preference were performed according to Lasek et al (Lasek et al., 2007) with slight modifications. Mice were individually housed in cages with continuous access to two tubes made from 10 mL polycarbonate pipettes connected to stainless steel sipper tubes containing ball-bearings to prevent leakage (Rhodes et al., 2005). Fluid consumption was measured every day for 4 days at each ethanol concentration by measuring the volume of fluid in the tubes. The position of the bottles (left or right) was changed every day to control for side preference. Mice drank water from both bottles for the first 4 days and then were given a choice between ethanol and water, starting with 3% (v/v) ethanol and increasing to 6%, 10%, 14%, and 20% ethanol. Ethanol consumption was calculated as g ethanol consumed per kg body weight per day. Preference ratios were calculated as the volume of ethanol consumed over the total volume of fluid consumed (water plus ethanol). Nine male Mdk +/+, 7 male Mdk −/−, 6 female Mdk +/+, and 9 female Mdk −/− mice were tested in this experiment. A subgroup of mice was tested for saccharin (0.03 and 0.06%) and quinine (15 and 30 μM) consumption using the same procedure eight days after the ethanol testing was completed.
Drinking in the dark (DID)
Binge-like drinking was measured using the one-bottle DID procedure as previously described using 20% ethanol (Dutton et al., 2016, Rhodes et al., 2005). Briefly, mice were individually housed in a 12 h reversed light/dark cycle room (lights off at 10 a.m. and on at 10 p.m.) for 2 weeks before being tested. For the Mdk shRNA experiments (details below), mice were given access to the ethanol solution 3 h into the dark cycle for a period of 2 h on the first 3 days of testing (Monday, Tuesday, and Wednesday). On the fourth day, mice were given access to the ethanol solution for 4 h and the volume consumed was measured at 2 and 4 h. For testing the Mdk −/− mice, animals were given access to the ethanol solution for 4 h instead of 2 h on all four days. In addition, the DID procedure was performed for 4 cycles (i.e. for 4 weeks on Monday through Thursday). We tested 6 male Mdk +/+, 5 male Mdk −/−, 4 female Mdk +/+, and 4 female Mdk −/− mice.
Lentiviral production and delivery to the ventral tegmental area (VTA)
Replication-deficient lentiviruses expressing a short-hairpin RNA (shRNA) targeting Mdk (shMdk) or the non-targeting shRNA control (shScr) were produced as described previously using the lentiviral vector pLL3.7, which expresses the shRNA from the U6 promoter and enhanced green fluorescent protein (EGFP) from the CMV promoter (Lasek et al., 2007). The 19-nucleotide targeting sequence for Mdk was 5′-GAGCCGACTGCAAATACAA-3′, corresponding to positions 602–620 on the Mdk transcript (Genbank accession number NM_010784.5). Stereotaxic delivery of virus (1 μl of ~3 × 107 pg Gag antigen/ml) bilaterally into the VTA was performed as described previously (Lasek et al., 2007). Briefly, 8-week old male C57BL/6J mice were anesthetized with ketamine and xylazine and placed in a stereotaxic alignment instrument for viral injections (David Kopf Instruments, Tujunga, CA, USA). Coordinates for bilateral infusion of virus were anterior/posterior (in reference to bregma), −3.2 mm; medial/lateral, +/− 0.5 mm; and dorsal/ventral −4.7 mm. Mice recovered for 3 weeks prior to being tested for DID. We tested 16 mice infused with shScr and 17 mice infused with shMdk virus. After behavioral testing, mice were euthanized with pentobarbital (Somnasol) and perfused transcardially with 4% paraformaldehyde. Brains were removed and processed for immunohistochemistry using a GFP antibody to verify injection sites as described previously (Lasek et al., 2007).
Validation of lentiviral vector efficacy in vitro and in vivo
To test in vitro knockdown efficacy, Neuro-2a cells (ATCC, Manassas, VA, USA) were cultured according to ATCC recommendations and transfected with lentiviral plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. Forty-eight hours after transfection, RNA was isolated, subjected to first strand cDNA synthesis, and quantitative real-time PCR (qPCR) using the GeneJet RNA Purification Kit, Maxima First Strand cDNA Synthesis Kit for RT-qPCR, and the Maxima Probe qPCR Master Mix, respectively (Thermo Fisher Scientific). Taqman primer-probe mixes for Mdk and Actb were purchased from Thermo Fisher Scientific and reactions were performed using the PikoReal Real-Time PCR System (Thermo Fisher Scientific).
To test in vivo knockdown efficacy, 8-week old male C57BL/6J mice were injected in the VTA with lentivirus as described above. We used 9 mice infused with shScr virus and 11 mice infused with shMdk virus. Three weeks after injection, mice were euthanized by CO2 followed by rapid decapitation. Brains were removed and flash frozen on dry ice. Sections (300 μm) were collected through the VTA and transferred to pre-cooled glass slides on dry ice. Micro-punch sampling of fluorescent tissue was performed on dry ice using a Dual Fluorescent Protein Flashlight (Nightsea, Bedford, MA, USA) and 1 mm-diameter biopsy punches (Integra LifeSciences, Plainsboro, NJ, USA). RNA was isolated and qPCR was performed as described above, except we used Hprt as a normalization control instead of Actb. Primer sequences are listed in Table S1.
Gene expression
To measure expression of Ccl2, Tnf, and Il1b in the VTA of mice expressing shMdk or shScr in the VTA, the same tissue samples used for verifying Mdk knockdown were used (dissection described above). To measure expression of these genes in the VTA of Mdk −/− mice, the VTA was dissected from frozen brain sections of male and female mice (12 Mdk +/+ and 15 Mdk −/− mice), RNA isolated, and cDNA synthesized as indicated above. qPCR was performed with Maxima SYBR green qPCR master mix (Thermo Fisher Scientific). Primers for Ccl2, Tnf, and Il1b, and Th are listed in Table S1.
Statistical analysis
Prism software (version 6, Graphpad, La Jolla, CA, USA) was used for statistical analysis. Behavioral tests were analyzed by two-way analysis of variance (ANOVA). Gene expression data was analyzed by student’s t-test.
Results
Increased voluntary ethanol consumption by Mdk −/− mice compared with controls
We first tested whether mice deficient in MDK show altered voluntary ethanol consumption using the two-bottle choice test. Male and female Mdk +/+ and Mdk −/− mice were tested for 4 days at each ethanol concentration, starting with 3% and escalating to 6, 10, 14, and 20% ethanol. There was a significant sex difference in wild type mice, with female mice consuming more ethanol than males (sex: F1, 65 = 81.77, P < 0.0001; concentration: F4, 65 = 50.15, P < 0.0001; interaction: F4, 65 = 6.37, P = 0.0002). As a result, we conducted statistical analyses and show the data separately for each sex. We found that both male and female Mdk −/− mice consumed significantly more ethanol than wild type controls (Fig. 1a, b). A two-way ANOVA within each sex demonstrated significant effects of ethanol concentration and genotype (males, genotype: F1, 70 = 9.58, P = 0.0028; males, concentration: F4, 70 = 26.47, P < 0.0001; females, genotype: F1, 65 = 13.14, P = 0.0006; females, concentration: F4, 65 = 58.57, P < 0.0001). Mdk −/− mice weighed slightly less than Mdk +/+ mice (male Mdk +/+, 21.6 ± 0.6 g; male Mdk −/−, 20.2 ± 0.8 g; female Mdk +/+, 20.7 ± 0.9 g; female Mdk −/−, 19.6 ± 0.9 g), however this difference in body weight between genotypes was not significant. The body weight differences between genotypes did not account for the increased ethanol consumption by Mdk −/− mice, since volumes of ethanol consumed (not corrected for body weight) were still significantly higher in Mdk −/− mice compared with Mdk +/+ mice (data not shown).
Figure 1. Midkine knockout mice consume more ethanol than controls in a 2-bottle choice ethanol consumption test.

Male (a, c, e) and female (b, d, f) wild type (Mdk +/+, filled circles) and homozygous midkine knockout (Mdk −/−, open squares) mice underwent a 2-bottle choice test for water or 3, 6, 10, 14, and 20% (v/v) ethanol with four days of 24 h access at each ethanol concentration. (a, b) ethanol consumed in g ethanol/kg body weight/day. There was a significant effect of genotype and ethanol concentration in both sexes. (c, d) Ethanol preference, calculated as the ratio of the volume of ethanol consumed over the volume of total fluid consumed. There was a significant effect of genotype and ethanol concentration in males, and a significant effect of ethanol concentration in females. (e, f) Water consumed during the ethanol consumption test at each concentration of ethanol. Data is presented as ml water consumed/kg body weight/day. No significant genotype effect was observed for water consumption in either sex. Male Mdk +/+, n = 9; male Mdk −/−, n = 7; female Mdk +/+, n = 6; female Mdk −/−, n = 9. * P < 0.05, ** P < 0.01, *** P < 0.001 by two-way ANOVA. Data are presented as the mean ± SEM.
In addition to consuming more ethanol than wild type mice, Mdk −/− mice also showed increased ethanol preference in the two-bottle choice test (Fig. 1c, d). Specifically, male Mdk −/− had a higher ethanol preference compared with male Mdk +/+ mice (genotype: F1, 70 = 4.013, P = 0.049; concentration: F4, 70 = 13.52, P < 0.0001), while there was a trend towards increased ethanol preference by female Mdk −/− mice (genotype: F1, 65 = 3.03, P = 0.086; concentration: F4, 65 = 4.44, P = 0.0031). There were no significant genotype differences in water consumption at each ethanol concentration tested in either sex (Fig. 1e, f). Together, these data indicate that a MDK deficiency leads to increased voluntary ethanol consumption.
Normal sweet and bitter taste preference in Mdk −/− mice compared with controls
To determine if the increased ethanol consumption exhibited by Mdk −/− mice might be due to a difference in taste preference, we tested Mdk −/− and +/+ mice for consumption of sweet (saccharin) and bitter (quinine) solutions. A two-bottle choice test was conducted with 0.03 and 0.06% saccharin vs. water and 15 and 30 μM quinine vs. water. In the saccharin preference test, both male and female mice consumed significantly more of the 0.06% saccharin solution compared with 0.03% saccharin, but there were no differences between genotypes (Fig. 2a, b; males, genotype: F1, 18 = 0.36, P = 0.56; males, concentration: F1, 18 = 17.77, P = 0.0005; females, genotype: F1, 16 = 0.30, P = 0.59; females, concentration: F1, 16 = 13.83, P = 0.0019). In the quinine taste test, there were no significant differences in preference between Mdk −/− and +/+ mice, nor were there differences in consumption between the two quinine concentrations (Fig. 2c, d). This taste test data demonstrates that the increased ethanol consumption by Mdk −/− mice is not likely due to inherent changes in sweet or bitter taste preference.
Figure 2. Midkine knockout mice do not differ in saccharin or quinine intake compared with controls.
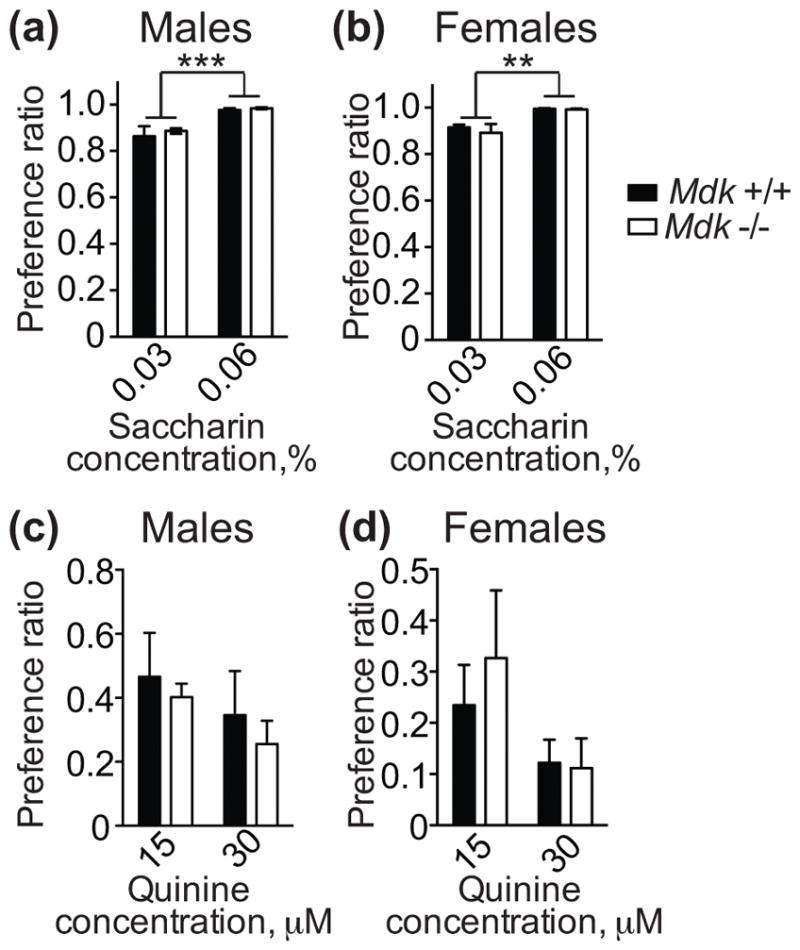
Male (a, c) and female (b, d) wild type (Mdk +/+, black bars) and homozygous midkine knockout (Mdk −/−, white bars) mice underwent a 2-bottle choice test for water and saccharin (0.03 or 0.06%, w/v) or water and quinine (15 or 30 μM). (a, b) Saccharin consumption plotted as the ratio of the volume of saccharin solution consumed over total fluid consumed. There were no significant effects of genotype, but there were significant main effects of saccharin concentration for both sexes. Male Mdk +/+, n = 9; male Mdk −/−, n = 7; female Mdk +/+, n = 6; female Mdk −/−, n = 9. ** P < 0.01, *** P < 0.001 by two-way ANOVA. (c, d) Quinine consumption plotted as the ratio of the volume of quinine solution consumed over total fluid consumed. There were no significant differences between genotypes or quinine concentration. Male Mdk +/+, n = 6; male Mdk −/−, n = 5; female Mdk +/+, n = 4; female Mdk −/−, n = 6. Data are presented as the mean ± SEM.
Mdk −/− mice consume more ethanol in a binge-drinking test compared with controls
We next tested whether MDK regulates binge-like ethanol consumption using the DID procedure. Male and female Mdk +/+ and −/− mice were given access to one bottle of 20% ethanol for 4 hours, 3 hours into the dark cycle, on Monday-Thursday. Male and female Mdk −/− mice consumed significantly more ethanol overall compared with wild type controls during the first 4 days of ethanol exposure (Fig. 3a, b; males, genotype: F1, 64 = 4.0, P = 0.050; males, day: F3, 64 = 1.45, P = 0.23; females, genotype: F1, 48 = 4.28, P = 0.044; females, day: F3, 48 = 3.26, P = 0.029), similar to what we observed in the two-bottle choice experiment, although the magnitude of the change was higher in the two-bottle choice experiment.
Figure 3. Midkine knockout mice consume more ethanol than controls in the drinking in the dark (DID) test.

Male (a, c) and female (b, d) wild type (Mdk +/+, filled circles) and homozygous midkine knockout (Mdk −/−, open squares) mice underwent a modified DID test, in which they were provided 20% ethanol in water (v/v) for 4 h per day, 4 days per week for 4 weeks. (a, b) Ethanol consumed in g ethanol/kg body weight/4 h during the first week of the DID test. There were significant effects of genotype for males and females and a significant effect of day for females. (c, d) Average ethanol consumed each week, expressed as g ethanol/kg body weight/4 h. There was a significant effect of genotype in males. Male Mdk +/+, n = 9; male Mdk −/−, n = 9; female Mdk +/+, n = 7; female Mdk −/−, n = 7. * P < 0.05 by two-way ANOVA. Data are presented as the mean ± SEM.
We decided to continue the DID experiment for an additional 3 cycles (3 weeks) to determine if Mdk deletion might alter ethanol consumption after repeated intermittent exposure to ethanol. Interestingly, there was still a small but significant main effect of genotype over the 4 weeks of DID in male mice (Fig. 3c; genotype: F1, 64 = 4.9, P = 0.031; week: F3, 64 = 1.9, P = 0.14). However, the genotype effect in females was eliminated after the first week, primarily because it appeared that wild type female mice escalated their intake by the second week of ethanol exposure (Fig. 3d; genotype: F1, 48 = 2.81, P = 0.10; week: F3, 48 = 1.83, P = 0.15). A two-way ANOVA within each genotype by sex and week indicated that wild-type mice escalated their drinking over the 4-week period (week: F3, 56 = 3.65, P = 0.018; sex: F1, 56 = 30.43, P < 0.0001), but Mdk −/− mice did not (week: F3, 56 = 0.43, P = 0.74; sex: F1, 56 = 30.1, P < 0.0001). This may be due to a “ceiling effect”, such that Mdk −/− mice drink the maximum amount of ethanol possible during the 4 hour session initially and therefore can’t subsequently increase drinking. Wilcox et al has shown that C57BL6/J males escalate DID over several weeks (Wilcox et al., 2014). Together, these data indicate that MDK may be involved in limiting binge ethanol consumption during the initial exposure to ethanol.
Mdk acts in the VTA to limit binge-like ethanol consumption
MDK promotes the survival of dopamine neurons and Mdk −/− mice have decreased levels of striatal dopamine and dopamine receptors (Kikuchi et al., 1993, Ohgake et al., 2009). Since dopamine neurons in the VTA regulate ethanol consumption (Gonzales et al., 2004), we hypothesized that MDK might act in this brain region to regulate ethanol drinking behavior. To test this hypothesis, we developed a lentiviral vector that expresses an shRNA to attenuate Mdk expression (Fig. 4a, shMdk). Validation of lentiviral plasmids in cell culture was performed by transfecting plasmids expressing shMdk or the control shRNA (shScr) into Neuro-2a cells. Cells expressing shMdk exhibited a 67% decrease in Mdk mRNA levels as measured by qPCR compared with cells expressing shScr (Fig. 4b, t4 = 11.27, P = 0.0004). To validate the shMdk lentiviral vector in vivo, we injected lentiviruses into the VTA and dissected infected VTA (as visualized by GFP fluorescence) three weeks after infection. Mdk expression by qPCR in mice expressing shMdk in the VTA was reduced by 48% compared with Mdk expression in mice expressing shScr (Fig. 4c, t18 = 5.43, P < 0.0001).
Figure 4. Mdk knockdown by RNAi in the ventral tegmental area (VTA) of male mice increases binge-like ethanol consumption.

C57BL/6J mice were stereotaxically injected in the VTA with a lentivirus expressing a short hairpin RNA (shRNA) targeting Mdk (shMdk) or a control shRNA (shScr) and green fluorescent protein (GFP). Mice were allowed to recover for three weeks and tested for ethanol consumption in the drinking in the dark protocol. (a) Schematic diagram showing the location of the 19 nucleotide targeting sequence on the Mdk transcript (transcript variant 1). The blue box indicates the open reading frame. The arrow points to a vertical black line indicating the location of the targeting sequence. Numbers indicate the nucleotide position on the transcript. (b) Demonstration of knockdown efficacy of shMdk in Neuro-2a cells transfected with the lentiviral plasmid encoding shMdk compared with cells transfected with shScr. RNA was isolated from cells 48 h after transfection and analyzed by quantitative real-time PCR (qPCR, n = 3) (c) Demonstration of knockdown efficacy of shMdk in mouse VTA three weeks after lentiviral infection. Infected VTA was dissected from mice 3 weeks after injection, RNA isolated and analyzed by qPCR. shScr, n = 9; shMdk, n = 11. (d) Representative image of lentiviral infection in the VTA. Immunohistochemistry with a GFP antibody was performed on brain sections containing the VTA. IPR, interpeduncular nucleus, rostral. Scale bar, 200 μM. (e) Ethanol consumption in the DID test by mice infected in the VTA with lentivirus expressing shMdk (open squares) or shScr (filled circles). Shown is the amount of ethanol consumed in g ethanol/kg body weight during the 2 h drinking sessions each day. There were significant main effects of shRNA and day. (f) Ethanol consumption in mice infected in the VTA with lentivirus expressing shMdk (white bar) or shScr (black bar) during the 4 h drinking session on day 4 in the DID test, expressed as g ethanol/kg body weight. shScr, n = 16; shMdk, n = 17. *P < 0.05 by two-way ANOVA. **P < 0.01, ***P < 0.001, and ****P < 0.0001 by student’s t-test. Data are presented as the mean ± SEM.
Having demonstrated the efficacy of the lentiviral vector, we next performed the 4-day DID test with male mice expressing shMdk or shScr in the VTA. Mice expressing shMdk in the VTA consumed more ethanol than controls during the 2-hour drinking sessions on days 1–4 (Fig. 4e; shRNA: F1, 124 = 4.35, P = 0.039; day: F3, 124 = 3.43, P = 0.019). In addition, there was a 16% increase in ethanol consumption by mice expressing shMdk compared to shScr during 4-hours of drinking on day 4 (Fig. 4f; t31 = 3.3, P = 0.0025). Consistent with the increased ethanol consumption by mice expressing shMdk in the VTA after the 4-hour drinking session, blood ethanol concentrations trended to be higher in mice expressing shMdk (156 ± 14.1 mg/dl) compared with mice expressing shScr (122.4 ± 17.9 mg/dl), although this effect was not statistically significant (t31 = 1.48, P = 0.149). We also performed an experiment in which we injected lentiviruses expressing shScr or shMdk into the prefrontal cortex of male mice and tested them in the DID procedure. There was no difference in ethanol consumption between mice expressing shScr and shMdk in the prefrontal cortex, demonstrating anatomical specificity in the effect of the shMdk-expressing lentivirus (Fig. S1). Together, these data indicate that MDK acts in the VTA to limit binge-like ethanol consumption.
Increased expression of innate immune response genes in the VTA of mice with reduced Mdk
Levels of the dopamine synthesizing enzyme tyrosine hydroxylase (TH) are decreased in the striatum of Mdk −/− mice as measured by immunohistochemistry, and expression of the Th gene is higher in aorta from Mdk −/− mice compared with wild type mice (Ezquerra et al., 2006, Prediger et al., 2011). This suggests that a deficiency in Mdk leads to dysregulation of the dopamine system. To test if Th expression is altered in mice with knockdown of Mdk, we examined Th expression by qPCR in mice expressing shScr or shMdk in the VTA. The same samples used in Fig. 4c were tested. Surprisingly, we found no difference in Th expression between mice expressing shScr or shMdk (Fig. 5a).
Figure 5. Increased expression of Ccl2 in the ventral tegmental area (VTA) of mice deficient in midkine.

(a–d) VTA RNA samples from mice expressing shScr (black bars, n = 9) or shMdk (white bars, n = 11) in the VTA were analyzed by qPCR for expression of (a) Th, (b) Ccl2, (c) Il1b, and (d) Tnf. The same samples from Fig. 4c that were used to test knockdown of Mdk in the VTA were used in this experiment. There were significant increases in Ccl2 and Il1b expression in VTA samples from mice expressing shMdk compared with shScr. (e–h) VTA RNA samples from wild type (+/+, black bars, n = 12) and Mdk knockout (−/−, white bars, n = 15) mice were analyzed by qPCR for expression of (e) Th, (f) Ccl2, (g) Il1b, and (h) Tnf. There was a significant increase in Ccl2 expression in the VTA of Mdk −/− compared with Mdk +/+ mice. *P < 0.05 by student’s t-test. Data are presented as the mean ± SEM.
We next tested the expression of select cytokine and chemokine genes in the VTA of mice expressing shScr and shMdk. MDK is involved in a number of inflammatory conditions and may regulate expression of cytokines and chemokines (Weckbach et al., 2011). Importantly, innate immune response genes are induced in the brain after chronic ethanol exposure and are relevant for ethanol consumption (Blednov et al., 2005, Blednov et al., 2012, Crews & Vetreno, 2015). We examined expression of genes encoding the pro-inflammatory cytokines interleukin 1 beta (Il1b) and tumor necrosis factor (Tnf), and the chemokine (C-C motif) ligand 2 (Ccl2) in the VTA of mice expressing shScr and shMdk. Ccl2 expression was significantly elevated by 98% in the VTA of mice with knockdown of Mdk (Fig. 5b; t17 = 2.51, P = 0.023). In addition, Il1b expression was significantly increased by 85% in the VTA of mice with knockdown of Mdk (Fig. 5c, t18 = 2.2, P = 0.041). There was no difference in the expression of Tnf between mice expressing shScr and shMdk in the VTA (Fig. 5d).
To confirm these results using an alternative approach, we examined expression of Th, Ccl2, Il1b, and Tnf in the VTA of Mdk −/− and +/+ mice. Expression of Ccl2 was increased by 58% in the VTA of Mdk −/− compared with Mdk +/+ mice (Fig. 5f, t25 = 2.31, P = 0.030), similar to what we observed with shRNA-mediated knockdown of Mdk in the VTA. Expression levels of Th, Il1b, and Tnf were similar between Mdk −/− and +/+ mice (Fig. 5). Collectively, these data indicate that MDK expression in the VTA may limit transcription of Ccl2. The increase in Ccl2 expression in the VTA is potentially one factor responsible for the increased ethanol consumption by Mdk −/− mice and mice with Mdk knockdown in the VTA (Blednov et al., 2005).
Discussion
Here we provide the first direct demonstration that MDK regulates ethanol consumption. We show that mice with a genetic deletion of Mdk consume more ethanol in a test of moderate consumption (two-bottle choice) and in a binge-drinking test (DID) compared with control mice. The results that we obtained with Mdk −/− mice are supported by our data showing that mice with lentiviral-mediated RNA interference of Mdk in the VTA also consume more ethanol than controls. Together these data strongly suggest that MDK functions in the brain to limit ethanol intake. The effects of Mdk deletion on ethanol consumption are not due to alterations in taste sensitivity, as evidenced by normal quinine and saccharin preference in Mdk −/− mice compared with Mdk +/+ mice. Prior to this study, Mdk −/− mice were tested for the rewarding properties of ethanol and found to be more sensitive to ethanol reward in the conditioned place preference test (Vicente-Rodriguez et al., 2014). It is possible that Mdk gene knockout mice consume more ethanol because they find ethanol more rewarding than mice with normal levels of Mdk.
One site in which MDK may act to limit ethanol consumption is the VTA. Our data demonstrate that reducing the expression of Mdk in the VTA causes increased binge-like ethanol consumption. We also tested mice expressing shMdk in the prefrontal cortex and did not see any difference in ethanol consumption compared with controls, indicating some anatomical specificity in the site of MDK action with respect to alcohol drinking. Interestingly, post-mortem analysis of the frontal cortex of human alcoholics demonstrated that MDK mRNA and protein are overexpressed in alcoholics vs. non-alcoholic controls (Flatscher-Bader & Wilce, 2006, Flatscher-Bader & Wilce, 2008). The VTA was not examined in those studies, so it is possible that MDK expression is also higher in the VTA of alcoholics. Although MDK is highly expressed in the frontal cortex of alcoholics, its expression in this brain region may not be important for regulating alcohol intake, or ethanol has different effects on MDK expression in the VTA compared with the frontal cortex. Alternatively, it is possible that MDK is produced by VTA neurons and secreted into the prefrontal cortex from VTA afferents, where it then acts to regulate ethanol consumption. VTA neurons also project to the nucleus accumbens, a key area of the brain involved in ethanol intake and reward (Gonzales et al., 2004). MDK may also be released into the nucleus accumbens to limit ethanol drinking and reward. Finally, MDK could act in an autocrine fashion through interactions with its receptors, such as ALK, RPTPβ/ζ, or others expressed on VTA neurons, resulting in decreased ethanol consumption. Our shRNA study does not discriminate between the above possibilities, but does indicate that MDK produced by VTA neurons limits ethanol consumption.
We previously found that treatment of neuroblastoma cells in culture with ethanol induces the expression of Mdk (He et al., 2015). Since Mdk is an ethanol-responsive gene and Mdk deletion causes increased intake of ethanol, we hypothesize that the increased expression of MDK in the brain of alcoholics might be an adaptive response to chronic ethanol exposure, meant to maintain homeostasis and curb excessive ethanol consumption. However, it is interesting that mice predisposed to drink high amounts of alcohol (HAP) have higher brain expression of Mdk compared with low-alcohol preferring mice (LAP). This would suggest that increased expression of Mdk is a predisposing factor for excessive drinking. We measured Mdk mRNA expression in the VTA in wild type mice immediately after the fourth drinking session in the DID test and did not observe any difference in Mdk expression compared with water-drinking control mice (Chen and Lasek, unpublished results). This suggests that, at least under these ethanol exposure conditions, Mdk expression in the VTA does not change. It will be important to determine the expression levels of Mdk in the VTA and other brain regions after chronic ethanol exposure and during withdrawal, since the increase in MDK in the frontal cortex of alcoholics may be due to an effect of chronic drinking or withdrawal from alcohol. Additionally future studies will test if increasing the expression of MDK leads to higher vs. lower levels of ethanol intake by directly infusing recombinant MDK protein into the VTA or other brain regions (such as the prefrontal cortex or nucleus accumbens) of ethanol-naïve and ethanol-experienced mice and testing ethanol consumption.
Dopamine neurons in the VTA are important for the rewarding and reinforcing effect of ethanol (Gonzales et al., 2004). MDK is a survival factor for dopamine neurons in vitro (Kikuchi et al., 1993) and Mdk −/− mice have decreased levels of dopamine and its metabolites in the striatum (Ohgake et al., 2009, Prediger et al., 2011). One study has also shown reduced numbers of TH-positive neurons in the substantia nigra pars compacta of Mdk −/− compared with Mdk +/+ mice, indicating that the loss of MDK impairs the survival or development of dopamine neurons (Prediger et al., 2011). However, Gramage et al did not observe any difference between Mdk −/− and Mdk +/+ mice in the innervation of the striatum by dopamine neurons using immunohistochemical staining for TH (Gramage et al., 2011). Here, we did not find any differences in Th mRNA expression in the VTA of Mdk −/− mice or in mice expressing shMdk in the VTA compared with controls. We also counted the number of TH-positive cells in the VTA in Mdk null mice and in mice expressing shMdk and did not find any changes in cell numbers (Chen and Lasek, unpublished data). It is possible, though, that Mdk −/− mice do have decreased levels of dopamine in the nucleus accumbens, which could be responsible for the increased ethanol consumption by Mdk −/− mice.
In this study we found that expression of Il1b and Ccl2 are increased in the VTA of mice expressing shMdk, and that Ccl2 expression is increased in the VTA of Mdk −/− mice. One caveat to the lentiviral injections is that the lentivirus could potentially activate glia at the site of injection, causing increased cytokine expression (Kunitsyna et al., 2016). Since we found that Ccl2 expression increased in both the Mdk knockout mice and in mice injected with Mdk lentivirus, it is likely that this increase is due to a specific effect of decreased Mdk levels. Although it is difficult to compare between different qPCR experiments, we found that the qPCR Cq values for Ccl2 did not differ between wild type, non-injected mice and mice injected with the control shScr lentivirus (data not shown). However, we did find that the Cq values for Il1b were significantly lower in the VTA of mice injected with the control lentivirus compared with wild type mice, suggesting that Il1b expression may be induced upon lentiviral infection and that the increase of Il1b in the VTA of mice expressing shMdk may be due to a non-specific effect of the shRNA or lentivirus.
Neuroimmune signaling has emerged as a potentially important contributor to excessive ethanol consumption (Robinson et al., 2014). Chronic ethanol exposure not only induces genes involved in neuroimmune signaling pathways in the brain, but some of these genes have been validated as important for alcohol consumption using gene knockout mice and pharmacological tools (Blednov et al., 2015, Blednov et al., 2005, Blednov et al., 2012). MDK expression is correlated with the expression of cytokines and chemokines in several different tissues types and plays a role in inflammatory diseases (Weckbach et al., 2011). Increased and decreased CCL2 (also known as MCP-1) protein expression is observed in the kidneys of Mdk −/− mice in models of glomerulonephritis and diabetic nephropathy, respectively (Kojima et al., 2013). Here, we found that Ccl2 gene expression is elevated in Mdk −/− mice and in mice expressing shMdk in the VTA. CCL2 is a potent chemoattractant that recruits monocytes and microglia to sites of tissue injury (Yao & Tsirka, 2014) and is induced by chronic ethanol exposure and withdrawal (Harper et al., 2015, Kane et al., 2013, Kane et al., 2014, Knapp et al., 2016, Lippai et al., 2013). CCL2 is also increased in the serum and brain of human alcoholics (He & Crews, 2008, Manzardo et al., 2016).
Female Ccl2 knockout mice consume less ethanol than control females (Blednov et al., 2005), so an increase in CCL2 expression in Mdk −/− mice could potentially be responsible for increased ethanol consumption by these mice. In addition, ethanol-preferring P rats have elevated CCL2 levels in the central nucleus of the amygdala and the VTA compared with non-preferring (NP) rats (June et al., 2015). Neuronal knockdown of Ccl2 in the VTA of P rats decreases binge drinking (June et al., 2015), and intracerebroventricular infusion of CCL2 protein increases operant ethanol self-administration in rats (Valenta & Gonzales, 2016). Future experiments are necessary to determine whether increased CCL2 in the VTA of Mdk −/− mice is responsible for increased ethanol consumption by knocking down Ccl2 in this brain region and testing drinking behavior.
Interestingly, there is a relationship between CCL2 and dopamine neurotransmission. CCL2 is expressed in dopamine neurons in the adult rat substantia nigra and VTA (Banisadr et al., 2005a, Banisadr et al., 2005b) and promotes dopamine neuron survival and neurite outgrowth in midbrain neuron cultures (Edman et al., 2008). Injection of CCL2 unilaterally into the rat substantia nigra decreases striatal dopamine content and its metabolites 1 hour after injection as measured by high-pressure liquid chromatography, and also rapidly increases dopamine release in the striatum as measured by microdialysis (Guyon et al., 2009). In addition, CCL2 treatment of brain slices containing the substantia nigra increases dopamine neuron firing rates (Guyon et al., 2009). Direct infusion of CCL2 into the VTA also increases the amount of phosphorylated TH (Wakida et al., 2014), which is an active form of the enzyme. Together, these studies provide evidence that CCL2 regulates dopamine neurotransmission. It is possible, then, that the increase in Ccl2 expression in the VTA of Mdk −/− mice impacts both dopamine levels and release in the nucleus accumbens, leading to enhanced ethanol consumption.
How might MDK signal to limit Ccl2 expression and alcohol drinking? MDK is a ligand for several receptors that are expressed in the central nervous system, such as anaplastic lymphoma kinase (ALK) and the protein tyrosine phosphatase β/ζ (Weckbach et al., 2011). We have found that shRNA-mediated knockdown of Alk in the mouse VTA decreases binge-like ethanol consumption (Dutton et al., 2016), which suggests that MDK in the VTA may not be acting through ALK to regulate alcohol drinking, since knockdown of Mdk in the VTA increases drinking. It is possible that MDK is a ligand for another receptor in the VTA whose downstream signaling pathways regulate the transcription of Ccl2 and ethanol drinking behavior. Further work is needed to determine the functional receptor for MDK in the VTA and the downstream signaling pathways activated by MDK.
In addition to MDK playing a role in ethanol reward and drinking, it may also be involved in addiction to other drugs of abuse. Mdk −/− mice are more sensitive to the anxiolytic effects of diazepam (Vicente-Rodriguez et al., 2014) and show reduced extinction to cocaine conditioned place preference (Gramage et al., 2013). Mdk expression is also induced in the VTA, the nucleus accumbens, and the hippocampus in response to morphine administration (Ezquerra et al., 2007, Garcia-Perez et al., 2015, Garcia-Perez et al., 2014). MDK seems to play an important role in immune responses in the brain in response to amphetamine because Mdk −/− mice show alterations in amphetamine-induced striatal astrocytosis and microgliosis (Vicente-Rodriguez et al., 2016). Thus, targeting MDK and its signaling pathways may be a novel strategy for reducing neuroinflammation and addiction to multiple substances, including alcohol (Vicente-Rodriguez et al., 2016).
Supplementary Material
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism, Integrative Neuroscience Initiative on Alcoholism (INIA, U01 AA016654 & U01 020912) and the Center for Alcohol Research in Epigenetics (P50 AA022538). The authors declare no conflict of interest.
References
- Banisadr G, Gosselin RD, Mechighel P, Kitabgi P, Rostene W, Parsadaniantz SM. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol. 2005a;489:275–292. doi: 10.1002/cne.20598. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Rostene W, Kitabgi P, Melik Parsadaniantz S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J Comp Neurol. 2005b;492:178–192. doi: 10.1002/cne.20729. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Ferguson LB, Schoenhard GL, Goate AM, Edenberg HJ, Wetherill L, Hesselbrock V, Foroud T, Harris RA. Peroxisome proliferator-activated receptors alpha and gamma are linked with alcohol consumption in mice and withdrawal and dependence in humans. Alcohol Clin Exp Res. 2015;39:136–145. doi: 10.1111/acer.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA. Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res. 2005;165:110–125. doi: 10.1016/j.bbr.2005.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol. 2012;17:108–120. doi: 10.1111/j.1369-1600.2010.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl) 2015 doi: 10.1007/s00213-015-3906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton JW, 3rd, Chen H, You C, Brodie MS, Lasek AW. Anaplastic lymphoma kinase regulates binge-like drinking and dopamine receptor sensitivity in the ventral tegmental area. Addict Biol. 2016 doi: 10.1111/adb.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman LC, Mira H, Arenas E. The beta-chemokines CCL2 and CCL7 are two novel differentiation factors for midbrain dopaminergic precursors and neurons. Experimental cell research. 2008;314:2123–2130. doi: 10.1016/j.yexcr.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Ezquerra L, Herradon G, Nguyen T, Silos-Santiago I, Deuel TF. Midkine is a potent regulator of the catecholamine biosynthesis pathway in mouse aorta. Life sciences. 2006;79:1049–1055. doi: 10.1016/j.lfs.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Ezquerra L, Perez-Garcia C, Garrido E, Diez-Fernandez C, Deuel TF, Alguacil LF, Herradon G. Morphine and yohimbine regulate midkine gene expression in the rat hippocampus. European journal of pharmacology. 2007;557:147–150. doi: 10.1016/j.ejphar.2006.11.024. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, van der Brug M, Hwang JW, Gochee PA, Matsumoto I, Niwa S, Wilce PA. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. Journal of neurochemistry. 2005;93:359–370. doi: 10.1111/j.1471-4159.2004.03021.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Chronic smoking and alcoholism change expression of selective genes in the human prefrontal cortex. Alcoholism, clinical and experimental research. 2006;30:908–915. doi: 10.1111/j.1530-0277.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Impact of alcohol abuse on protein expression of midkine and excitatory amino acid transporter 1 in the human prefrontal cortex. Alcoholism, clinical and experimental research. 2008;32:1849–1858. doi: 10.1111/j.1530-0277.2008.00754.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez D, Laorden ML, Milanes MV. Regulation of Pleiotrophin, Midkine, Receptor Protein Tyrosine Phosphatase beta/zeta, and Their Intracellular Signaling Cascades in the Nucleus Accumbens During Opiate Administration. Int J Neuropsychopharmacol. 2015:19. doi: 10.1093/ijnp/pyv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez D, Luisa Laorden M, Nunez C, Victoria Milanes M. Glial activation and midkine and pleiotrophin transcription in the ventral tegmental area are modulated by morphine administration. Journal of neuroimmunology. 2014;274:244–248. doi: 10.1016/j.jneuroim.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther. 2004;103:121–146. doi: 10.1016/j.pharmthera.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Gramage E, Martin YB, Ramanah P, Perez-Garcia C, Herradon G. Midkine regulates amphetamine-induced astrocytosis in striatum but has no effects on amphetamine-induced striatal dopaminergic denervation and addictive effects: functional differences between pleiotrophin and midkine. Neuroscience. 2011;190:307–317. doi: 10.1016/j.neuroscience.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Gramage E, Perez-Garcia C, Vicente-Rodriguez M, Bollen S, Rojo L, Herradon G. Regulation of extinction of cocaine-induced place preference by midkine is related to a differential phosphorylation of peroxiredoxin 6 in dorsal striatum. Behavioural brain research. 2013;253:223–231. doi: 10.1016/j.bbr.2013.07.026. [DOI] [PubMed] [Google Scholar]
- Guyon A, Skrzydelski D, De Giry I, Rovere C, Conductier G, Trocello JM, Dauge V, Kitabgi P, Rostene W, Nahon JL, Melik Parsadaniantz S. Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience. 2009;162:1072–1080. doi: 10.1016/j.neuroscience.2009.05.048. [DOI] [PubMed] [Google Scholar]
- Harper KM, Knapp DJ, Breese GR. Withdrawal from Chronic Alcohol Induces a Unique CCL2 mRNA Increase in Adolescent But Not Adult Brain--Relationship to Blood Alcohol Levels and Seizures. Alcohol Clin Exp Res. 2015;39:2375–2385. doi: 10.1111/acer.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Chen H, Muramatsu H, Lasek AW. Ethanol activates midkine and anaplastic lymphoma kinase signaling in neuroblastoma cells and in the brain. Journal of neurochemistry. 2015;135:508–521. doi: 10.1111/jnc.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D, Puche A, Aurelian L. CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology. 2015;40:1549–1559. doi: 10.1038/npp.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Drew PD. Effects of ethanol on immune response in the brain: region-specific changes in aged mice. J Neuroinflammation. 2013;10:66. doi: 10.1186/1742-2094-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Phelan PS, Drew PD. Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol Clin Exp Res. 2014;38:384–391. doi: 10.1111/acer.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Muramatsu H, Muramatsu T, Kim SU. Midkine, a novel neurotrophic factor, promotes survival of mesencephalic neurons in culture. Neuroscience letters. 1993;160:9–12. doi: 10.1016/0304-3940(93)90904-y. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Harper KM, Whitman BA, Zimomra Z, Breese GR. Stress and Withdrawal from Chronic Ethanol Induce Selective Changes in Neuroimmune mRNAs in Differing Brain Sites. Brain sciences. 2016:6. doi: 10.3390/brainsci6030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Kosugi T, Sato W, Sato Y, Maeda K, Kato N, Kato K, Inaba S, Ishimoto T, Tsuboi N, Matsuo S, Maruyama S, Yuzawa Y, Kadomatsu K. Deficiency of growth factor midkine exacerbates necrotizing glomerular injuries in progressive glomerulonephritis. Am J Pathol. 2013;182:410–419. doi: 10.1016/j.ajpath.2012.10.016. [DOI] [PubMed] [Google Scholar]
- Kunitsyna TA, Ivashkina OI, Roshchina MA, Toropova KA, Anokhin KV. Lentiviral Transduction of Neurons in Adult Brain: Evaluation of Inflammatory Response and Cognitive Effects in Mice. Bull Exp Biol Med. 2016;161:316–319. doi: 10.1007/s10517-016-3404-4. [DOI] [PubMed] [Google Scholar]
- Lasek AW, Janak PH, He L, Whistler JL, Heberlein U. Downregulation of mu opioid receptor by RNA interference in the ventral tegmental area reduces ethanol consumption in mice. Genes, brain, and behavior. 2007;6:728–735. doi: 10.1111/j.1601-183X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- Lippai D, Bala S, Csak T, Kurt-Jones EA, Szabo G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS One. 2013;8:e70945. doi: 10.1371/journal.pone.0070945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo AM, Poje AB, Penick EC, Butler MG. Multiplex Immunoassay of Plasma Cytokine Levels in Men with Alcoholism and the Relationship to Psychiatric Assessments. Int J Mol Sci. 2016;17:472. doi: 10.3390/ijms17040472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, Finn DA, Hoffman PL, Iyer VR, Koob GF, Bergeson SE. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Harris RA, Ponomarev I. Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcoholism, clinical and experimental research. 2011;35:659–670. doi: 10.1111/j.1530-0277.2010.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura E, Kadomatsu K, Yuasa S, Muramatsu H, Mamiya T, Nabeshima T, Fan QW, Ishiguro K, Igakura T, Matsubara S, Kaname T, Horiba M, Saito H, Muramatsu T. Disruption of the midkine gene (Mdk) resulted in altered expression of a calcium binding protein in the hippocampus of infant mice and their abnormal behaviour. Genes to cells : devoted to molecular & cellular mechanisms. 1998;3:811–822. doi: 10.1046/j.1365-2443.1998.00231.x. [DOI] [PubMed] [Google Scholar]
- Ohgake S, Shimizu E, Hashimoto K, Okamura N, Koike K, Koizumi H, Fujisaki M, Kanahara N, Matsuda S, Sutoh C, Matsuzawa D, Muramatsu H, Muramatsu T, Iyo M. Dopaminergic hypofunctions and prepulse inhibition deficits in mice lacking midkine. Progress in neuro-psychopharmacology & biological psychiatry. 2009;33:541–546. doi: 10.1016/j.pnpbp.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Prediger RD, Rojas-Mayorquin AE, Aguiar AS, Jr, Chevarin C, Mongeau R, Hamon M, Lanfumey L, Del Bel E, Muramatsu H, Courty J, Raisman-Vozari R. Mice with genetic deletion of the heparin-binding growth factor midkine exhibit early preclinical features of Parkinson’s disease. Journal of neural transmission. 2011;118:1215–1225. doi: 10.1007/s00702-010-0568-3. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Robinson G, Most D, Ferguson LB, Mayfield J, Harris RA, Blednov YA. Neuroimmune pathways in alcohol consumption: evidence from behavioral and genetic studies in rodents and humans. Int Rev Neurobiol. 2014;118:13–39. doi: 10.1016/B978-0-12-801284-0.00002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta JP, Gonzales RA. Chronic Intracerebroventricular Infusion of Monocyte Chemoattractant Protein-1 Leads to a Persistent Increase in Sweetened Ethanol Consumption During Operant Self-Administration But Does Not Influence Sucrose Consumption in Long-Evans Rats. Alcohol Clin Exp Res. 2016;40:187–195. doi: 10.1111/acer.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Rodriguez M, Fernandez-Calle R, Gramage E, Perez-Garcia C, Ramos MP, Herradon G. Midkine Is a Novel Regulator of Amphetamine-Induced Striatal Gliosis and Cognitive Impairment: Evidence for a Stimulus-Dependent Regulation of Neuroinflammation by Midkine. Mediators Inflamm. 2016;2016:9894504. doi: 10.1155/2016/9894504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Rodriguez M, Perez-Garcia C, Haro M, Ramos MP, Herradon G. Genetic inactivation of midkine modulates behavioural responses to ethanol possibly by enhancing GABA(A) receptor sensitivity to GABA(A) acting drugs. Behavioural brain research. 2014;274:258–263. doi: 10.1016/j.bbr.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Wakida N, Kiguchi N, Saika F, Nishiue H, Kobayashi Y, Kishioka S. CC-chemokine ligand 2 facilitates conditioned place preference to methamphetamine through the activation of dopamine systems. J Pharmacol Sci. 2014;125:68–73. doi: 10.1254/jphs.14032fp. [DOI] [PubMed] [Google Scholar]
- Weckbach LT, Muramatsu T, Walzog B. Midkine in inflammation. The Scientific World Journal. 2011;11:2491–2505. doi: 10.1100/2011/517152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, Lovinger DM, Alvarez VA. Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology. 2014;39:579–594. doi: 10.1038/npp.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. Monocyte chemoattractant protein-1 and the blood-brain barrier. Cell Mol Life Sci. 2014;71:683–697. doi: 10.1007/s00018-013-1459-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.