

Abstract
Dural cerebral veins (CV) are required for cerebrospinal fluid reabsorption and brain homeostasis, but mechanisms that regulate their growth and remodeling are unknown. We report molecular and cellular processes that regulate dural CV development in mammals, and describe venous malformations in humans with craniosynostosis and TWIST1 mutations that are recapitulated in mouse models. Surprisingly, Twist1 is dispensable in endothelial cells but required for specification of osteoprogenitor cells that differentiate into preosteoblasts that produce Bone Morphogenetic Proteins. Inactivation of Bmp2 and Bmp4 in preosteoblasts and periosteal dura cause skull and CV malformations, similar to humans harboring TWIST1 mutations. Notably, arterial development appears normal, suggesting morphogens from the skull and dura establish optimal venous networks independent from arterial influences. Collectively, our work establishes a paradigm whereby CV malformations result from primary or secondary loss of paracrine BMP signaling from preosteoblasts and dura, highlighting unique cellular interactions that influence tissue-specific angiogenesis in mammals.
TOC image
By characterizing dural cerebral vein malformations in TWIST1 mutation-positive humans and mouse models with craniosynostosis, Tischfield et al. report that cerebral vein angiogenesis requires paracrine BMP signaling from skull preosteoblasts and periosteal dura. The effects are independent from arterial influences and highlight unique cellular interactions that pattern tissue-specific vascular networks.
Introduction
Arteries and veins generally run adjacent to one another and similar growth and guidance cues influence their development (Kidoya et al., 2015). A notable exception, however, is found in the mammalian head. At maturity, the major cerebral arteries enter the skull base medially and traverse the subarachnoid space. By contrast, the major cerebral veins (CV), also known as the dural venous sinuses, lie within the dura mater beneath the skull and exit the skull base posteriorly beneath the cerebellum (Figure S1A,B). This anatomical distinction suggests the head has evolved unique signaling mechanisms that independently regulate the growth of these vessels.
Unlike arterial development, little is known about CV development beyond anatomical studies published 60–100 years ago (Padget, 1956; Streeter, 1921). This is surprising because CV malformations can have a significant impact on human health. Many pediatric cerebrovascular anomalies result from defects in venous development, alter hydrostatic pressure within the dural venous sinuses, perturb cerebrospinal fluid (CSF) reabsorption, and lead to elevated intracranial pressure (ICP) (Raets et al., 2015; Wilson, 2016). Elevated ICP causes severe headaches and can lead to vision loss, and potential longer-term health effects of elevated ICP and altered CSF reabsorption remain poorly defined.
Multiple forms of craniosynostosis, a skull malformation caused by abnormal development or premature fusion of the cranial sutures, put patients at risk for elevated ICP (Abu-Sittah et al., 2016; Twigg and Wilkie, 2015). Elevated ICP secondary to craniosynostosis is often assumed to result from a small fused skull (Johnson and Wilkie, 2011), but patients can have chronic ICP after skull expansion, and evidence suggests their elevated ICP may result, at least in part, from venous malformations (Hayward, 2005; Liesegang, 2001; Stevens et al., 2007). Individuals with craniosynostosis secondary to activating mutations in FGFR2 and FGFR3 have CV malformations that include aplasia or hypoplasia of the transverse (TVs) and sigmoid (SgS) sinuses, and stenosis of the internal jugular veins (iJV) (Figure S1B) (Johnson and Wilkie, 2011; Robson et al., 2000). Individuals with TWIST1 haploinsufficiency, another common cause of syndromic craniosynostosis, are also reported to have chronically elevated ICP (Twigg and Wilkie, 2015; Woods et al., 2009), suggesting they also harbor CV malformations. The overall prevalence and nature of CV abnormalities in the craniosynostoses are, however, unclear. Thus, these and other venous malformation syndromes underscore a need to identify genes, pathways, and cellular events that influence CV development and physiology.
We now report a spectrum of dural CV malformations in humans with TWIST1 mutation-positive craniosynostosis, and use mouse genetics to characterize cellular and molecular mechanisms that regulate CV growth and remodeling in development and disease. Modeling CV development in wild-type and mutant mice, we find that key growth and remodeling events that optimize venous circulation in the head are independent from arterial angiogenesis. Instead, Twist1 coordinates CV growth, remodeling, and refinement in conjunction with skull and dura development in a non-cell autonomous fashion, in part, through the specification of mesoderm-derived osteoprogenitor cells (osteo-PC) and their sequential differentiation into preosteoblasts (pre-OB) that express Bone Morphogenetic Protein (BMP). By conditionally ablating Bmp2 and Bmp4 in coronal suture (corSuture) derived pre-OB and periosteal dura (periDura), we also show that cross-talk between pre-OB derived BMPs and the underlying CVs is critical for venous growth and remodeling. Collectively, our findings provide mechanistic insight into human vascular disease and how tissue-specific vascular networks grow and remodel in mammals.
Results
Humans harboring heterozygous TWIST1 mutations have a spectrum of CV anomalies
To explore a connection between TWIST1 mutations and CV malformations, we examined cerebral vessel magnetic resonance venograms (MRV) and arteriograms (MRA), and skull computed tomography (CT) scans, of two controls (C1–C2) and six individuals with moderate to severe craniosynostosis (Tl – T6) harboring five unique heterozygous TWIST1 mutations. Five individuals harboring TWIST1 mutations (T1–T5) had segmental aplasia or hypoplasia of one TVS and/or SgS, and four had jugular foraminal stenosis (Figure 1A, Table S1). T3–T5 had enlarged optic nerve sheaths, T4 had inconspicuous cerebral sulci, and T5 had papilledema, all suggestive of elevated ICP (Table S1). Skull examination revealed that four had moderate to severe bilateral (T1, T2) or unilateral (siblings T3 and T4) coronal synostosis (Figure 1B,C). Notably, T3 and T4 had right-sided CV anomalies and right-sided coronal synostosis. T5, who had right-sided TVS aplasia and SgS and iJV hypoplasia, also had stenosis of the distal left TVS and iJV (Figure 1A). Only individual T6, who had a normal skull at age seven, had a normal MRV (Figure 1A,C). Magnetic resonance angiograms (MRA) of individuals T1–T4 and T6 showed normal cerebral arteries (Figure 1D), with only minor arterial variations at the base of the brain (Table S1). These findings demonstrate that humans with TWIST1 mutations have a spectrum of CV malformations associated with elevated ICP, but normal arterial development.
Fig. 1. CV malformations segregate with cranial deformities and coronal synostosis in TWIST1 mutation-positive humans.
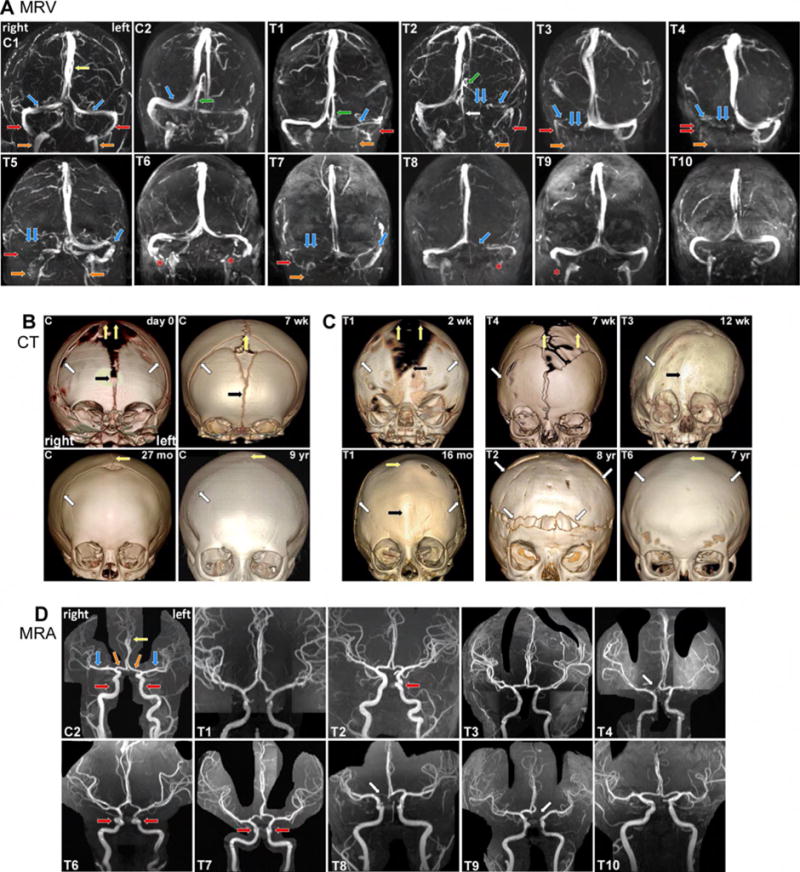
(A) Coronal MRV images depicting the dural CV in control (C1 and C2) and affected individuals (T1–T10). Yellow and green arrows denote the SSS and straight sinus (SS), and the paired blue, red, and orange arrows denote the paired TVS, SgS, and iJV, respectively. Cl has co-dominant TVS. C2 has a dominant right TVS and a SS, which drains into the minor TVS. T1–T5, T7, T8 have segmental hypoplasia (single arrow) and aplasia or stenosis (double arrows) and of the TVS, SgS, and iJV. Tl, T2 and T5 have unilateral and bilateral iJV stenosis, respectively, and T5 and T7 have stenosis of the distal left TVS. Compensatory vessels are noted, including a prominent occipital sinus (white arrow) in T2 that drains blood from the SS. T6, T9, and T10 have normal CV anatomy, and artefactual signal loss (asterisks) that was not present on source images. (B and C) Skull CT images in control and TWIST1 mutation-positive individuals. (B) Prominent coronal (white arrow), metopic (black arrow), and sagittal sutures (yellow arrows) narrow in control newborns by 7 weeks. The metopic suture is fused at 27 months, but the coronal and sagittal sutures remain patent. (C) Suture and cranial deformities in TWIST1 mutation-positive individuals T1–T4 and T6. T1: At week 2 (top), has bilateral coronal synostosis (white arrows), smaller parietal bones, and abnormally widened metopic and sagittal sutures (black and yellow arrowheads). At 16 months, there are postoperative changes from bilateral coronal craniotomies (white arrows), and premature fusion of the metopic and sagittal sutures (yellow and black arrows). T4: At seven weeks, the right corSuture (white arrow) has prematurely fused. The sagittal suture (yellow arrows) and anterior fontanel are abnormally wide. T3: At 12 weeks, right coronal craniosynostosis causes right frontal plagiocephaly. T2: At eight years has brachycephaly, bilateral coronal synostosis, and postoperative changes from frontal and parietal osteotomies (white arrows). T6: At seven years shows bilateral coronal and sagittal (white and yellow arrow) synostosis without significant deformity, suggestive of late postnatal fusion. (D) Coronal MIP 3D TOF MRA images depict normal arterial anatomy. C2 has normal anatomy of the cavernous internal carotid arteries (iCA, red arrows), anterior cerebral arteries (aCA, yellow arrow) and the paired aCA A1 segments (orange arrows), and middle cerebral (blue arrows) arteries, T1–T4 and T6–T10 have no significant anatomic changes. Individuals T6 and T7, who are related, have localized bilateral and T2 has unilateral tortuosity of the cavernous iCA (red arrows). T4, T8, and T9 are related and have unilateral hypoplasia/aplasia of the aCA A1 segment (white arrow), See also Table S1.
To further define the spectrum of CV malformations in TWIST1 mutation-positive humans, we evaluated MRV and MRA scans from four additional individuals, none of whom required early interventional surgery for craniosynostosis, had symptoms of elevated ICP, or had CT imaging (T7–T10). Skull shapes were relatively normal on MRI, although some had minor skull base abnormalities (Table S1). T9 and T10 had normal MRVs, and T8 had segmental hypoplasia of the left TVS (Figure 1A). T7, however, had segmental aplasia of the right TVS, hypoplasia of the right SgS and iJV, and stenosis of the distal left TVS; prominent optic nerve sheaths were also noted (Figure 1A and Table S1). MRA findings were similar to the first group (Figure 1D and Table S1). Thus, CV malformations in TWIST1 mutation-positive humans generally correlate with skull malformations and early-onset craniosynostosis, suggesting normal skull development is required for venous growth and remodeling.
CV growth and remodeling is compromised in conditional Twist1 mutant mice
Although Twist1 has been shown in mouse to regulate cranial suture and skull patterning (Goodnough et al., 2015; Ting et al., 2009), cerebral blood vessel development has not been examined. To explore the etiology of CV abnormalities in TWIST1 mutation-positive humans, we first asked if human CV development could be modeled in mouse. We compared CV growth in mouse to wax-plate reconstructions of serially sectioned human fetuses, published in 1921 by George Streeter (Streeter, 1921). Our results establish that critical growth and remodeling events, including the formation and remodeling of the anterior, middle, and posterior venous plexuses (antPlexus, midPlexus, and posPlexus, respectively) as described below and depicted in Figure 2, are conserved between the two species and occur between E12.5–E14.5 in mouse. Thus, mouse is an excellent model for human CV development (Figure 2A–F, Figure S1B,C).
Fig. 2. CV growth and remodeling is temporally and spatially coupled with skull development.
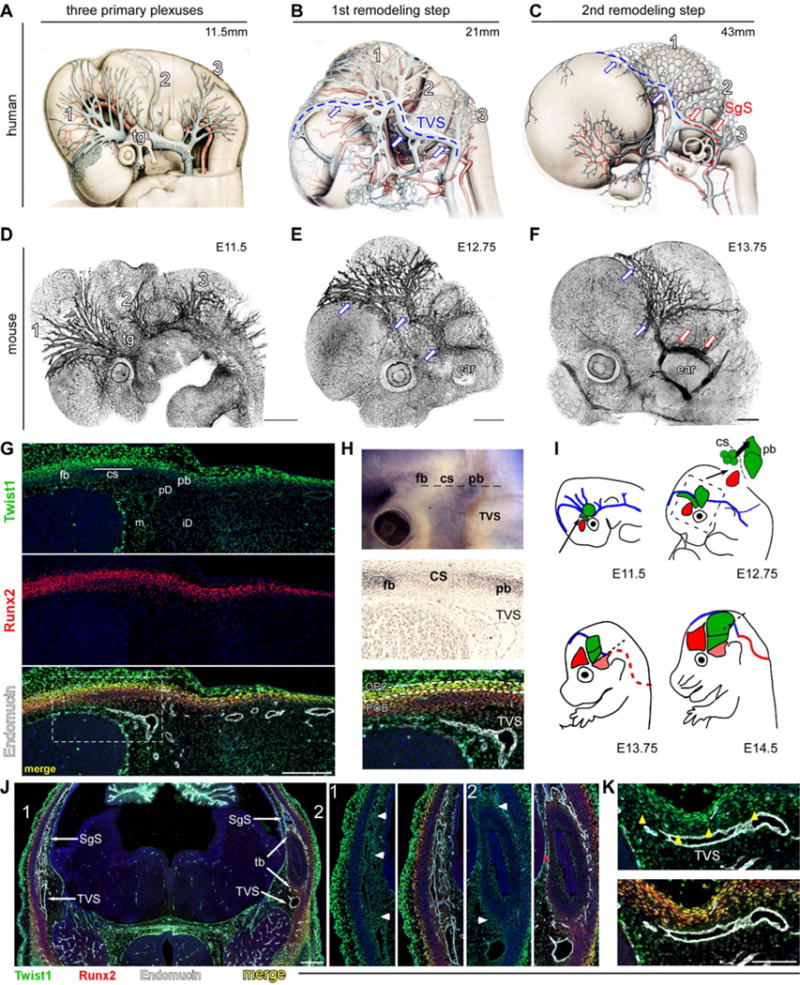
(A–C) Wax-plate reconstructions of human fetuses depicting growth and remodeling of the dural CV (Streeter, 1921). (A) Three plexuses (l=anterior [antPlexus], 2=middle [midPlexus], 3=posterior [posPlexus]) drain into the primitive head vein (PHV) adjacent to the eye and trigeminal ganglia (tg). (B) First remodeling step results from an anastomosis between the antPlexus and midPlexus to form the transverse sinus (TVS, blue lines and arrows). The PHV regresses. (C) Second remodeling step results from an anastomosis between the midPlexus and posPlexus to form the sigmoid sinus (SgS, red arrows), located above the temporal bone (temBone, tb). (D–F) Z-stacks of E11.5 (D), E12.75 (E), and E13.75 (F) mouse embryos stained with endomucin. Growth and remodeling of the CV are highly conserved with humans. (G) E12.5 transverse section through the head at the level indicated in the top panel of H. High levels of Twist1 expression co-localize with Runx2 in the frontal and parietal bone periosteum (frBone [fb] and parBone [pb]) and in the coronal suture (corSuture [CS]). Weaker expression is seen in periosteal dura (periDura [pd]) and inner dura (iD), and the arachnoid mater/pia (meninges, m). TVS and antPlexus grow and remodel in periDura underlying the corSuture and parBone. (H) (Top) E12.75 mouse head showing endogenous ALP activity in frBone and parBone pre-OB. (Middle) Transverse section, adjacent to G. The TVS remodels in proximity to the corSuture and osteogenic front of the parBone. (Bottom) High levels of Twist1 expression occur in osteo-PCs within the corSuture and periosteum (white dashes), but protein levels are low in Runx2/ALP+ Pre-OB. (I) CV remodeling and skull development. E11.5: frBone- and parBone osteo-PCs (red and green circles, respectively) develop within supraorbital mesenchyme adjacent to the antPlexus and PHV (blue). The corSuture (green, indicated by black arrow) develops from mesoderm. E12.75: TVS remodels proximal to pre-OB (green circles) that migrate perpendicularly from the corSuture to the parBone. Boxed region corresponds to inset. E13.75: the SgS (red dotted line) remodels above the temBone (pink). E14.5: parBone Pre-OB have reached the apex of the head. TVS remains juxtaposed to the parBone in periDura. (J) E13.75 transverse section at the level indicated by dotted line in l (E13.75). The right side (1, magnified in middle two panels), is slightly above the left (2, magnified in right two panels), and shows the SgS has remodeled in Twist1+ dura (arrowheads,panel 1) around Twistl/Runx2+ temBone-periosteum (arrow head, panel 2). (K) E14.5 transverse section, indicated by black line in I (E14.5). Twist1 is expressed in periDura (yellow arrowheads) underneath Runx2highTwistllow parBone pre-OB. Scale bar=5OOum (A–F), 200, 500, 100um (G, J, K respectively). See also Fig. S1, S7.
We next examined Twist1 expression in the developing mouse skull and cranial blood vessels between E12.5–E14.5. Consistent with a role for Twist1 in skull development, Twist1 protein was high in Runx2+ osteo-PCs in the corSuture and along the periosteum of the developing frontal and parietal bones (frBone and parBone, respectively) (Figure 2G). Protein levels were weaker in Runx2/Alkaline Phosphatase (ALP)+ pre-OB that differentiate from these osteo-PCs, in Runx2+ chondral progenitors along the temporal bone (temBone) periosteum, and in the periDura and meninges (Figure 2H,J). At later ages (E13.5–14.5), Twist1 expression was similar; it remained high in osteo-PCs, low in differentiating pre-OB, and present throughout the periDura (Figure 2K). Surprisingly, Twist1 was absent in endothelial cells, and removal of Twist1 with a pan-endothelial Cre-recombinase, Tie2-Cre, yielded normal cranial and retinal blood vessel development (Figure S2A–C). Thus, any influence of Twist1 on CV development is non-cell autonomous.
Interestingly, dural CV growth and remodeling exhibited tight temporal and spatial coupling with skull development between E12.5–E14.5 (Figure 2I). Notably, the TVS remodeled within Twist1+ periDura underlying the corSuture and osteogenic front of the parBone (Figure 2H), and vessels in the antPlexus were located in periDura juxtaposed to parietal Runx2/ALP+ pre-OB and differentiating Runx2+ chondrocytes (Figure 2G). Furthermore, the SgS also remodeled in Twistl+ dura along the periosteal surface of the temBone (Figure 2J). These anatomical relationships suggest Twist1 may modulate paracrine signaling cues from the skull and/or dura to influence CV growth and remodeling.
To investigate paracrine signaling between the skull, dura, and underlying CV, we characterized recombinase tools that would allow for specific spatial manipulation of gene pathways across these different cells types. Pdgfrb-Cre;R26LSL-tdTomato(Ai14) reporter mice showed diffuse labeling in the head at E11.75, including neural crest and mesoderm-derived supraorbital dermal mesenchyme, which gives rise to the frBone and parBone, respectively (Figure S2D,E). By E12.5, labeling was seen in Runx2+ osteo-PCs in the corSuture and along the parBone periosteum, in Runx2/ALP+ parBone pre-OB, and throughout the dura and meninges (Figure S2H,I). Sm22a-Cre;Ai14 reporter mice showed similar but more restricted cell labeling (Figure S2F). Sm22a-Cre (Sm-Cre) activity was sparse in frBone osteo-PCs, but diffuse throughout Runx2+ osteo-PCs in the corSuture, parBone periosteum, periDura, and differentiating Runx2/ALP+ parBone pre-OB. Also, Sm-Cre activity appeared weaker and more scattered in the meninges (Figure S2F–I) and, as seen for Pdgfrb-Cre (Pd-Cre), was absent from the venous endothelium (Figure S2J). Thus, we used these alleles to inactivate Twist1 in discrete and overlapping populations of cells that give rise to the skull, dura, and meninges.
Conditional loss of Twist1 perturbed CV growth and remodeling in several ways. First, although the three venous plexuses formed by E11.75 in Twist1CK0/−; Pd-Cre and Twist1CK0/−;Sm-Cre embryos, branching and overall complexity of the antPlexus was diminished (Figure 3A). Also, cross-sections at E12.75 revealed small and poorly lumenized vessels (Figure 3D). The CV were properly specified, however, as mutants expressed EphB4 and endomucin on the venous endothelium (Figure 3E).
Fig. 3. Twist1 mutants have perturbed CV development despite normal arterial growth and remodeling.
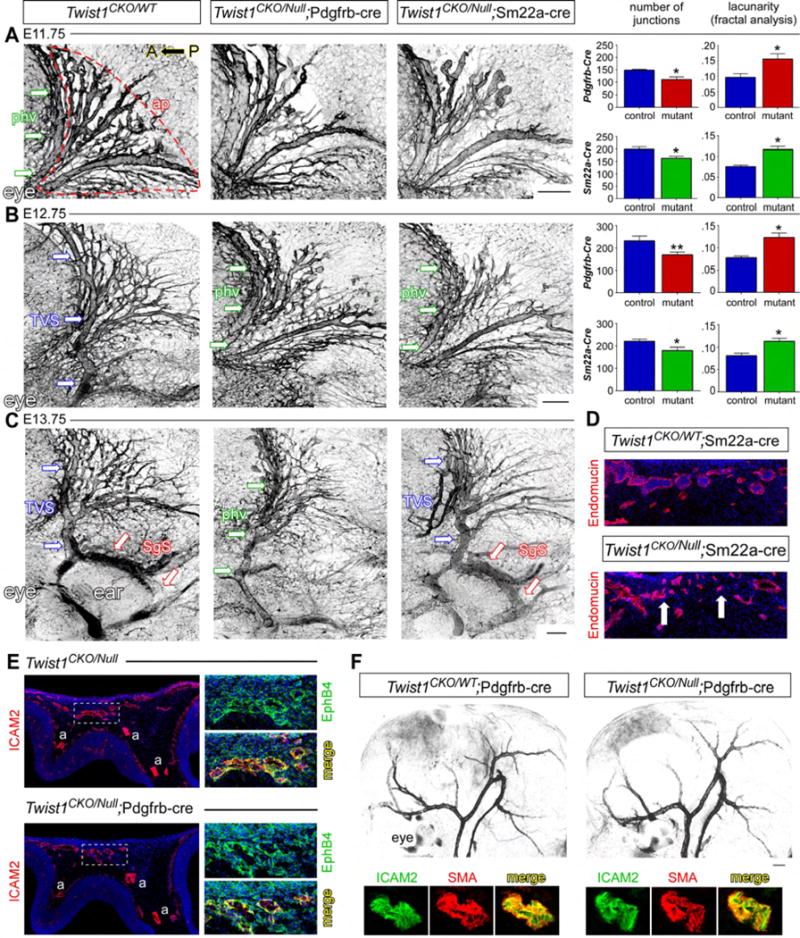
(A–C) Z-stacks of the venous plexuses stained with endomucin detailing the growth and remodeling of the CV. (A) Primitive head vein (PHV, green arrows) and antPlexus (ap, outlined) in an E11.75 control. The antPlexus in Twist1CKO/−;Pd-Cre (n=4) and Twist1CKO/−;Sm-Cre (n=5) embryos have a significant reduction in vessel branching and more empty space (lacunarity, right graphs). (B) By E12.75, the TVS has remodeled in controls, but the PHV is still present in Twist1CKO/−;Pd-Cre (n=5) and Twist1CKO/−;Sm-Cre (n=6) embryos. antPlexus branching remains reduced. (C) By E13.75, the SgS remodels in control and Twist1CKO/−;Sm-Cre embryos (n=5, red arrows), while the TVS shows poor remodeling in both mutants. The PHV is still present in Twist1CKO/−;Pd-Cre embryos and SgS remodeling is absent (n=5). (D) Cross sections through the antPlexus in E12.75 control and Twist1CKO/−;Sm-Cre embryos. Many vessels are poorly developed with collapsed lumens compared to heterozygous controls (n=6). (E) E11.75 transverse sections through the head, boxed regions are magnified on right. Venous endothelial cells are properly specified and express Ephb4 in Twist1CKO/−;Pd-Cre mutants (n=3). a=arterial. (F) E12.75 wholemount smooth muscle actin staining of the arterial head vasculature. Arterial growth and remodeling and smooth muscle coverage appear normal in Twist1CKO/−;Pd-Cre embryos (n=5). Scale bar=250um. *P<0.05, **P<0.01, unpaired student t test. See also Fig. S2, S7.
Second, the midPlexus of all Twist1CKO/−; Pd-Cre embryos failed to anastomose with the antPlexus, which is the first remodeling step that forms the nascent TVS. Instead, Twist1CK0/−; Pd-Cre mutants retained the primitive head veins (PHV) bilaterally (Figure 3B). While the nascent TVS also failed to form by E12.75 in Twist1CK0/−;Sm-Cre embryos (Figure 3B and refer to Figure 6A), TVS remodeling was eventually observed by E13–13.5. Despite remodeling, however, the TVS appeared tortuous and hypoplastic compared with controls (Figure 3C). Overall, Twist1CK0/− ;Sm-Cre embryos were less severely affected than Pd-Cre embryos as the midPlexus exhibited delayed and aberrant–rather than absent–growth and remodeling.
Fig. 6. BMP2 and BMP4 are required for CV growth and remodeling.

(A) Z-stack images of the TVS (blue open arrows) and antPlexus at E12.75. Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre and Bmp2CKO/CKO;Bmp4CKO/CKO;Pd-Cre mutants have delayed remodeling of the TVS (arrow) and poor development of the antPlexus, best seen from dorsal views of the head (bottom row). Bmp2CKO/CKO;Bmp4CKO/CKO;Sm-Cre mutants can be indistinguishable from Twist1CKO/−;Sm-Cre embryos. (B) Color-coded genotypes as annotated in (C). Branching is highly variable and not statistically significant in mutants versus controls, but lacunarity is significantly increased in Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre and Bmp2CKO/CKO;Bmp4CKO/CKO;Pd-Cre embryos. (C) Top panel: E18.5 dorsal views of the head depicting the SSS and paired TVS and SgS in controls and three and four allele loss Bmp2/4;Pd-Cre embryos. Bottom panel: The mean width of the dominant TVS is significantly smaller in all Bmp2/4;Pd-Cre mutant genotypes, and the minor vessel is hypoplastic or absent. The SSS is often hypoplastic, truncated, and/or split (arrows). C′: The distribution of the right versus left TVS width ratios in control and Bmp2/4;Pd-Cre mutants; color-coded genotypes as annotated in (C). Population medians are skewed towards unilateral hypoplasia or aplasia in Bmp2/4 two, three, and four allele loss mutants. (D) Bmpr1b and Bmpr2 are expressed in wild-type Cdh5+ venous endothelium. (E) E18.5 Bmpr2CKO/CKO; Tie2-Cre mutants show mild and moderate hypoplasia of the dominant and minor TVS, respectively. Scale bar=250um. *P<0.05, **P<0.01, ***P<0.001, one-way Anova with Dunnett’s post-test. See also Fig. S4–S7.
Third, by E13.75, none of the Twist1CK0/−; Pd-Cre embryos executed the second major remodeling step that forms the SgS, and continued to retain a hypoplastic PHV in place of a proper TVS and SgS (Figure 3C). By contrast, the SgS remodeled normally in less affected Twist1CKO/−;Sm-Cre embryos (Figure 3C). Interestingly, Pd-Cre, but not Sm-Cre, is expressed in the temBone and dura adjacent to the remodeling SgS, demonstrating that venous growth and remodeling can be variably perturbed by inactivating Twist1 in different non-vascular cell populations (Figure S2E,F, S3A). Notably, conditional Twist1CKO/− mutants showed normal arterial growth, remodeling, and smooth muscle coverage by E12.5–13.5 (Figure 3F and data not shown), suggesting that the venous malformations arose independently.
Finally, all Twist1CKO/−; Pd-Cre and Sm-Cre embryos developed complete unilateral regression of the PHV or TVS, respectively (Figure 4A). Following normal TVS remodeling in E12.75 controls, we observed a broad distribution of vessel caliber differences between the right and left TVS that were still present at birth, similar to humans (Figure 4B,C). In a subset (~7%), one TVS was significantly smaller, more frequently the left, but complete regression was never observed (Figure 4B). By contrast, the PHV or TVS regressed unilaterally between E13.5–E14.5 in all Twist1CK0/−; Pd-Cre and Sm-Cre embryos, respectively (Figure 4A). Cleaved caspase-3 staining did not show signs of cell death before and during TVS regression (Figure 4D). This suggests that TVS remodeling defects, and poor development of the antPlexus that drains into this vessel, might have altered hemodynamic forces and caused deregulated vessel pruning (Korn and Augustin, 2015). In Twist1CK0/−;Sm-Cre embryos, the TVS completely regressed on the right side in 67% (29/43), the side also more frequently affected in humans (Figure 4E and Table S1). The remaining TVS was hypoplastic and tortuous and the SgS was often hypoplastic and received a prominent occipital sinus (Figure 4E). Thus, loss of Twist1 significantly skewed the normal TVS size distribution curve from right-sided dominance to right-sided aplasia.
Fig. 4. Wild-type mice have TVS morphogenetic variation that is skewed towards unilateral regression in Twist1 mutant mice.

(A) Top: Superior sagittal sinus (SSS, paired arrows) and paired TVS (arrows) meeting at the sinus confluence (SC) on the dorsal surface of control and mutant E13.75 brains. The antPlexus is boxed. Middle: Magnified images of the boxed areas detail TVS regression (single arrows) and poor development of the antPlexus (double arrows) in Twist1CK0/−;Pd-Cre and Sm-Cre mutants. Bottom: Unilateral regression of the TVS (arrow) is fully penetrant and complete by E15.5 in Twist1CK0/−;Pd-Cre and Sm-Cre mutants. (B) SSS (double arrow), paired TVS (single arrow), and SC development in wild-type embryos with magnifications on right. E11.5 (top): primitive head veins (PHV, white arrows) are equal in size and independently drain the cortices. E12.5 (middle): the PHV form an anastomosis with the SSS at the SC. TVS remodeling is concurrent with SC development and the vessel fuses with the PHV near the confluence. The side that remodels first can form a dominant vessel continuous with the SSS. E13.5 (bottom): the SC has matured and caliber differences may appear between the paired TVS. (C) Dorsal images (inverted) of the head and skull depicting the SSS (double arrows) and paired TVS (single arrow) in newborn mice. Box and whisker plots demonstrate the width ratio distribution of the paired TVS in controlss. The right TVS is generally larger than the left (median ratio 1.18 right/left TVS). In 50% of neonatal mice, TVS width ratios were between ~0.92 to 1.36, and in ~20% the dominant vessel was >1.6× the width of the minor vessel. Among these ~20%, right-sided dominance was more common (22/34). Extreme asymmetry (dominant /minor vessel width >2.0) was found in ~7% (11/150). (D) Z-stacks from the side of E13.0 (top and middle) and top of E13.75 (bottom) embryos. Cell death is seen in the trigeminal ganglia in heterozygous control and Twist1CK0/−; Sm-Cre mutants, but is not observed in the antPlexus or TVS at either age. (E) Top: E18.5 Twist1CK0/−;Sm-Cre embryo. The left TVS is tortuous and hypoplastic and the right TVS has regressed (asterisk). A persistent occipital vein is present and drains into the SgS (arrows). Middle and bottom: The remaining TVS is hypoplastic in E18.5 Twist1CK0/−;Sm-Cre embryos and the right side is more frequently absent. **** P<0.0001, unpaired student T test. Scale bar=300um (a) and 500um (c, d). See also Fig. S3.
CV malformations in conditional Twist1 mutants coincide with deficient osteoblast differentiation
In Twist1CK0/−; Pd-Cre mutants, CV remodeling failed to occur altogether and Twist1 was largely absent throughout the head mesoderm and in subpopulations of neural crest. This pattern of Twist1 loss caused severe reduction of Runx2+ osteo- and chondral progenitor cells, corresponding loss of Runx2/ALP+ pre-OB, and failure to form the skull vault and cartilage (Figure 5A–C). By contrast, in Twist1CK0/−; Sm-Cre mutants where TVS remodeling is delayed and aberrant, Twist1 loss was primarily restricted to the corSuture, portions of the developing parBone, and the underlying periDura (Figure 5A). This caused loss of Runx2+ osteo-PCs in the corSuture and along the parBone periosteum, a corresponding reduction of Runx2/ALP+ pre-OB, and severe hypoplasia of the parBone, more pronounced towards the apex (Figure 5A–C). Also, the corSuture was abnormally wide and ectopic differentiation of ALP+ pre-OB was sometimes seen in the mid-sutural mesenchyme (Figure 5B and data not shown). Thus, specific loss of Twist1 in the corSuture, parBone, and periDura is sufficient to perturb growth and remodeling of the underlying CV, particularly the TVS and antPlexus.
Fig. 5. Twist1 mutant mice have compromised skull and cranial suture development.
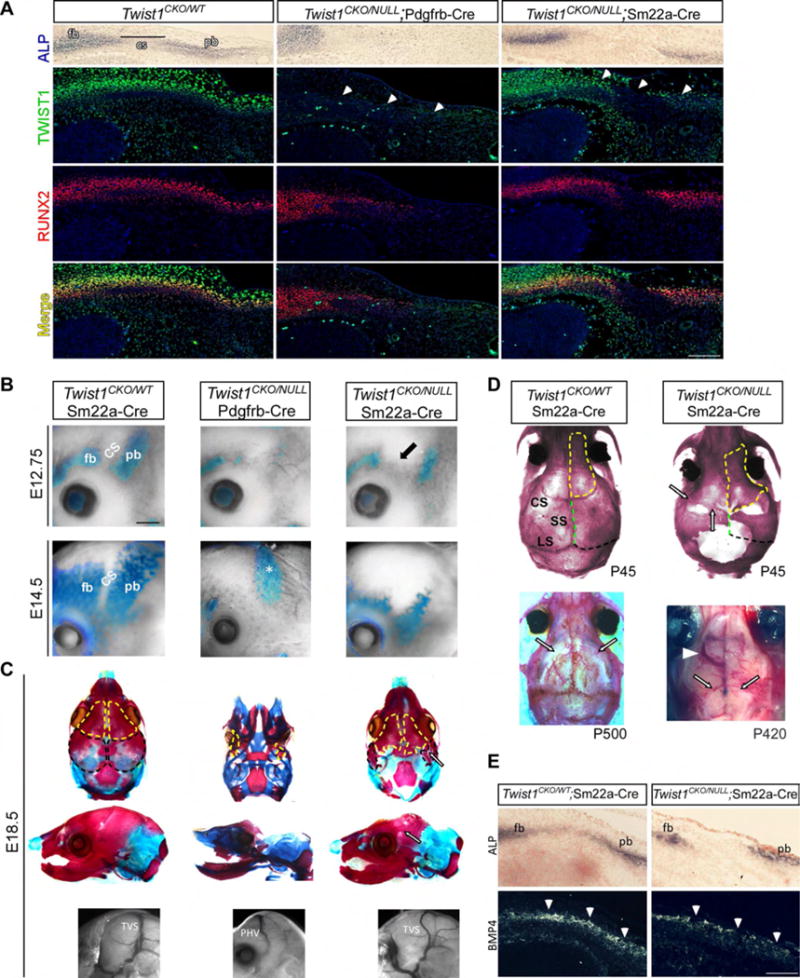
(A) E12.5 transverse sections showing Twist1, Runx2, and ALP expression in supraorbital mesenchyme. In Twist1CK0/−;Pd-Cre mutants, Twist1 is absent in cranial mesoderm (white arrows) and Runx2+ osteo-PCs and ALP+ parBone pre-OB are lost. Some Runx2/ALP+ Pre-OB are present at the frBone (fb) base, despite the loss of Twist1 expression. In Twist1CK0/−; Sm-Cre mutants, Twist1 expression is lost in the corSuture (CS) and parBone (pb) periosteum (white arrows) and in periDura. Runx2+ osteo-PCs are lost within the sutural mesenchyme and parBone periosteum, more pronounced towards the apex of the head. (B) Differentiation of ALP+ frBone and parBone pre-OB is absent or reduced in Twist1CK0/−;Pd-Cre (n=5) and Twist1CK0/−;Sm-Cre (n=5) mutants, respectively. The corSuture fails to properly develop (black arrow). The dura in Twist1CK0/−;Pd-Cre mutants (E14.5, asterisk) shows elevated ALP. (C) Top and middle rows: E18.5 skulls stained with alizarin red (bone) and alcian blue (cartilage). The skull vault is absent in Twist1CK0/−; Pd-Cre mutants. Twist1CK0/−;Sm-Cre mutants have hypoplastic parBone (black outlines in top row) and interparietal bones, and mildly hypoplastic frBone (yellow outlines) and temBone. Single white arrows denote fusion between the frBone and parBone. Bottom row: a PHV (Twist1CK0/−;Pd-Cre) and a tortuous TVS (Twist1CK0/−;Sm-Cre) are mispositioned in respect to the parBone/interparietal bone boundary. (D) (Top) Alizarin-red stained P45 skulls. Twist1CK0/− ;Sm-Cre mice have poorly developed corSuture, sagittal (SS, green dotted line), and lambdoid sutures (LS, black dotted line). Fusion between frBone (yellow dotted outline) and parBone rudiments is seen (arrows) and the neural crest-mesoderm boundary is shifted caudally. (Bottom left) Some Twist1CK0/WT;Sm-Cre heterozygotes (3/6) have juvenile/adult corSuture fusion (black open arrow). (Bottom right) P420 adult has bilateral absence of the TVS (black open arrow) and a large ectopic vein that drains into the olfactory sinus (white arrowhead). (E) (Top and bottom) Adjacent E12.5 transverse cross sections through the head. In Twist1CK0/−;Sm-Cre embryos, Bmp4 expression is patchy and reduced in the corSuture and in ALP+ parBone pre-OB (arrow heads). Scale bar A,E=250um B=700um. See also Fig. S3, S7.
In contrast to other conditional Twist1 mouse mutants (Goodnough et al., 2015), approximately one-third of Twist1CK0/−;Sm-Cre neonates survived to adults. Strikingly, the coronal, sagittal, and lambdoid sutures were poorly developed and/or absent, large gaps of tissue persisted between the skull bones, and fusion was evident between the remaining frBone and parBone rudiments (Figure 5D). These adults had persistent CV malformations (Figure 5D). By contrast, suture morphology was normal in some Twist1CKO/WT;Sm-Cre heterozygous mice, while others had juvenile/adult onset fusion of the coronal and lambdoid sutures (Figure 5D). Notably, CV anatomy was normal in these animals (data not shown), similar to our findings in TWIST1 mutation-positive humans with late-onset suture fusion and mild skull abnormalities (Figure 1A,C). This implies that earlier-onset skull patterning defects have a greater impact on CV development.
The meninges influence TVS refinement but not its initial growth and remodeling
To determine if Twist1 activity is also required in the meninges for growth and remodeling of the dural CV, and to parse out the effects from Osteo-PCs and periDura, we examined Twist1CK0/−;Myf5-Cre embryos; Myf5-Cre was active in the dermis, inner dura, and arachnoid matter, but sparsely throughout the periDura, corSuture, and Runx2/ALP+ osteogenic mesoderm (Figures S1F, S2H,I). By contrast to Twist1CK0/−; Pd-Cre and Twist1CK0/−; Sm-Cre embryos, development of the antPlexus and remodeling of both TVS were normal in Twist1CK0/− ;Myf5-Cre embryos (Figure S3B,C). Furthermore, none of the Twist1CK0/−;Myf5-Cre mutants had complete regression of the TVS at E14.5. Approximately one third (6/17), however, did have unilateral TVS hypoplasia or tortuosity at E18.5, with the right side more frequently affected (4/6) (Figure S3D). These phenotypes did not result from parBone malformations and the skull vault appeared normal (Figure S3E). However, dura mater was diminished and the arachnoid mater and subarachnoid space more compact. These findings were similar to, but less severe than, those in Twist1CK0/−; Pd-Cre and Twist1CK0/−;Sm-Cre embryos (Figure S1F, S3F). This implies that paracrine signaling from skull progenitor cells is required for initial growth and remodeling of the TVS and SgS, whereas signaling within the dura may contribute to subsequent vessel refinement and maturation (Figure S7).
Bmp2 and Bmp4 expression in skull progenitor cells and dura is perturbed in conditional Twist1 mutants
Our results reveal that restricted loss of Twist1 in osteo-PCs and periDura attenuates the differentiation of pre-OB with simultaneous CV growth and remodeling defects. These findings support a model whereby the growth and remodeling of CV are influenced by paracrine signaling from pre-OB and periDura. Consistent with this model, BMP2/4 have been implicated in skull development and suture patency, and loss of Bmp2 in zebrafish perturbs tail-vein angiogenesis despite normal arterial development (Bonilla-Claudio et al., 2012; Justice et al., 2012; Wiley et al., 2011). Thus, we examined Bmp2 and Bmp4 expression in the corSuture, parBone pre-OB, and periDura in controls and Twist1 mutants between E12.5–14.5, coinciding with onset of CV remodeling and vessel regression.
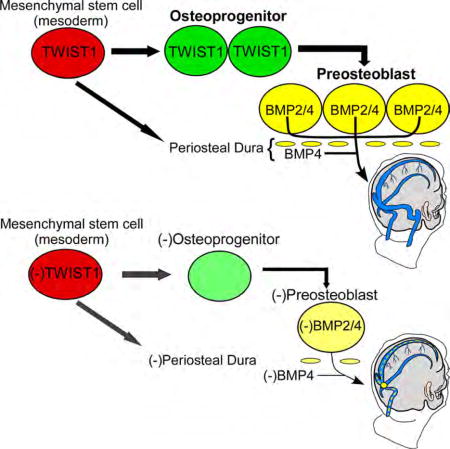
Bmp2 was expressed in ALP+ frBone and parBone pre-OB, and expression was weak and/or absent in the corSuture, dura, and meninges (Figure S4A). Bmp4 was more widely expressed; it was present in ALP+ frBone- and parBone pre-OB, the corSuture and overlying periosteum, periDura, and the meninges (Figure S4A). Expression appeared faint or absent in Twist1-expressing osteo-PCs along the parBone periosteum and within the corSuture. The exception was a subset of cells in the corSuture that were positive for both Twist1 and Bmp4, suggesting these cells were in the process of differentiating from a Twist1/Runx2+ osteo-PC to a Runx2/ALP/BMP4+ pre-OB (Figure S4A′). Thus, Bmp2/4 expression is upregulated once Twist1 levels decline, suggesting high levels of Twist1 are required for the maintenance of the osteoprogenitor state, whereas lower levels of Twist1 permit BMP expression and pre-OB differentiation.
Next, we examined if BMP expression was perturbed in conditional Twist1CKO/−;Sm-Cre mice due to the loss of osteo-PCs and pre-OB. Indeed, normal Bmp2/4 expression in the head was altered. Bmp4 was patchy and reduced in the corSuture, weak and/or absent in dermal mesoderm and pre-OB forming the parBone, and mainly absent in the periDura (Figure 5E, S4C). Bmp2 expression was also absent in dermal mesoderm and parBone pre-OB (Figure S4C). However, Bmp2/4 expression was normal in pre-OB forming the frBone and Bmp4 was relatively normal in the meninges, although it appeared weaker in some sections (Figure S4C,D). Thus, Twist1CKO/−;Sm-Cre embryos show perturbations to Bmp expression that are specific to parBone pre-OB and periDura, consistent with their attenuated production of these mesoderm-derived tissues.
BMP2 and BMP4 regulate dural CV development in mammals
We tested whether loss of BMPs in pre-OB and periDura can cause CV malformations. Conditional inactivation of three (Bmp2CKO/WT;Bmp4CKO/CKO or Bmp2CKO/CKO;Bmp4CKO/WT) or four (Bmp2CKO/CKO;Bmp4CKO/CKO) floxed Bmp2/4 alleles in Pd-Cre and Sm-Cre embryos resulted in tortuous antPlexus vessels with abnormal, non-uniform branching patterns and increased lacunarity (Figure 6A,B and data not shown). By comparison with controls and Twist1CKO/−;Sm-Cre embryos, however, vessel density and branching were variable and not significantly reduced in all. However, TVS remodeling was delayed in most Bmp2CKO/CKO;Bmp4CKO/CKO; Pd-Cre and Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre embryos (Figures 6A, S1G, S5B). A smaller fraction of Bmp2CKO/CKO;Bmp4CKO/WT;Pd-Cre mutants had delayed TVS development and the antPlexus was less affected, suggesting BMP4 has a more pronounced effect on CV growth and remodeling (Figure S5A,B). Very similar results were obtained in Bmp2/4;Sm-Cre three and four allele-loss embryos, and SgS remodeling was normal in both crosses (Figure 6A and data not shown). Finally, cerebral artery anatomy was normal in all Bmp2/4 mutant embryos, demonstrating that similar to conditional Twist1 mutants, venous malformations did not result from arterial growth and remodeling defects (Figure S5C and data not shown).
Unilateral TVS regression was also observed by E14.5 in Bmp2CKO/CKO;Bmp4CKO/CKO;Pd-Cre and Sm-Cre embryos and, by E18.5, all embryos had unilateral absence, segmental aplasia, or severe hypoplasia of the TVS, more frequently on the right (21/34) (Figure 6C, S5D). In many cases, there was secondary SgS hypoplasia on the corresponding side, similar to the phenotype in TWIST1 mutation-positive humans and mice (Figure S6A). The contralateral TVS was also hypoplastic, but not tortuous as seen in conditional Twistl mutant embryos. This may be attributed to normal meningeal patterning in Bmp2/4 embryos with loss of three or four alleles (Figure S5E and data not shown). Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre and Bmp2CKO/CKO;Bmp4CKO/WT;Pd-Cre embryos were similar in appearance, but the overall phenotypes were sometimes milder, and ~50% of Bmp2CKO/CKO;Bmp4CKO/WT;Pd-Cre embryos had normal development of the paired TVS and SgS (Figure 6C). Thus, inactivating Bmp2/4 in parBone pre-OB and periDura, via Pd-Cre or Sm-Cre, similarly skews normal right-sided TVS dominance found in controls towards unilateral absence in mutants (Figure 6C′).
Venous malformations in either Pd-Cre or Sm-Cre;Bmp2/4 mutants with loss of three or four alleles were highly similar. Also, they most closely converged with Twist1CKO/−; Sm-Cre mutants that display loss of Twist1 and Bmp2/4 in the corSuture, parBone, and periDura. Inactivation of Bmp2 and Bmp4 with Pd-Cre resulted in loss of Bmp2/4 expression in frBone and parBone pre-OB, and Bmp4 expression was mostly absent in the corSuture, periDura, and meninges (Figure S4E). By contrast, inactivation of Bmp2 and Bmp4 with Sm-Cre restricted the loss of Bmp2/4 expression to parBone pre-OB and, in addition, Bmp4 expression to the corSuture and periDura (Figure S4B, B′, E). Thus, loss of paracrine BMP signaling from parBone pre-OB and periDura is sufficient to perturb venous growth and remodeling.
To determine if paracrine BMP signaling from pre-OB and/or periDura could act through type I and II receptors on venous endothelium, we analyzed expression of Bmpr1a/b, Bmpr2, and Acvr2a/b. Bmpr1b, Bmpr2, and weak levels of Acvr2b were co-expressed with Cdh5 on venous endothelium, whereas Bmpr1a and Acvr2a appeared absent (Figure 6D and data not shown). We inactivated Bmpr2 in the venous endothelium with Tie2-Cre to determine if attenuated cell-autonomous BMP signaling affects CV development. Bmpr2CKO/CKO;Tie2-Cre neonates had variable hypoplasia of the paired TVS, most similar to Bmp2CKO/CKO;Bmp4CKO/WT;Pd-Cre mutants (Figure 6E). Thus, loss of BMPR2 signaling in the venous endothelium affects normal TVS development.
BMP signaling in mesoderm regulates cranial suture, skull, and CV development downstream of Twist1
We next asked if conditional Bmp2/4 mutants had skull malformations similar to Twist1CKO/−;Sm-Cre embryos. Indeed, the differentiation and expansion of Runx2/ALP+ parBone pre-OB was diminished in Bmp2CKO/CKO;Bmp4CKO/CKO;Pd-Cre and Sm-Cre mutants between E12.5–E14.5 (Figure 7A, B). Four allele loss mutants died at birth and had severely hypoplastic parBone; temBone were unaffected, consistent with normal SgS remodeling in these mutants (Figure 7C, C′ and data not shown). More subtle differences were seen in Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre and Sm-Cre embryos; at birth, the paired parBone showed mild hypoplasia with enlarged fontanelles, reminiscent of humans with TWIST1 haploinsufficiency and moderate/severe craniosynostosis (Figure 7C, C′, 1B, and data not shown). The frBone and parBone did not properly overlap, and less Runx2+ staining was seen within and along the parBone surface (Figure 7D). Three-allele loss mutants survived, adults had small misshapen heads, and the corSuture was dysmorphic and located caudal to the normal neural crest-mesoderm boundary, similar to Twist1CKO/−; Sm-Cre mutants (Figure 7E, 5C,D). Also, we observed a gap at the boundary between the parBone and interparietal bones which, interestingly, overlies the paired TVS (Figure 7E). Thus, these mutants have skull and suture deformities that are similar to human craniosynostosis and conditional Twist1 mutant mice.
Figure 7. BMP2 and BMP4 are required for cranial suture patterning, osteoblast differentiation, and skull vault development.
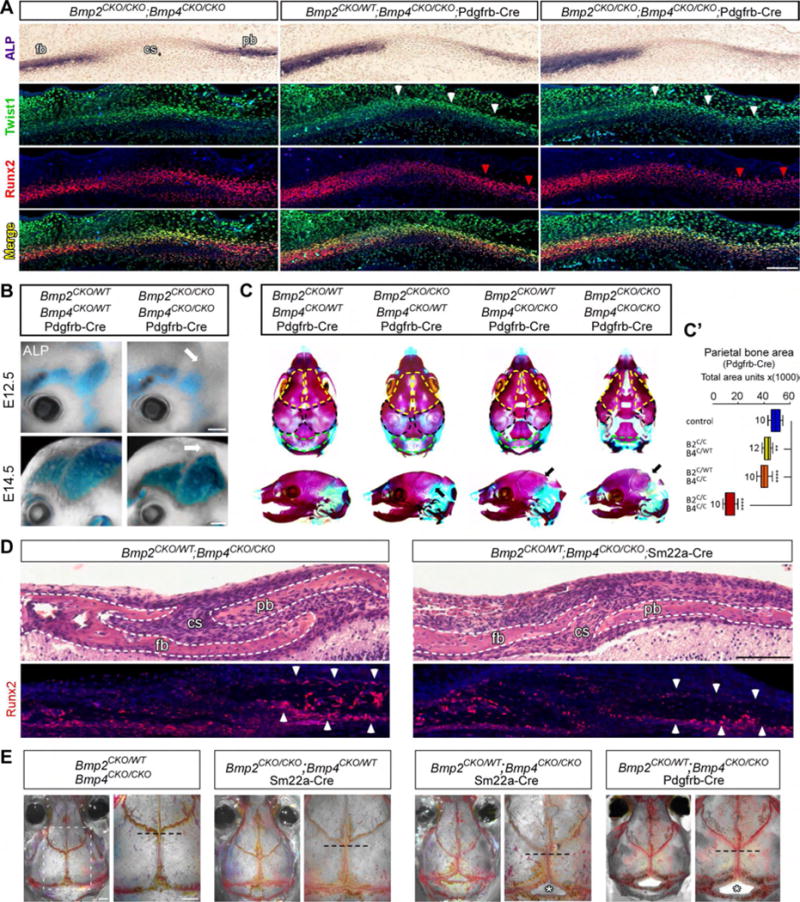
(A) E12.75 transverse adjacent sections showing ALP staining in supraorbital mesenchyme, and Twist1 and Runx2 expression on adjacent sections. There is a mild/moderate loss of Runx2/ALP+ parBone (pb) pre-OB in three and four allele loss Bmp2/4;Pd-Cre mutants, respectively (red arrowheads), but Twist1 levels are normal in the corSuture (CS) and along the parBone-periosteum (white arrowheads). (B) ALP staining in E12.75 and E14.5 heads. Differentiation and expansion of ALP+ parBone pre-OB (arrow) are reduced in Bmp2CKO/CKO/Bmp4CKO/CKO;Pd-Cre mutants. (C) E18.5 alizarin red and alcian blue stained skulls. The boundary between the frBone (fb, yellow outlines) and parBone (black outlines) has shifted caudally in Bmp2/4;Pd-Cre three and four allele loss embryos. The frBone, parBone (black arrow) and interparietal bones (iparBone, green outline) are hypoplastic and the fontanelles are enlarged in Bmp2CKO/WT;Bmp4CKO/CKO;Pd-Cre and Bmp2CKO/CKO;Bmp4CKO/CKO;Pd-Cre embryos. (C′) Quantifications of parBone area at E18.5. (D) P2 H&E stained transverse sections (top), with adjacent Runx2 expression (bottom), through the frBone, corSuture, and parBone. The corSuture is narrower, the frBone and parBone do not properly overlap, and less Runx2 staining is seen in and around the parBone (arrowheads) in Bmp2CKO/WT;Bmp4CKO/CKO;Sm-Cre embryos. (E) Alizarin-red stained P90 skulls. Background colors have been subtracted to highlight the cranial sutures. Right magnified images correspond to the white boxed region in the control. Mutant skulls are smaller and the corSuture (dotted line indicates midline origin) boundary is shifted caudally in three allele-loss Sm-Cre and Pd-Cre adults. The sagittal suture is foreshortened and bone loss is seen at the parBone/iparBone border (bottom asterisk). *P<.05, ** P<0.0l*** P<0.00l **** P<0.000l, one-way Anova with Dunnett’s post-test. Scale bar A=250um, B, E=600um. See also Fig. S4, S6–7.
Bmp4 is also expressed in the mesoderm-derived meninges (Figure S4C,D), so we generated Bmp2CKO/WT;Bmp4CKO/CKO;Myf5-Cre mutants to determine the requirement of BMP4 signaling from the meninges versus that of parBone pre-OB and periDura. Bmp2CKO/WT;Bmp4CKO/CKO;Myf5-Cre mutants had normal skull and CV development (Figure 5SF). This provides further evidence that BMP signaling from mesoderm-derived pre-OB and periDura is necessary for growth and remodeling of the TVS.
To determine whether BMP signaling is upstream or downstream of Twist1 in mesoderm-derived skull bones and periDura, we assayed Twist1 expression in three and four allele-loss Bmp2/4;Pd-Cre and Sm-Cre mutants. In all E12.5 mutants, Twist1 was appropriately expressed in the periosteum of the corSuture and parBone, in Runx2+ osteo-PCs within the corSuture, and in periDura (Figure 7A, S4B). This indicates BMP signaling is downstream of Twist1 expression in mesoderm-derived skull progenitor cells and periDura. Collectively, our results support a model whereby Twist1 is required for the specification of parBone osteo-PCs, and thus the sequential differentiation of pre-OB that regulate TVS growth and remodeling via paracrine BMP signaling (Figure S7). Also, our findings suggest that CV anomalies in human craniosynostosis syndromes may result from deregulated BMP signaling secondary to skull malformations.
Paracrine BMP4 signaling from neural crest-derived dura is required for the development of the superior sagittal sinus and sinus confluence
We next examined the development of the superior sagittal sinus (SSS), which is embedded in neural crest-derived dura underneath the sagittal suture and is the major vessel that drains blood from both cortical hemispheres into the paired TVS through their connection at the sinus confluence (SC) (Figure S1E). The SSS was often absent, hypoplastic, or truncated and replaced by smaller cortical veins in Bmp2CKO/CKO; Bmp4CKO/CKO;Pd-Cre and Sm-Cre embryos (Figure 6C, S5D). Because Bmp4 expression was detected in dura as well as pre-OB at E12.5 when the SSS and SC are developing, we tested whether paracrine BMP4 signaling from neural-crest derived dura is also required (Figure S6D). We used a neural crest-specific Wnt1-Cre driver (Figure S2I) to generate Bmp2CKO/WT;Bmp4CKO/CKO;Wntl-Cre embryos, with Bmp4 selectively inactivated in neural crest-derived dura (Figure S6D).
By E12.75, the majority of Bmp2CKO/WT;Bmp4CKO/CKO;Wnt1-Cre embryos had a hypoplastic or truncated SSS and a spectrum of SC malformations, phenotypes that appeared distinct from conditional Twist1 and Bmp2/4;Sm-Cre and Pd-Cre embryos (Figure S6A, A’, C, F). These phenotypes did not resolve and, by E18.5, 78% (11/14) also had TVS hypoplasia that was more severe proximal to the SC (Figure S6A, A’, B). However, at E12.75, Bmp2CKO/WT;Bmp4CKO/CKO;Wntl-Cre mutants had normal development of the antPlexus, TVS, and differentiation and expansion of parBone pre-OB (Figure S6E, E’, G). This implies that TVS hypoplasia in Bmp2CKO/WT;Bmp4CKO/CKO;Wntl-Cre mutants is likely secondary to abnormal development of the SC and preferential blood flow to one side. Therefore, the development of venous networks in the head can be differentially regulated by tissue types; paracrine BMP4 signaling from neural crest-derived dura is necessary for primary development of the SSS and the SC and, secondarily, the TVS, whereas BMP2/4 signaling from mesoderm-derived pre-OB and periDura is required principally for the growth and remodeling of the TVS.
Discussion
CV growth and remodeling is influenced by cross-talk from the skull and dura under the control of Twist1 and BMP signaling
Using mouse genetics, we provide the first description of cellular and molecular mechanisms required for CV angiogenesis. We show that paracrine BMP signaling from skull progenitor cells and dura is required for normal dural CV growth and remodeling, likely through activation of type I and II BMP receptors on the venous endothelium. Thus, gene mutations that affect skull and/or dura development alter BMP signaling from these tissues and disrupt normal venous angiogenesis. This paradigm can explain the link between bone and vein malformations present in human craniosynostosis. As such, we now propose that individuals with TWIST1 haploinsufficiency, and possibly those with activating mutations in FGFR2 and FGFR3, develop CV malformations as a secondary consequence of their skull malformations.
It is still debated how skull malformations result from TWIST1 haploinsufficiency or other types of syndromic craniosynostosis, primarily because osteoblast differentiation is a complex and poorly understood process that involves several different lineage decisions. Moreover, the activating and repressing activities of transcription factors and signaling molecules can change as these lineage decisions arise (Huang, 2007). Previous work has shown that Twist1 is required both for the specification of osteo-PCs from uncommitted mesenchyme, and for the repression of downstream pre-OB differentiation through antagonism of Runx2, BMP, and Smad1 signaling (Hayashi et al., 2007; Miraoui and Marie, 2010). While appearing paradoxical, this is necessary to maintain a pool of osteo-PCs that can support balanced rates of osteoblast differentiation. Interestingly, Twist1/E-protein heterodimers have high affinity for tandem e-box elements in the Bmp4 promoter (Chang et al., 2015), and we find that high levels of Twist1 coincide with low and/or undetectable levels of Bmp2/4 expression in osteo-PCs. This suggests that Twist1 maintains an osteoprogenitor state, in part, through the repression of BMP signaling. Furthermore, pre-OB differentiation ensues once Twist1 levels decline and Bmp2/4 expression increases, possibly due to E-protein sequestration via BMP mediated Id1 expression (Hayashi et al., 2007). Under this model, Twist1 is upstream of BMP signaling in mesoderm-derived skull progenitor cells and, thus, also CV growth and remodeling.
Although it is still unclear if Twist1 directly regulates BMP expression in osteo-PCs and dura, our data strongly indicate that insults which either directly and/or indirectly alter BMP expression in osteogenic mesoderm and dura—and coincide with critical vein growth and remodeling events—have the potential to perturb CV development. Thus, direct inactivation of Bmp2/4 in pre-OB and periDura or secondary loss of BMP signaling due, for example, to loss of Twist1 expression leading to a reduction of pre-OB/dura, can deleteriously affect dural CV growth and remodeling. Subsequently, hemodynamic forces within venous networks may be altered, resulting in abnormal vessel pruning, regression, and loss or hypoplasia of the CV. These effects could be worse in those who have severe craniosynostosis with multiple sutures affected, because loss of paracrine BMP signaling from neural crest and mesoderm-derived tissues can cause distinct (SSS and SC) and overlapping (TVs) vein malformations. Further studies are necessary to determine the relationships between the types of cranial deformities present in craniosynostosis and their effects on venous morphogenetic variation and elevated ICP.
The evolution of skull development and CV growth and remodeling in mammals
Our findings demonstrate that Twist1 regulates venous growth and remodeling in mammals, at least in part, because it is required for the specification of osteo-PCs within the corSuture and parBone periosteum, and these progenitor cells differentiate into pre-OB (Figure S7). Pre-OB express BMPs that signal in an autocrine fashion to stimulate both their differentiation and expansion, while also signaling in a paracrine fashion to the underlying CV to regulate venous angiogenesis (Figure S7) (Phimphilai et al., 2006). From an evolutionary standpoint, this seems logical because CV anatomy is highly conserved among mammals—versus birds, reptiles, and fish—and appears to have evolved in parallel with the brain, meninges, and skull (Aurboonyawat et al., 2007; Streeter, 1921). In particular, signaling pathways within the corSuture evolved in mammals to mediate parBone enlargement in response to the explosive growth of the cerebral cortex (Jiang et al., 2002; Kim et al., 1998). Considering the dural CV are tightly attached to the skull within periDura (Figure S1A,E,F), our results suggest these vessels coevolved with the corSuture and skull in mammals because they share similar developmental cues. This observation may explain the anatomic decoupling of the venous sinuses from cerebral arteries, the requirement of specific morphogens for venous growth and remodeling, and the presence of CV malformations in humans and mice with craniosynostosis.
BMPs can function as vein-specific angiogenic cues in fish and mammals
The ability of the venous vasculature to grow and remodel in response to BMP signaling is also evolutionarily conserved. Zebrafish BMP2b is a vein-specific angiogenic cue that activates type I/II BMP receptors on the tail vein to influence sprouting angiogenesis via R-Smad and Erk signaling (Wiley et al., 2011). Also, the ability of BMP ligands to induce tip cell formation and blood vessel branching is dependent upon the levels of Notch and SMAD6 (Mouillesseaux et al., 2016). High levels of Notch activity, found in arterial endothelium, antagonize BMP signaling by activating Smad6 and inhibiting nuclear pSMAD1/5 translocation. By contrast, veins have low Notch activity and Smad6 expression and are thus intrinsically primed to respond to BMPs (Mouillesseaux et al., 2016). Interestingly, in mouse embryos, crosstalk between BMP and Notch signaling regulates endothelial cell migration, tip-cell polarity, and sprouting angiogenesis from both arteries and veins (Moya et al., 2012). Conditional ablation of Smad1/5 (three or four allele loss) in endothelial cells via Tie2-cre results in cardiac, angiogenesis, and lymphangiogenesis defects with bleeding, edema, and death between E10–E14.5. By contrast, these phenotypes were absent upon conditional removal of Bmp2/4 with four different Cre recombinase alleles that target different populations of cells in the head, and we found that loss of Bmpr2 in endothelial cells affects TVS development in the absence of other severe malformations. Notably, loss of Smadl/5 can also interfere with ALK1/TGFB signaling, and BMP9/10 signals through the ALK1 receptor to regulate embryonic and postnatal angiogenesis via cross-talk with Notch signaling (Chen et al., 2013; Larrivée et al., 2012; Pardali et al., 2010). Collectively, these data suggest that dural CV growth and remodeling may be decoupled from other vascular processes at the level of the ligand and, possibly, the receptor as well. This could result in the activation of distinct combinations of type I/II BMP receptors and/or downstream signaling pathways in the venous endothelium, similar to what has been proposed in the retina (Lee et al., 2017).
Human skull malformations can skew normal venous morphogenetic variation and cause disease
Our results in craniosynostosis mouse models show that dural CV angiogenesis is a pliable process that is strongly dependent upon the development of neighboring tissues and, likely, upon the different growth environments of each fetus. This is clinically relevant because natural population variation, particularly concerning the anatomy of the TVS, has confounded the clinical definition of what constitutes “normal” CV anatomy versus morphogenetic variations that cause cerebrovascular disease (Zouaoui and Hidden, 1988). In extreme circumstances, gene mutations may predispose fetuses to morphogenetic variants that are suboptimal and result in vascular disease, elevated ICP, and neurological dysfunction. In particular, humans with craniosynostosis and skull malformations likely demonstrate increased venous morphogenetic variation that could heighten their overall risks for these complications. Therefore, it is more accurate, at least in these individuals, to classify and treat CV “anatomical variants” as developmental malformations. Finally, because CV malformations in individuals with severe craniosynostosis are likely present by birth and may even worsen over time, routine MRV imaging may be beneficial.
METHOD DETAILS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects were enrolled according to the ethical guidelines of the Declaration of Helsinki and with approval of the Institutional Review Board at Boston Children’s Hospital (research, MRI, and retrospective review protocols 05-03-036R, P00000492, and P00019970, respectively). Informed consent was obtained from all subjects. MR images were examined from four males and six females, ranging from 7–66 years of age. Skull and radiology findings did not show any bias between males and females, including a sibling pair, and thus detailed analyses based on gender-specific differences were not performed. Additional details can be found in Table SI. The following transgenic mouse alleles were used in mixed-background mice: TwistCKO (Chen et al., 2007), Pdgfrb-Cre (Cuttler et al., 2011), Sm22a-Cre (Jax #004746), Myf5-Cre (Jax #007893), Wnt1-Cre (Jax#022137), CAG-R26 LxL-tdTomato Ail4 (Jax#007908), CAG-R26 LxL-Sun1/sfGFP (Jax#021039), Bmp2CKO;Bmp4CKO (Maatouk et al., 2009), Tie2-Cre (Jax#004128), Bmpr2CKO (Beppu et al., 2009). A Twistl− allele was generated by mating male Twist1CKO mice with female Sox2Cre mice (JAX#014094). For all experiments, both male and female mice were included. The CV phenotypes were fully penetrant on mixed background strains and thus were not dependent upon strain specific modifying effects. All animal work was approved and performed in compliance with Boston Children’s Hospital Institutional Animal Care and Use Committee protocols (16-10-3247R).
Antibodies
Rat anti-endomucin (Santa Cruz, sc-65495), goat anti-GFP FITC (Abcam, ab6662), rabbit anti-DsRed (Clontech 632496), rat anti-mouse CD102/ICAM2 (BD Biosciences, 553326), mouse anti-Twist1 (Santa Cru z, sc-81417), rat anti-mouse CD31/PECAM-1 (BD pharmingen 553370), rabbit anti-Sox17 (Zhou et al., 2015), mouse anti-smooth muscle actin (Sigma-Aldrich; C6198), rabbit anti-Runx2 (Abeam abl92256), rabbit cleaved caspase-3 (Cell Signaling 9661) and goat anti-EphB4 (R&D systems AF446). Alex Fluor-labeled secondary antibodies were from Invitrogen. See STAR Methods chart.
Radiology
Nine of ten participants were scanned on a 3 Tesla (3T) magnetic resonance (MR) imaging system and one was scanned on a 1.5T MR unit. MR technical parameters for 3T were as follows: sagittal T1 magnetization prepared rapid acquisition gradient echo (MPRAGE): TR/TE, 15202/2.27 ms; NEX, 1; matrix, 256 × 256; field of view (FOV), 27 × 22 cm; coronal 2D time of flight (TOF) MR venography (MRV): TR/TE, 27/7.2 ms; NEX, 1; matrix, 256 × 256; FOV, 30.8 × 25 cm; axial 3D TOF MR angiography (MRA) with maximum intensity projection images: TR/TE, 22/3.88 ms; NEX, 1; matrix, 230 × 512, FOV, 24.7 × 20 cm; axial and coronal T2: TR/TE, 5500/89 ms; NEX, 1 – 2; matrix, 302-314 × 448-512 FOV, 24.7 × 20 cm; axial T2 fluid attenuated inversion recovery (FLAIR): TR/TE, 8000/137 ms; NEX, 1; matrix 288 × 320; FOV, 24.7 × 20 cm; axial susceptibility weighted imaging (SWI): TR/TE 28/20 ms; NEX, 1; matrix, 200 × 256; FOV, 24.7 × 20 cm; and axial echoplanar imaging (EPI) diffusion weighted imaging (DWI): TR/TE, 4800/89 ms; NEX, 1; matrix, 128 × 128; FOV, 27.1 × 22 cm; B value 1000. CT was available for review in 6/10 patients. CT exams were clinical exams performed for the assessment of craniosynostosis. Exams prior to 2006 utilized the following parameters: helical axial slice thickness 2.5 – 3 mm; KVp 120; mA 350; pitch 0.562:1; rotation 0.6. Exams from 2006 utilized a low dose technique: helical axial slice thickness 2.5 – 3 mm; KVp 100 – 120; mA 50 – 175; pitch 0.562:1; rotation 0.6. The following parameters were assessed on MR: craniocervical junction anatomy, position of the cerebellar tonsils, deformity of the calvarial bones, morphology of the dural venous sinuses and internal jugular veins on MR venography source images (MRV), arterial anatomy on MR angiography (MRA), anatomy of the corpus callosum, and the appearance of the brain parenchyma, ventricles and sulci. The inter carotid distance (ICD) was measured on coronal T2 weighted images as the smallest distance between the internal carotid artery (ICA) signal voids at the level at the level of the cavernous sinuses. Results were compared with age-related norms (Banu et al., 2014). Optic nerve sheath measurements were made 4mm behind the globe. CT images were assessed for calvarial anatomy, the combined area of the right and left jugular foramina (JF) at the base of the skull as well as the presence of enlarged occipitomastoid and/or occipital venous emissary foramina (EF) (Robson et al., 2000).
Immunohistochemistry
For wholemount immunohistochemistry, E10.5–14.5 embryos were fixed in 2% paraformaldehyde (PFA) in PBS at 4°C for 18–24 hours. After fixation and a brief wash in PBS, embryos were fixed in Dent's fix (80% Methanol/20% DMSO) overnight. Embryos were incubated at 4°C for four days in primary antibodies diluted in PBST (.5% Tritonx-100, 10% NGS) and then washed extensively in blocking buffer (PBST). Secondary antibodies were diluted in PBST and incubated at 4°C for three days. After washing with PBS, the embryos were dehydrated through a series of methanol washes (25%, 50%, 75%, 100%) and clarified in benzyl benzoate:benzyl alcohol (BBBA, 2 parts benzyl benzoate and 1 part benzyl alcohol). Fluorescent reporter staining in the temporal bone and middle ear was performed using 125um-thick vibratome (Leica Vtl200s) sections that were immunostained with primary and secondary antibodies at 4 degrees Celsius overnight in PBST, followed by clearing in BBBA. 12um-thick z-stack images were captured using a Zeiss LSM700 confocal microscope with a 10×, 0.45 NA ApoChromat lens and processed with Zen, Image J, and Adobe Canvas. For older wholemount embryo preparations, the laser power was manually increased during image acquisition to enhance the visualization of deeper and smaller blood vessels. For frozen tissue sections, embryos were washed in PBS and fixed overnight in 4% PFA. After washing, the embryos were sunk in 30% sucrose overnight and embedded in O.C.T compound (Tissue-Tek), and 10um-thick sections were cut. Immunohistochemistry on paraffin embedded sections was performed using 10um-thick sections and sodium-citrate (pH 6.0) antigen retrieval for 30 minutes in a commercial vegetable steamer prior to antibody incubation. The sections were imaged using a Zeiss LSM700 confocal microscope with a 20×, 0.8 NA ApoChromat lens. Wholemount retina staining was performed as previously described (Zhou et al., 2014). For wholemount fluorescent reporter staining of E18.5 skulls, the dermis was removed and the skulls were photographed under native fluorescence using a Nikon SMZ18 fluorescent stereomicroscope, a Nikon DS-Ri1 camera, and Nikon Elements imaging software. Post-processing was done using Canvas version 12 (ACD Systems).
In situ hybridization
In situ hybridization (ISH) was performed on 20μm thick sections cut from fresh frozen E12.5–14.5 WT and mutant mouse heads embedded in O.C.T. compound (Tissue-Tek), Templates used to produce Digoxigenin-labeled or fluorescently labeled mRNA antisense probes against Bmp2, Bmp4, and Twist1 were generous gifts from Se-Jin Lee (Jackson Laboratory), For double fluorescence ISH, probes against Bmp2, Bmp4, Twist1, Cdh5, Bmpr1a, Bmprlb, Bmpr2, and Acvr2a/b were generated using the templates described above, or using oligonucleotide primers provided by Allen Brain Atlas and/or Gene Paint, The probe for Bmp4 corresponds to exon 4, ISH was performed by the RNA In Situ Hybridization Core at Baylor College of Medicine using an automated robotic platform as previously described (Yaylaoglu et al,, 2005). Bmpr1a, Bmpr1b, Bmpr2, and Acvr2a/b were hybridized to the tissue simultaneously with Cdh5, (Bmpr1a/Cdh5, Bmpr1b /Cdh5, Bmpr2/Cdh5, Acvr2a/Cdh5, Acvr2b/Cdh5), Bmp2 or Bmp4 were hybridized simultaneously with Twistl, Adjacent frozen sections were used for endogenous ALP staining to demarcate frontal and parietal bone preosteoblasts and the intervening coronal suture, After the previously described washes and blocking steps (Yaylaoglu et al., 2005), the DIG-labeled probes were visualized using tyramide-Cy3 Plus (1/50 dilution, 15 minute incubation, Perkin Elmer), After washes in TNT buffer, the remaining HRP-activity was blocked by a 10 minute incubation in 0,2M HCl, The tissue was then washed in TNT, blocked in TNB for 15 minutes before a 30 minute room temperature incubation with HRP-labeled sheep anti-FITC antibody (1/500 in TNB, Roche), After washes in TNT the FITC-labeled probe was visualized using tyramide-FITC Plus (1/50 dilution, 15 minute incubation, Perkin Elmer), Following washes in TNT the slides were removed from the machine and mounted in ProLong Diamond with DAPI, Digoxigenin-labeled slides were photographed using either 10× 0,45 NA Plan Apo or 20× 0,75NA Pan Apo objectives and a Nikon Eclipse 80i microscope equipped with a color SPOT Flex camera, Fluorescently labeled slides were imaged using a Zeiss LSM700 confocal microscope with a 20×, 0.8 NA ApoChromat objective. For the high magnification images in Figures S4 A′ and B′, a 63×, 1.4 NA ApoChromat objective was used.
Endogenous ALP staining
Wholemount endogenous ALP staining was performed on E12.5–14.5 embryos freshly dissected in HBSS and fixed overnight in 80% ethanol at room temperature, followed by incubation overnight in 95% ethanol at room temperature. Embryo heads were removed and washed three times in NMTT buffer (100mM NaCl, 50mM MgCl2, 100mM Tris-HCl, pH 9.5, 0.1% Tween 20). Endogenous ALP activity was detected using the BM Purple AP substrate precipitation kit (Roche #11442074001) and incubation times of 20–30 minutes at room-temperature. The reaction was stopped by washing several times in water. All embryos were photographed using a Nikon SMZ18 stereo microscope. Images of control and mutant embryos were post-processed in an identical fashion in Adobe Canvas version 12 using the color equalization algorithm to manually subtract out background colors to produce a “greyscale” image that retained visualization of endogenous ALP staining in the blue/purple color spectrum. Endogenous ALP staining was also performed on 4% PFA fixed tissue that was embedded in paraffin (tissue was deparaffinized using three 100% xylene washes and a series of ethanol washes consisting of 100%, 95%, 70%, and 50% ethanol diluted in water) or 4% PFA fixed frozen tissue sections that were washed in NMTT buffer before application of the BM Purple AP substrate for 20 minutes. The sections were counterstained with Nuclear Fast Red (Vector labs, H3403) and images were captured using either 10× 0.45 NA Plan Apo or 20× 0.75NA Plan Apo objectives and a Nikon Eclipse 80i microscope equipped with a color SPOT Flex camera. Images were post-processed in Canvas.
Alizarin Red and Alcian Blue staining
Embryonic and postnatal skulls were carefully dissected to remove the dermis, muscle, and connective tissue, and were fixed overnight in 80% ethanol followed by additional overnight fixation in 95% ethanol. The skulls were then incubated in acetone at room temperature for three days prior to staining with 0.1% Alizarin red and 0.3% Aldan blue (in ethanol). The skulls were incubated in the staining solution at 37°C for at least three days on a rotator. The skulls were then washed for one hour in 95% ethanol, and cleared in a 1–2% KOH solution for several days. The skulls were hardened with a series of overnight glycerol washes (25%, 50%, and 75% in a 1% KOH solution) and photographed in 100% glycerol using a Nikon SMZ18 stereo microscope equipped with a DS-Ri1 camera. Images were post-processed in Canvas by adjusting the color balance and/or using the color equalization algorithm.
Visualization of the dural cerebral veins in E18.5 embryos and newborn mice
PO pups and E18.5 mouse embryos were included in the analysis. E18.5 embryos were obtained from sacrificed dams, cleaned, and placed on a 37°C incubator until the embryos turned pink and were breathing normally. Newborn pups and embryos were then placed on ice for 5–10 minutes until the animal was fully anesthetized. The dermis was carefully removed to avoid damaging any superficial blood vessels and, following cold exposure, the cerebral veins were very apparent and engorged with blood underneath the skull cap. Photographs with identical magnifications were obtained using a Nikon SMZ18 stereo microscope, and images were post-processed in Canvas using greyscale and image inversion. The vessel widths were measured using the measurement function in ImageJ. The widths of both the paired TVS and SSS were calculated from averaging the measurements of three equidistant points along the lengths of the vessels. Aplasia and/or segmental aplasia were scored as zero and averaged with the other points.
Wholemount quantifications of the anterior plexus in mouse embryos
Three-dimensional image representations of wholemount embryo heads were obtained and the contrast of each individual Z stack was increased by 4%. A maximum intensity projection of no more than 30 z-stacks with 10um spacing (300 microns in depth) was created. The primary plexus of each embryo was traced such that it was enclosed in a polygonal area mimicking the convex hull containing the plexus and with an area no more than 250,000 pixels. Once this area was selected, the plexus was thresholded and binarized. The image was then imported into Angiotool (Zudaire et al., 2011) and assayed with the following experimentally standardized parameters: vessel diameter (4,6), threshold (40–200), remove small particles (80), fill holes (no).
Statistical Analysis
Statistical analysis was performed as indicated using GraphPad Prism software for analysis and graphical representations. Unpaired student T-tests were performed when assessing a single mutant genotype compared to control littermates, and one-way ANOVA with Dunnett’s posttest was used when comparing multiple mutant genotypes to control littermates. Error bars represent mean +/− SEM. For TVS ratio analysis, box and whisker plots show, at their centers, the median (horizontal line) and the 25th and 75th percentiles (boxed region). The whiskers denote the minimum and maximum data points for each set, and all values that fall below or above the 25th and 75th percentile, respectively.
Supplementary Material
100 word email alert.
Dural cerebral veins (CV) are required for blood flow and cerebrospinal fluid reabsorption, but mechanisms that regulate their development are unknown. We characterize dural CV malformations in TWIST1 mutation-positive humans with craniosynostosis and in mouse models. We find that Twist1 is dispensable in endothelial cells for CV development, but is required for the specification of osteoprogenitor cells that differentiate into preosteoblasts. Preosteoblasts express Bone Morphogenetic Proteins, and inactivation of Bmp2/Bmp4 in preosteoblasts and periosteal dura causes similar skull and CV malformations. Arterial development appears normal, demonstrating morphogens from the skull and dura pattern optimal venous networks independent from arterial influences.
Bullet Points.
Humans with TWIST1 mutations and craniosynostosis have dural cerebral vein defects
Twist1 loss in osteoprogenitor cells and dura causes skull and vascular defects
Twist1 mouse mutants show loss of Bmp2/4 expression in osteogenic mesoderm and dura
Bmp2/4 loss from mesoderm-derived preosteoblasts/dura causes skull and vein defects
Acknowledgments
We thank the research participants who enrolled in the study; Hannah Marudzinski and Cecilia Ljungberg of the RNA In Situ Hybridization Core facility at Baylor for technical support; Volkhard Lindner, Brian Harfe, Kendra Mckee, Vicki Rosen, and Akiko Mammoto for sharing mouse reagents; Victoria Abraira, Jay Tischfield, Lisa Goodrich, Amit Agarwal, Yanshu Wang, Amir Rattner, Phil Smallwood, John Williams, and Arastoo Vossough for advice and discussions. Funding provided by NEI NRSA 1F32EY022274-01 (MAT), HHMI (JN, ECE), NIH IDDRC P30 HD018655 (Boston Children’s Hospital), and NIH IDDRC 1U54 HD083092 and NIH 1S10 OD016167 (Baylor College of Medicine).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Conceived and designed experiments (MAT); performed experiments (MAT with assistance from FAS, NG, AG); enrolled and clinically assessed human participants (BJB, SM, LRD, ECE); interpreted human radiology (CDR); assisted with mouse crosses (MMD, SMC); analyzed data (MAT, JN, ECE); wrote and edited manuscript (MAT, JN, ECE).
References
- Abu-Sittah GS, Jeelani O, Dunaway D, Hayward R. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. J Neurosurg Pediatr. 2016;17:469–475. doi: 10.3171/2015.6.PEDS15177. [DOI] [PubMed] [Google Scholar]
- Aurboonyawat T, Suthipongchai S, Pereira V, Ozanne A, Lasjaunias P. Patterns of cranial venous system from the comparative anatomy in vertebrates. Part I, introduction and the dorsal venous system. Interv Neuroradiol. 2007;13:335–344. doi: 10.1177/159101990701300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu MA, Guerrero-Maldonado A, McCrea HJ, Garcia-Navarro V, Souweidane MM, Anand VK, Heier L, Schwartz TH, Greenfield JP. Impact of skull base development on endonasal endoscopic surgical corridors. Journal of neurosurgery Pediatrics. 2014;13:155–169. doi: 10.3171/2013.10.PEDS13303. [DOI] [PubMed] [Google Scholar]
- Beepu H, Malhotra R, Beppu Y, Lepore JJ, Parmacek MS, Block KD. BMP type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Developmental Biology. 2009;331:167–175. doi: 10.1016/j.ydbio.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla-Claudio M, Wang J, Bai Y, Klysik E, Selever J, Martin JF. Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development. 2012;139:709–719. doi: 10.1242/dev.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AT, Liu Y, Ayyanathan K, Benner C, Jiang Y, Prokop JW, Paz H, Wang D, Li HR, fu XD, et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015;29:603–616. doi: 10.1101/gad.242842.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Brady Ridgway J, Sai T, Lai J, Warming S, Chen H, Roose-Girma M, Zhang G, Shou W, Yan M. Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc Natl Acad Sci U S A. 2013;110:11887–11892. doi: 10.1073/pnas.1306074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Akinwunmi PO, Deng JM, Tam OH, Behringer RR. Generation of a Twist1 conditional – allele in the mouse. Genesis. 2007;45:588–592. doi: 10.1002/dvg.20332. [DOI] [PubMed] [Google Scholar]
- Cuttler AS, LeClair RJ, Stohn JP, Wang Q, Sorenson CM, Liaw L, Lindner V. Characterization of Pdgfrb-Cre transgenic mice reveals reduction of ROSA26 reporter activity in remodeling arteries. Genesis. 2011;49:673–680. doi: 10.1002/dvg.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeraerts T, Newcombe VF, Coles JP, Abate MG, Perkes IE, Hutchinson PJ, Outtrim JG, Chatfield DA, Menon DK. Use of T2-weighted magnetic resonance imaging of the optic nerve sheath to detect raised intracranial pressure. Critical care. 2008;12:R114. doi: 10.1186/cc7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnough LH, Dinuoscio GJ, Atit RP. Twistlcontributes to cranial bone initiation and dermal condensation by maintaining wnt signaling responsiveness. Dev Dyn. 2015;245:144–156. doi: 10.1002/dvdy.24367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Nimura K, Kashiwagi K, Harada T, Takaoka K, Kato H, Tamai K, Kaneda Y. Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J Cell Sci. 2007;120:1350–1357. doi: 10.1242/jcs.000067. [DOI] [PubMed] [Google Scholar]
- Hayward R. Venous hypertension and craniosynostosis. Childs Nerv Syst. 2005;21:880–888. doi: 10.1007/s00381-004-1114-0. [DOI] [PubMed] [Google Scholar]
- Huang W. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci. 2007;12:3068. doi: 10.2741/2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Johnson D, Wilkie AOM. Craniosynostosis. Eur J Hum Genet. 2011;19:369–376. doi: 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice CM, Yagnik G, Kim Y, Peter I, Jabs EW, Erazo M, Ye X, Ainehsazan E, Shi L, Cunningham ML, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet. 2012;44:1360–1364. doi: 10.1038/ng.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidoya H, Naito H, Muramatsu F, Yamakawa D, Jia W, Ikawa M, Sonobe T, Tsuchimochi H, Shirai M, Adams RH, et al. APJ Regulates Parallel Alignment of Arteries and Veins in the Skin. Dev Cell. 2015;33:247–259. doi: 10.1016/j.devcel.2015.02.024. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rice DP, Kettunen PJ, Thesleff I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125:1241–1251. doi: 10.1242/dev.125.7.1241. [DOI] [PubMed] [Google Scholar]
- Korn C, Augustin HG. Mechanisms of Vessel Pruning and Regression. Dev Cell. 2015;34:5–17. doi: 10.1016/j.devcel.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Larrivée B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell. 2012;22:489–500. doi: 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Chong DC, Ola R, Dunworth WP, Meadows S, Ka J, Kaartinen VM, Qyang Y, Cleaver O, Bautch VL, et al. Alk2/ACVR1 and Alk3/BMPR1A Provide Essential Function for Bone Morphogenetic Protein-Induced Retinal Angiogenesis. Arterioscler Thromb Vasc Biol. 2017;37:657–663. doi: 10.1161/ATVBAHA.116.308422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatouk DM, Choi KS, Bouldin CM, Harfe BD. In the limb AER Bmp2 and Bmp4 are required for dorsal-ventral patterning and interdigital cell death but not limb outgrowth. Developmental biology. 2009;327:516–523. doi: 10.1016/j.ydbio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Miraoui H, Marie PJ. Pivotal role of Twist in skeletal biology and pathology. Gene. 2010;468:1–7. doi: 10.1016/j.gene.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Mouillesseaux KP, Wiley DS, Saunders LM, Wylie LA, Kushner EJ, Chong DC, Citrin KM, Barber AT, Park Y, et al. Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat Commun. 2016;7:1–12. doi: 10.1038/ncomms13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, Umans L, Maas E, Pereira PNG, Beets K, Francis A, Sents W, Robertson EJ, Mummery CL, Huylebroeck D, et al. Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev Cell. 2012;22:501–514. doi: 10.1016/j.devcel.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padget DH. The cranial venous system in man in reference to development, adult configuration, and relation to the arteries. Am J Anat. 1956;98:307–355. doi: 10.1002/aja.1000980302. [DOI] [PubMed] [Google Scholar]
- Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–567. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Miner Res. 2006;21:637–646. doi: 10.1359/JBMR.060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raets M, Dudink J, Raybaud C, Ramenghi L, Lequin M, Govaert P. Brain vein disorders in newborn infants. Dev Med Child Neurol. 2015;57:229–240. doi: 10.1111/dmcn.12579. [DOI] [PubMed] [Google Scholar]
- Robson CD, Mulliken JB, Robertson RL, Proctor MR, Steinberger D, Barnes PD, McFarren A, Müller U, Zurakowski D. Prominent basal emissary foramina in syndromic craniosynostosis: correlation with phenotypic and molecular diagnoses. AJNR Am J Neuroradiol. 2000;21:1707–1717. [PMC free article] [PubMed] [Google Scholar]
- Stevens SA, Previte M, Lakin WD, Thakore NJ, Penar PL, Hamschin B. Idiopathic intracranial hypertension and transverse sinus stenosis: a modelling study. Math, Med Biol. 2007;24:85–109. doi: 10.1093/imammb/dql025. [DOI] [PubMed] [Google Scholar]
- Streeter GL. The Developmental Alterations in the Vascular System of the Brain of the Human Embryo. Contributions to Embryology. 1921;8:7–38. [Google Scholar]
- Taylor WJ, Hayward RD, Lasjaunias P, Britto JA, Thompson DNP, Jones BM, Evans RD. Enigma of raised intracranial pressure in patients with complex craniosynostosis: The role of abnormal intracranial venous drainage. J Neurosurg. 2001;94:377–385. doi: 10.3171/jns.2001.94.3.0377. [DOI] [PubMed] [Google Scholar]
- Ting M-C, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, Maxson RE., Jr EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136:855–864. doi: 10.1242/dev.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SRF, Wilkie AOM. New insights into craniofacial malformations. Hum Mol Genet. 2015;24:R50–R59. doi: 10.1093/hmg/ddv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW. Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol. 2011;13:686–692. doi: 10.1038/ncb2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MH. Monro-Kellie 2.0: The dynamic vascular and venous pathophysiological components of intracranial pressure. J Cereb Blood Flow Metab. 2016;36:1338–1350. doi: 10.1177/0271678X16648711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RH, Ul-Haq E, Wilkie AOM, Jayamohan J, Richards PG, Johnson D, Lester T, Wall SA. Reoperation for intracranial hypertension in TWIST1-confirmed Saethre-Chotzen syndrome: a 15-year review. Plast Reconstr Surg. 2009;123:1801–1810. doi: 10.1097/PRS.0b013e3181a3f391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Developmental dynamics: an official publication of the American Association of Anatomists. 2005;234:371–386. doi: 10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, Taketo MM, Nathans J. Canonical WNT signaling components in vascular development and barrier formation. The Journal of clinical investigation. 2014;124:3825–3846. doi: 10.1172/JCI76431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouaoui A, Hidden G. Cerebral venous sinuses: anatomical variants or thrombosis? Acta Anat. 1988;133:318–324. doi: 10.1159/000146661. [DOI] [PubMed] [Google Scholar]
- Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. Plos one. 2011;6:e27385. doi: 10.1371/journal.pone.0027385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.