

Abstract
Huntington’s disease (HD) is a fatal, neurodegenerative movement disorder that has no cure and few treatment options. In these preclinical studies, we tested the effects of chronic treatment of glatiramer acetate (GA; Copaxone®), an FDA-approved drug used as first-line therapy for MS, in two different HD mouse models, and explored potential mechanisms of action of drug efficacy. Groups of CAG140 knock-in and N171-82Q transgenic mice were treated with GA for up to 1 year of age (CAG140 knock-in mice) or 20 weeks (N171-82Q mice). Various behavioral assays were measured over the course of drug treatment whereby GA treatment delayed the onset and reduced the severity of HD behavioral symptoms in both mouse models. The beneficial actions of GA were associated with elevated levels of promoter I- and IV-driven brain-derived neurotrophic factor (Bdnf) expression and reduced levels of cytokines, in particular, interleukins IL4 and IL12, in the brains of HD mice. In addition, the GA-induced effects on BDNF, IL4 and IL12 levels were detected in plasma from drug-treated mice and rats, suggesting utility as a peripheral biomarker of treatment effectiveness. These preclinical studies support the use of GA as a relevant clinical therapy for HD patients.
Introduction
Huntington’s disease (HD) is caused by a CAG repeat expansion mutation in the Huntingtin (HTT) gene, which leads to progressive movement dysfunction, cognitive impairment and behavioral abnormalities (Group 1993). Clinical signs of HD typically emerge in adult ages (30–40s), but juvenile-onset and late-onset cases are also observed, with death following approximately 15 years after disease onset. Since the identification of the HTT gene in 1993, HD has been the focus of extensive preclinical and clinical research in efforts to find a treatment for this devastating disorder, and although there are many promising candidates on the horizon, few are clinically available.
Glatiramer acetate (GA; copolymer 1; Copaxone®) is an FDA-approved drug used as first-line treatment for relapsing-remitting multiple sclerosis (MS). It is a random polymer composed of four amino acids that are found in myelin basic protein, namely glutamic acid, lysine, alanine, and tyrosine. The mechanisms of action of GA in MS are not fully understood, but are thought to involve immunomodulatory effects, via a downregulation of proinflammatory cytokines (Neuhaus et al. 2001; Blanco et al. 2006) and/or neuroprotective effects, via increased release of brain-derived neurotrophic factor (BDNF) from immune cells (Aharoni et al. 2005; Chen, Valenzuela, and Dhib-Jalbut 2003; Ziemssen et al. 2002). Neuroprotective and immunomodulatory mechanisms have relevance not only for the treatment of MS, but have also been implicated as target pathways for HD. For example, reduced BDNF expression is thought to play a crucial role in HD pathogenesis, whereby decreased expression and transport of BDNF has been observed in brain tissue from human HD patients (Ferrer et al. 2000; Zuccato et al. 2001) and in several different mouse models transgenic for mutant huntingtin, including R6/2, YAC72 and N171-82Q transgenic mice (Duan et al. 2003; Zhang et al. 2003; Zuccato et al. 2001). Accordingly, restoring striatal BDNF levels has been shown to have therapeutic effects in HD mouse models (Gharami et al. 2008; Giampa et al. 2013) (Xie, Hayden, and Xu 2010).
Additionally, data are emerging to implicate inflammation and immune dysfunction as playing an important role in the pathogenic mechanisms of cell death in HD (Ellrichmann et al. 2013; Silvestroni et al. 2009). Although inflammation is not an initiating factor in the pathology of HD, growing evidence indicates that inflammatory responses involving astrocytes, microglia, as well as the peripheral immune system contributes to disease progression (Dalrymple et al. 2007; Bjorkqvist et al. 2008; Silvestroni et al. 2009). For example, specific increases in proinflammatory cytokines, including interleukin 6 (IL-6), interleukin 8 (IL-8), and tumor necrosis factor alpha (TNFα), have been found in post-mortem striatum from HD patients and mouse models (Dalrymple et al. 2007; Bjorkqvist et al. 2008; Silvestroni et al. 2009), as well as in plasma from human HD patients (Dalrymple et al. 2007; Chang et al. 2015). Given these overlapping disease mechanisms between MS and HD, we have investigated whether GA might be a relevant treatment option for HD.
In our early studies, we demonstrated that GA can increase BDNF levels in cultured striatal cells and in striatal tissue after short-term (5-day) in vivo administration (Corey-Bloom et al. 2014). In the current study, we further carried out preclinical studies to assess the efficacy of chronic GA administration in two different HD mouse models, CAG140 knock-in (KI) mice and N171-82Q transgenic mice, and to explore potential mechanisms of action for GA’s beneficial effects. Our results demonstrate that GA treatment improved disease phenotypes in both HD mouse models. The beneficial effects of GA were associated with modifying levels of BDNF and interleukins, not only in brain tissue, but also in plasma. These findings strongly suggest that GA might represent a relevant clinical therapy for HD patients and the blood measurements of BDNF and IL4/IL12 might serve as markers for drug effectiveness.
Materials and Methods
Animals and drug treatments
All animals were housed n=3–4 per cage, and maintained on a reverse 12-h light/dark cycle with lights on at 9:00 p.m. and free access to food (normal rodent chow) and water. CAG140 KI mice contain a chimeric mouse/human exon 1 with 140 CAG repeats inserted into the mouse gene by homologous targeting (Menalled et al. 2003), and were maintained by breeding of heterozygote pairs. CAG140 KI mice were genotyped at 4 weeks of age to determine homozygosity for the CAG140 mutation. The CAG repeat lengths in these mice has been verified by commercial genotyping (Laragen Inc, Culver City, CA) and found to be 130 ± 3 CAGs (reduced from the original description of 140 CAGs) (Menalled et al. 2003). Previous studies on these mice have reported climbing deficits in these mice as early as 6 weeks of age and rotarod deficits by 4 months of age (Hickey et al. 2008; Hickey et al. 2012). Transgenic N171-82Q HD mice were maintained by breeding heterozygous N171-82Q males with C3B6F1 females (Jackson Laboratories). At the age of 4 weeks, mice were genotyped according to the Jackson Laboratories protocols. The CAG repeat length in these mice is 82 ± 1 CAGs (Laragen, Los Angeles, CA). The lifespan of the N171-82Q mice is ~20 weeks in our colony, with HD-like symptoms beginning at ~8 weeks of age. Litters of these mice were assigned randomly to the various experimental groups to achieve a minimum of n=8 per group for behavioral testing. To avoid litter effects, mice from the same litter were evenly split into vehicle- and drug-treated groups. Power calculations show that groups of 8 mice are sufficient to have a 90% chance of detecting a 25% improvement in rotarod behavior, which is a minimal expected level of improvement [n=log(0.1)/log(0.75)=8]. All other tests are expected to reveal >25% improvement as a result of drug treatment.
Groups of CAG140 KI mice (n=17–20 per genotype and drug condition; 50:50, M:F) were injected s.c. with GA (0.625 mg/mouse) or an equal amount of vehicle (40% mannitol) once a day, 3× per week, beginning at 3 months of age. One half of the mice were sacrificed at 7 months of age, with the remaining mice (n=8–10 per group) sacrificed at 1 year of age. Mice were sacrificed using isofluarane overdose in a bell jar. For the N171-82Q transgenic line, groups of mice (n=8–10 per genotype and drug treatment; 50:50, M:F) were injected with 1 mg/mouse GA, 5×/week, beginning at 8 weeks of age until 20 weeks of age. Body weight was recorded at each injection. The doses used for injection were based on previous studies in mice where ranges of 0.15 to 2 mg/mouse have been used (Moore et al. 2014; Poittevin et al. 2013; Smirnov, Walsh, and Kipnis 2013; Teitelbaum et al. 2004). Further, extrapolation of the dose from animals to humans requires consideration of body surface area, which is related to metabolic rate of an animal (Nair and Jacob 2016). Considering body surface, our mouse dose correlates to 75 mg/m2, which is ~3-fold higher than a typical human dose of 23.8 mg/m2 (Nair and Jacob 2016). Mice were sacrificed 4 hours after the final injection, brains rapidly removed and trunk blood collected into heparin-coated tubes. For rat studies, Sprague-Dawley rats (Charles River) were used. At the age of 6 weeks, four groups of rats (n=4 per group) received daily injections of GA (i.p.) for 5 days. Rats were sacrificed 4 hours after the final injection by isofluorane overdose, brains rapidly removed and trunk blood collected into heparin-coated tubes. All procedures were in strict accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Behavioral tests
All mice were tested in the following behavioral paradigms at the time points indicated: Open field activity (OFA) test: OFA was measured in a square plexiglass chamber (27.3 cm × 27.3 cm) (Med Associates INC). The test chamber is divided into 16 squares (12 outer and 4 inner) of equal areas and includes three 16 photobeam I/R arrays to automatically record movement. Eight behavioral parameters (ambulatory counts, ambulatory time, stereotypic counts, stereotypic time, vertical counts, vertical time, resting time and jumping time) were automatically recorded during a 10 minute observation period. Mice were tested in the open field at 18 weeks of age (N171-82Q transgenic mice) or 6 and 8.5 months (CAG140 KI mice). Rotarod test: Animals were tested on an AccuRotor rotarod (AccuScan Instruments) during the dark phase of the 12h light-dark cycle using an accelerating rotation paradigm (4–40 rpm over 10 min). The time of fall was recorded. During the course of the experiment, mice were tested in a set of four trials each day for four consecutive days. Mice were tested at 15 weeks (N171-82Q transgenic mice) or 6, 9 and 12 months (CAG140 KI mice). Climbing test: To assess climbing activity, mice were placed on the floor of a wire cylinder (8″ height×4″ diameter) for 5 min. Climbing was recorded when two or four paws of the mouse are off the floor of the testing bench. Mice were tested in climbing at 11 and 17 weeks (N171-82Q transgenic mice) or 5.5 and 7 months (CAG140 KI mice). Alternating T-maze. The alternating T-maze test was performed as described previously (Jia et al. 2015) at 8 months of age. The T-maze is made of transparent Plexiglas with a central arm (75 cm long × 12 cm wide × 20 cm high) and two lateral arms (32 cm long × 12 cm wide × 20 cm high) positioned at a 90° angle relative to the central arm. Forced alternation was used for the T-maze training. For T-maze testing, mice were provided an initial free choice for either arm of the maze, and the percentage of alternation over the next nine trials was determined. Grip strength. Grip strength was determined at 1 year of age using a Grip Strength Meter consisting of a baseplate, a stainless-steel grip, and a portable force sensor (Bioseb). Mice were lifted over the baseplate by the tail so that their forepaws were allowed to grasp onto the steel grip. The mouse was gently pulled backward by the tail until the grip released. Five trials were performed for each mouse with a 1-minute resting period between trials and the best trial used for statistical analysis. All behavioral tests were conducted between 10 am and 2 pm, by individuals blind to genotype and treatment. At the end of the drug treatments, trunk blood was collected, brains were removed, and striata and cortex dissected out and immediately frozen.
Statistical analyses for OFA and rotarod tests were performed using Two-way analysis of variance (ANOVA) (GraphPad Prism, San Diego, CA), testing the effects of genotype and drug treatment on each behavior measured. For climbing, T-maze and grip strength data, One-way ANOVA was utilized to determine significant differences among groups, followed by Dunnett’s post-test comparing all groups vs. the vehicle-treated CAG140 KI group (GraphPad Prism, San Diego, CA). Significance was accepted at P<0.05. We did not observe significant differences between males and females in (GraphPad Prism, San Diego, CA), any of these tests, with the exception of rotarod for the N171-82Q mice, as reported previously (Jia et al. 2015). Hence, data from male and females were combined for all graphs and statistical analyses). For rotarod data on N171-82Q mice, only male mice are shown.
Real-Time PCR Analysis
Real-time PCR experiments were performed using the ABI StepOne Detection System (Applied Biosystems, Foster City, CA) as described previously (Jia et al. 2016). Amplification was performed on a cDNA amount equivalent to 25 ng total RNA with 1 × SYBR® Green universal PCR Master mix (Applied Biosystems) containing deoxyribonucleotide triphosphates, MgCl2, AmpliTaq Gold DNA polymerase, and forward and reverse primers designed against exons I, IV and IX of the Bdnf gene as used in our previous studies (Tang et al. 2011). Sequences for these primers are shown in Supplementary Table 1. Efficiencies for these primers sets were 96–104%. The amount of cDNA in each sample was calculated using SDS2.1 software by the comparative threshold cycle (Ct) method and expressed as 2exp(Ct) using hypoxanthine guanine phosphoribosyl transferase (Hprt) as an internal control. One-way ANOVA with Dunnett’s post-test was used to determined significant effects GA on Bdnf expression levels in WT and HD mice. Statistical tests were performed using GraphPad software (GraphPad Prism). Significance was accepted at P<0.05.
ELISA Assays
Striatal and cortical samples were homogenized in lysis buffer at a 1:10 (mg tissue:ml buffer) ratio. Serum samples were used at a 1:25 dilution. For the BDNF ELISA, tissue and rat plasma levels of BDNF were determined by ELISA using an adaptation from a method reported previously (Mandel, Ozdener, and Utermohlen 2011). For the Cytokine ELISA, The Mouse Inflammatory Cytokines & Chemokines Multi-Analyte ELISArray Kit was used (Qiagen) and assayed according to the manufacturers’ protocols. The cytokines & chemokines represented by this array were IL1A, IL1B, IL2, IL4, IL6, IL10, IL12, IL17A, IFNγ, TNFα, G-CSF, and GM-CSF. Individual BDNF and cytokine levels were normalized by the total protein in each sample, determined by the BCA protein assay reagent (ThermoScientific). One-way ANOVA with Dunnett’s post-test was used to determined significant difference in levels of each protein among the different groups. Due to low sample availability to evenly match sexes, only male mice were used for the cytokines analysis. Statistical tests were performed using GraphPad software (GraphPad Prism), with significance accepted at P<0.05.
Results
Behavioral effects of GA administration
The CAG140 KI mouse line shows a prolonged disease time course with a near-normal lifespan (Menalled et al. 2003)(Hickey et al. 2008). These mice were treated with GA (0.625 mg/mouse; s.c.; 3× per week) for up to 1 year of age, beginning at 3 months of age, and the effects of GA administration on several disease phenotypes, including body weight, rotarod performance, climbing, and open field exploratory behavior, were measured. GA administration resulted in a small, but significant increase in the body weight of CAG140 KI female mice, but no effects in male CAG140 KI mice or WT mice of either sex (Fig. S1). In the rotarod test, vehicle-treated CAG140 KI mice showed a progressive decline in rotarod function at 9 and 12 months of age (Fig. 1A). In contrast, CAG140 KI mice treated with GA showed improved motor performance at 9 and 12 months of age (Fig. 1A), with GA-treated mice performing nearly at the same level at WT mice (Fig. 1A). GA showed no significant effects on rotarod performance of WT mice (Fig. 1A).
Fig. 1. The effects of GA treatment on motor behavior in CAG140 KI mice.
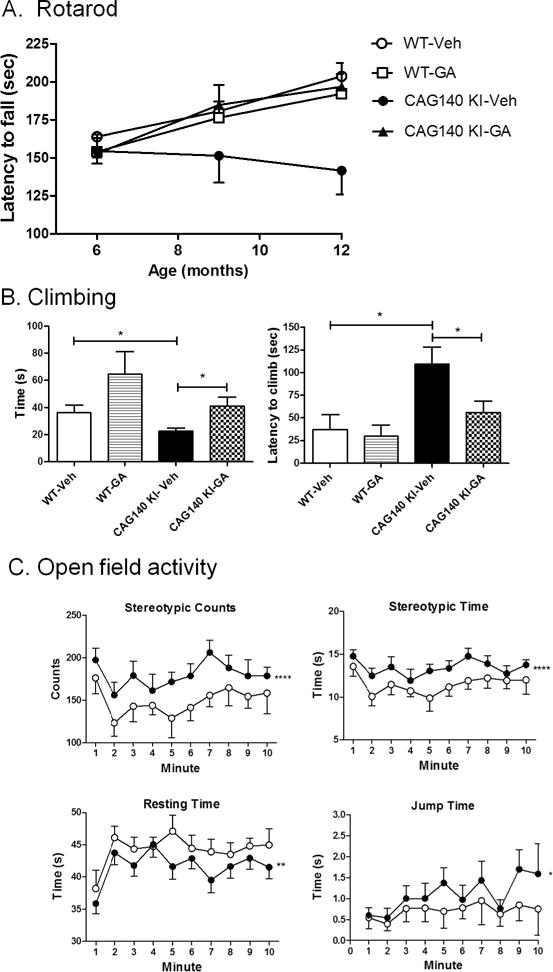
Panel A shows the effects of GA on mean rotarod performance at 6 months (n=17–20 per group), 9 months (n=8–10 per group) and 12 months of age (n=8–10 per group). Values depict the mean initial performance of the test at the time point indicated. *, P=0.011; (Two-way ANOVA; F(1,126)=23.5). Panel B shows climbing behavior, assessed at 8 months of age, in vehicle- and drug-treated WT mice (n=8) and CAG140 KI mice (n=10). Data show mean +/− S.E.M. time for the indicated measure for each group. Significant differences among groups were determined by One-way ANOVA, followed by Dunnett’s post-test to compare against the vehicle-treated CAG140 KI group, and are indicated by asterisks. *, P<0.05. Panel C shows significant differences between GA-treated CAG140 KI mice vs. vehicle-treated CAG140 KI mice on jump time (*, P=0.029; F(1,150)=4.8), stereotypic time (****, P<0.0001; F(1,150)=16.5), stereotypic counts (****, P<0.0001; F(1,150)=17.1) and resting time (**, P=0.003; F(1,150)=9.0) at 8.5 months of age, measured over a 10 minute time frame. Open symbols represent mean +/− S.E.M. performance of vehicle-treated CAG140 KI mice (n=8), while closed circles depict mean +/− S.E.M. performance of GA-treated CAG140 KI mice (n=9). Significant effects of GA were determined by Two-way ANOVA.
In the climbing test, CAG140 KI mice showed significantly reduced climbing time at 7 months of age and increased latency to climb when compared to WT littermates (Fig. 1B). GA significantly reduced climbing latency and improved climbing time of CAG140 KI mice while having no significant effects on WT mice (Fig 1B). In the open field activity test, CAG140 KI mice showed significantly worse performance compared to WT mice at 8.5 months of age in four open field measures: stereotypic time, stereotypic counts, jumping time and resting time (data not shown). GA-treated CAG140 KI mice showed significantly improved performance in each of these four measures compared to vehicle-treated CAG140 KI mice (Fig. 1C). Similarly, in the alternating T-maze test, CAG140 KI mice showed fewer correct choices for a food reward compared to WT mice at 8 months of age (Fig. S2). However, this deficit was prevented by treatment with GA (Fig. S2). There was no significant difference between CAG140 KI and WT mice in grip strength, tested at 1 year of age, and there were no significant effects of GA treatment on grip strength performance in either WT or CAG140 KI mice (data not shown).
We also tested the effects of GA treatment in a second HD mouse model, the N171-82Q transgenic mouse line, which exhibits a more rapidly progressing disease course. We treated groups of mice with GA (1 mg/mouse; s.c.; 5 × week) beginning at 8 weeks of age and continuing until 20 weeks of age, which is near the age of death due to the disease. GA treatment elicited improved performance on several motor function measures. GA treatment significantly improved the performance of N171-82Q transgenic mice at 15 weeks of age in the rotarod test measured over the course of 4 days (Fig. 2A). In other measures of motor function, GA-treated N171-82Q mice showed improved climbing behavior at 18 weeks of age. Specifically, the GA-treated N171-82Q mice showed a reduced latency to begin climbing compared to vehicle-treated N171-82Q mice (Fig. 2B). GA treatment also had an effect to improve climbing time of WT mice, although did not show significant effects in HD mice (Fig. 2B. Also at 18 weeks of age, N171-82Q transgenic mice showed a significant deficit in vertical activity measured in the open field compared to WT mice (Fig. 2C). GA-treated N171-82Q transgenic mice showed a significant increase in this measure compared to vehicle-treated N171-82Q mice (Fig. 2C).
Fig. 2. The effects of GA treatment on motor behavior in N171-82Q transgenic mice.
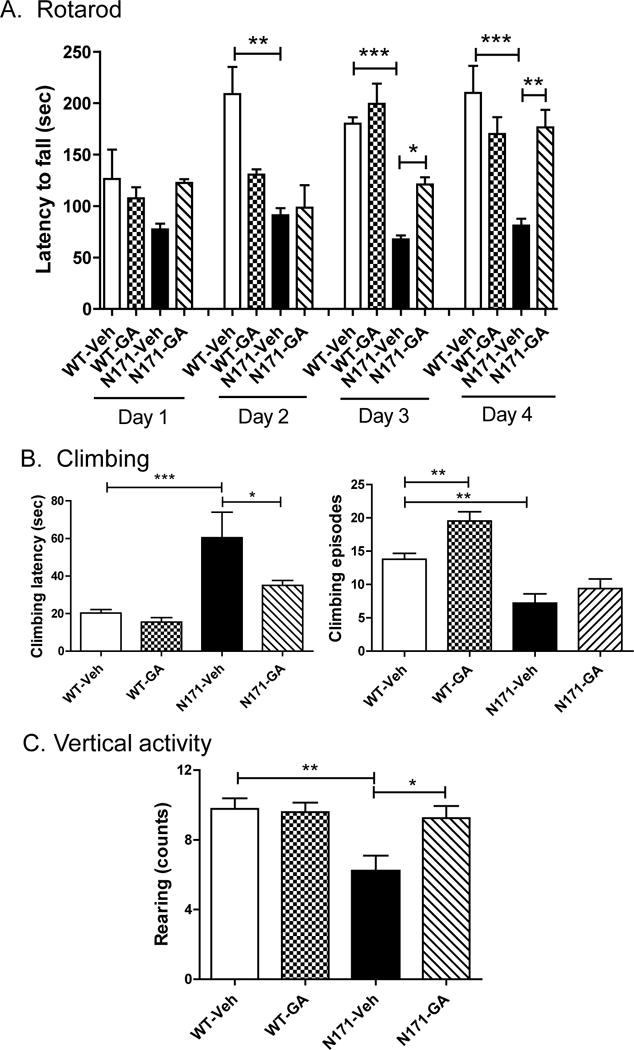
A). Rotarod was measured over four days at 15 weeks of age using an accelerating paradigm. The mean ± S.E.M. level of performance for each group of WT and N171-82Q transgenic mice (n=5–6 per group, males only) on each day is shown. Panel B shows the effects of GA treatment on climbing activity of WT and N171-82Q transgenic mice 17 weeks of age. Bar graphs depict mean +/− S.E.M. performance from n=8–10 mice per group. Panel C depicts the effects of GA on vertical activity of WT and N171-82Q transgenic mice (n=8–10 per group) at 18 weeks of age. Bar graphs depict mean +/− S.E.M. performance from n=8−10 mice per group. Significant differences were determined by One-way ANOVA, followed by Dunnett’s post-test. *, P<0.05; **, P<0.01; ***, P<0.001.
Effects of GA on Bdnf gene expression
Our previous studies implicated have implicated BDNF as a potential target for GA therapy in HD (Corey-Bloom et al. 2014). Therefore, in this study, we measured expression of Bdnf mRNA in response to chronic GA treatment. Multiple promoters control Bdnf transcription, leading to the existence of distinct mRNA species, however Bdnf-I and Bdnf-IV promoter-driven species are most widely studied in relation to brain function. Hence, we used primer sets recognizing the Bdnf-I, Bdnf-IV species and primers directed at the 3′ exon IX to recognize all forms of Bdnf in order to assess the effects of GA on Bdnf expression in the cortex of 1 year old CAG140 KI mice. We found that GA treatment elicited significant increases in promoter I and IV-driven Bdnf mRNA forms compared to vehicle-treated mice, but had no significant effect on Bdnf IX expression (Fig. 3A). Interestingly, all three forms of Bdnf mRNA were found to be significantly decreased in 1 year-old vehicle-treated CAG140 KI mice compared to age-matched WT mice (Fig. 3A). Similar effects of GA were observed in the cortex of N171-82Q transgenic mice, assessed at 20 weeks of age, where GA treatment elicited increases only in Bdnf-I and Bdnf-IV mRNA species, but not exon IX Bdnf (Fig. 3B).
Fig. 3. The effects of GA treatment on Bdnf expression in CAG140 KI and N171-82Q transgenic mice.
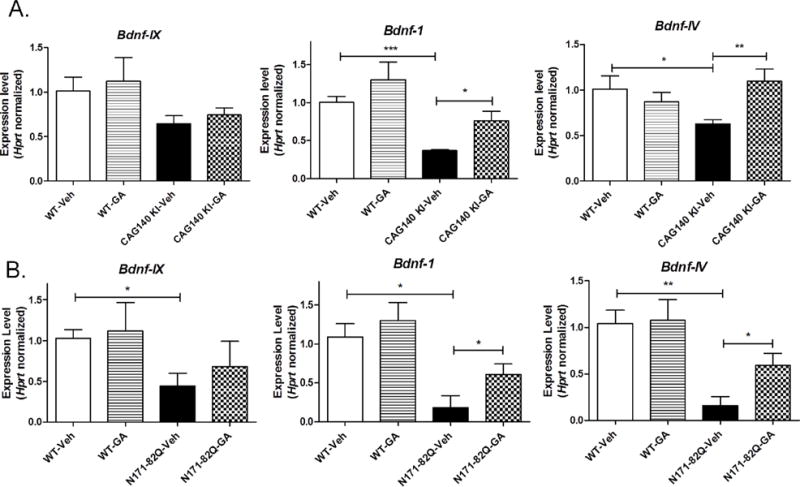
Real-time qPCR analysis showing the effects of GA on the expression of promoter I (“Bdnf-1”), promoter IV (“Bdnf-4”) and exon IX (“Bdnf-IX”) forms of Bdnf expression in the cortex of CAG140 KI mice at 12 months of age (Panel A), and N171-82Q transgenic mice at 20 weeks of age (Panel B). Values shown are the mean ± S.E.M. expression values (n=6 mice per group). Significant differences were determined by Student’s t-tests for the indicated genes. *, p<0.05; **, p<0.01; ***, p<0.001. Panel C show the effects of GA on BDNF protein levels measured in rat plasma at the indicated doses of GA. Bars show the mean ± S.E.M. BDNF level (n=6 mice per group). Significant differences were determined by Oneway ANOVA, followed by Dunnett’s post-test. *, P<0.05; **, P<0.0. Panel D shows the positive correlation observed between BDNF levels measured in rat plasma and striatum levels of BDNF as determined by Pearson Correlation analysis (r=0.626, **, P=0.0078).
We then tested whether GA-elicited changes in BDNF expression could be assessed in blood, which could have relevance for biomarker identification. Consistent with other reports on mouse blood (Klein et al. 2011), we found that levels of BDNF protein in mouse serum were undetectable using our BDNF ELISA assay. Therefore, we treated groups of normal Sprague-Dawley rats with different doses of GA (25–100 mg/kg; s.c.) for 5 days, followed by measurements of BDNF protein in plasma and striatum. Our results showed that GA treatment resulted in significantly elevated levels of serum BDNF at 25 and 100 mg/kg doses (p<0.05, Student’s t test) (Fig. 3C). Striatal levels of BDNF were also elevated by GA treatment in normal rats, and, interestingly, the striatal levels of BDNF were found to be significantly positively correlated with plasma levels from the same animals (Spearman r=0.626; P=0.006) (Fig. 3D).
Effects of GA on cytokines
Because GA has previously been shown to downregulate proinflammatory cytokines in the context of MS (Neuhaus et al. 2001; Blanco et al. 2006), we tested whether GA treated targets cytokine levels in WT and CAG140 KI HD mice using a multianalyte ELISA, which measures the protein expression levels of 12 cytokines: IL1A, IL1B, IL2, IL4, IL6, IL10, IL12, IL17A, IFNγ, TNFα, G-CSF, and GM-CSF, several of which have been previously implicated in HD. Comparing CAG140 KI mice to WT controls, we found that nearly all of the cytokines tested were found to be significantly elevated in the HD mouse brains, including IL1A, IL1B, IL4, IL6 and 1L17A (Fig. 4A; Table 1). GA treatment did not exhibit any significant effects on cytokine levels in WT mice, however, GA treatment reduced the levels of IL1A, IL4 and IL12 in the cortex of CAG140 KI mice (Fig. 4B; Table 1). Given previous studies reporting effects of GA on IL4 and IL12 in MS, we further focused on measuring levels of these two cytokines in plasma samples from drug-treated CAG140 KI and N171-82Q transgenic mice using ELISA. We found remarkably similar patterns of regulation of these cytokines in the plasma compared to the cortex. Levels of both IL4 and IL12 were higher in plasma from CAG140 KI mice compared to WT mice, and GA treatment caused significant reductions of these interleukins in this mouse model (Fig. 4C). In N171-82Q transgenic mice, levels of IL12 showed the same pattern of regulation due to GA treatment as CAG140 KI mice (Fig. 4C). However, GA did not elicit changes in the levels of IL4 in the plasma of N171-82Q transgenic mice (Fig. 4C).
Fig. 4. Measures of cytokine levels in the cortex of vehicle- and GA-treated CAG140 KI mice.
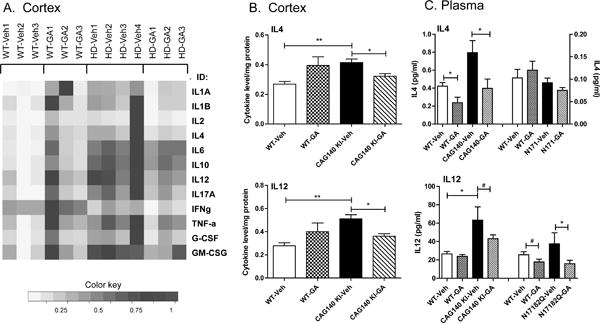
Panel A shows a heatmap depiction to summarize the expression changes of all 12 cytokines in the cortex of male WT and CAG140 KI (“HD”) mice. Cytokine levels were determined by multianalyte ELISA, with yellow indicating decreased expression and blue indicating increased expression as shown by the color key. Panel B shows the significant effects of GA on cortical levels of IL4 and IL12 in a bar graph format depicting mean +/− S.E.M. level from n=4 mice per group. Panel C shows the effects of GA on IL4 and IL12 levels in plasma from CAG140 KI and N171-82Q transgenic mice. Levels depict mean +/− S.E.M. level of the indicated cytokine from n=8 mice per group. Significant differences were determined by Student’s t-tests for the indicated genes. #, p<0.08; *, p<0.05; **, p<0.01.
Table 1.
The effects of GA treatment on cytokine levels in WT and CAG140 KI mice.
| HD Veh vs. WT Veh: | WT GA vs. WT Veh | HD GA vs. HD Veh: | ||||
|---|---|---|---|---|---|---|
| Cytokine: | FC: | p-value: | FC: | p-value: | FC: | p-value: |
| IL1A | 1.48 | 0.0007 | 1.78 | 0.180 | 0.81 | 0.048 |
| IL1B | 1.05 | 0.0284 | 1.38 | 0.213 | 0.84 | 0.073 |
| IL2 | 1.32 | 0.0205 | 1.53 | 0.100 | 0.81 | 0.061 |
| IL4 | 1.55 | 0.0074 | 1.48 | 0.103 | 0.76 | 0.036 |
| IL6 | 1.81 | 0.0004 | 1.42 | 0.170 | 0.94 | 0.144 |
| IL10 | 2.06 | 0.0004 | 1.36 | 0.149 | 0.87 | 0.111 |
| IL12 | 1.76 | 0.0084 | 1.45 | 0.197 | 0.72 | 0.013 |
| IL17A | 1.70 | 0.0086 | 1.41 | 0.198 | 0.79 | 0.094 |
| IFNg | 0.92 | 0.2412 | 1.31 | 0.090 | 0.85 | 0.154 |
| TNF-a | 1.37 | 0.0178 | 1.37 | 0.155 | 0.88 | 0.224 |
| G-CSF | 1.37 | 0.0140 | 1.31 | 0.232 | 0.85 | 0.188 |
| GM-CSF | 1.52 | 0.0078 | 1.29 | 0.241 | 0.80 | 0.070 |
These data reflect cortical levels of the indicated cytokines after GA treatment (50 mg/kg; s.c. 5× week; 4 months duration). Significant differences are indicated in bold. P-values reflect significant differences as determined by Student’s t test (unpaired; two-tailed) from male mice only (n=4 mice per group).
Discussion
In this study, we carried out chronic drug treatment paradigms with GA using two different HD mouse models: N171-82Q transgenic and CAG140 KI mice. The CAG140 KI mice have a prolonged disease course, which allows for capturing truly chronic effects of drug treatment. We treated this line of mice from 3 months until 1 year of age, which, to our knowledge, is the longest duration of drug treatment tested in any HD mouse model. We found that chronic GA treatment improved several disease phenotypes, including rotarod, climbing behavior and locomotor effects, in both CAG140 KI and N171-82Q transgenic mice. These findings are consistent with recent studies demonstrating beneficial effects of GA immunization on motor function and survival in other HD transgenic mouse lines (YAC128 and R6/2 transgenic mice) (Reick et al. 2016).
GA has been hypothesized to act via neuroprotective and immune-related pathways in MS. With regards to neuroprotective pathways, one study found reduced levels of BDNF in the serum and CSF of relapsing-remitting MS patients, which were reversed by treatment with GA (Azoulay et al. 2005). Similar findings were shown in immune cells where GA treatment increased BDNF levels in cultured peripheral blood mononuclear cells (Blanco et al. 2006) and other studies that demonstrated GA-reactive T cells can release brain-derived neurotrophic factor (BDNF) (Aharoni et al. 2003; Ziemssen et al. 2002; Chen, Valenzuela, and Dhib-Jalbut 2003). We have previously shown similar findings in tissues relevant to HD, where we demonstrated that GA increased BDNF protein levels in cultured STHdh striatal cells and in striatal tissue after short-term (5-day) in vivo administration in N171-82Q mice (Corey-Bloom et al. 2014). Consistent with this finding, in the current study, we detected increases in striatal BDNF in rats treated with GA for 5 days. Although STHdh striatal cells are of neuronal origin, striatal tissue contains a mixture of cell types, any of which might be contributing to BDNF production. A recent study demonstrated that GA increased the expression of BDNF in astrocyte cultures and in astrocytes of GA treated HD mice (Reick et al. 2016). Hence, it is possible that multiple cell types are contributing to the elevated levels of BDNF elicited by GA.
A major question remains as to the mechanism by which GA alters BDNF levels. To address this question, we tested whether GA-elicited regulation of BDNF expression occurred at the level of gene transcription by measuring Bdnf mRNA species using real-time PCR analysis. There are as many as nine differentially regulated 5′ promoters that control spatial and temporal transcription of the Bdnf gene (Aid et al. 2007). Bdnf forms I and IV are the most widely studied, with the latter being strongly induced in response to neuronal activity. We showed that chronic treatment with GA elicited highly significant increases in Bdnf I and IV gene expression in the cortex of both HD mouse models, with the biggest changes, i.e. >3-fold increases, induced by GA, in N171-82Q mice. However, whether GA exerts direct or indirect effects on Bdnf transcription remains to be determined. Previous high-throughput transcriptome studies have identified several transcription factors, such as Forkhead box P3 and Transcription factor 8, as gene targets for GA (Achiron, Feldman, and Gurevich 2009; Bakshi et al. 2013). Alterations in these factors could provide a possible mechanism for the effect of GA on Bdnf transcriptional changes.
Whether BDNF levels are also altered in peripheral tissues is an important question. Previous studies comparing BDNF levels in brain and blood support the view that measures of plasma BDNF levels can reflect those in brain tissue (Klein et al. 2011). Although we could not detect blood levels of BDNF in mouse, consistent with previous studies (Klein et al. 2011), BDNF could readily be measured in rat serum and brain tissue, where we observed that GA treatment could modulate BDNF levels and further, that serum levels correlated with those levels detected in the striatum. This finding is consistent with previous studies showing positive correlations between whole-blood BDNF levels and hippocampal BDNF levels in rats, and between plasma BDNF and hippocampal BDNF in pigs (Klein et al. 2011). These data support the view that blood and plasma BDNF levels reflect brain BDNF levels, suggesting that serum BDNF levels may represent a peripheral biomarker for GA effectiveness in the brain, although additional studies on larger numbers of animals would be needed to confirm this idea.
In the context of MS, GA is also thought to exhibit beneficial effects via immunomodulatory mechanisms. GA has been shown to induce a shift from a “pro-inflammatory” Th1 state in T cells, to an “anti-inflammatory” Th2 state, both in vitro and in vivo (Tselis, Khan, and Lisak 2007). This conversion is accompanied by a change in the expression of different cytokines (Begum-Haque et al. 2010). In particular, GA treatment was found to reverse the up-regulation of IL-6 and IL-12 levels in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS and in plasma from MS patients (Begum-Haque et al. 2010)(Begum-Haque, 2008)(Losy J, 2002). Another study showed that IL4 levels were elevated in untreated MS patients, an effect that was reversed with GA treatment (Dressel A, 2006). Additionally, GA has been shown to decrease IL4 levels in serum from a hepatic fibrogenesis mouse model (Horani A, 2007). Consistent with these studies, we found that levels of IL4 and IL12 were elevated in both the cortex and plasma of HD mice compared to WT controls and that GA treatment significantly reversed the observed increases in these cytokines. Importantly, the similar effects observed in brain and plasma suggest that HD and its potential treatments affect the whole body, and supports the existence of relevant communication or crosstalk between central and peripheral tissues. Additionally, we found significant increases in the levels of other cytokines, including IL-1A, IL-6, IL-17 and TNF-alpha in the cortex of CAG140 KI mice compared to WT littermate controls, indicative of an increased immune activation/inflammation state. These results are consistent with growing evidence suggesting that inflammation plays an important role in the ongoing brain pathology associated with HD (Ellrichmann et al. 2013; Silvestroni et al. 2009; Dalrymple et al. 2007; Bjorkqvist, Wild, and Tabrizi 2009).
Finally, we must consider the dose used in this study, as compared to the current human dose used for MS. We based our doses (0.625 or 1.0 mg/mouse) on previous studies in mice, where ranges of 0.15 to 2 mg/mouse were used (Moore et al. 2014; Poittevin et al. 2013; Smirnov, Walsh, and Kipnis 2013; Teitelbaum et al. 2004). Extrapolation of our dose from mice to humans requires consideration of body surface area, which is related to metabolic rate of the mouse (Nair and Jacob 2016). Hence, our extrapolated mouse dose correlates to 75 mg/m2, which is approximately 3-fold higher than dose typically used in humans with MS (23.8 mg/m2). This represents a limitation of our study, in that typical human dose was not used. However, we did not observe any toxicity of the higher doses, which testifies to the safety and tolerability of GA as a therapeutic.
Overall, we show that GA treatment improves HD disease phenotypes in two different mouse models, suggesting that GA could be a useful therapeutic for HD patients. The beneficial actions of GA likely include a dual mechanism that includes both an elevation of Bdnf expression (variants I and IV), and a reduction of proinflammatory cytokines. Glatiramer acetate received FDA approval in 1996 and numerous studies over the last 10–15 years have characterized the clinical efficacy and safety of GA (Johnson 2012). This excellent safety and tolerability record of GA makes it an ideal candidate for drug repurposing efforts for HD.
Supplementary Material
Table S1. Primer sequences used for real-time PCR analysis.
Fig. S1. The effects of chronic GA administration on body weight of WT and CAG140 KI.
Fig. S2. The effects of chronic GA treatment on the alternating T-maze in CAG140 KI mice.
Highlights.
-
-
GA improves disease phenotypes in N171-82Q transgenic and CAG140 knock-in mice.
-
-
Beneficial effects of GA were associated with elevated Bdnf-I and Bdnf-IV expression and reduced IL4 and IL12 levels in the brains of HD mice.
-
-
The effects of GA on BDNF and cytokine levels was also detected in plasma from drug treated animals.
-
-
These findings support the existence of relevant communication between central and peripheral tissues.
Acknowledgments
Funding
This study was funded by grants from the National Institutes of Health (NS087986 to E.A.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures of all authors
None.
References
- Achiron A, Feldman A, Gurevich M. Molecular profiling of glatiramer acetate early treatment effects in multiple sclerosis. Dis Markers. 2009;27:63–73. doi: 10.3233/DMA-2009-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005;102:19045–50. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R, Kayhan B, Eilam R, Sela M, Arnon R. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc Natl Acad Sci U S A. 2003;100:14157–62. doi: 10.1073/pnas.2336171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–35. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay D, Vachapova V, Shihman B, Miler A, Karni A. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol. 2005;167:215–8. doi: 10.1016/j.jneuroim.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Bakshi S, Chalifa-Caspi V, Plaschkes I, Perevozkin I, Gurevich M, Schwartz R. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets. 2013;17:351–62. doi: 10.1517/14728222.2013.778829. [DOI] [PubMed] [Google Scholar]
- Begum-Haque S, Sharma A, Christy M, Lentini T, Ochoa-Reparaz J, Fayed IF, Mielcarz D, Haque A, Kasper LH. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;219:47–53. doi: 10.1016/j.jneuroim.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Tabrizi SJ. Harnessing immune alterations in neurodegenerative diseases. Neuron. 2009;64:21–4. doi: 10.1016/j.neuron.2009.09.034. [DOI] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205:1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco Y, Moral EA, Costa M, Gomez-Choco M, Torres-Peraza JF, Alonso-Magdalena L, Alberch J, Jaraquemada D, Arbizu T, Graus F, Saiz A. Effect of glatiramer acetate (Copaxone) on the immunophenotypic and cytokine profile and BDNF production in multiple sclerosis: a longitudinal study. Neurosci Lett. 2006;406:270–5. doi: 10.1016/j.neulet.2006.07.043. [DOI] [PubMed] [Google Scholar]
- Chang KH, Wu YR, Chen YC, Chen CM. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immun. 2015;44:121–7. doi: 10.1016/j.bbi.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Chen M, Valenzuela RM, Dhib-Jalbut S. Glatiramer acetate-reactive T cells produce brain-derived neurotrophic factor. J Neurol Sci. 2003;215:37–44. doi: 10.1016/s0022-510x(03)00177-1. [DOI] [PubMed] [Google Scholar]
- Corey-Bloom J, Jia H, Aikin AM, Thomas EA. Disease Modifying Potential of Glatiramer Acetate in Huntington’s Disease. J Huntingtons Dis. 2014;3:311–6. doi: 10.3233/JHD-140110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Bjorkqvist M, Petersen A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP, Leavitt BR, Keir G, Ward M, Tabrizi SJ. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–40. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. 2003;100:2911–6. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrichmann G, Reick C, Saft C, Linker RA. The role of the immune system in Huntington’s disease. Clin Dev Immunol. 2013;2013:541259. doi: 10.1155/2013/541259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–61. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J Neurochem. 2008;105:369–79. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampa C, Montagna E, Dato C, Melone MA, Bernardi G, Fusco FR. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS ONE. 2013;8:e64037. doi: 10.1371/journal.pone.0064037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group, Huntington’s Disease Collaborative Research. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience. 2008;157:280–95. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Zhu C, Medvedeva V, Lerner RP, Patassini S, Franich NR, Maiti P, Frautschy SA, Zeitlin S, Levine MS, Chesselet MF. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol Neurodegener. 2012;7:12. doi: 10.1186/1750-1326-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Morris CD, Williams RM, Loring JF, Thomas EA. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc Natl Acad Sci U S A. 2015;112:E56–64. doi: 10.1073/pnas.1415195112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang Y, Morris CD, Jacques V, Gottesfeld JM, Rusche JR, Thomas EA. The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s Disease Mice. PLoS ONE. 2016;11:e0152498. doi: 10.1371/journal.pone.0152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KP. Glatiramer acetate for treatment of relapsing-remitting multiple sclerosis. Expert Rev Neurother. 2012;12:371–84. doi: 10.1586/ern.12.25. [DOI] [PubMed] [Google Scholar]
- Klein AB, Williamson R, Santini MA, Clemmensen C, Ettrup A, Rios M, Knudsen GM, Aznar S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol. 2011;14:347–53. doi: 10.1017/S1461145710000738. [DOI] [PubMed] [Google Scholar]
- Mandel AL, Ozdener H, Utermohlen V. Brain-derived neurotrophic factor in human saliva: ELISA optimization and biological correlates. J Immunoassay Immunochem. 2011;32:18–30. doi: 10.1080/15321819.2011.538625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Moore S, Khalaj AJ, Patel R, Yoon J, Ichwan D, Hayardeny L, Tiwari-Woodruff SK. Restoration of axon conduction and motor deficits by therapeutic treatment with glatiramer acetate. J Neurosci Res. 2014;92:1621–36. doi: 10.1002/jnr.23440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus O, Farina C, Wekerle H, Hohlfeld R. Mechanisms of action of glatiramer acetate in multiple sclerosis. Neurology. 2001;56:702–8. doi: 10.1212/wnl.56.6.702. [DOI] [PubMed] [Google Scholar]
- Poittevin M, Deroide N, Azibani F, Delcayre C, Giannesini C, Levy BI, Pocard M, Kubis N. Glatiramer Acetate administration does not reduce damage after cerebral ischemia in mice. J Neuroimmunol. 2013;254:55–62. doi: 10.1016/j.jneuroim.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Reick C, Ellrichmann G, Tsai T, Lee DH, Wiese S, Gold R, Saft C, Linker RA. Expression of brain-derived neurotrophic factor in astrocytes – Beneficial effects of glatiramer acetate in the R6/2 and YAC128 mouse models of Huntington’s disease. Exp Neurol. 2016;285:12–23. doi: 10.1016/j.expneurol.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Moller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009;20:1098–103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- Smirnov I, Walsh JT, Kipnis J. Chronic mild stress eliminates the neuroprotective effect of Copaxone after CNS injury. Brain Behav Immun. 2013;31:177–82. doi: 10.1016/j.bbi.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Di Lena P, Schaffer L, Head SR, Baldi P, Thomas EA. Genome-wide identification of Bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS ONE. 2011;6:e23691. doi: 10.1371/journal.pone.0023691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum D, Aharoni R, Klinger E, Kreitman R, Raymond E, Malley A, Shofti R, Sela M, Arnon R. Oral glatiramer acetate in experimental autoimmune encephalomyelitis: clinical and immunological studies. Ann N Y Acad Sci. 2004;1029:239–49. doi: 10.1196/annals.1309.055. [DOI] [PubMed] [Google Scholar]
- Tselis A, Khan O, Lisak RP. Glatiramer acetate in the treatment of multiple sclerosis. Neuropsychiatr Dis Treat. 2007;3:259–67. doi: 10.2147/nedt.2007.3.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708–18. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li M, Drozda M, Chen M, Ren S, Sanchez RO Mejia, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM. Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem. 2003;87:101–6. doi: 10.1046/j.1471-4159.2003.01980.x. [DOI] [PubMed] [Google Scholar]
- Ziemssen T, Kumpfel T, Klinkert WE, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain. 2002;125:2381–91. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–8. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences used for real-time PCR analysis.
Fig. S1. The effects of chronic GA administration on body weight of WT and CAG140 KI.
Fig. S2. The effects of chronic GA treatment on the alternating T-maze in CAG140 KI mice.