

Summary
Mitophagy is thought to be a critical mitochondrial quality control mechanism in neurons and has been extensively studied in neurological disorders such as Parkinson's disease. However, little is known about how mitochondria are maintained in the lengthy neuronal axons in the context of physiological aging. Here, we utilized the unique Drosophila wing nerve model and in vivo imaging to rigorously profile changes in axonal mitochondria during aging. We revealed that mitochondria became fragmented and accumulated in aged axons. However, lack of Pink1 or Parkin did not lead to the accumulation of axonal mitochondria or axonal degeneration. Further, unlike in in vitro cultured neurons, we found that mitophagy rarely occurred in intact axons in vivo, even in aged animals. Furthermore, blocking overall mitophagy by knockdown of the core autophagy genes Atg12 or Atg17 had little effect on the turnover of axonal mitochondria or axonal integrity, suggesting that mitophagy is not required for axonal maintenance; this is regardless of whether the mitophagy is PINK1‐Parkin dependent or independent. In contrast, downregulation of mitochondrial fission–fusion genes caused age‐dependent axonal degeneration. Moreover, Opa1 expression in the fly head was significantly decreased with age, which may underlie the accumulation of fragmented mitochondria in aged axons. Finally, we showed that adult‐onset, neuronal downregulation of the fission–fusion, but not mitophagy genes, dramatically accelerated features of aging. We propose that axonal mitochondria are maintained independently of mitophagy and that mitophagy‐independent mechanisms such as fission–fusion may be central to the maintenance of axonal mitochondria and neural integrity during normal aging.
Keywords: aging, axonal integrity, fission–fusion, in vivo imaging, mitochondria, mitophagy
Introduction
Healthy mitochondria are critical for maintaining normal bioenergetically demanding activities of neurons. Such energy demand in neuronal axons is likely to be especially extreme due to the activities such as synaptic transmission, generating and propagating action potentials, and transporting biomaterials over a long distance. Deleterious mitochondrial changes such as a decrease in mitochondrial integrity and function are associated with aging and neurodegenerative diseases (Bratic & Larsson, 2013; López‐Otín et al., 2013). Mitophagy is an important mitochondrial quality control mechanism that selectively eliminates damaged mitochondria by autophagy (Wang and Klionsky, 2014; Wei et al., 2015). The process is regulated by the PTEN‐induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin (Pickrell & Youle, 2015), whose mutations can cause familial forms of Parkinson's disease (PD).
It is widely assumed that damaged mitochondria accumulate in neurons if mitophagy fails, which causes PD in PINK1‐Parkin deficiency. For axons, the studies based on in vitro cultured neurons have been controversial, especially regarding whether mitophagy occurs locally in axons. PINK1‐Parkin‐mediated mitophagy was reported to occur in neurons but restricted to the somatodendritic regions (Seibler et al., 2011; Cai et al., 2012). However, Maday et al. (2012); Maday & Holzbaur (2014) showed that autophagosomes were generated at neurite tips, which fused with axonal mitochondria and together were transported to the neuronal soma for lysosome‐mediated degradation. By contrast, another study reported that autophagosomes were readily available in axons and were recruited to damaged mitochondria in a PINK1‐Parkin‐dependent manner followed by local turnover in axons (Ashrafi et al., 2014). Other than by inference, evidence in vivo supporting an essential role of PINK1‐Parkin in regulating mitochondrial quality control in axons during aging is lacking. As axonal mitochondria may behave differently in neurons cultured in vitro (Sung et al., 2016), it is important to elucidate how mitochondria are maintained in the lengthy axons under physiological conditions in vivo.
Mitochondria are dynamic organelles that undergo constant fission and fusion. The fission–fusion balance is critical for determining mitochondrial morphology, distribution, abundance, and function (Chan, 2012), and allows mitochondria to quickly adapt to different metabolic needs in different subcellular compartments or upon different energetic states (Wai & Langer, 2016). Moreover, fission–fusion is also an important means to control mitochondrial quality, via removing the damaged portion of a mitochondrion by fission or via refreshing a dysfunctional mitochondrion by fusion to a healthy one (Schrepfer & Scorrano, 2016). The key regulators of mitochondrial dynamics are an evolutionarily conserved family of dynamin‐related GTPases (Chan, 2012). Specifically, mitofusin‐1 (Mfn1) and ‐2 (Mfn2) mediate fusion of the mitochondrial outer membrane and optic atrophy 1 (Opa1) mediates that of the inner membrane; fission is mainly controlled by dynamin‐related protein 1 (Drp1) (Chan, 2012; Bertholet et al., 2016). However, the role of mitochondrial fission–fusion, or its relative contribution compared to that of mitophagy, in axonal mitochondrial quality control in vivo is unknown.
Dissecting the mechanism maintaining axonal mitochondria in animals during their lifetime has been challenging, which is at least in part due to the limitation of classical sample fixation and staining processes that grossly alter mitochondrial morphology. In this regard, the Drosophila wing model that we developed provides a unique system (Fang et al., 2012, 2013; Fang & Bonini, 2015) that allows direct visualization of the morphology of axonal mitochondria under near physiological conditions in vivo. Using this system, we critically evaluate the role of PINK1‐Parkin‐dependent and PINK1‐Parkin‐independent mitophagy as well as mitochondrial fission–fusion in maintaining axonal integrity during aging. We find that fragmented mitochondria accumulate in aged axons, which is consistent with the general idea that mitochondrial quality and function decline with age (Bratic & Larsson, 2013; López‐Otín et al., 2013). However, our study reveals a striking scarcity and dispensability of axonal mitophagy in neuronal aging, whether PINK1‐Parkin dependent or independent. Instead, our findings raise the possibility that mitophagy‐independent mechanisms such as fission–fusion play a central role in the maintenance of axonal mitochondria during normal aging.
Results
In vivo neuroimaging reveals morphological alterations of axonal mitochondria during aging
To characterize the dynamic morphological changes in neuronal mitochondria during aging, we used the GAL4/UAS system to label neuronal mitochondria with mitochondria‐localized GFP (mitoGFP) in the Drosophila wing nerve. The flies were aged to different time points and the axons were imaged in live fly wings without fixation by confocal microscopy. Mitochondria in the neuronal soma and axons of the costal wing nerve (the boxed area in Fig. 1A) were directly visualized in vivo (Fig. 1B). The axonal mitochondria displayed a mixed population of morphology, from round or almost round (1.0 ≤ length/width < 1.5), intermediate (1.5 ≤ length/width < 2.0), tubular (2.0 ≤ length/width < 5.0), to hyperfused (length/width ≥ 5) (Fig. 1C,D). We quantified the length and width of each mitochondrion in the costal wing nerve, and calculated the average length/width ratio at different ages in Fig. 1E. It is evident that both the average length/width ratio and the proportion of long mitochondria (tubular and hyperfused) were decreased, whereas those of short mitochondria (intermediate and round) were increased in aged flies (D30 and D50). Concurrently, the number of axonal mitochondria was increased with age (Fig. 1F). These data suggest that mitochondria became fragmented and accumulated in aged axons.
Figure 1.
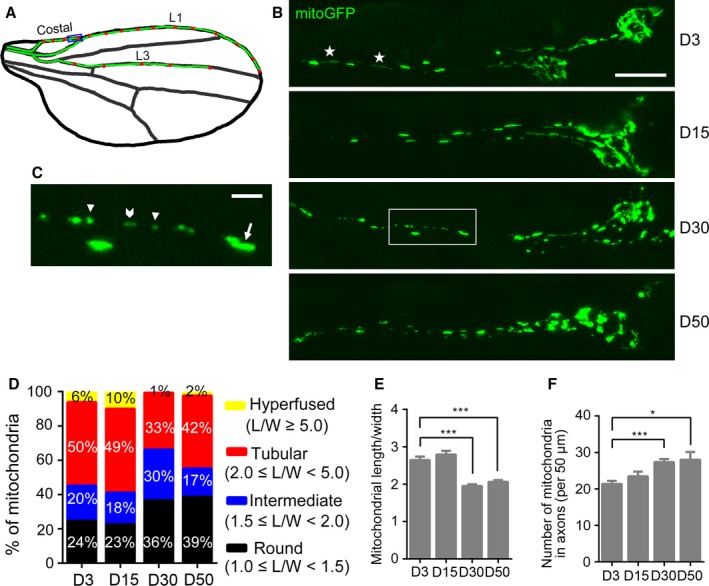
In vivo neuroimaging reveals the accumulation of fragmented mitochondria in aged axons. (A) A cartoon illustration of the Drosophila wing. The green line highlights the wing nerve in the costal, L1, and L3 wing veins, and the red dots denote the neuronal soma. (B, C) Representative confocal images of mitochondria in the axons of the Drosophila costal wing nerve (the blue box in A) at days D3, D15, D30, and D50. Mitochondria are labeled by mitoGFP using a dpr‐Gal4 driver. (C) A higher magnification of the area in the white box in B. Arrowheads: short; chevron: intermediate; arrow: tubular; and asterisks: hyperfused axonal mitochondria. (D) The axonal mitochondria imaged in (B) are classified into four groups based on the length/width ratio as specified. (E, F) The average length/width ratio (E) and number (F) of axonal mitochondria at indicated ages. Data are shown as mean ± SEM, n = 9–16 wings per group. *P < 0.05, ***P < 0.001. Scale bars, 10 μm in (B) and 2 μm in (C).
The PINK1‐Parkin pathway is dispensable for axonal maintenance in both sensory and motor neurons in adult Drosophila
The accumulation of fragmented mitochondria in axons might be a result of declined mitochondrial turnover during aging. Damaged axonal mitochondria in cultured neurons could be cleared by local mitophagy, which requires the function of PINK1 and Parkin (Ashrafi et al., 2014). Hence, we first investigated the contribution of the PINK1‐Parkin pathway to the age‐associated accumulation of axonal mitochondria in vivo. The Pink1 loss‐of‐function (LOF)‐mutant flies, Pink1 B9 and Pink1 5, exhibited collapsed thorax and abnormal wing posture (Fig. S1A,B, Supporting information), which was consistent with the mitochondrial defects in muscles as previously reported (Clark et al., 2006; Park et al., 2006). Unlike in muscles, the morphology of axonal mitochondria in the wing axons of Pink1 B9 and Pink1 5 flies looked mostly normal (Fig. 2A). Neither the average mitochondrial length/width ratio nor the number of mitochondria showed a significant difference between Pink1 mutants and w 1118 control flies (Fig. 2B,C). Moreover, the axonal integrity of the Pink1 mutants was well maintained and no axonal degeneration was observed during aging (Fig. 2D).
Figure 2.
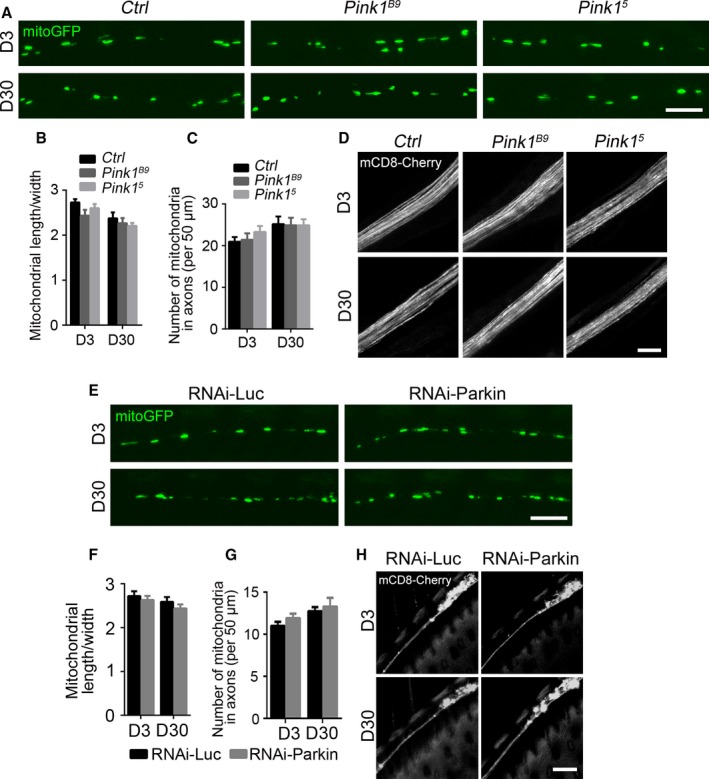
Flies with Pink1‐Parkin deficiency exhibit normal mitochondrial morphology and intact axonal integrity. (A) Axonal mitochondria labeled by mitoGFP in the costal wing nerve of control (w 1118), Pink1 B9 , and Pink1 5 flies at D3 or D30. (B‐C) Quantification of length/width ratio (B) and the number (C) of the axonal mitochondria. (D) Representative in vivo images of distal axons of the L1 wing nerve of control (w 1118), Pink1 B9 , and Pink1 5 flies at D3 or D30. (E) Representative images of axonal mitochondria of RNAi‐Luc (control) and RNAi‐Parkin flies at D3 or D30, the average length/width ratio, and the number of axonal mitochondria are quantified in (F, G). Data are shown as mean ± SEM, n = 8–10 wings per group. Two‐way ANOVA shows no statistically significant difference between any of the mutant or RNAi groups and their respective controls, whereas the mitochondrial changes with age are significant (P < 0.05) except for (F). (H) Representative in vivo images of neuronal soma and proximal axons of the L1 nerve of indicated genotypes at D3 and D30. No axonal degeneration is detected. Scale bars, 5 μm in (A) and (E), and 10 μm in (D) and (H).
Similarly, despite an age‐associated decrease in Parkin expression (Fig. S1C, Supporting information), downregulation of Parkin in the wing nerve did not result in the accumulation of fragmented mitochondria (Fig. 2E–G) or axonal degeneration (Fig. 2H). The knockdown (KD) efficiency was examined by quantitative RT–PCR (qPCR) (Fig. S1D, Supporting information). In contrast, downregulation of Parkin or Pink1 in muscles using the same transgenic RNAi strains caused muscle degeneration and collapsed thoraces (Fig. S1E, Supporting information), confirming their essential role in the muscle cells. As the fly wing nerve consists of only sensory neurons (Nakamura et al., 2002), we asked whether the lack of axonal degeneration in PINK1‐Parkin deficiency was sensory neuron‐specific. We then examined the axons of the motor neurons in the adult Drosophila leg (Fig. S2A, Supporting information). Similar to the sensory wing nerve, the motor neurons of the Pink1 LOF mutants showed no axonal degeneration, even in aged flies (Fig. S2B, Supporting information).
As overexpression of Parkin in adult neurons was shown to enhance mitochondrial activity and extend longevity in Drosophila (Rana et al., 2013), we were keen to test whether upregulation of PINK‐Parkin could promote turnover of fragmented mitochondria and improve axonal maintenance during aging. Unexpectedly, overexpression of Pink1 or Parkin in the wing neurons led to extremely fragmented mitochondria (Fig. S3A,B, Supporting information). Expression of the enzymatically inactive mutants, Pink1 L464P (Song et al., 2013), Parkin T240R , or Parkin R275W (Lee et al., 2010; Kim et al., 2013) did not show such effect, indicating that the impact of upregulating PINK1‐Parkin on axonal mitochondrial morphology requires their enzymatic activity. Furthermore, in striking contrast to LOF and KD of Pink1 and Parkin (Fig. 2D,H), the wing nerve of Pink1‐ or Parkin‐overexpressing flies degenerated rapidly with age (Fig. S3C,E, Supporting information). This is likely due to a mitophagy‐independent function of Parkin in regulating Marf, the Drosophila homologue of mammalian mitofusins that promote mitochondria fusion (Chan, 2012; Schrepfer & Scorrano, 2016; Fig. S3A, Supporting information). Parkin was known to ubiquitinate Marf for proteasome‐mediated degradation and resulted in shortened mitochondria in insect cells (Poole et al., 2010; Ziviani et al., 2010). Consistently, we found that both mitochondrial morphology (Fig. S3A,B, Supporting information) and axonal integrity (Fig. S3D,E, Supporting information) of Parkin‐overexpressing neurons were significantly improved by co‐expression of Marf. This result is in line with the idea that mitophagy‐independent PINK1‐Parkin functions may cause or contribute to the pathogenesis of PD (Pryde et al., 2016).
Mitophagy rarely occurs in intact axons in vivo during normal aging
In addition to the PINK1‐Parkin‐dependent mitophagy, mitophagy can take place independently of PINK1‐Parkin (Wei et al., 2015). Hence, the accumulation of axonal mitochondria might also be due to an age‐associated decline of autophagic clearance of damaged mitochondria that is independent of PINK‐Parkin. To test this possibility, we generated the fly strains to express mCherry‐labeled Atg8a (an homologue of mammalian LC3) in the wing nerve to visualize and investigate autophagy by in vivo imaging. Induction of autophagy leads to the cleavage and translocation of Atg8a to the autophagosome membrane, which transforms mCherry‐Atg8a signal from diffuse to punctate. Formation of Atg8a/LC3 puncta is widely used as an autophagosome marker (Klionsky et al., 2016). As illustrated in Fig. 3A, in the neuronal soma, autophagosomes (mCherry‐Atg8a puncta) were frequently observed in both young (D3) and aged (D30) flies. The neuronal soma maintained relatively constant basal levels of autophagy during aging, as neither the number nor the size of mCherry‐Atg8a puncta was significantly changed in the soma during aging (Fig. 3B,C). In contrast, autophagosome puncta were sparse in young axons. With age, an increasing number of mCherry‐Atg8a puncta were detected in axons (Fig. 3A,F); the size of axonal autophagosomes showed a trend to increase although this was not statistically significant (Fig. 3G). These results suggested an increase in autophagy induction and/or a decrease in autophagic flux in axons during aging.
Figure 3.
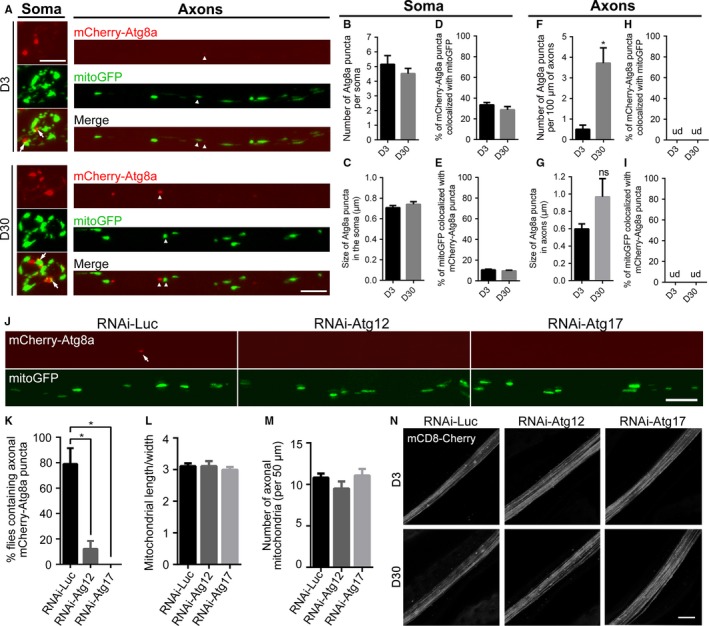
Mitophagy is both rare and dispensable for intact axons in vivo during aging. (A–I) Axonal mitophagy rarely occurs in vivo under physiological conditions. (A) Representative images showing neuronal soma (left) and axons (right) with autophagosomes (AP) labeled by mCherry‐Atg8a and mitochondria labeled by mitoGFP. mCherry‐Atg8a puncta are commonly seen in the soma of both young and aged flies; axonal AP are rarely observed in young flies but significantly increase with age. Arrows, colocalization of mitoGFP and mCherry‐Atg8a puncta in the soma; arrowheads, nearby but not colocalized mitochondria and autophagosome in axons. The number and size of somatic AP (B‐C) and axonal AP (F‐G) as well as colocalization of mitoGFP and mCherry‐Atg8a in the soma (D, E) are quantified. Colocalization of mitochondria and autophagosomes is almost never detected in axons in vivo even in aged animals; hence, the colocalization occurrence is essentially zero in (H, I). Data are shown as mean ± SEM, n = 7–12 wings per group. *P < 0.05; ns, not significant; ud, undetected. (J–N) Blocking axonal autophagy has little effect on the maintenance of axonal mitochondria or axonal integrity in vivo. (J) Representative images showing AP (mCherry‐Atg8a, arrow) and mitochondria (mitoGFP) in the costal wing axons of RNAi‐Luc, RNAi‐Atg12, and RNAi‐Atg17 flies. (K) The percentage of flies containing axonal AP in the entire wing nerve. (L, M) Quantification of the length/width ratio (L) and the number (M) of the axonal mitochondria. Data are shown as mean ± SEM, n = 11–14 wings per group. *P < 0.05. (N) Representative images of the distal wing nerve of the indicated genotypes at D3 and D30. No sign of axonal degeneration is observed in the RNAi‐Atg12 or RNAi‐Atg17 flies. Scale bars, 5 μm in (A) and (J), and 10 μm in (N).
As the numbers of fragmented mitochondria and autophagosomes were both increased in aged axons, we asked whether this concurrence represented an increase in mitophagy in axons. A previous study showed that depolarized axonal mitochondria became colocalized with autophagosomes, leading to local mitophagy in axons (Ashrafi et al., 2014). Hence, we carefully examined colocalization of mitoGFP and mCherry‐Atg8a puncta in the wing nerve. Unexpectedly, however, we found mitoGFP and mCherry‐Atg8a puncta were essentially not colocalized in vivo, even in aged flies (Fig. 3A, arrowheads). Quantification showed that neither the proportion of mCherry‐Atg8a puncta containing mitochondria nor the percentage of mitochondria colocalized with mCherry‐Atg8a changed significantly during aging (Fig. 3H–I). Of note, among over 110 fly wings examined in this study, we only detected two individual cases of a mitochondrion colocalized with mCherry‐Atg8a puncta in the entire wing nerve, which is composed of more than 200 axons (Fang et al., 2013). As such, the occurrence of axonal mitophagy under physiological conditions in vivo is <1 of 10 000 axons, suggesting that in axons virtually no mitophagy occurs, regardless of age. Similarly, McWilliams et al. (2016) reported that the majority of mitochondrial turnover occurred in the soma of Purkinje neurons with minimal mitophagy observed in mouse axons in vivo.
Blocking autophagy in the Drosophila wing nerve has a minimal effect on mitochondrial turnover or axonal integrity during aging
Although axonal mitophagy rarely occurred in vivo, it did not exclude the possibility that occasional axonal mitophagy played an essential role. As both PINK1‐Parkin‐dependent and PINK1‐Parkin‐independent mitophagy pathways rely on the autophagy machinery for the final clearance of mitochondria, we sought to examine the effect of blocking autophagy on mitochondrial turnover and axonal integrity during aging.
Autophagy is controlled by a family of evolutionarily conserved ATG genes. One of the core ATG genes is Atg12, required for the formation of autophagosomes in promoting the conjugation of ATG8 protein to the lipid phosphatidylethanolamine (Ktistakis & Tooze, 2016). As the aging context limited the use of the null mutants in this study, we could only assess the impact of these genes in a partial compromise situation. Nevertheless, in the RNAi‐Atg12 flies, both the number and size of mCherry‐Atg8a puncta in the soma were significantly decreased; meanwhile, diffused mCherry‐Atg8a signal was increased (Fig. S4A–C, Supporting information). The KD efficiency of RNAi‐Atg12 and RNAi‐Atg17 was examined in Fig. S4D (Supporting information). Importantly, despite the low basal level of autophagy in axons (Fig. 3J, control flies, RNAi‐luc), Atg12 KD caused a further decrease in axonal autophagy (Fig. 3J, RNAi‐Atg12), which was seen as a drastic reduction in flies with detectable mCherry‐Atg8a puncta in axons (Fig. 3K, from ~80% in the control to only ~10% in the RNAi‐Atg12). As such, axonal autophagy was inhibited in the wing nerve in the majority of RNAi‐Atg12 flies. Nonetheless, the maintenance of axonal mitochondria was not impaired, as RNAi‐Atg12 flies showed no difference in either the average length/width ratio or the density of axonal mitochondria (Fig. 3J,L,M).
The lack of a deleterious effect on axonal mitochondrial turnover was further confirmed by manipulation of another core ATG gene Atg17 (required for the autophagosome assembly, Ktistakis & Tooze, 2016) in the wing nerve (Fig. 3J–M). In nearly 100% of RNAi‐Atg17 flies, an axonal autophagosome was never found in over two hundred axons examined in the entire wing nerve (Fig. 3K). Nevertheless, no degeneration of a single axon was observed, even in aged RNAi‐Atg17 flies (Fig. 3N, D30). These results further support the conclusion that axonal mitochondria do not rely on mitophagy for their maintenance during normal aging.
Mitophagy‐independent mechanisms may regulate mitochondrial maintenance and axonal integrity during aging
Neuronal integrity and functions rely on mitochondria for ATP production. Because we revealed that mitophagy was dispensable for axons under physiological conditions in vivo, neurons must use other means to maintain axonal mitochondria and axonal integrity. Mitochondrial quality and abundance is also controlled by mitochondrial transport and fission–fusion (Saxton & Hollenbeck, 2012; Schrepfer & Scorrano, 2016). Indeed, we and others previously showed that proper mitochondrial transport and distribution are essential for the integrity of Drosophila axons (Avery et al., 2012; Fang et al., 2012). As fission–fusion also regulates the morphology and turnover of mitochondria (Chan, 2012; Schrepfer & Scorrano, 2016), we next investigated the possibility that the accumulation of fragmented mitochondria in aged axons resulted from an age‐associated change in the fission–fusion dynamics. Interestingly, we found that Opa1 expression in the fly head became reduced with age (Fig. 4A, D30). However, as a similar expression decrease was detected with Parkin (Fig. S1C, Supporting information), but KD of Parkin did not impair mitochondrial turnover or axonal integrity (Fig. 2E–H), it was yet to be proven that fission–fusion was indeed essential for axonal maintenance during aging.
Figure 4.
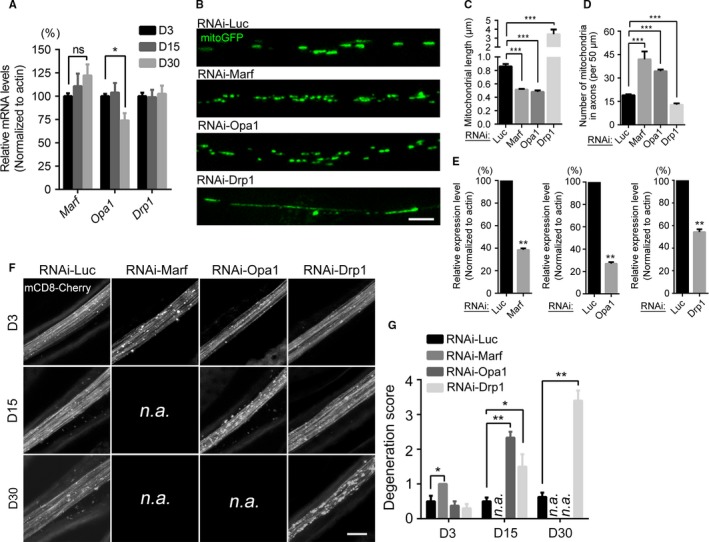
The fission–fusion balance is critically required for the maintenance of axonal mitochondria and axonal integrity during aging. (A) The relative mRNA levels of mitochondrial fission–fusion genes in the fly head at the indicated time points. Opa1 expression is significantly decreased at D30. (B–D) Representative images of axonal mitochondria of indicated genotypes, and the axonal mitochondrial length and number are quantified in (C) and (D), respectively. Data are shown as mean ± SEM, n = 8–14 wings per group except for RNAi‐Marf, of which only three adult flies were available for imaging due to early lethality. (E) The knockdown efficiency of the above RNAi lines was examined by qPCR. (F, G) Age‐dependent axonal degeneration by downregulation of the fission–fusion genes in the adult fly wing nerve. The representative images of the L1 wing nerve of indicated genotypes at D3, D5, and D30 are shown in (F) and the degeneration scores are quantified in (G) as described in the Methods. Data are shown as mean ± SEM, n = 6–8 wings per genotype per time point except for RNAi‐Marf (n = 3). n.a., not available; because KD of Marf or Opa1 by the dpr‐Gal4 driver (which is highly expressed in the adult wing neurons that facilitates the in vivo imaging but is also expressed in neurons elsewhere) causes early lethality in young adults, the imaging data at later time points for RNAi‐Marf and RNAi‐Opa1 flies are unavailable. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Scale bars, 2 μm in (B) and 10 μm in (F).
As shown in Fig. 4B–D, downregulation of Marf or Opa1 significantly decreased, whereas KD of Drp1 increased, the length of axonal mitochondria in the wing nerve. We then examined the impact on axonal integrity. RNAi‐Marf flies showed remarkable axonal degeneration as early as D3, RNAi‐Opa1 flies at D15, and RNAi‐Drp1 flies at D30 (Fig. 4E). Of note, because KD of Marf or Opa1 using the dpr‐Gal4 driver (which is highly expressed in the adult wing neurons to facilitate in vivo imaging, but is also expressed in neurons elsewhere) caused early lethality of young adults, the axonal imaging data at later time points for RNAi‐Marf (D15 and D30) and RNAi‐Opa1 flies (D30) were unavailable. Nevertheless, downregulation of the fission–fusion genes by genetic manipulations caused age‐dependent, progressive axonal degeneration. This result suggests that the change in their expression during normal aging should not be overtly large; otherwise, aging itself would cause spontaneous axonal degeneration. Indeed, the small (~30%) but significant reduction in Opa1 expression in aged neurons (Fig. 4A) is consistent with the moderate alteration of mitochondrial morphology in axons during normal aging.
Inhibition of fission–fusion but not mitophagy in adult neurons significantly accelerates aging in Drosophila
It was possible that disruption of the PINK1‐Parkin pathway or the autophagy machinery led to functional deficits before morphological changes were detectable. The climbing assay is widely used to evaluate the function of neurons and muscles in Drosophila models of neurodegenerative diseases including PD. As the function of mitophagy/autophagy in neural development might compromise the analysis, we used an inducible, pan‐neuronal driver elav‐GeneSwitch (elav‐GS, Osterwalder et al., 2001) and induced the expression of the RNAi transgenes at adulthood only. As shown in Fig. 5A, no significant difference in the climbing ability was found in RNAi‐Pink1 or RNAi‐Parkin flies. In contrast, KD of Pink1 or Parkin in muscles (Mef2‐Gal4) caused age‐dependent climbing defects as early as D24 in RNAi‐Parkin and D36 in RNAi‐Pink1 flies (Fig. 5B). In addition, KD of the core ATG gene Atg12 or Atg17 in adult neurons also led to accelerated decline of climbing ability (Fig. 5C). This was even more dramatic when the flies were raised at an elevated environmental temperature of 29 °C (Fig. S4E, Supporting information). Heat stress is known to disturb the proteostasis and require the function of autophagy to maintain the homeostasis (Dokladny et al., 2015). Thus, although the autophagy machinery is dispensable for the turnover of axonal mitochondria (Fig. 3J), these results confirmed the essential role of autophagy in neuronal aging likely due to the important functions in regulating the proteostasis in neurons.
Figure 5.
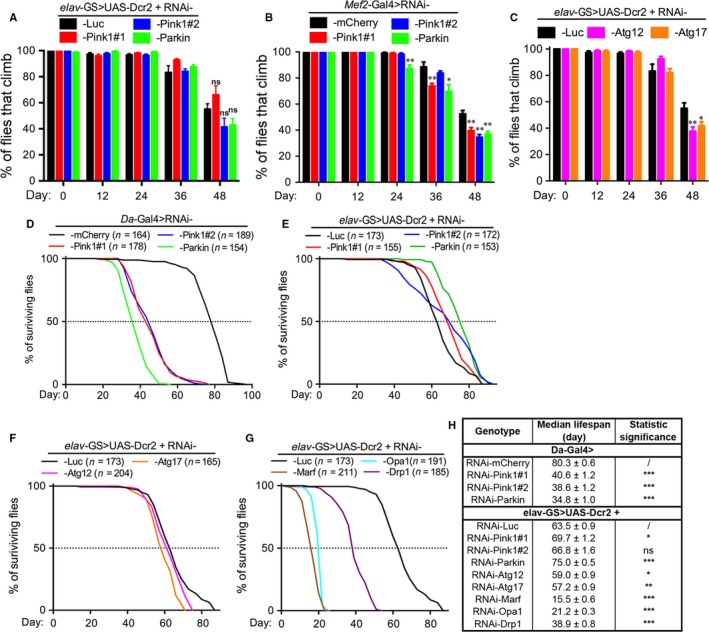
Inhibition of fission–fusion but not mitophagy in adult neurons significantly accelerates aging. (A‐C) Climbing assays of flies with (A) Pink1 or Parkin downregulated in adult neurons by elav‐ GS, (B) in muscles by Mef2‐Gal4 (B), or (C) Atg12 or Atg17 downregulated in adult neurons. The locomotive ability was assessed as the average percentage of flies climbing over 5 cm within 15 s. Data are shown as mean ± SEM, n = 20 flies per vial and 6–8 vials each group. *P < 0.05, **P < 0.01; ns, not significant. (D) Knockdown of Pink1 or Parkin in all cells by the Da‐Gal4 driver dramatically reduces the longevity. (E‐G) Lifespan assays of the flies with adult‐onset, neuronal downregulation (elav‐GS) of Pink1 or Parkin (E), Atg12 or Atg17 (F), or Marf, Opa1, or Drp1 (G). A copy of UAS‐Dcr2 was co‐expressed to boost the RNAi knockdown efficiency in neurons (Ni et al., 2008), but was not needed with the Mef2‐Gal4 or Da‐Gal4 drivers, in which cases the RNAi‐mCherry was used as a control. n, the number of flies tested for each genotype is indicated. (H) Summary of the median lifespans, shown as mean ± SEM, *P < 0.01, **P < 0.001, ***P < 0.0001.
Finally, as an attempt to compare the overall involvement of mitophagy and fission–fusion in neuronal aging, we examined the effect of their downregulation on longevity. Similar to their LOF mutants (Greene et al., 2003; Cha et al., 2005; Clark et al., 2006; Park et al., 2006), ubiquitous KD of Pink1 or Parkin in all cells using a Daughterless‐Gal4 (Da‐Gal4) driver dramatically shortened lifespan (Fig. 5D,H). To our great surprise, however, the adult‐onset, neuronal downregulation of Pink1 or Parkin (elav‐GS) did not decrease the longevity; instead, the RNAi‐Parkin and one of the RNAi‐Pink1 lines showed a puzzling extension of the lifespan (Fig. 5E,H). In addition, KD of the core autophagy genes Atg12 or Atg17 in adult neurons led to a modest reduction in the lifespan (Fig. 5F,H). In contrast, downregulation of the fission–fusion genes in adult neurons all dramatically reduced the longevity (Fig. 5G,H). Of note, disruption of fission–fusion consequently leads to impaired mitochondrial functions (Pich et al., 2005), which may add to the deleterious effects on axonal integrity and longevity. Nevertheless, our data demonstrate that mitophagy is dispensable for axonal integrity and physiological aging in neurons, whereas mitophagy‐independent mechanisms such as fission–fusion play an essential role in the maintenance of axonal mitochondria and neural integrity (Fig. 6).
Figure 6.
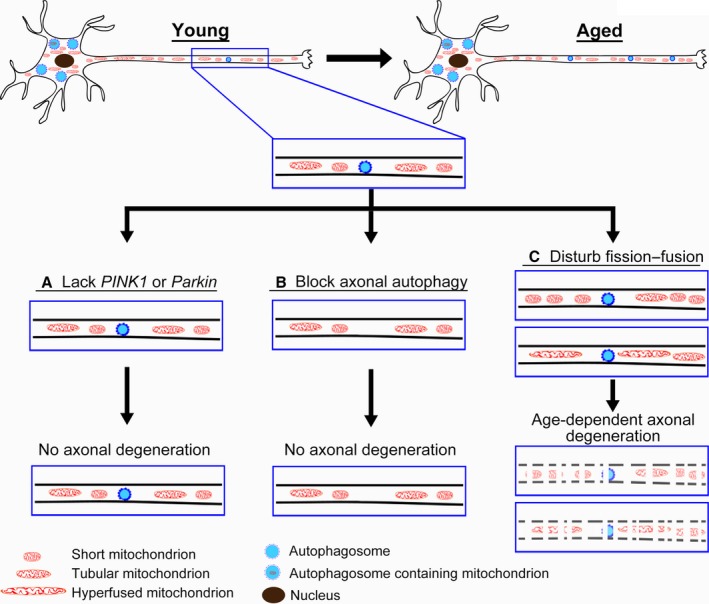
A schematic model of differential requirements of mitophagy and fission–fusion in the maintenance of axonal mitochondria and axonal integrity during aging. During aging, fragmented mitochondria accumulate in axons and the basal levels of axonal autophagy increase. However, mitophagy (evident by the colocalization of mitochondria and autophagosomes) is predominantly observed in the soma but rarely in axons in vivo, even in aged neurons. Further, disrupting the PINK1‐Parkin pathway (A) or blocking axonal autophagy by knockdown of the core autophagy genes Atg12 or Atg17 (B) does not impair mitochondrial turnover or axonal integrity. Instead, knockdown of the fission–fusion genes Opa1 Marf or Drp1 (C) causes profound changes to the morphology and abundance of axonal mitochondria, which leads to age‐dependent, progressive axonal degeneration and significantly shortens lifespan. Together, our study indicates that mitophagy is dispensable for axonal maintenance in vivo, whereas mitophagy‐independent mechanisms such as mitochondrial fission–fusion may play an essential and central role in the maintenance of axonal mitochondria and axonal integrity during normal aging.
Discussion
Neuronal aging is known to be associated with deleterious changes in mitochondria including a decrease in mitochondrial biogenesis, the respiratory chain efficacy and ATP generation, an increase in the production of reactive oxygen species, accumulation of mitochondrial DNA mutations, and reduction in mitochondrial transport and turnover (Green et al., 2011; López‐Otín et al., 2013; Bratic & Larsson, 2013). The development of the in vivo imaging paradigm of the Drosophila wing nerve has enabled the systematic characterization of mitochondrial morphology in axons during aging in this study. Moreover, taking the advantage of vast genetic tools in Drosophila, we have manipulated mitochondrial quality control genes in a spatially and temporally controlled manner, and directly visualized the consequence on mitochondria and axonal integrity in vivo. The imaging data show a clear change in mitochondrial heterogeneity from long, tubular mitochondria in young axons toward short, round mitochondria in aged axons. This change is concurrent with an increased number of axonal mitochondria. Thus, aging is associated with an accumulation of fragmented mitochondria in axons. Our finding is consistent with a recent study showing that the axonal mitochondria size decreased in aged C. elegans (Morsci et al., 2016). This is in addition to the reported decline of axonal mitochondrial transport during aging in nematode neurons (Morsci et al., 2016) and mouse retinal ganglion cells (Takihara et al., 2015).
Unlike in the neuronal soma where lysosomes are abundant and mitochondria can be readily cleared by mitophagy, mitochondria in distal axons face unique challenges. It remains debated whether damaged axonal mitochondria are transported back to the soma for turnover (Maday et al., 2012; Maday & Holzbaur, 2014) or are degraded locally in axons by PINK1‐Parkin‐dependent mitophagy (Ashrafi et al., 2014). In a recent study, Sung et al. (2016) elegantly demonstrated that the appearance and the regulation of axonal mitochondria and autophagy differ between in vivo conditions and in vitro setups – autophagosomes and mitochondria did not colocalize in axons of Drosophila motor neurons in vivo; however, when these neurons were cultured in vitro, numerous autophagosomes were colocalized with mitochondria in axons, even in the absence of any drug‐induced mitochondrial damage. In this study, we provide in vivo evidence that the PINK1‐Parkin pathway is not required for mitochondrial turnover or axonal integrity in either sensory (wing) or motor (leg) neurons of the adult Drosophila during aging. Certainly, we cannot rule out the possibility that PINK1‐Parkin deficiency may still compromise the ultrastructure or function of axonal mitochondria. Interestingly, Devireddy et al. (2015) showed that Pink1 deletion led to a slight decrease in mitochondrial membrane potential and abnormal mitochondrial morphology in the soma of Drosophila larval neurons but did not change mitochondrial density or length in axons. Furthermore, the results of climbing and lifespan assays strongly suggest that the PINK1‐Parkin pathway is dispensable for adult neurons at the animal level. And the reduced longevity of the LOF mutants of Pink1 and Parkin (Greene et al., 2003; Cha et al., 2005; Clark et al., 2006; Park et al., 2006) is likely due to their essential role in non‐neuronal cells such as muscles. As the difference ‘between neurons and muscles’ may represent a cell type‐specific expression and/or requirement of PINK1‐Parkin, our data do not exclude the possibility that the PINK1‐Parkin pathway plays an essential role in select subtypes of neurons, such as dopaminergic neurons, and thus contributes to the pathogenesis of PD.
The autophagy machinery is required for the final turnover of mitochondria in all mitophagy pathways (Pickrell & Youle, 2015; Wei et al., 2015). Our in vivo imaging data reveal that, accompanying the accumulation of axonal mitochondria, the number of axonal autophagosomes increases with age. However, mitochondria and autophagosomes are essentially not colocalized in axons in vivo, even in aged neurons. This is in sharp contrast to the neuronal cell bodies where colocalization of mitochondria and autophagosomes is frequently observed, lending support to an important role of mitophagy in the soma (Cai et al., 2012; Sung et al., 2016). Moreover, knockdown of Atg12 or Atg17 inhibits axonal autophagy but does not affect mitochondrial turnover or axonal integrity. As all mitophagy pathways rely on the autophagy machinery, our data demonstrate that neither PINK1‐Parkin‐dependent nor PINK1‐Parkin‐independent mitophagy is required for maintaining axonal mitochondria in vivo. Although the function of PINK1‐Parkin in axons has been debated (Ashrafi et al., 2014; Devireddy et al., 2015; Sung et al., 2016), our study is the first to show that overall mitophagy is dispensable for axonal maintenance during normal aging (to the best of our knowledge). Nevertheless, blocking autophagy in adult Drosophila neurons results in accelerated aging and slightly shortened lifespan, signifying the importance of autophagy in neuronal aging.
Mitochondrial fission–fusion is required for proper axonal projections and synaptic development and is involved in human neurological disorders (Burté et al., 2015; Bertholet et al., 2016). However, whether fission–fusion constitutes an essential quality control mechanism for mitochondria in axons and how it impacts on neuronal aging compared to mitophagy are unclear. We show that the proper levels of the fission–fusion genes are critically required in axons during aging and that disturbance of the fission–fusion balance in adult neurons dramatically reduces the longevity. When comparing mitophagy and fission–fusion, it is important to bear in mind that axons can be hundreds or even thousands of times longer than the size of the soma. Should neurons routinely turnover axonal mitochondria by mitophagy, the amount of energy it would cost to constantly transport mitochondria from the soma to distal axons and then back to the soma for lysosome‐mediated degradation would be unmanageably enormous and wasteful. Fission–fusion provides an important alternative mechanism to control mitochondrial quality by ‘repairing and reusing’ mitochondria (Schrepfer & Scorrano, 2016). As such, maintaining axonal mitochondria by fission–fusion can be much more ‘cost‐effective’ than mitophagy. This alternative model is especially attractive as damage to axonal mitochondria under physiological conditions during aging is likely mild and gradual.
Together, our in vivo data strongly suggest that maintenance of axonal mitochondria does not require mitophagy, whereas mitophagy‐independent mechanisms such as fission–fusion may play a pivotal role in maintaining axonal mitochondria and neural integrity during normal aging. Finally, it is important to point out that there are multiple levels of mitochondrial quality control, including mitochondrial proteases, ubiquitin‐dependent turnover, vesicle transport to lysosomes, and the new emerging mechanisms of mitochondrial ‘expulsion’ that have been recently reported as an alternative means of autophagy and PINK1‐Parkin‐independent turnover (Melentijevic et al., 2017). Each of them may be active or inactive under certain conditions, in select types of cells, or at restricted subcellular compartments. Thus, it will be of great interest to investigate the role of mitophagy‐independent mechanisms of mitochondrial quality control in aging in the future.
Experimental procedures
Fly stocks
Flies tested in this study were raised on standard cornmeal media and maintained at 25 °C and 60% relative humidity. The following strains were obtained from the Bloomington Drosophila Stock Center (BDSC): w 1118 (5905), Pink1 B9 (34749), Pink1 5 (51649), Da‐Gal4 (5460), elav‐GS (43642), UAS‐mCherry‐Atg8a (37750), UAS‐LacZ (8529), UAS‐Pink1 (51648), UAS‐Parkin (51651), UAS‐RNAi‐Parkin (38333), UAS‐RNAi‐Atg12 (34675), UAS‐RNAi‐Atg17 (36918), and UAS‐RNAi‐Marf (55189). The following strains were obtained from the Tsinghua Fly Center (TFC): UAS‐RNAi‐Opa1 (THU0811), UAS‐RNAi‐Drp1 (TH02258.N), Mef2‐Gal4 (THJ0244). For neuronal expression (Dpr‐Gal4, D42‐Gal4, and elav‐GS) of long hairpin RNAi lines used in this study, the RNAi‐luciferase (31603) was used as a control and a copy of UAS‐Dcr2 was co‐expressed to boost the knockdown efficiency (Ni et al., 2008); for ubiquitous expression (Da‐Gal4) or expression in muscles (Mef2‐Gal4), no additional UAS‐Dcr2 was needed and the RNAi‐mCherry (35785) was used as a control. The UAS‐Pink1 L464P, UAS‐Parkin T240R , and UAS‐Parkin R275W flies are kind gifts from J. Chung, K.L. Lin, and C.H. Chen.
For simultaneous in vivo imaging of axonal mitochondria and axon membrane integrity, the following stable fly strain containing multiple transgenes was generated by chromosomal recombination: yw;dpr‐Gal4,UAS‐mitoGFP/CyO;UAS‐mCD8‐mCherry/TM6B.Tb.
For simultaneous in vivo imaging of axonal mitochondrial and axonal autophagosomes, the following stable fly line containing multiple transgenes was generated by chromosomal recombination: yw; dpr‐Gal4, UAS‐mCherry‐Atg8a, UAS‐mitoGFP/Cyo.
Statistical analysis
Unless otherwise noted, statistical significance in this study is determined by unpaired, two‐tailed Student's t‐test with unequal variance at *P < 0.05, **P < 0.01, and ***P < 0.001. Error bars represent standard error of mean (SEM).
Additional experimental procedures are available in Supporting Information.
Funding
This study is supported by the National Key R&D Program of China (No. 2016YFA0501902), the National Natural Science Foundation of China (No. 31471017 and No. 81671254), and the State High‐Tech Development Plan of China (the ‘863 Program’, No. 2014A020526) to Y.F.
Author contributions
X.C. planned and performed experiments, analyzed data, and wrote the manuscript. H.W. and Q.W. performed experiments. X.C., H.W., and Z.W. contributed critical reagents. S.Z. and Y.D. provided technical assistance. Y.F. designed and supervised research and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Fig. S1 Disruption of the PINK1‐Parkin pathway leads to overt abnormal muscle phenotypes.
Fig. S2 The PINK1‐Parkin pathway is also dispensable in Drosophila motor neurons.
Fig. S3 Upregulation of the PINK1‐Parkin pathway is detrimental to axons in vivo.
Fig. S4 The core ATG gene Atg12 and Atg17 are required for neuronal autophagy.
Acknowledgments
We thank J. Chung, K.L. Lin, and C.H. Chen for providing the Pink1 L464P, Parkin T240R , and Parkin R275W flies, the BDSC and the TFC for providing fly strains, and A. Li, C. Liu, and J. Yuan for helpful discussion and critical reading of the manuscript.
References
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR (2012) WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr. Biol. 22, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune‐Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P (2016) Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 90, 3–19. [DOI] [PubMed] [Google Scholar]
- Bratic A, Larsson NG (2013) The role of mitochondria in aging. J. Clin. Invest. 123, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burté F, Carelli V, Chinnery PF, Yu‐Wai‐Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. [DOI] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH (2012) Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS (2005) Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila . Proc. Natl Acad. Sci. USA 102, 10345–10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. [DOI] [PubMed] [Google Scholar]
- Devireddy S, Liu A, Lampe T, Hollenbeck PJ (2015) The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J. Neurosci. 35, 9391–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokladny K, Myers OB, Moseley PL (2015) Heat shock response and autophagy–cooperation and control. Autophagy 11, 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Bonini NM (2015) Hope on the (fruit) fly: the Drosophila wing paradigm of axon injury. Neural Regen. Res. 10, 173–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Soares L, Teng X, Geary M, Bonini NM (2012) A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 22, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Soares L, Bonini NM (2013) Design and implementation of in vivo imaging of neural injury responses in the adult Drosophila wing. Nat. Protoc. 8, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy‐inflammation‐cell death axis in organismal aging. Science. 333, 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA 100, 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635. [DOI] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP (2010) Disease‐causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6‐dependent mitophagy. J. Cell Biol. 189, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur EL (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG (2016) mito‐QC illuminates mitophagy and mitochondrial architecture in vivo . J. Cell Biol. 214, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, Taub D, Parker JA, Neri C, Gabel CV, Hall DH, Driscoll M (2017) C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsci NS, Hall DH, Driscoll M, Sheng ZH (2016) Age‐related phasic patterns of mitochondrial maintenance in adult Caenorhabditis elegans neurons. J. Neurosci. 36, 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Baldwin D, Hannaford S, Palka J, Montell C (2002) Defective proboscis extension response (DPR), a member of the Ig superfamily required for the gustatory response to salt. J. Neurosci. 22, 3463–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J‐Q, Markstein M, Binari R, Pfeiffer B, Liu L‐P, Villalta C, Booker M, Perkins L, Perrimon N (2008) Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster . Nat. Methods 5, 49–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H (2001) A conditional tissue‐specific transgene expression system using inducible GAL4. Proc. Natl Acad. Sci. USA 98, 12596–12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. [DOI] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A (2005) The Charcot‐Marie‐Tooth type 2A gene product, Mfn2, up‐regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 14, 1405–1415. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion‐promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5, e10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde KR, Taanman JW, Schapira AH (2016) A LON‐ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep. 17, 2522–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW (2013) Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl Acad. Sci. USA 110, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ (2012) The axonal transport of mitochondria. J. Cell Sci. 125, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrepfer E, Scorrano L (2016) Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694. [DOI] [PubMed] [Google Scholar]
- Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Jang S, Park J, Bang S, Choi S, Kwon KY, Zhuang X, Kim E, Chung J (2013) Characterization of PINK1 (PTEN‐induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila . J. Biol. Chem. 288, 5660–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Tandarich LC, Nguyen K, Hollenbeck PJ (2016) Compartmentalized regulation of parkin‐mediated mitochondrial quality control in the Drosophila nervous system in vivo . J. Neurosci. 36, 7375–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, Takamura Y, Sakamoto H, Satoh K, Kondo M, Sakamoto T, Goldberg JL, Nabekura J, Tanihara H (2015) In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc. Natl Acad. Sci. USA 112, 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Wang K, Klionsky DJ (2014) Mitochondria removal by autophagy. Autophagy. 7, 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Liu L, Chen Q (2015) Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 1853, 2784–2790. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl Acad. Sci. USA 107, 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Disruption of the PINK1‐Parkin pathway leads to overt abnormal muscle phenotypes.
Fig. S2 The PINK1‐Parkin pathway is also dispensable in Drosophila motor neurons.
Fig. S3 Upregulation of the PINK1‐Parkin pathway is detrimental to axons in vivo.
Fig. S4 The core ATG gene Atg12 and Atg17 are required for neuronal autophagy.