

Summary
Excessive production of mitochondrial reactive oxygen species (mROS) is strongly associated with mitochondrial and cellular oxidative damage, aging, and degenerative diseases. However, mROS also induces pathways of protection of mitochondria that slow aging, inhibit cell death, and increase lifespan. Recent studies show that the activation of the mitochondrial permeability transition pore (mPTP), which is triggered by mROS and mitochondrial calcium overloading, is enhanced in aged animals and humans and in aging‐related degenerative diseases. mPTP opening initiates further production and release of mROS that damage both mitochondrial and nuclear DNA, proteins, and phospholipids, and also releases matrix NAD that is hydrolyzed in the intermembrane space, thus contributing to the depletion of cellular NAD that accelerates aging. Oxidative damage to calcium transporters leads to calcium overload and more frequent opening of mPTP. Because aging enhances the opening of the mPTP and mPTP opening accelerates aging, we suggest that mPTP opening drives the progression of aging. Activation of the mPTP is regulated, directly and indirectly, not only by the mitochondrial protection pathways that are induced by mROS, but also by pro‐apoptotic signals that are induced by DNA damage. We suggest that the integration of these contrasting signals by the mPTP largely determines the rate of cell aging and the initiation of cell death, and thus animal lifespan. The suggestion that the control of mPTP activation is critical for the progression of aging can explain the conflicting and confusing evidence regarding the beneficial and deleterious effects of mROS on health and lifespan.
Keywords: aging, calcium, mitochondria, NAD, permeability transition, reactive oxygen species
Introduction
Oxidative stress in animals is strongly correlated with aging and lifespan, as predicted by the free radical theory of aging (FRTA; Harman, 1956; Sohal & Allen, 1985; Beckman & Ames, 1998; Barja, 2014). Because most reactive oxygen species are generated in the mitochondria (mROS), in close proximity to mtDNA and the mitochondrial oxidative phosphorylation system, it was suggested that oxidative damage to mtDNA, mitochondrial proteins, and phospholipids is the direct cause of aging and determines lifespan (Harman, 1972). This more specific version of FRTA was named the mitochondrial free radical theory of aging (mFRTA; Barja, 2014; Dai et al., 2014). The evidence supporting mFRTA is extensive (reviewed in Dai et al., 2014). For example, accumulation of mtDNA mutations in the polymerase G‐deficient mice accelerated cardiac aging and this was mitigated by catalase, specifically expressed in the mitochondria (Dai et al., 2010), whereas in wild‐type, overexpression of catalase, specifically in the mitochondria, extended mouse lifespan (Dai et al., 2017). As predicted by mFRTA, maximal lifespan of vertebrate homeotherms is inversely correlated with mROS production (Lambert et al., 2007). In addition, many age‐dependent degenerative diseases appear to be driven by excess production of mROS (Dai et al., 2014). However, recent evidence suggests that it is the oxidative damage to other cellular components, nuclear DNA in particular, that largely determines the rate of aging and lifespan of animals (Schaar et al., 2015; Fang et al., 2016; Ma et al., 2016). Oxidative damage to nuclear DNA triggers the DNA damage response, DDR, which induces both pro‐apoptotic signaling (Nicolai et al., 2015) and protective pathways (Fang et al., 2016; Sun et al., 2016) in postmitotic cells, and leads to senescence in mitotic cells (Campisi & Robert, 2014). Among the most important protection pathways are those that depend on induction of PARP1, which repairs damaged DNA in an NAD+‐dependent manner (Golia et al., 2015), and on the induction of NAD‐dependent deacetylases, sirtuins (Merksamer et al., 2013), in particular sirt 1 (Mouchiroud et al., 2013 ; Imai & Guarente, 2014 ; Fang et al., 2016). Histone demethylases also contribute to stress‐induced protection (Merkwirth et al., 2016). Moreover, it is now evident that mROS or mROS‐induced mitochondrial damage initiate signals that activate several pathways that protect mitochondria from stress, slow aging, inhibit cell death, and may result in lifespan extension (Sun et al., 2016; Desjardins et al., 2017). The mitochondrial sirtuins, particularly sirt3, are critical in the protection of mitochondria (Ansari et al., 2017). Among the protective pathways are the mitochondrial unfolded protein response, UPR(mt), which enhances mitochondrial homeostasis (Pellegrino et al., 2013; Tian et al., 2016), and the nrf2 antioxidant response, which protects against mROS‐induced mitochondrial damage and cell death (Strom et al., 2016). Mitochondria that have been damaged induce the PINK1/Parkin pathway, which initiates mitophagy to remove damaged mitochondria (Durcan & Fon, 2015) and stimulates induction of the PGC‐1α pathway to initiate mitochondrial biogenesis and replace damaged mitochondria (Austin & St‐Pierre, 2012). Manipulations of a large number of antioxidant enzymes did not impact mouse lifespan, despite moderate nuclear and cellular oxidative damage (Pérez et al., 2009), apparently because the induction of DDR by moderate oxidative damage is sufficient to protect the cells. By contrast, knockout of SOD1, which resulted in more extensive oxidative damage to DNA, did shorten lifespan significantly (Pérez et al., 2009). It was recently suggested that the effect on lifespan of SOD1 knockout resulted from the extensive nuclear DNA damage that drove a large number of cells to senescence (Zhang et al., 2017). In regenerating tissues, progenitor cells and stem cells, aging‐associated mitochondrial stress and particularly excess mROS also lead to senescence (Ziegler et al., 2015). Senescent cells may release inflammatory factors that lead to inflammation (Campisi & Robert, 2014). Chronic inflammation is associated with aging and contributes to a number of age‐associated diseases, and is an important determinant of lifespan (De Martinis et al., 2005). Therefore, even if organismal aging, degenerative disease, or lifespan is largely dependent on senescence and not on the aging and death of postmitotic cells (as believed by many), mitochondrial stress appears to be the driving force in these processes as well.
The mitochondrial permeability transition pore (mPTP) is an inner membrane protein complex that can be induced to form a nonselective channel. The channel is voltage‐gated, activated by matrix calcium overloading and reactive oxygen species (ROS), and controlled additionally by a number of associated proteins and the post‐translational modifications and binding of ions to these proteins and to the channel itself (Bernardi et al., 2006). The channel exhibits several conducting states that can open for short (ms) or long (s) periods, and with different permeabilities (Biasutto et al., 2016). Full opening of the mPTP results in increased production of mROS and release of most matrix metabolites (up to 1500 kDa) including mROS, calcium, NAD+, and glutathione. As a result, the mitochondrial membrane potential collapses, oxidative phosphorylation and mitochondrial metabolism are inhibited, the matrix swells, and on prolonged opening the outer membrane ruptures, releasing intermembrane space proteins. Moreover, the release to the cytosol of ROS, calcium, NAD+, glutathione, and other metabolites disrupts cellular homeostasis and increases oxidative damage to proteins, nuclear DNA, ion channels, transporters, and membrane phospholipids (Bernardi et al., 2006; Zorov et al., 2014). Prolonged pore opening in a large number of mitochondria in the cell can lead to cell death by necrosis or similar pathways (Di Lisa et al., 2001; Petronilli et al., 2001; Kim et al., 2003; Vaseva et al., 2012; Hou et al., 2016; Izzo et al., 2016). Several mitochondrial proteins were found to control pore opening including cyclophilin D (CypD), the adenine nucleotide translocator (ANT), the outer membrane anion channel (VDAC), and the phosphate translocator (PiC), and these, together or in various combination, were suggested to form the mPTP (cf. Bernardi et al., 2006). However, more recent studies, and particularly knockout models of each of these proteins, suggest that all these proteins are not indispensable for mPTP activity. More recently, it was suggested that the conducting core of mPTP is subunit c of ATP synthase (Alavian et al., 2014; Bonora et al., 2015), but it appears more likely that it is the large ATP synthase (FoF1) dimer that forms an alternative structure that constitutes the conducting core of the mPTP (Bernardi et al., 2015 ). Nevertheless, the precise identity of the pore components has not yet been elucidated (Biasutto et al., 2016; Izzo et al., 2016). It cannot be excluded that in addition to a core‐conducting structure that is necessary for pore opening, several alternative structures that include additional components exist under some conditions or in some types of cells.
Frequent and extended opening of the mPTP, with its associated bursts of mROS, can overwhelm the cell's antioxidant systems resulting in extensive DNA damage. As a result, the greatly enhanced PARP1 activity leads to a decline in the concentration of its substrate NAD+, and because NAD+ is also required for deacetylation reactions catalyzed by the sirtuins, the balance is shifted from sirtuin‐dependent protective mechanisms to pro‐apoptotic pathways (Gomes et al., 2013; Imai & Guarente, 2014; Fang et al., 2016). Moreover, because NAD+ exits the mitochondrial matrix and is hydrolyzed by CD38 in the intermembrane space when the mPTP is activated (Di Lisa et al., 2001), the mitochondrial protection that is provided by the mitochondrial sirtuins, particularly sirt 3 (Brown et al., 2013), is also diminished. This mPTP‐dependent hydrolysis of NAD+ contributes directly to the loss of cellular NAD+. A more moderate ROS production by mitochondria may not lead to strong pro‐apoptotic signals but is sufficient to trigger various mechanisms that adjust cellular processes and protect the mitochondria and the cell from damage (Patterson et al., 2015; Reczek & Chandel, 2015; Wei & Kenyon, 2016), without causing indiscriminate oxidative damage. This level of ROS formation is mostly contained by antioxidant systems (Forkink et al., 2015; Araki et al., 2016). When their capacity is exceeded, the increased oxidative stress activates the mPTP. While short, infrequent, opening of the mPTP also triggers protective pathways (Hou et al., 2014a), increasing the frequency and duration of the mPTP is associated with more persistent oxidative damage that may result in aging and even cell death. Damage by mROS to mitochondrial matrix proteins, DNA, and phospholipids results in inhibition of oxidative phosphorylation and a further increase in production of mROS (Barja, 2014; Dai et al., 2014; Zorov et al., 2014). However, this damage, while extensive, is partially mitigated by the numerous mechanisms that protect the mitochondria and replace damaged mitochondria with new mitochondria as described above. By contrast, oxidative damage to cytosolic enzymes, calcium transporters, and nuclear DNA in postmitotic cells is more cumulative, increasing cell aging and eventually leading to cell death (Schaar et al., 2015; Fang et al., 2016). While stem cells that can replace damaged cells are emerging as important for healthy aging (Goodell & Rando, 2015), they are also constrained by aging‐induced mitochondrial dysfunction (Min‐Wen et al., 2016). mPTP opening mediates toxic insults on stem cells (Wang et al., 2015; Chen et al., 2016), inhibits proliferation of progenitor cells (Hou et al., 2014b), and inhibits liver regeneration (Antony et al., 2016). Sirt3, which inhibits both mROS production and mPTP activation, is critical for proliferation of stem cells (Shin et al., 2015), and therefore, the inhibition of sirt3 in aging (Brown et al., 2013) as well as depletion of its substrate NAD+ in stem cells (Zhang et al., 2016) is predicted to enhance mPTP activation in aging stem cells and inhibit proliferation. This is likely to also drive the aging‐induced conversion of proliferating cells to senescent cells (Campisi & Robert, 2014). This process is driven by excess mROS and by several additional mitochondrial dysfunction drivers (e.g., calcium overloading, loss of membrane potential; Ziegler et al., 2015), suggesting that this effect, too, is mediated by the mPTP.
Because it is difficult to untangle the protective effects of mROS from its deleterious effects, the concept of FRTA has not been widely accepted (cf. Lapointe & Hekimi, 2010; Stuart et al., 2014). Instead, a consensus is emerging in which the balance between mROS‐induced protective pathways and cell damage‐induced apoptotic pathways is somehow integrated in the mitochondria to determine the progression of aging and ultimately cell death (Wang & Hekimi, 2015; Bhola & Letai, 2016; Min‐Wen et al., 2016; Riera et al., 2016; Sun et al., 2016). Here, we propose that these contrasting signals are integrated at the level of the mPTP, which largely determines the rate of aging and ultimately lifespan by the frequency and duration of pore openings. This is illustrated schematically in Fig. 1.
Figure 1.
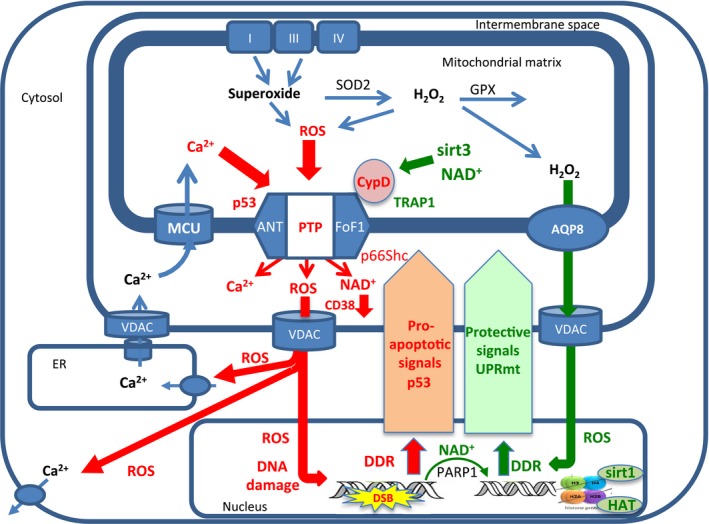
mPTP integration of protective and apoptotic signals determines the rate of cell aging. In cells from young animals, the opening of the mPTP is infrequent and most metabolism‐related increase in mROS production induces mROS signaling to the nucleus, largely through AQP8 (green arrow from mitochondria to nucleus), activating the DNA damage response (DDR) that triggers PARP1 and a number of mitochondria protective pathways (green arrow from nucleus to mitochondria), such as the antioxidant defenses (Nrf2), the mitochondrial unfolded protein response (UPRmt), and sirtuins (e.g., Sirt1, Sirt3), all of which inhibit the mPTP directly and indirectly and prevent more frequent opening of mPTP. Nevertheless, with time, oxidative damage resulting from the frequent and prolonged activity of mPTP (red arrows) damages both mitochondrial electron transport complexes and calcium transporters, particularly in the ER, resulting in increased mROS production and mitochondrial calcium overloading that further enhance mPTP opening. Moreover, the increased release of mROS by the mPT also increases oxidative damage to nuclear DNA resulting in increased pro‐apoptotic signaling (red arrows from nucleus to mitochondria) inducing transfer of P53 and p66sch and other pro‐apoptotic proteins to the mitochondria where they enhance mPTP opening. As aging progresses, the increased mPTP opening also depletes mitochondrial NAD +, inhibiting the protective effect of Sirt3 and further stimulating mPTP opening. Finally, the increased oxidative damage to nuclear DNA increases PARP1 activity leading to nuclear NAD + depletion, which inhibits the nuclear sirtuins (e.g., Sirt1), weakening the mitochondrial protective pathways and further enhancing mPTP activation. This cycle continues, leading to longer and more frequent mPTP activation events, weaker and weaker protection signaling and stronger and stronger pro‐apoptotic signaling, eventually leading to irreversible opening of mPTP, mitochondrial swelling, rupture of the outer membrane, and cell death. The mROS damaging effects that enhance mPTP activation are in red and mROS signaling and the protective pathways are in green (see text for more details).
The hypothesis that mPTP is the driver of aging can be considered a refinement of mFRTA as it is proposed that much of the oxidative damage to the mitochondria itself results from the activation of mPTP and that most of the effects of ‘mitochondrial dysfunction’ and mROS on aging and lifespan are mediated through activation of the mPTP. By controlling both the depletion of cellular NAD+ and the induction of a strong DDR, mPTP can drive aging and death of postmitotic cells as well as senescence in mitotic cells. Moreover, it is likely that mPTP opening also mediates mROS‐driven inflammation (Rimessi et al., 2016), because the formation of the NPLR3 inflammasome appears to depend on opening of the mPTP (Murakami et al., 2012; Iyer et al., 2013) and chronic activation of the mPTP (by deletion of MICU1) was found to extend the pro‐inflammatory response to liver resection (Antony et al., 2016).
In this review, we describe the evidence that the mPTP is enhanced in aging and aging‐driven degenerative diseases, and discuss the mechanisms by which aging enhances mPTP activation and the mechanisms by which the mPTP drives the aging process.
mPTP activation is enhanced by aging and in aging‐dependent degenerative diseases
We first reported two decades ago that the activation of the mPTP is enhanced in lymphocytes from old mice (Rottenberg & Wu, 1997). In these lymphocytes, the mitochondrial membrane potential and respiration rates were lower than in young mice but could be restored to normal values by the mPTP inhibitor cyclosporine A (see below). We later showed that most of the lymphocytes with activated mPTP are memory T cells (Mather & Rottenberg, 2002). It was also reported that the calcium‐induced swelling of isolated liver mitochondria (which follows extended opening of the mPTP) is faster in old mice than in young mice (Goodell & Cortopassi, 1998). Similarly, in isolated mitochondria from both liver and brain of old mice, the calcium‐induced calcium release (CICR), which results from the calcium‐induced opening of the mPTP, can be triggered by lower amounts of calcium than in mitochondria from young mice (Mather & Rottenberg, 2000). Aging was also reported to impair calcium loading in rat heart mitochondria (Jahangir et al., 2001). These early observations suggested that mPTP activation is enhanced by aging in several tissues of mice and rats. Indeed, more recently, many more investigators reported similar observations in different tissues of humans, mice, rats, C. elegans, and fungi. Some of these studies were reviewed previously and the possible role of mPTP in aging was discussed (Crompton, 2004; Di Lisa & Bernardi, 2005; Toman & Fiskum, 2011; Paradies et al., 2013). There are differences in the properties of the mPTP and the effects of aging between mitochondria from different organisms, different tissues (cf. Mather & Rottenberg, 2000), different types of cells from the same tissue (cf. LaFrance et al., 2005; Bambrick et al., 2006; Picard et al., 2011; Lores‐Arnaiz et al., 2016), and even between mitochondria from different locations in the cell (Brown et al., 2006). This variability probably results from the large differences in the expression and activity of the proteins that control activation of the mPTP such as ANT1 (Stepien et al., 1992), CyPD (Hazelton et al., 2009), the MCU complex (Paillard et al., 2017), and probably other proteins. Nevertheless, there is strong evidence that in mitochondria from cells that play a role in aging (e.g., intrafibrillar heart mitochondria, brain cortex synaptic mitochondria), activation of the mPTP is enhanced by aging. A sampling of the evidence for the enhanced activation of mPTP by aging in different tissues and organisms is presented in Table 1.
Table 1.
mPTP activation is enhanced by aging
| Species | System | References |
|---|---|---|
| Mouse | Lymphocytes | Rottenberg & Wu (1997), Mather & Rottenberg (2000) |
| Mouse | T cells | Mather & Rottenberg (2002) |
| Mouse | Liver mitochondria | Goodell & Cortopassi (1998), Mather & Rottenberg (2000) |
| Mouse | Brain mitochondria | Mather & Rottenberg (2000), Lores‐Arnaiz et al. (2016) |
| Rat | Brain mitochondria | Brown et al. (2004), LaFrance et al. (2005), Krestinina et al. (2015) |
| Mouse | Heart mitochondria | Fernandez‐Sanz et al. (2015) |
| Rat | Heart mitochondria | Petrosillo et al. (2010), Escobales et al. (2014) |
| Rat | Skeletal muscle mitochondria | Marzetti et al. (2008), Picard et al. (2011) |
| Rat | Myocytes | Liu et al. (2011) |
| Human | Permeabilized myofibrils | Gouspillou et al. (2014) |
| Mouse | Osteocytes | Shum et al. (2016) |
| P. anserine (Fungi) | Organism | Brust et al. (2010), Kramer et al. (2016) |
| C. elegans | Pharyngeal muscle cells | Shen et al. (2014) |
Most of the studies that demonstrated enhanced activation of the mPTP in aged animals were conducted with isolated mitochondria or cell suspensions and did not allow observation of discrete, individual mPTP opening events, in situ. Moreover, using one specific assay, for example, calcium‐induced activation of the mPTP (CICR), measures only one attribute of the mPTP, namely the calcium threshold, and may not reflect other important changes in the susceptibility of the mPTP to opening (e.g., sensitivity to oxidants, membrane potential threshold). Also, as a control, most assays rely on cyclosporine, which inhibits activation of the mPTP by CypD (see below). However, cyclosporine is only a partial inhibitor of the mPTP (Novgorodov et al., 1992). There are several techniques that facilitate measurement of mPTP activation in living cells (cf. Bonora et al., 2016) and these should be preferred in future studies of the relationship between aging and mPTP. Recent studies of the phenomenon of ‘mitoflashes’, which are observed by fluorescence microscopy in individual cells and are interpreted as short openings of the mPTP (Wang et al., 2008, 2016a; Hou et al., 2013, 2014b; Wu et al., 2016), may provide a better way to correlate directly the frequency and duration of mPTP opening with the progression of aging. Although the interpretation of the signal of the mitochondria‐directed fluorescence probe cpYFP is controversial (Schwarzlander et al., 2014; Wang et al., 2016a,b), there is little doubt that these and other mitoflashes are associated with mPTP opening. A recent study with C. elegans, utilizing cpYFP, demonstrated that the frequency of mPTP opening is age dependent (Shen et al., 2014).
There are numerous reports that mPTP is activated in aging‐dependent degenerative diseases and contributes to their pathology (reviewed in Paradies et al., 2013). Indeed, it is well established that redox stress is a major driver of most aging‐dependent degenerative disease (cf. Dai et al., 2014). mPTP opening plays a major role in ischemic heart damage (discussed below) and also contributes to other aging‐dependent cardiac diseases (Zulian et al., 2016), diabetes (Riojas‐Hernandez et al., 2015), and multiple neurodegenerative diseases (Martin, 2012). A sampling of the evidence for the role of mPTP in aging‐dependent degenerative diseases is listed in Table 2. The fact that inhibition of the mPTP slows the progression of many aging‐dependent degenerative diseases (Table 2) supports the conclusion (see below) that mPTP opening accelerates the progression of aging. To the extent that disease pathology depends on the death of particular types of cell (e.g., Parkinson's disease), the critical role of the mPTP in oxidative stress‐induced cell death confers a decisive role for mPTP opening in the pathology of that disease. Moreover, the effects of physical exercise on improving health and protection from age‐dependent disease, which depends largely on the activation of mitochondrial biogenesis by PGC‐1α (Austin & St‐Pierre, 2012), were shown to result in inhibition of mPTP (cf. Marcil et al., 2006; Goncalves et al., 2016). Whether there are degenerative disease‐specific modulations of mPTP is not yet known.
Table 2.
mPTP is activated in aging‐dependent degenerative disease
| Alzheimer | Du & Yan (2010), Valasani et al. (2016), Chen et al. (2016) |
| Parkinson's disease | Thomas et al. (2012), Martin et al. (2014a), Rasheed et al. (2017) |
| Huntington's disease | Brustovetsky et al. (2005), Quintanilla et al. (2013, 2017) |
| Amyotrophic lateral sclerosis (ALS) | Kawamata & Manfredi (2010), Martin et al. (2014b) |
| Multiple sclerosis | Warne et al. (2016), Savino et al. (2013) |
| Diabetes mellitus | Taddeo et al. (2013), Riojas‐Hernandez et al. (2015), Yan et al. (2016) |
| Heart disease | Hafner et al. (2010), Gordan et al. (2016), Zulian et al. (2016) |
| Osteoporosis | Zhen et al. (2014), Shum et al. (2016) |
Aging is also the most important risk factor for carcinogenesis, and aging‐associated excess mROS appear to play a major role in this process (Park et al., 2011). It is likely that the aging‐associated increase in DNA mutations drives the evolution of cancer in the old (Hanahan & Weinberg, 2011). However, once a mutated cell has converted to a neoplastic phenotype, its antiapoptotic signaling capacity is greatly activated and the mPTP becomes resistant to activation by calcium and ROS (Rasola & Bernardi, 2014; Bonora & Pinton, 2014). This is then the conundrum that confronts anticancer intervention: For cancer prevention, it should be helpful to inhibit the mPTP before any cell in the body has converted to a cancer cell, but it is necessary to activate the mPTP in order to kill cells that already have converted to the neoplastic phenotype. Below, we discuss how mPTP opening may accelerate aging and how aging, in turn, can accelerate mPTP activation.
mPTP opening contributes critically to age‐dependent NAD depletion, which in turn further enhances the activation of mPTP
The frequent and prolonged mPTP opening in aged cells, as described above, contributes to aging‐dependent depletion of cellular NAD+ by the release of matrix NAD+ and its hydrolysis by the mitochondrial NADase, CD38, as observed in postischemia–reperfusion of myocytes (Di Lisa et al., 2001). In addition, in myocytes, mROS released by mPTP opening induce NAD+ depletion by activation of PARP1, and this prolongs the opening of the mPTP, leading to further damage and NAD+ depletion (Schriewer et al., 2013). It was also shown that in cortical neurons the depletion of NAD+ during anoxia and/or glucose deprivation depends on both mPTP opening and PARP activation (Kahraman et al., 2015). It was recently shown that aging‐related NAD+ depletion results largely from the age‐dependent increase in both the expression and activity of CD38, as the effect of aging on cellular NAD+ concentration was absent in the CD38‐knockout mouse (Camacho‐Pereira et al., 2016). Although most of the CD38 is located on the cell surface, a fraction has been found to be localized to the mitochondria and the nucleus (Di Lisa et al., 2001; Camacho‐Pereira et al., 2016). This finding supports the suggestion that the hydrolysis of NAD+ by the mitochondrial CD38 following mPTP opening contributes critically to aging‐induced depletion of cellular NAD+. It is possible that the increased expression of CD38 results from an aging‐induced chronic inflammation (Lee et al., 2012), which also partially depends on mPTP activation. Perhaps as critical as the decrease in cellular NAD+ is the direct effect of loss of NAD+ from the mitochondrial matrix during mPTP opening. The Km for NAD+ of the mitochondrial sirt 3 and sirt 5 is one order of magnitude higher than that of the nuclear sirtuins and in the same range as matrix concentrations of NAD+ of 3–5 mm (Canto et al., 2015). This suggests that even a modest loss of NAD+ from the mitochondrial matrix could decrease the activity of mitochondrial sirtuins and thus inhibit the protection afforded by these sirtuins against oxidative stress and mPTP activation (see below).
The exit of excess mROS from mitochondria to cytosol may depend on mPTP opening
While much has been learned in recent years about the sites, mechanisms, and rates of mitochondrial ROS production under different metabolic conditions (Figueira et al., 2013; Zorov et al., 2014; Goncalves et al., 2015 Brand, 2016), little effort was devoted to elucidating the routes by which mROS leave the mitochondria and reach the cytosol. It is generally accepted that the inner mitochondrial membrane is impermeable to all ions and small solutes except for very lipophilic molecules that can dissolve in the phospholipid membrane. Although a number of ROS and RNS are formed within the mitochondrial matrix, only a few reactive species are known to be transported out of the mitochondria. Quantitatively, the most important are H2O2 and superoxide. H2O2 is a highly polar molecule (D = 84.2), while superoxide is both polar and charged and neither of these readily permeates phospholipid membranes. The mPTP is permeable to all solutes smaller that ~1500 kDa and therefore to all ROS and RNS. The only inner membrane channel that transports ROS is aquaporin 8 (AQP8), which can transport H2O2 (Bienert & Chaumont, 2014; Chauvigne et al., 2015). Little is known about the relative contributions of mPTP and AQP8 to H2O2 release from the matrix. It is likely that in young cells, AQP8‐mediated diffusion is the dominant pathway. However, as the frequency of mPTP opening increases with age, the mPTP contribution probably becomes more significant. It was reported recently that oxidative stress inhibits the transport of H2O2 through AQP8 (Medrano‐Fernandez et al., 2016), which suggests that in aged cells most of the matrix‐produced H2O2 is released through the mPTP. This suggestion is compatible with the observation that ablation of AQP8 enhances the activity of the mPTP (Marchissio et al., 2012; Chauvigne et al., 2015). AQP8 releases H2O2 into the mitochondrial intermembrane space (IMS), where additional mitochondrial superoxide is produced directly (Brand, 2016). Most of the superoxide in the intermembrane space is consumed by oxidized cytochrome c (Zhao et al., 2003) and some is converted to H2O2 by SOD1. It has been shown that the outer membrane anion channel, VDAC, releases superoxide that is generated by complex III in the IMS (Han et al., 2003) and this presumably also applies to H2O2 and other ROS/RNS species, because VDAC also allows for transport of solutes up to ~1500 kDa (Shoshan‐Barmatz et al., 2010). Most of the superoxide that exits the matrix to the cytosol also passes through VDAC as the VDAC inhibitor DDI inhibited the exit of matrix‐produced superoxide from isolated mitochondria (Lustgarten et al., 2012). Although VDAC is not required for mPTP opening (Baines et al., 2007), there are many reports of VDAC involvement in mPTP opening (cf. Tomasello et al., 2009; Hou et al., 2016). It is possible that VDAC associates with the mPTP to form a continuous pore from the matrix to the cytosol. If the mPTP can open either directly to the cytosol or to the intramembrane space, depending on the circumstances, it would allow an additional level of control of the outcome of mPTP opening.
In aging cells, mPTP opening enhances the production of mROS that in turn further enhances mPTP activation
Excessive production of mROS triggers the opening of the mPTP (Bernardi et al., 2006), and there is little doubt that aging increases the production of mROS (Barja, 2014; Dai et al., 2014). Inhibition of electron transport can result in increased production of ROS by complexes I, II, and III (cf. Forkink et al., 2015). Therefore, the increased mROS production with age was attributed to oxidative damage to electron transport complexes and/or to mtDNA, which codes for core peptides of complexes I, III, and IV, resulting in the production of defective, ROS‐producing complexes. The latter process was suggested to create a ‘vicious cycle’ that results in increasingly damaged electron transport complexes with age (Dai et al., 2014). Additional ‘vicious cycles’ can be suggested in which the aging‐dependent enhancement of mPTP‐induced mROS production (Zorov et al., 2014), mainly by complex I (Batandier et al., 2004) and Krebs cycle enzymes (Bonke et al., 2016), causes much of the increased mitochondrial oxidative damage observed in aged cells (Takeyama et al., 1993). Moreover, recent evidence suggests that the protective signaling induced by mROS, which normally prevents sustained mROS production and protects OX PHOS enzymes, is inhibited in aged cells, largely due to NAD depletion, as discussed above. Therefore, the age‐dependent decrease in this protection may be another reason for excess mROS production resulting in more persistent damage to OX PHOS enzymes and mtDNA in aged cells. While inhibition of mitochondrial electron transport in aged cells was reported by many investigators, it should be emphasized that it is not always clear whether this observation is not simply a reflection of an enhanced activation of the mPTP (which inhibits electron transport). In lymphocytes from old mice, electron transport rates were inhibited but were completely restored to those of young mice by the addition of cyclosporine A (Rottenberg & Wu, 1997). Thus, in this case, at least, aging did not lead to inhibition of electron transport and the apparent inhibition was simply the result of enhanced activation of the mPTP.
Most of the proteins that are known to control mPTP opening and the protein complex that forms the pore itself (presumably ATP synthase) carry ‐SH groups, and some of these groups are oxidized in aged animals (Tajeddine, 2016) as the matrix redox balance is shifted toward a more oxidative state (Dan Dunn et al., 2015, Sies et al., 2017). However, it is still not clear which of these sulfhydryl sites control the activity of the mPTP. Because mROS also cause oxidative damage to membrane phospholipids, it has been suggested that oxidized phospholipids, and in particular oxidized cardiolipin, activates the mPTP (Petrosillo et al., 2006). Indeed, it has been argued that oxidation of membrane phospholipids is critical for the progression of aging (Pamplona, 2008; possibly through activation of the mPTP). In addition, oxidative stress triggers the transfer of proteins that enhance mPTP activation such as P53, which is transported to the mitochondrial matrix, or P66Shc, which is transported to the mitochondrial intermembrane space where its cytochrome c‐dependent ROS production, in turn, activates the mPTP (Savino et al., 2013; Priami et al., 2015; Di Lisa et al., 2017). Moreover, oxidized pyridine nucleotides also activate the mPTP (Chernyak & Bernardi, 1996; Ronchi et al., 2015) and oxidative stress is manifested in increased oxidation of pyridine nucleotides (Sies et al., 2017). In summary, oxidative stress, which increases in aged cells (Dai et al., 2014), enhances mPTP opening by a variety of mechanisms and is, in turn, further increased by the enhanced activation of mPTP.
Disruption of calcium homeostasis in aging increases the frequency of calcium‐triggered mPTP opening
Calcium overloading of mitochondria is an essential trigger of mPTP opening (Bernardi et al., 2006; Tajeddine, 2016; Hurst et al., 2017). Mitochondrial calcium accumulation mediated by the calcium uniporter (MCU) depends critically on the elevation of cytosolic calcium concentration (cf. Rottenberg & Marbach, 1990; Antony et al., 2016), although often in restricted spatial domains, such as those maintained between the ER and mitochondrial outer membranes. It is well established that aging disrupts calcium homeostasis (cf. Tsai et al., 1997; Mattson, 2007) and may disrupt the local association between ER and mitochondria (Fernandez‐Sanz et al., 2014). While calcium signaling pathways may be modulated by aging in different ways, depending on the tissue and cell type, a general observation is that calcium buffering power in the cytosol is diminished (cf. Tsai et al., 1997; Gant et al., 2015; Pandya et al., 2015) and this could lead to an increase in mitochondrial calcium accumulation. The effect of aging on calcium homeostasis is believed to be mostly due to oxidative damage to calcium transporters and channels (cf. Andersson et al., 2011; Cooper et al., 2013), which increases the leak of calcium into the cytosol and increases calcium overload of mitochondria. Calcium overload of the matrix is also reported to be enhanced by increased direct transfer of calcium from ER to mitochondria (Calvo‐Rodriguez et al., 2016). In addition, aging downregulates the expression of the calcium buffering proteins regucalcin (Vaz et al., 2015) and FKBP1b (Gant et al., 2015). MICU1 is a subunit of the mitochondrial calcium transport complex located in the intermembrane space that controls opening of MCU. Under normal conditions, MICU1 limits mitochondrial calcium accumulation to concentrations above the normal resting cytosolic calcium range, thus preventing Ca overloading and activation of the mPTP (Antony et al., 2016; Liu et al., 2016). It was also reported that another MCU subunit, MCUR1, directly controls the calcium‐dependent activation of mPTP (Chaudhuri et al., 2016). Very recently, it was reported that MCU itself is modified under oxidative stress by S‐glutathionylation of cysteine 97, which enhances calcium overload‐induced cell death (Dong et al., 2017). In aging cells, the cytosolic free calcium is frequently above the MICU1‐set threshold for calcium uptake, and the calcium threshold for mPTP activation is below the normal threshold (Mather & Rottenberg, 2000: and see Table 1); therefore, more frequent calcium‐induced mPTP openings are likely to be observed in aged cells.
Mitochondrial membrane potential and the potential threshold for mPTP activation may be lower in aged cells
The mPTP is voltage‐gated and lowering of the mitochondrial membrane potential enhances the activation of the mPTP (Bernardi, 1992). There are many reports that the mitochondrial membrane potential is lower in aged cells (cf. Sugrue & Tatton, 2001). Oxidative damage is probably the most important factor that contributes to lowering the mitochondrial membrane potential in aged cells. Oxidative damage to membrane phospholipids is known to increase the membrane permeability to ions (cf. Runas & Malmstadt, 2015), and it is well documented that aging increases oxidative damage to mitochondrial phospholipids (Pamplona, 2008). However, as mPTP opening collapses the membrane potential, some of the reported effects of aging on membrane potential probably reflect the fact that aging enhances mPTP activation. In these cases, the mitochondrial membrane potential can be restored by the inhibition of mPTP by cyclosporine (cf. Rottenberg & Wu, 1997). Aging most probably also lower the potential threshold for mPTP opening. It was recently shown that this threshold depends on the expression of the adenine nucleotide translocator ANT1 (Doczi et al., 2016) and that ANT1 overexpression inhibits the mPTP (Klumpe et al., 2016). Inhibition of the mPTP by ANT depends on locking ANT in its matrix‐facing M configuration by ADP (Rottenberg & Marbach, 1990; Bernardi et al., 2006). This effect was suggested to result from the increase in the negative surface charge on the matrix face of the inner membrane (Rottenberg & Marbach, 1990), which is compatible with the suggestion that ANT determines the voltage threshold for mPTP activation. GSK‐3β also controls the mPTP and it was suggested that phosphorylation of ANT by GSK‐3β may contribute to aging‐dependent activation of the mPTP (Zhu et al., 2013). ANT was reported to be oxidized in aged cells (Le Bras et al., 2005), and oxidation of SH resides was reported to lower the threshold for mPTP opening (Petronilli et al., 1994). Therefore, it is very likely that the potential threshold for mPTP activation is lowered in aged cells.
The enhancement of mPTP opening by aging is mediated by aging‐induced modifications of cyclophilin D
Cyclophilin D (CypD) interacts with the mPTP complex (presumably FoF1) to sensitize the mPTP to activation by ROS and Ca2+ (Bernardi et al., 2006). This activation depends on masking an mPTP inhibitory phosphate binding site by CypD (Basso et al., 2008). Several proteins that are known to regulate mPTP, as well as CypD itself, are subject to numerous post‐translational modifications that modulate their activity. Increased expression of CypD enhances activation of the mPTP (Lam et al., 2015) and knockout of the gene inhibits the mPTP (Shum et al., 2016). It was reported recently that the concentration of CypD increases in the brain of old mice (Gauba et al., 2017) and this effect alone should enhance mPTP opening. In young animals, CypD is largely inhibited as a result of a number of post‐translational modifications (Elrod & Molkentin, 2013). CypD also interacts with inhibitory proteins such as HSP90 (TRAP1 in humans), which was reported to aggregate CypD (Lam et al., 2015) and prevent its interaction with activating proteins (e.g., P53) and the mPTP (Lebedev et al., 2016). In aged cells, the UPRmt is suppressed, inhibiting HSP90 synthesis and its translocation to the matrix, while P53 is translocated to the matrix to further enhance CypD activation (Vaseva et al., 2012; Priami et al., 2015). Aging reverses several of the post‐translational modifications that inhibit CypD resulting in a more active CypD. Inhibition of the deacetylation of a lysine residue by sirt3 (Hafner et al., 2010) is, most likely, the strongest effect of aging on CypD activity as the concentration of NAD+ is lower in old animals, it being lost both through PARP1 activation and through mPTP opening, as discussed above. Moreover, the concentration of sirt 3 itself is also lower in old animals (Kwon et al., 2015). Sirt 3 also inhibits ROS production so that the weaker activity of sirt 3 with age enhances mPTP activation, both directly and indirectly (Brown et al., 2013; Kincaid & Bossy‐Wetzel, 2013; Ansari et al., 2017). The thioredoxin/glutathione system is in equilibrium with SH groups on CYPD, so that the increased redox stress in old animals will oxidize the SH groups of CypD (Folda et al., 2016). This is possibly one of the redox sites that activate the mPTP (Nguyen et al., 2011). It is also believed that in aged animals, GSK‐3β phosphorylates CypD, thereby further enhancing CypD activity (Zhu et al., 2010, 2013). Other proteins that control mPTP opening may also work, at least partially, through their interaction with CypD. Thus, ANT was reported to interact with CYPD and control the activation of the mPTP (Crompton et al., 1988; Woodfield et al., 1998). CypD can be inhibited directly by the drug cyclosporine A, which is the most frequently used inhibitor of mPTP (Crompton et al., 1988). There are marked differences between mitochondria from different organisms, tissues, and ages in the extent to which cyclosporine inhibits the mPTP (cf. Bambrick et al., 2006; Liu et al., 2011). It was observed that cyclosporine A does not inhibit ANT‐dependent stimulation of the mPTP in heart mitochondria of aged animals (Garcia et al., 2009; Liu et al., 2011). This probably reflects the independent activation of mPTP by ANT, as discussed above. Similarly, in the presence of ADP, mouse brain mitochondria are more resistant to Ca2+‐induced mPTP opening than liver mitochondria and require a larger amount of calcium to trigger mPTP opening (Mather & Rottenberg, 2000). Thus in brain mitochondria, but not in liver, locking the ANT in the M configuration is sufficient to strongly inhibit mPTP. As a result, in brain mitochondria, cyclosporine A only slightly increases the Ca2+ loading threshold (~20%), while in liver mitochondria cyclosporine A increases the threshold to a much larger extent (~400%; Mather & Rottenberg, 2000). In aged mice, the cyclosporine effect was larger than in young mice (by 25–30%) both in liver and in brain mitochondria, indicating more active CypD in both brain and liver of old mice. Because aging in general decreases antiapoptotic pathways and increases pro‐apoptotic pathways, other mitochondrial proteins that are known to inhibit mPTP are probably also reduced by aging; these may include HSP75 (Wang et al., 2015), HSP60 and HSP90 (TRAP1; Altieri, 2013), which are induced by the UPR(mt), and antiapoptotic proteins, such as BCL‐2 and BCL‐xL (Jonas et al., 2014). Moreover, an increasing number of metabolites were reported to control the mPTP directly or indirectly, for example, mitochondrial cAMP and cyclic nucleotide phosphodiesterase (CNP; Baburina et al., 2015), polyphosphate (Baev et al., 2017), spermine (Wei et al., 2016), and H2S (Li et al., 2015). These effects are probably also modulated by aging. For instance, it was reported that the enhanced activation of the mPTP in aged neurons partially results from a reduction in the activity of CNP and increased cAMP (Krestinina et al., 2015; Wang et al., 2016a,b).
Preconditioning in ischemia–reperfusion (IRPC) is greatly diminished by old age
Ischemic heart damage (as well as ischemia in brain, kidney, and other tissues) is largely the result of the reperfusion that follows ischemia. Reperfusion floods the cells with calcium, which triggers massive mPTP opening leading to extensive cell death (Bernardi & Di Lisa, 2015). Aging is associated with vascular damage, which increases the incidence of ischemia in the old and the aging‐dependent activation of mPTP results in increased ischemic damage in the old (Petrosillo et al., 2010). In the young, mild ischemic events protect the tissues from further ischemic damage by triggering the mitochondrial protection pathways—a process called ischemia–reperfusion preconditioning (IRPC). The mitochondrial protection pathways increase the resistance of the mPTP to opening, both directly and indirectly, although it is not clear which mechanism is dominant in IRPC (Bernardi & Di Lisa, 2015; Halestrap & Richardson, 2015). Aging greatly attenuates IRPC (Boengler et al., 2009; Fernandez‐Sanz et al., 2015). Similarly, cyclosporine A protects from ischemia–reperfusion damage in the young but not in the old (Liu et al., 2011; Pottecher et al., 2016). It was suggested that the deacetylation of CypD by sirt3 is an important contributor to IRPC (Bochaton et al., 2015) and this protection is lost in aged cells because of NAD+ depletion, as discussed above. However, the loss of IRPC in old age appears to be mediated by other mechanisms as well. One suggestion is the age‐dependent activation of phosphorylation of ANT (and perhaps other proteins) by GSK‐3β (Zhu et al., 2013). Aerobic exercise also induces protection of myocytes from mPTP opening (cf. Marcil et al., 2006; Lumini‐Oliveira et al., 2011). As the health benefits of aerobic exercise largely depend on reducing the risk of ischemia, and to the extent that activation of the mPTP causes the damage of IR, the increased risk of heart disease in the aged may reflect the aging‐dependent activation of mPTP.
Lifespan extension by mutations and caloric restriction most likely depends on pathways that inhibit mPTP opening, directly or indirectly
The fact the lifespan can be extended experimentally in several animal models of aging (e.g., C. elegans, Drosophila, and mice), and the findings that in many cases lifespan extension appears to depend on mROS signaling are often cited as the strongest evidence against mFRTA, as discussed in the introduction. Evidently, in these cases, mROS initiate the mitochondria protection pathways at an early age and this leads to lifespan extension. The mitochondrial protection pathways invariably lead to inhibition of the mPTP, whether indirectly by inhibition of mROS production, increased antioxidant protection, increased mitophagy, and increased mitochondrial biogenesis, or by direct inhibition of mPTP activation, as discussed above. It has been shown that lifespan extension by caloric restriction is associated with inhibition of the mPTP (cf. Kristal & Yu, 1998; Hofer et al., 2009; Amigo et al., 2017) and that lifespan extension by genetic manipulations is often associated with the induction of the UPRmt (cf. Bennett & Kaeberlein, 2014; Pulliam et al., 2014). A recent study on lifespan extension in C. elegans by germline loss shows that lifespan extension in this case depends on two independent pathways, one of which is mROS dependent (UPRmt) and the other not (the H2S pathway; Wei & Kenyon, 2016). However, H2S, similar to UPRmt, also inhibits the mPTP (Li et al., 2015). In a study of a very large number of C. elegans lifespan modulations by mutations and environmental manipulations, it was shown that lifespan correlates negatively with the frequency of ‘mitoflashes’ at an early adult age (Shen et al., 2014). If one accepts the interpretation that ‘mitoflashes’ signal the opening of the mPTP (Wang et al., 2016a), it could be argued that in all these cases lifespan extension is the result of inhibition of mPTP opening in early adulthood. Metformin, the first drug approved for clinical trials for retarding the progress of human aging, was shown to inhibit the mPTP (Guigas et al., 2004; Bhamra et al., 2008). Thus, it is likely that in most, if not all, manipulations that extend animal lifespan, the mPTP is inhibited, directly or indirectly.
Conclusions
Although ROS has been suspected for more than half a century to be the driving force of aging, as rationalized first by FRTA, and more recently by mFRTA, and although the association between ROS, aging, age‐related degenerative disease, and lifespan was proven to be robust, it has been more difficult to prove that ROS actually drives the progression of aging. The recent discoveries that mROS signaling triggers a large number of pathways that protect the cell, and mitochondria in particular, against oxidative damage, inhibit mROS production, slow aging and even increase lifespan, appear to directly contradict mFRTA. Nevertheless, because mROS signaling originates in the mitochondria and most of the protection pathways triggered by mROS are directed at the mitochondria, it became evident that the control of the progression of aging must reside in the mitochondria. These organelles must, somehow, integrate the protection signals as well as the stress‐induced pro‐apoptotic signals to determine the progression of aging. It is well accepted that oxidative stress‐induced cell death is driven by massive opening of the mPTP, but the cumulative effects of a more moderate opening of the mPTP have not been fully appreciated. Reviewing the large number of recent studies that show that the mPTP is enhanced in aging and in aging‐associated degenerative disease, and that inhibition of the mPTP can slow aging and degenerative diseases, we suggest that the mPTP itself is the elusive site of integration of the contrasting pro‐ and antiapoptotic signals that determine the rate of progression to aging. While many processes upstream of the mPTP (e.g., oxidative phosphorylation, electron transport, mROS production, mitochondrial antioxidant defense, mitophagy, mitochondrial biogenesis) are also affected by the various protection mechanisms, it is likely that these upstream processes affect aging largely through their effects on mPTP activation. There is still much to be learned about the composition and structure of the mPTP, the mechanisms that control mPTP opening, the various activation states of the mPTP, the extent and types of ions and metabolites that are released, and how the progression of aging affects these processes. The progression of aging to death does not follow a uniformly shaped curve in all animals (Jones et al., 2014). An animal's lifespan can be determined by the failure of one particular critical organ, by either postmitotic or mitotic cells, and differences between the control of the mPTP in different organs, and different types of cells, may account for some of the differences between species. Further studies of the control of mPTP in aging can open the door to a much better understanding of the determinants of longevity.
Funding
This work was supported by NIH Grant AA018873 to JB Hoek.
Conflict of interest
Both authors report no conflict of interest.
References
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA (2014) An uncoupling channel within the c‐subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl Acad. Sci. USA 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC (2013) Hsp90 regulation of mitochondrial protein folding: from organelle integrity to cellular homeostasis. Cell. Mol. Life Sci. 70, 2463–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amigo I, Menezes‐Filho SL, Luevano‐Martinez LA, Chausse B, Kowaltowski AJ (2017) Caloric restriction increases brain mitochondrial calcium retention capacity and protects against excitotoxicity. Aging Cell 16, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 14, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH (2017) Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 16, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, Hajnoczky G (2016) MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 7, 10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Kusano H, Sasaki N, Tanaka R, Hatta T, Fukui K, Natsume T (2016) Redox sensitivities of global cellular cysteine residues under reductive and oxidative stress. J. Proteome Res. 15, 2548–2559. [DOI] [PubMed] [Google Scholar]
- Austin S, St‐Pierre J (2012) PGC1α and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 125, 4963–4971. [DOI] [PubMed] [Google Scholar]
- Baburina Y, Azarashvili T, Grachev D, Krestinina O, Galvita A, Stricker R, Reiser G (2015) Mitochondrial 2’, 3’‐cyclic nucleotide 3’‐phosphodiesterase (CNP) interacts with mPTP modulators and functional complexes (I‐V) coupled with release of apoptotic factors. Neurochem. Int. 90, 46–55. [DOI] [PubMed] [Google Scholar]
- Baev AY, Negoda A, Abramov AY (2017) Modulation of mitochondrial ion transport by inorganic polyphosphate – essential role in mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 49, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage‐dependent anion channels are dispensable for mitochondrial‐dependent cell death. Nat. Cell Biol. 9, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambrick LL, Chandrasekaran K, Mehrabian Z, Wright C, Krueger BK, Fiskum G (2006) Cyclosporin A increases mitochondrial calcium uptake capacity in cortical astrocytes but not cerebellar granule neurons. J. Bioenerg. Biomembr. 38, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G (2014) The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 127, 1–27. [DOI] [PubMed] [Google Scholar]
- Basso E, Petronilli V, Forte MA, Bernardi P (2008) Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem. 283, 26307–26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batandier C, Leverve X, Fontaine E (2004) Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 279, 17197–17204. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol. Rev. 78, 547–581. [DOI] [PubMed] [Google Scholar]
- Bennett CF, Kaeberlein M (2014) The mitochondrial unfolded protein response and increased longevity: cause, consequence, or correlation? Exp. Gerontol. 56, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P (1992) Modulation of the mitochondrial cyclosporin A‐sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 267, 8834–8839. [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 78, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly‐Dyson E, Di Lisa F, Forte MA (2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F, Fogolari F, Lippe G (2015) From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circ. Res. 116, 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, Yellon DM (2008) Metformin protects the ischemic heart by the Akt‐mediated inhibition of mitochondrial permeability transition pore opening. Basic Res. Cardiol. 103, 274–284. [DOI] [PubMed] [Google Scholar]
- Bhola PD, Letai A (2016) Mitochondria‐judges and executioners of cell death sentences. Mol. Cell 61, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasutto L, Azzolini M, Szabo I, Zoratti M (2016) The mitochondrial permeability transition pore in AD 2016: an update. Biochim. Biophys. Acta 1863, 2515–2530. [DOI] [PubMed] [Google Scholar]
- Bienert GP, Chaumont F (2014) Aquaporin‐facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 1840, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Bochaton T, Crola‐Da‐Silva C, Pillot B, Villedieu C, Ferreras L, Alam MR, Thibault H, Strina M, Gharib A, Ovize M, Baetz D (2015) Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3‐mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 84, 61–69. [DOI] [PubMed] [Google Scholar]
- Boengler K, Schulz R, Heusch G (2009) Loss of cardioprotection with ageing. Cardiovac. Res. 83, 247–261. [DOI] [PubMed] [Google Scholar]
- Bonke E, Siebels I, Zwicker K, Drose S (2016) Manganese ions enhance mitochondrial H2O2 emission from Krebs cycle oxidoreductases by inducing permeability transition. Free Radic. Biol. Med. 99, 43–53. [DOI] [PubMed] [Google Scholar]
- Bonora M, Pinton P (2014) The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol. 4, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P (2015) Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34, 1475–1486. [DOI] [PubMed] [Google Scholar]
- Bonora M, Morganti C, Morciano G, Giorgi C, Wieckowski MR, Pinton P (2016) Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence‐imaging‐based techniques. Nat. Protoc. 11, 1067–1080. [DOI] [PubMed] [Google Scholar]
- Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31. [DOI] [PubMed] [Google Scholar]
- Brown MR, Geddes JW, Sullivan PG (2004) Brain region‐specific, age‐related, alterations in mitochondrial responses to elevated calcium. J. Bioenerg. Biomembr. 36, 401–406. [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Geddes JW (2006) Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem. 281, 11658–11668. [DOI] [PubMed] [Google Scholar]
- Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D (2013) SIRT3 reverses aging‐associated degeneration. Cell Rep. 3, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust D, Daum B, Breunig C, Hamann A, Kuhlbrandt W, Osiewacz HD (2010) Cyclophilin D links programmed cell death and organismal aging in Podospora anserina . Aging Cell 9, 761–775. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM (2005) Age‐dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington's disease. J. Neurochem. 93, 1361–1370. [DOI] [PubMed] [Google Scholar]
- Calvo‐Rodriguez M, Garcia‐Durillo M, Villalobos C, Nunez L (2016) In vitro aging promotes endoplasmic reticulum (ER)‐mitochondria Ca2+ cross talk and loss of store‐operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 1863, 2637–2649. [DOI] [PubMed] [Google Scholar]
- Camacho‐Pereira J, Tarrago MG, Chini CC, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN (2016) CD38 dictates age‐related NAD decline and mitochondrial dysfunction through an SIRT3‐dependent mechanism. Cell Metab. 23, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Robert L (2014) Cell senescence: role in aging and age‐related diseases. Interdiscip. Top. Gerontol. 39, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J (2015) NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE (2016) Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl Acad. Sci. USA 113, E1872–E1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvigne F, Boj M, Finn RN, Cerda J (2015) Mitochondrial aquaporin‐8‐mediated hydrogen peroxide transport is essential for teleost spermatozoon motility. Sci. Rep. 5, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Wang K, Jiang D, Wang Y, Xiao X, Zhu N, Li M, Jia S, Wang Y (2016) Blocking mPTP on neural stem cells and activating the nicotinic acetylcholine receptor alpha7 subunit on microglia attenuate Aβ‐induced neurotoxicity on neural stem cells. Neurochem. Res. 41, 1483–1495. [DOI] [PubMed] [Google Scholar]
- Chernyak BV, Bernardi P (1996) The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur. J. Biochem. 238, 623–630. [DOI] [PubMed] [Google Scholar]
- Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D (2013) Redox modification of ryanodine receptors by mitochondria‐derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J. Physiol. 591, 5895–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M (2004) Mitochondria and aging: a role for the permeability transition? Aging Cell 3, 3–6. [DOI] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A (1988) Inhibition by cyclosporin A of a Ca2+‐dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 255, 357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM (1998) Cyclophilin‐D binds strongly to complexes of the voltage‐dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258, 729–735. [DOI] [PubMed] [Google Scholar]
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS (2010) Age‐dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9, 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS (2014) Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chiao YA, Martin GM, Marcinek DJ, Basisty N, Quarles EK, Rabinovitch PS (2017) Mitochondrial‐targeted catalase: extended longevity and the roles in various disease models. Prog. Mol. Biol. Transl. Sci. 146, 203–241. [DOI] [PubMed] [Google Scholar]
- Dan Dunn J, Alvarez LA, Zhang X, Soldati T (2015) Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox. Biol. 6, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martinis M, Franceschi C, Monti D, Ginaldi L (2005) Inflamm‐ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 579, 2035–2039. [DOI] [PubMed] [Google Scholar]
- Desjardins D, Cacho‐Valadez B, Liu JL, Wang Y, Yee C, Bernard K, Khaki A, Breton L, Hekimi S (2017) Antioxidants reveal an inverted U‐shaped dose‐response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans . Aging Cell 16, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P (2005) Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc. Res. 66, 222–232. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 276, 2571–2575. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Giorgio M, Ferdinandy P, Schulz R (2017) New aspects of p66Shc in ischemia reperfusion injury and cardiovascular diseases. Br. J. Pharmacol. 174, 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doczi J, Torocsik B, Echaniz‐Laguna A, Mousson de Camaret B, Starkov A, Starkova N, Gal A, Molnar MJ, Kawamata H, Manfredi G, Adam‐Vizi V, Chinopoulos C (2016) Alterations in voltage‐sensing of the mitochondrial permeability transition pore in ANT1‐deficient cells. Sci. Rep. 6, 26700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jaña F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M (2017) Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell 65, 1014–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Yan SS (2010) Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid β. Biochim. Biophys. Acta 1802, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Fon EA (2015) The three ‘P's of mitophagy: PARKIN, PINK1, and post‐translational modifications. Genes Dev. 29, 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Molkentin JD (2013) Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 77, 1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobales N, Nunez RE, Jang S, Parodi‐Rullan R, Ayala‐Pena S, Sacher JR, Skoda EM, Wipf P, Frontera W, Javadov S (2014) Mitochondria‐targeted ROS scavenger improves post‐ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 77, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Scheibye‐Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Sanz C, Ruiz‐Meana M, Miro‐Casas E, Nunez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez‐Sinovas A, Vazquez J, Garcia‐Dorado D (2014) Defective sarcoplasmic reticulum‐mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5, e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Sanz C, Ruiz‐Meana M, Castellano J, Miro‐Casas E, Nunez E, Inserte J, Vazquez J, Garcia‐Dorado D (2015) Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb. Haemost. 113, 441–451. [DOI] [PubMed] [Google Scholar]
- Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, Sluse FE, Souza‐Pinto NC, Vercesi AE (2013) Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 18, 2029–2074. [DOI] [PubMed] [Google Scholar]
- Folda A, Citta A, Scalcon V, Cali T, Zonta F, Scutari G, Bindoli A, Rigobello MP (2016) Mitochondrial thioredoxin system as a modulator of cyclophilin D redox state. Sci. Rep. 6, 23071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkink M, Basit F, Teixeira J, Swarts HG, Koopman WJ, Willems PH (2015) Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox. Biol. 6, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Chen KC, Kadish I, Blalock EM, Thibault O, Porter NM, Landfield PW (2015) Reversal of aging‐related neuronal Ca2+ dysregulation and cognitive impairment by delivery of a transgene encoding FK506‐binding protein 12.6/1b to the hippocampus. J. Neurosci. 35, 10878–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia N, Zazueta C, Martinez‐Abundis E, Pavon N, Chavez E (2009) Cyclosporin A is unable to inhibit carboxyatractyloside‐induced permeability transition in aged mitochondria. Comp. Biochem. Physiol. Toxicol. Pharmacol. 149, 374–381. [DOI] [PubMed] [Google Scholar]
- Gauba E, Guo L, Du H (2017) Cyclophilin D promotes brain mitochondrial F1FO ATP synthase dysfunction in aging mice. J. Alzheimers Dis. 55, 1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golia B, Singh HR, Timinszky G (2015) Poly‐ADP‐ribosylation signaling during DNA damage repair. Front Biosci. 20, 440–457. [DOI] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA (2013) Declining NAD+ induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 155, 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey‐Mogensen M, Brand MD (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 290, 209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves IO, Passos E, Diogo CV, Rocha‐Rodrigues S, Santos‐Alves E, Oliveira PJ, Ascensao A, Magalhaes J (2016) Exercise mitigates mitochondrial permeability transition pore and quality control mechanisms alterations in nonalcoholic steatohepatitis. Appl. Physiol. Nutr. Metab. 41, 298–306. [DOI] [PubMed] [Google Scholar]
- Goodell S, Cortopassi G (1998) Analysis of oxygen consumption and mitochondrial permeability with age in mice. Mech. Ageing Dev. 101, 245–256. [DOI] [PubMed] [Google Scholar]
- Goodell MA, Rando TA (2015) Stem cells and healthy aging. Science 350, 1199–1204. [DOI] [PubMed] [Google Scholar]
- Gordan R, Fefelova N, Gwathmey JK, Xie LH (2016) Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: evidence from cyclophilin D knockout mice. Cell Calcium 60, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M, Hepple RT (2014) Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Guigas B, Detaille D, Chauvin C, Batandier C, De Oliveira F, Fontaine E, Leverve X (2004) Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem. J. 382, 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA (2010) Regulation of the mPTP by SIRT3‐mediated deacetylation of CypD at lysine 166 suppresses age‐related cardiac hypertrophy. Aging 2, 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Richardson AP (2015) The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 78, 129–141. [DOI] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E (2003) Voltage‐dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. [DOI] [PubMed] [Google Scholar]
- Harman D (1972) The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 20, 145–147. [DOI] [PubMed] [Google Scholar]
- Hazelton JL, Petrasheuskaya M, Fiskum G, Kristian T (2009) Cyclophilin D is expressed predominantly in mitochondria of γ‐aminobutyric acidergic interneurons. J. Neurosci. Res. 87, 1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, Wohlgemuth SE, Leeuwenburgh C (2009) Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech. Ageing Dev. 130, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Mattson MP, Cheng A (2013) Permeability transition pore‐mediated mitochondrial superoxide flashes regulate cortical neural progenitor differentiation. PLoS One 8, e76721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T, Wang X, Ma Q, Cheng H (2014a) Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J. Physiol. 592, 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Ghosh P, Wan R, Ouyang X, Cheng H, Mattson MP, Cheng A (2014b) Permeability transition pore‐mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid β1‐42 on neural progenitor cell proliferation. Neurobiol. Aging 35, 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Zhou Z, Qin J, Liu W, Wang B, Gu Y (2016) Erastin disrupts mitochondrial permeability transition pore (mPTP) and induces apoptotic death of colorectal cancer cells. PLoS One 11, e0154605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst S, Hoek J, Sheu SS (2017) Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 49, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper‐Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo V, Bravo‐San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 26, 655–667. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A (2001) Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech. Ageing Dev. 122, 1073–1086. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Porter GA, Alavian KN (2014) Bcl‐xL in neuroprotection and plasticity. Front. Physiol. 5, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones OR, Scheuerlein A, Salguero‐Gomez R, Camarda CG, Schaible R, Casper BB, Dahlgren JP, Ehrlen J, Garcia MB, Menges ES, Quintana‐Ascencio PF, Caswell H, Baudisch A, Vaupel JW (2014) Diversity of ageing across the tree of life. Nature 505, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman S, Siegel A, Polster BM, Fiskum G (2015) Permeability transition pore‐dependent and PARP‐mediated depletion of neuronal pyridine nucleotides during anoxia and glucose deprivation. J. Bioenerg. Biomembr. 47, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G (2010) Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 131, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, He L, Lemasters JJ (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 304, 463–470. [DOI] [PubMed] [Google Scholar]
- Kincaid B, Bossy‐Wetzel E (2013) Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpe I, Savvatis K, Westermann D, Tschope C, Rauch U, Landmesser U, Schultheiss HP, Dorner A (2016) Transgenic overexpression of adenine nucleotide translocase 1 protects ischemic hearts against oxidative stress. J. Mol. Med. 94, 645–653. [DOI] [PubMed] [Google Scholar]
- Kramer P, Jung AT, Hamann A, Osiewacz HD (2016) Cyclophilin D is involved in the regulation of autophagy and affects the lifespan of P. anserina in response to mitochondrial oxidative stress. Front. Genet. 7, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krestinina O, Azarashvili T, Baburina Y, Galvita A, Grachev D, Stricker R, Reiser G (2015) In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2’,3’‐cyclic nucleotide‐3’‐phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 80, 41–50. [DOI] [PubMed] [Google Scholar]
- Kristal BS, Yu BP (1998) Dietary restriction augments protection against induction of the mitochondrial permeability transition. Free Radic. Biol. Med. 24, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Kim J, Lee CY, Kim H (2015) Expression of SIRT1 and SIRT3 varies according to age in mice. Anat. Cell Biol. 48, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFrance R, Brustovetsky N, Sherburne C, Delong D, Dubinsky JM (2005) Age‐related changes in regional brain mitochondria from Fischer 344 rats. Aging Cell 4, 139–145. [DOI] [PubMed] [Google Scholar]
- Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G, Kranias EG (2015) HAX‐1 regulates cyclophilin‐D levels and mitochondria permeability transition pore in the heart. Proc. Natl Acad. Sci. USA 112, E6466–E6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD (2007) Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell 6, 607–618. [DOI] [PubMed] [Google Scholar]
- Lapointe J, Hekimi S (2010) When a theory of aging ages badly. Cell. Mol. Life Sci. 67, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras M, Clement MV, Pervaiz S, Brenner C (2005) Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 20, 205–219. [DOI] [PubMed] [Google Scholar]
- Lebedev I, Nemajerova A, Foda ZH, Kornaj M, Tong M, Moll UM, Seeliger MA (2016) A novel in vitro CypD‐mediated p53 aggregation assay suggests a model for mitochondrial permeability transition by chaperone systems. J. Mol. Biol. 428, 4154–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CU, Song EK, Yoo CH, Kwak YK, Han MK (2012) Lipopolysaccharide induces CD38 expression and solubilization in J774 macrophage cells. Mol. Cells 34, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang C, Sun W, Li L, Wu B, Bai S, Li H, Zhong X, Wang R, Wu L, Xu C (2015) Exogenous hydrogen sulfide restores cardioprotection of ischemic post‐conditioning via inhibition of mPTP opening in the aging cardiomyocytes. Cell Biosci. 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhu J, Brink PR, Glass PS, Rebecchi MJ (2011) Age‐associated differences in the inhibition of mitochondrial permeability transition pore opening by cyclosporine A. Acta Anaesthesiol. Scand. 55, 622–630. [DOI] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E, Finkel T (2016) MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16, 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lores‐Arnaiz S, Lombardi P, Karadayian AG, Orgambide F, Cicerchia D, Bustamante J (2016) Brain cortex mitochondrial bioenergetics in synaptosomes and non‐synaptic mitochondria during aging. Neurochem. Res. 41, 353–363. [DOI] [PubMed] [Google Scholar]
- Lumini‐Oliveira J, Magalhaes J, Pereira CV, Moreira AC, Oliveira PJ, Ascensao A (2011) Endurance training reverts heart mitochondrial dysfunction, permeability transition and apoptotic signaling in long‐term severe hyperglycemia. Mitochondrion 11, 54–63. [DOI] [PubMed] [Google Scholar]
- Lustgarten MS, Bhattacharya A, Muller FL, Jang YC, Shimizu T, Shirasawa T, Richardson A, Van Remmen H (2012) Complex I generated, mitochondrial matrix‐directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 422, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Upneja A, Galecki A, Tsai YM, Burant CF, Raskind S, Zhang Q, Zhang ZD, Seluanov A, Gorbunova V, Clish CB, Miller RA, Gladyshev VN (2016) Cell culture‐based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. Elife 5, e19130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchissio MJ, Frances DE, Carnovale CE, Marinelli RA (2012) Mitochondrial aquaporin‐8 knockdown in human hepatoma HepG2 cells causes ROS‐induced mitochondrial depolarization and loss of viability. Toxicol. Appl. Pharmacol. 264, 246–254. [DOI] [PubMed] [Google Scholar]
- Marcil M, Bourduas K, Ascah A, Burelle Y (2006) Exercise training induces respiratory substrate‐specific decrease in Ca2+‐induced permeability transition pore opening in heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 290, H1549–H1557. [DOI] [PubMed] [Google Scholar]
- Martin LJ (2012) Biology of mitochondria in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 107, 355–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Semenkow S, Hanaford A, Wong M (2014a) Mitochondrial permeability transition pore regulates Parkinson's disease development in mutant α‐synuclein transgenic mice. Neurobiol. Aging 35, 1132–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Fancelli D, Wong M, Niedzwiecki M, Ballarini M, Plyte S, Chang Q (2014b) GNX‐4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C (2008) Age‐related activation of mitochondrial caspase‐independent apoptotic signaling in rat gastrocnemius muscle. Mech. Ageing Dev. 129, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather M, Rottenberg H (2000) Aging enhances the activation of the permeability transition pore in mitochondria. Biochem. Biophys. Res. Commun. 273, 603–608. [DOI] [PubMed] [Google Scholar]
- Mather MW, Rottenberg H (2002) The inhibition of calcium signaling in T lymphocytes from old mice results from enhanced activation of the mitochondrial permeability transition pore. Mech. Ageing Dev. 123, 707–724. [DOI] [PubMed] [Google Scholar]
- Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6, 337–350. [DOI] [PubMed] [Google Scholar]
- Medrano‐Fernandez I, Bestetti S, Bertolotti M, Bienert GP, Bottino C, Laforenza U, Rubartelli A, Sitia R (2016) Stress regulates aquaporin‐8 permeability to impact cell growth and survival. Antioxid. Redox Signal. 24, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merksamer PI, Liu Y, He W, Hirschey MD, Chen D, Verdin E (2013) The sirtuins, oxidative stress and aging: an emerging link. Aging 5, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A (2016) Two conserved histone demethylases regulate mitochondrial stress‐induced longevity. Cell 165, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min‐Wen JC, Jun‐Hao ET, Shyh‐Chang N (2016) Stem cell mitochondria during aging. Semin. Cell Dev. Biol. 52, 110–118. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J (2013) The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl Acad. Sci. USA 109, 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E (2011) Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 286, 40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai S, Rossi A, Di Daniele N, Melino G, Annicchiarico‐Petruzzelli M, Raschella G (2015) DNA repair and aging: the impact of the p53 family. Aging 7, 1050–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP (1992) The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J. Biol. Chem. 267, 16274–16282. [PubMed] [Google Scholar]
- Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, Hajnóczky G (2017) Tissue‐specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 18, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona R (2008) Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim. Biophys. Acta 1777, 1249–1262. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Grondin R, Yonutas HM, Haghnazar H, Gash DM, Zhang Z, Sullivan PG (2015) Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 36, 1903–1913. [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2013) Changes in the mitochondrial permeability transition pore in aging and age‐associated diseases. Mech. Ageing Dev. 134, 1–9. [DOI] [PubMed] [Google Scholar]
- Park SH, Ozden O, Jiang H, Cha YI, Pennington JD, Aykin‐Burns N, Spitz DR, Gius D, Kim HS (2011) Sirt3, mitochondrial ROS, ageing, and carcinogenesis. Int. J. Mol. Sci. 12, 6226–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson HC, Gerbeth C, Thiru P, Vogtle NF, Knoll M, Shahsafaei A, Samocha KE, Huang CX, Harden MM, Song R, Chen C, Kao J, Shi J, Salmon W, Shaul YD, Stokes MP, Silva JC, Bell GW, MacArthur DG, Ruland J, Meisinger C, Lodish HF (2015) A respiratory chain controlled signal transduction cascade in the mitochondrial intermembrane space mediates hydrogen peroxide signaling. Proc. Natl Acad. Sci. USA 112, E5679–E5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Haynes CM (2013) Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A (2009) The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P (1994) The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation‐reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J. Biol. Chem. 269, 16638–16642. [PubMed] [Google Scholar]
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F (2001) The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276, 12030–12034. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G (2006) Interaction of peroxidized cardiolipin with rat‐heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett. 580, 6311–6316. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Moro N, Paradies V, Ruggiero FM, Paradies G (2010) Increased susceptibility to Ca2+‐induced permeability transition and to cytochrome c release in rat heart mitochondria with aging: effect of melatonin. J. Pineal Res. 48, 340–346. [DOI] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Thomas MM, Wright KJ, Hepple RT (2011) Alterations in intrinsic mitochondrial function with aging are fiber type‐specific and do not explain differential atrophy between muscles. Aging Cell 10, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Pottecher J, Kindo M, Chamaraux‐Tran TN, Charles AL, Lejay A, Kemmel V, Vogel T, Chakfe N, Zoll J, Diemunsch P, Geny B (2016) Skeletal muscle ischemia‐reperfusion injury and cyclosporine A in the aging rat. Fundam. Clin. Pharmacol. 30, 216–225. [DOI] [PubMed] [Google Scholar]
- Priami C, De Michele G, Cotelli F, Cellerino A, Giorgio M, Pelicci PG, Migliaccio E (2015) Modelling the p53/p66Shc aging pathway in the shortest living vertebrate Nothobranchius furzeri . Aging Dis. 6, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam DA, Deepa SS, Liu Y, Hill S, Lin AL, Bhattacharya A, Shi Y, Sloane L, Viscomi C, Zeviani M, Van Remmen H (2014) Complex IV‐deficient Surf1(‐/‐) mice initiate mitochondrial stress responses. Biochem. J. 462, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Jin YN, von Bernhardi R, Johnson GV (2013) Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener. 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Tapia C, Pérez MJ (2017) Possible role of mitochondrial permeability transition pore in the pathogenesis of Huntington disease. Biochem. Biophys. Res. Commun. 483, 1078–1083. [DOI] [PubMed] [Google Scholar]
- Rasheed MZ, Tabassum H, Parvez S (2017) Mitochondrial permeability transition pore: a promising target for the treatment of Parkinson's disease. Protoplasma 254, 33–42. [DOI] [PubMed] [Google Scholar]
- Rasola A, Bernardi P (2014) The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 56, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS (2015) ROS‐dependent signal transduction. Curr. Opin. Cell Biol. 33, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Merkwirth C, De Magalhaes Filho CD, Dillin A (2016) Signaling networks determining life span. Ann. Rev. Biochem. 85, 35–64. [DOI] [PubMed] [Google Scholar]
- Rimessi A, Previati M, Nigro F, Wieckowski MR, Pinton P (2016) Mitochondrial reactive oxygen species and inflammation: molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 81, 281–293. [DOI] [PubMed] [Google Scholar]
- Riojas‐Hernandez A, Bernal‐Ramirez J, Rodriguez‐Mier D, Morales‐Marroquin FE, Dominguez‐Barragan EM, Borja‐Villa C, Rivera‐Alvarez I, Garcia‐Rivas G, Altamirano J, Garcia N (2015) Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 141, 32–43. [DOI] [PubMed] [Google Scholar]
- Ronchi JA, Henning B, Ravagnani FG, Figueira TR, Castilho RF, dos Reis SF, Vercesi AE (2015) Increased susceptibility of Gracilinanus microtarsus liver mitochondria to Ca2+‐induced permeability transition is associated with a more oxidized state of NAD(P). Oxid. Med. Cell. Longev. 2015, e940627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, Marbach M (1990) Regulation of Ca2+ transport in brain mitochondria. II. The mechanism of the adenine nucleotides enhancement of Ca2+ uptake and retention. Biochim. Biophys. Acta 1016, 87–98. [DOI] [PubMed] [Google Scholar]
- Rottenberg H, Wu S (1997) Mitochondrial dysfunction in lymphocytes from old mice: enhanced activation of the permeability transition. Biochem. Biophys. Res. Commun. 240, 68–74. [DOI] [PubMed] [Google Scholar]
- Runas KA, Malmstadt N (2015) Low levels of lipid oxidation radically increase the passive permeability of lipid bilayers. Soft Matter 11, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino C, Pelicci P, Giorgio M (2013) The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxid. Med. Cell. Longev. 2013, 719407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar CE, Dues DJ, Spielbauer KK, Machiela E, Cooper JF, Senchuk M, Hekimi S, Van Raamsdonk JM (2015) Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11, e1004972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriewer JM, Peek CB, Bass J, Schumacker PT (2013) ROS‐mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia‐reperfusion. J. Am. Heart Assoc. 2, e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzlander M, Wagner S, Ermakova YG, Belousov VV, Radi R, Beckman JS, Buettner GR, Demaurex N, Duchen MR, Forman HJ, Fricker MD, Gems D, Halestrap AP, Halliwell B, Jakob U, Johnston IG, Jones NS, Logan DC, Morgan B, Muller FL, Nicholls DG, Remington SJ, Schumacker PT, Winterbourn CC, Sweetlove LJ, Meyer AJ, Dick TP, Murphy MP (2014) The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature 514, E12–E14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen EZ, Song CQ, Lin Y, Zhang WH, Su PF, Liu WY, Zhang P, Xu J, Lin N, Zhan C, Wang X, Shyr Y, Cheng H, Dong MQ (2014) Mitoflash frequency in early adulthood predicts lifespan in Caenorhabditis elegans . Nature 508, 128–132. [DOI] [PubMed] [Google Scholar]
- Shin J, Mohrin M, Chen D (2015) Reversing stem cell aging. Oncotarget 6, 14723–14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan‐Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N (2010) VDAC, a multi‐functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285. [DOI] [PubMed] [Google Scholar]
- Shum LC, White NS, Nadtochiy SM, Bentley KL, Brookes PS, Jonason JH, Eliseev RA (2016) Cyclophilin D knock‐out mice show enhanced resistance to osteoporosis and to metabolic changes observed in aging bone. PLoS One 11, e0155709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H, Berndt C, Jones DP (2017) Oxidative stress. Annu. Rev. Biochem. 86, 715–748. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Allen RG (1985) Relationship between metabolic rate, free radicals, differentiation and aging: a unified theory. Basic Life Sci. 35, 75–104. [DOI] [PubMed] [Google Scholar]
- Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC (1992) Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 267, 14592–14597. [PubMed] [Google Scholar]
- Strom J, Xu B, Tian X, Chen QM (2016) Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 30, 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JA, Maddalena LA, Merilovich M, Robb EL (2014) A midlife crisis for the mitochondrial free radical theory of aging. Longev. Healthspan 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue MM, Tatton WG (2001) Mitochondrial membrane potential in aging cells. Biol. Signals Recept. 10, 176–188. [DOI] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol. Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddeo EP, Laker RC, Breen DS, Akhtar YN, Kenwood BM, Liao JA, Zhang M, Fazakerley DJ, Tomsig JL, Harris TE, Keller SR, Chow JD, Lynch KR, Chokki M, Molkentin JD, Turner N, James DE, Yan Z, Hoehn KL (2013) Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 3, 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajeddine N (2016) How do reactive oxygen species and calcium trigger mitochondrial membrane permeabilisation? Biochim. Biophys. Acta 1860, 1079–1088. [DOI] [PubMed] [Google Scholar]
- Takeyama N, Matsuo N, Tanaka T (1993) Oxidative damage to mitochondria is mediated by the Ca2+‐dependent inner‐membrane permeability transition. Biochem. J. 294, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Banerjee R, Starkova NN, Zhang SF, Calingasan NY, Yang L, Wille E, Lorenzo BJ, Ho DJ, Beal MF, Starkov A (2012) Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxid. Redox Signal. 16, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A (2016) Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toman J, Fiskum G (2011) Influence of aging on membrane permeability transition in brain mitochondria. J. Bioenerg. Biomembr. 43, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, Thoraval D, Crouzet M, Ichas F, De Pinto V, De Giorgi F (2009) Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress‐induced apoptosis. Cell Res. 19, 1363–1376. [DOI] [PubMed] [Google Scholar]
- Tsai H, Hewitt CW, Buchholz JN, Duckles SP (1997) Intracellular calcium buffering declines in aging adrenergic nerves. Neurobiol. Aging 18, 229–233. [DOI] [PubMed] [Google Scholar]
- Valasani KR, Sun Q, Fang D, Zhang Z, Yu Q, Guo Y, Li J, Roy A, ShiDu Yan S (2016) Identification of a small molecule cyclophilin D inhibitor for rescuing Aβ‐mediated mitochondrial dysfunction. ACS Med. Chem. Lett. 7, 294–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz CV, Marques R, Maia CJ, Socorro S (2015) Aging‐associated changes in oxidative stress, cell proliferation, and apoptosis are prevented in the prostate of transgenic rats overexpressing regucalcin. Transl. Res. 166, 693–705. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hekimi S (2015) Mitochondrial dysfunction and longevity in animals: untangling the knot. Science 350, 1204–1207. [DOI] [PubMed] [Google Scholar]
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H (2008) Superoxide flashes in single mitochondria. Cell 134, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lin J, Chen QZ, Zhu N, Jiang DQ, Li MX, Wang Y (2015) Overexpression of mitochondrial Hsp75 protects neural stem cells against microglia‐derived soluble factor‐induced neurotoxicity by regulating mitochondrial permeability transition pore opening in vitro. Int. J. Mol. Med. 36, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gong G, Wang X, Wei‐LaPierre L, Cheng H, Dirksen R, Sheu SS (2016a) Mitochondrial flash: integrative reactive oxygen species and pH signals in cell and organelle biology. Antiox. Redox Signal. 25, 534–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu D, Varin A, Nicolas V, Courilleau D, Mateo P, Caubere C, Rouet P, Gomez AM, Vandecasteele G, Fischmeister R, Brenner C (2016b) A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 7, e2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne J, Pryce G, Hill JM, Shi X, Lennerås F, Puentes F, Kip M, Hilditch L, Walker P, Simone MI, Chan AW, Towers GJ, Coker AR, Duchen MR, Szabadkai G, Baker D, Selwood DL (2016) Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J. Biol. Chem. 291, 4356–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Kenyon C (2016) Roles for ROS and hydrogen sulfide in the longevity response to germline loss in Caenorhabditis elegans . Proc. Natl Acad. Sci. USA 113, E2832–E2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Li H, Wang Y, Peng X, Shao H, Li H, Bai S, Xu C (2016) Exogenous spermine inhibits hypoxia/ischemia‐induced myocardial apoptosis via regulation of mitochondrial permeability transition pore and associated pathways. Exp. Biol. Med. 241, 1505–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP (1998) Direct demonstration of a specific interaction between cyclophilin‐D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 336, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shamoto‐Nagai M, Maruyama W, Osawa T, Naoi M (2016) Rasagiline prevents cyclosporine A‐sensitive superoxide flashes induced by PK11195, the initial signal of mitochondrial membrane permeabilization and apoptosis. J. Neural. Transm. 123, 491–494. [DOI] [PubMed] [Google Scholar]
- Yan S, Du F, Wu L, Zhang Z, Zhong C, Yu Q, Wang Y, Lue LF, Walker DG, Douglas JT, Yan SS (2016) F1F0 ATP synthase‐cyclophilin D interaction contributes to diabetes‐induced synaptic dysfunction and cognitive decline. Diabetes 65, 3482–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Unnikrishnan A, Deepa SS, Liu Y, Li Y, Ikeno Y, Sosnowska D, Van Remmen H, Richardson A (2017) A new role for oxidative stress in aging: the accelerated aging phenotype in Sod1‐/‐ mice is correlated to increased cellular senescence. Redox. Biol. 11, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang ZB, Xu JX (2003) Effect of cytochrome c on the generation and elimination of O2*‐ and H2O2 in mitochondria. J. Biol. Chem. 278, 2356–2360. [DOI] [PubMed] [Google Scholar]
- Zhen YF, Wang GD, Zhu LQ, Tan SP, Zhang FY, Zhou XZ, Wang XD (2014) P53 dependent mitochondrial permeability transition pore opening is required for dexamethasone‐induced death of osteoblasts. J. Cell. Physiol. 229, 1475–1483. [DOI] [PubMed] [Google Scholar]
- Zhu J, Rebecchi MJ, Tan M, Glass PS, Brink PR, Liu L (2010) Age‐associated differences in activation of Akt/GSK‐3β signaling pathways and inhibition of mitochondrial permeability transition pore opening in the rat heart. J. Gerontol. A Biol. Sci. Med. Sci. 65, 611–619. [DOI] [PubMed] [Google Scholar]
- Zhu J, Rebecchi MJ, Glass PS, Brink PR, Liu L (2013) Interactions of GSK‐3β with mitochondrial permeability transition pore modulators during preconditioning: age‐associated differences. J. Gerontol. A Biol. Sci. Med. Sci. 68, 395–403. [DOI] [PubMed] [Google Scholar]
- Ziegler DV, Wiley CD, Velarde MC (2015) Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol. Rev. 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulian A, Schiavone M, Giorgio V, Bernardi P (2016) Forty years later: mitochondria as therapeutic targets in muscle diseases. Pharmacol. Res. 113, 563–573. [DOI] [PubMed] [Google Scholar]