

Abstract
Background and Purpose
Methoxetamine (MXE) is a novel psychoactive substance that is emerging on the Internet and induces dissociative effects and acute toxicity. Its pharmacological effects have not yet been adequately investigated.
Experimental Approach
We examined a range of behavioural effects induced by acute administration of MXE (0.5–5 mg·kg−1; i.p.) in rats and whether it causes rapid neuroadaptive molecular changes.
Key Results
MXE (0.5–5 mg·kg−1) affected motor activity in a dose‐ and time‐dependent manner, inducing hypermotility and hypomotility at low and high doses respectively. At low and intermediate doses (0.5 and 1 mg·kg−1), MXE induced anxious and/or obsessive–compulsive traits (marble burying test), did not significantly increase sociability (social interaction test) or induce spatial anxiety (elevated plus maze test). At a high dose (5 mg·kg−1), MXE induced transient analgesia (tail‐flick and hot‐plate test), decreased social interaction time (social interaction test) and reduced immobility time while increasing swimming activity (forced swim test), suggesting an antidepressant effect. Acute MXE administration did not affect self‐grooming behaviour at any dose tested. Immunohistochemical analysis showed that behaviourally active doses of MXE (1 and 5 mg·kg−1) increased phosphorylation of ribosomal protein S6 in the medial prefrontal cortex and hippocampus.
Conclusions and Implications
MXE differentially affected motor activity, behaviour and emotional states in rats, depending on the dose tested. As reported for ketamine, phosphorylation of the ribosomal protein S6 was increased in MXE‐treated animals, thus providing a ‘molecular snapshot’ of rapid neuroadaptive molecular changes induced by behaviourally active doses of MXE.
Abbreviations
- BLA
basolateral amygdala
- IL
infralimbic cortex
- MXE
methoxetamine
- NAcC
nucleus accumbens core
- NAcS
nucleus accumbens shell
- NMDA
N‐methyl D‐aspartate
- PrL
prelimbic cortex
- rpS6
ribosomal protein S6
Introduction
In recent years, ‘legal highs’ have emerged and proliferated as legal substitutes for controlled drugs of abuse. Among these, methoxetamine (MXE) is one of the newest compounds purposely designed and increasingly available on the Internet as ‘legal ketamine’ (European Monitoring Centre for Drugs and Drug Addiction, 2014). MXE (2‐(3‐methoxyphenyl)‐2‐(N‐ethylamino)cyclohexanone) is an arylcyclohexylamine derivative that shares similar chemical structure with ketamine and phencyclidine but with modifications that could confer (i) more potency and higher opioid receptor affinity than phencyclidine (Corazza et al., 2013; Adamowicz and Zuba, 2015) and (ii) weaker analgesic and anaesthetic effects but longer duration of action than ketamine (Morris and Wallach, 2014). Over the past few years, its use has been associated with sympathomimetic toxicity (Wood et al., 2012), accidental fatal intoxication (Wikström et al., 2013; Imbert et al., 2014) and acute neurological toxicity (Elian and Hackett, 2014; Fassette and Martinez, 2016), including a dissociative state (Ward et al., 2011) and reversible cerebellar toxicity (Shields et al., 2012). Recently, MXE was found to potently inhibit neuronal activity in vitro (Hondebrink et al., 2016), with an IC50 value (0.5 μM) that overlaps with serum concentrations (0.1–2 μM) expected in recreational users (Abe et al., 2013; Łukasik‐Głebocka et al., 2013).
MXE is a dissociative anaesthetic thought to act as a (i) non‐competitive antagonist of NMDA receptors (Coppola and Mondola, 2012), (ii) inhibitor of dopamine reuptake and (iii) agonist at muscarinic and 5‐HT2 receptors (Adamowicz and Zuba, 2015). On the basis of chemical similarities, MXE has been hypothesized to induce ketamine‐like effects (Hofer et al., 2012) and to produce a rapid antidepressant action (Coppola and Mondola, 2012). Preclinical studies have recently started investigating its pharmacokinetics, behavioural effects and underlying brain mechanisms (Hajkova et al., 2016; Horsley et al., 2016; Hondebrink et al., 2017). In rats, MXE was reported to induce conditioned place preference and maintain intravenous self‐administration behaviour (Botanas et al., 2015) and to substitute for ketamine in a drug self‐administration substitution study (Mutti et al., 2016). We recently showed that MXE fully generalize to ketamine interoceptive stimulus in a two‐lever operant drug discrimination paradigm in rats trained to discriminate ketamine from saline, similarly to the NMDA channel blocker MK‐801, thus exhibiting ketamine‐like discriminative stimulus properties (Chiamulera et al., 2016). In support of its abuse liability, following MXE administration we observed an enhanced level of dopamine in the rat nucleus accumbens shell (NAcS) and a dose‐dependent stimulation of the firing rate and burst firing of dopamine neurons in the ventral tegmental area projecting to the NAcS (Mutti et al., 2016). Intriguingly, MXE was recently reported to be used for self‐medication purposes to treat chronic foot pain (Maskell et al., 2016) and post‐traumatic stress disorder (Striebel et al., 2017), suggesting possible use as an analgesic and calming drug.
Based on the collected body of evidence and being aware of the gaps in our understanding of the pharmacological effects of MXE, this study evaluated the effects of acute doses of MXE (0.5–5 mg·kg−1) given i.p., on spontaneous locomotor activity, analgesia (hot‐plate and tail‐flick test), repetitive and obsessive–compulsive behaviour (marble burying test and self‐grooming behaviour), spatial (elevated plus maze test) and social anxiety (social interactions) and depression (forced swim test). Finally, we tested the possibility that MXE, similarly to ketamine, may increase the phosphorylation of the ribosomal protein S6 (rpS6) (Tedesco et al., 2013) in brain areas known to be involved in mood and reward, that is, prelimbic (PRL) and infralimbic (IL) cortices, nucleus accumbens core (NAcC) and NAcS, hippocampus (CA1, CA2 and CA3) and basolateral amygdala (BLA).
Methods
Animals
All animal care and experimental procedures were carried out in an animal facility according to Italian (D.L. 26/2014) and European Council directives (63/2010) and in compliance with the approved animal policies by the Ethical Committee for Animal Experiments (CESA, University of Cagliari) and the Italian Department of Health, and were approved by the local Animal Care Committees. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Adult male Sprague Dawley rats (250–275 g, Harlan‐Nossan, Italy) were housed 4 per cage under a 12/12 h light/dark cycle (light on: 07:00 h) with constant room temperature (22 ± 2°C) and humidity (60%) and free access to standard laboratory chow and tap water. Experiments were conducted during the light phase (10:00–13:00 h) in rooms illuminated with the same light intensity of the housing room. Animals were left undisturbed in each experimental room for 1 h acclimatation period before starting behavioural testing. All efforts were made to minimize animal suffering and discomfort and to reduce the number of animals used.
Locomotor activity test
To assess the effects of MXE administration on spontaneous motor activity, rats were divided into five experimental groups (n = 6–8 per group) and treated with MXE 0, 0.5, 1, 2.5 and 5 mg·kg−1. Horizontal and vertical activity, total distance, margin and centre time were measured using Digiscan Animal Activity Analyser (Omnitech Electronics, USA) every 10 min intervals for a total of 60 min as previously described (Castelli et al., 2014).
Tail‐flick and hot‐plate tests
As pain is processed through multiple pathways, to test potential analgesic effects of MXE, we used two different thermal assays, the hot‐plate and the tail‐flick test, that primarily measure supraspinal and spinal response to a stimulus respectively (Cannon et al., 2010). These assays have been extensively used for the assessment of analgesia in rodents (Gagliese and Melzack, 2000). In the tail‐flick test (Tail‐Flick Unit 37360; Ugo Basile, Comerio, Italy), the animal's tail was gently placed at the centre of a concentrated light beam (50 W) with an irradiation intensity of 55°C and the basal thermal reaction time (s), that is, flicking or removing the tail, was assessed in each animal twice every 15 min before receiving MXE i.p. (0, 1, 2.5 and 5 mg·kg−1; n = 8–10 per group). After drug treatment, tail‐flick latency was calculated at 15 min intervals for 90 min. In order to prevent tissue damage, a different patch of the tail skin was stimulated in each trial and a cut‐off time of 12 s was fixed. The hot‐plate apparatus consisted of an aluminium plate (10 × 20 cm) with the underlying heating system and a Plexiglas observation chamber (Socrel, model‐DS37; Ugo Basile, Comerio, Italy). The temperature of hot‐plate surface was set at 55 ± 1°C. The time between placement of the animal on the hot‐plate and the occurrence of jumping, double steps, licking or shaking of a paw was recorded as the reaction time (s). Twenty minutes after baseline measurement, rats were divided in four different groups (n = 8–10 per group) and injected i.p. with MXE 0, 1, 2.5 and 5 mg·kg−1. After drug treatment, reaction time (s) was calculated at 15 min intervals for 90 min, with a cut‐off time of 12 s.
Marble burying
The marble burying test, a cost‐effective animal model of compulsive activity with good face validity (Albelda and Joel, 2012), was conducted in open transparent plastic boxes (54 × 34.5 × 20 cm) with 5 cm of fresh hardwood chip bedding as previously described (Satta et al., 2016). Twenty‐four standard glass marbles (1.5 cm in diameter, arranged in six rows of four marbles each) were placed uniformly over the surface. Individual subjects were placed in the test cage in a marble‐free area (34.5 × 15 cm) 30 min after administration of MXE (0, 0.5, 1 and 2.5 mg·kg−1, i.p.; n = 7–11 per group.), and activity was monitored for 30 min by a video camera placed above the cage. At the end of the session, animals were gently removed from the Plexiglas boxes, and the number of marbles partially (≥67%) and totally (>95%) buried was counted (Anymaze Software, Stoelting, Illinois, USA). New bedding was used for each animal, and marbles were cleaned with soap and tap water.
Self‐grooming
Each animal was placed individually in open transparent plastic boxes (54 × 34.5 × 20 cm) and allowed to freely explore the box. After 10 min habituation, rats were injected with MXE (0, 0.5, 1, 2.5 and 5 mg·kg−1, i.p.; n = 6 per group), and self‐grooming behaviour was monitored twice for 5 min, after 30 and 60 min from drug administration. Two independent investigators, blind to treatments, sat approximately 1.5 m from the test cage and recorded in real time with a stopwatch the cumulative total time (s) spent in the following components of grooming behaviour: face and head washing, body grooming, genital and tail grooming, scratching and paw licking.
Elevated plus maze
The elevated plus maze test was used to evaluate possible spatial anxiety‐like effects of MXE. Apparatus and procedure were as previously described (Scherma et al., 2015). Each rat received MXE (0, 0.5, 1 and 5 mg·kg−1, i.p.; n ≥ 6 per group) 30 min before being placed on the central platform facing an open arm and left free to explore the maze for 5 min; after which, they were removed and the apparatus cleaned with H2O2 to eliminate olfactory traces. Tests were videotaped (Anymaze Software, Stoelting) and the animal's behaviour scored by a observer, blinded to treatments. The percentage of time spent in open arms (all four paws into an arm) [time spent in open arms/(total time) × 100] and the percentage of the number of open‐arm entries [open arms entries/(open arms entries + closed arms entries) × 100] were used as measures of an anxiety‐like state.
Social interaction
To evaluate MXE‐induced effects on sociability, the social interaction task was performed as previously described (Spano et al., 2010; Castelli et al., 2014). Each rat received MXE (0, 0.5, 1 and 5 mg·kg−1, i.p.; n = 6 per group) 30 min before being placed for 10 min into the arena, facing the opposite corner, with a novel unfamiliar rat of same sex and similar weight. Time spent in social interactions (sniffing, following or grooming the partner, boxing and wrestling) and number of interactions were measured using Anymaze Software (Stoelting). After each session, the box was cleaned with H2O2 and new bedding was used for each pair of animals.
Forced swim test
As recently described (Satta et al., 2016), the forced swim test was carried out in two different days, with a 15 min pre‐swim session conducted 24 h before the test to facilitate the rat to quickly adopt an immobile posture on the test day, thus easing the detection of a potential antidepressant effect. On the test day, each rat was treated with MXE (0, 0.5, 1 and 5 mg·kg−1, i.p.; n ≥ 6 per group) 30 min before being placed individually for 5 min in a clear Plexiglas cylinder (diameter: 20 cm) filled with 30 cm water (25 ± 2°C). At the end of the session, animals were removed and thoroughly dried under a heating red lamp (30°C). Each session was videotaped (Anymaze Software, Stoelting), and a investigator analysed videos, without knowledge of the treatments. A decrease in immobility (time spent floating in the water with front paws not moving) and an increase in swimming or in the latency to the first episode of immobility are considered indices of an antidepressant‐like effect.
Immunohistochemistry
Immunohistochemical assessment of rpS6 phosphorylation was performed as previously described (Tedesco et al., 2013). Briefly, rats were anaesthetized by an i.p. injection of 350 mg·kg−1·2 mL−1 chloral hydrate (Fluka, Italy) 60 min after drug treatment, then transcardially perfused with heparin 100 UI·L−1 (Sigma‐Aldrich, Milan, Italy) and paraformaldehyde 4% in PBS. This time point was selected as showed significant changes in rpS6 phosphorylation levels under our conditions (Tedesco et al., 2013). The brain was removed and post‐fixed for 2 h into paraformaldehyde 4% in PBS, then put in 30% sucrose, as a cryoprotective medium, and left over the weekend. Free‐floating sections (40 μm), obtained from a freezing microtome, containing (coordinated from the atlas of Paxinos and Watson, 2008) prefrontal cortices ,PrL and IL, corresponding to a bregma +2.70 section; accumbal areas shell (NacS) and core (NacC) (bregma +1.70 mm); hippocampal CA1, CA2 and CA3 (bregma −3.30 mm); and BLA (bregma −3.00 mm), were processed for phosphorylated rpS6. After washing in PBS, endogenous peroxidase was neutralized with 0.75% H2O2 for 10 min. Sections were blocked in PBS + 0.5% HS + 0.5% Triton X‐100 wash solution and then incubated overnight at 4°C in anti‐phospho‐ rpS6(Ser235/236)antibody (1:1000, rabbit polyclonal; Cell Signaling, EuroClone S.p.A., Milano, Italy) in wash solution. After washes, sections were incubated for 2 h at room temperature, with anti‐rabbit biotinylated antibody (1:1000, AmershamGE Healthcare Europe, Milano, Italy). Following washes in wash solution, and finally in PBS, tissue sections were visualized using the VectaStain ABC kit and developed in DAB peroxidase substrate. Sections were mounted on gelatinized slides, dehydrated, then closed with Entellan. Images of the sections were acquired using a light transmission microscope (Axioskop 2 Zeiss). Six images for each region (one for each hemisphere, three sections for each rat, that is 2 × 3 = 6 images per region per rat) were acquired by the connected video camera (Optikam B3) using a 10× objective (0.3 mm2). The number of neurons positive for phospho‐ rpS6(Ser235/236) was counted using the NIH software 74 ‘Image‐J’. Intensity threshold and the minimum and maximum cell size parameter values were initially determined in an empirical manner under blinded conditions. The dependent variable for the immunohistochemical experiments was the number of neurons staining positive for phospho‐rpS6.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as mean ± SEM. Locomotor activity and analgesia tests were analysed by repeated measures two‐way ANOVA. Where ANOVA reached a significant factor effect (P < 0.05), it was followed by the Bonferroni post hoc test for multiple comparisons. Data from elevated plus maze, marble burying test, social interaction and forced swim test were analysed by one‐way ANOVA. Where ANOVA reached a significant factor effect (P < 0.05), it was followed by Dunnett's multiple comparison test against the corresponding vehicle (VEH) group. For immunohistochemistry, the positive neuron count for rpS6 phosphorylation was carried out for each region and analysed by one‐way ANOVA. Where ANOVA reached a significant factor effect (P < 0.05), it was followed by Dunnett's test against the corresponding VEH group. All analyses were performed using the GraphPad software package for Windows (Prism version 6.01; GraphPad, San Diego, California, USA). A P value of less than 0.05 was considered statistically significant. All experiments included at least three doses of MXE and a minimum of six animals per group; sizes of samples differed according to inclusion of preliminary data from pilot experiments performed during the early phase of the study.
Materials
MXE was purchased from LGC Standards S.r.l. (Milan, Italy), dissolved in sterile physiological saline (0.9%) and injected i.p. at doses ranging from 0.5 to 5 mg·kg−1 (volume of injection: 5 mL·kg−1). MXE was administered immediately before starting the locomotor activity test, while in all other tests, it was administered 30 min before. Each animal was used in one test only.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d,e).
Results
Locomotor activity
MXE affected spontaneous motor activity in a dose‐ and time‐dependent manner. As shown in Figure 1A, the lowest dose of MXE tested (0.5 mg·kg−1) increased horizontal motor activity from 20 to 40 min after treatment with respect to control group. On the contrary, the highest dose of MXE (5 mg·kg−1) reduced horizontal activity during the first 20 min (two‐way ANOVA; significant main effect of drug treatment × time interaction: F (20,145) = 4.15, P < 0.05). No significant differences were found in the groups treated with the two intermediate doses of MXE (1 and 2.5 mg·kg−1).
Figure 1.
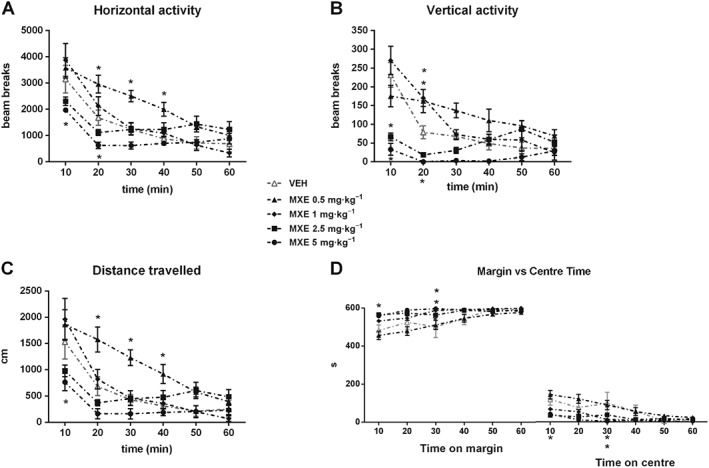
Spontaneous motor activity in MXE‐treated rats. Spontaneous horizontal (A) and vertical (B) activity, expressed as mean counts of photobeam interruptions, total distance (cm) travelled (C) and time (s) spent on margin or centre (D) after treatment with VEH (n = 8), MXE 0.5 (n = 8) or MXE 1, 2.5 and 5 mg·kg−1 (n = 6 per group). Data are shown as means (±SEM) during 60 min test. *P < 0.05, significantly different from corresponding VEH‐treated groups; two‐way ANOVA for repeated measures followed by Bonferroni post hoc test for multiple comparisons.
As for vertical activity (Figure 1B), two‐way ANOVA showed a significant main effect of drug treatment × time interaction: (F (20,145) = 6.21, P < 0.05). Specifically, with respect to control group, rats treated with MXE( 0.5 and 1 mg·kg−1) showed a higher activity 20 min after drug administration, while those treated with MXE (2.5 and 5 mg·kg−1) displayed a significantly lower activity during the first 10 min (both doses) and 20 min (MXE 5 mg·kg−1 only) of the test. Moreover, animals treated with MXE (0.5 mg·kg−1) displayed higher distance travelled from 20 to 40 min after administration (Figure 1C), while rats treated with MXE (5 mg·kg−1) displayed lower values during the first 10 min (two‐way ANOVA significant main effect of drug treatment × time interaction: F (20,145) = 3.55, P < 0.05). No significant differences were found in rats treated with the intermediate doses of MXE (1 and 2.5 mg·kg−1). Finally, significant differences were found between VEH‐ and MXE‐treated rats in the time spent on margin and on centre (Figure 1D). Indeed, MXE (1 and 2.5 mg·kg−1) significantly increased the time spent on margin 30 min after treatment, while MXE (5 mg·kg−1) induced this effect 10 min after treatment. A significant reduction in the time spent on centre, which suggests an anxiety‐like state, was observed 10 min after administration of MXE (5 mg·kg−1) and 30 min after MXE (1 and 2.5 mg·kg−1 ; two‐way ANOVA significant main effect of drug treatment × time interaction: F (20,145) = 2.11, P < 0.05).
Tail‐flick and hot‐plate assays
Two‐way ANOVA did not revealed significant variations in the thermal threshold (i.e. analgesia) in the tail‐flick test following MXE treatment (Figure 2A). However, in the hot‐plate test (Figure 2B), animals injected with MXE (5 mg·kg−1) displayed a significantly increased thermal threshold from 30 to 60 min after MXE administration with respect to VEH‐treated animals (two‐way ANOVA significant main effect of drug treatment × time interaction: F (18,180) = 1.82, P < 0.05).
Figure 2.
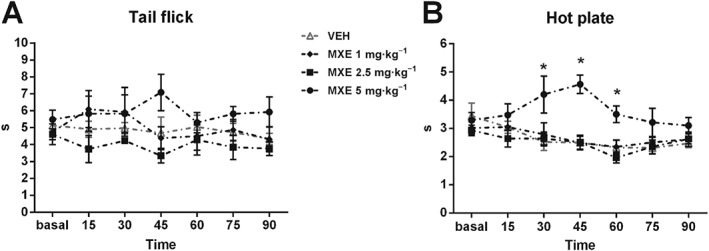
Analgesic effects of MXE (1, 2.5 and 5 mg·kg−1) during tail‐flick (A) and hot‐plate (B) tests. Data are shown as means (±SEM) during 90 min test (VEH: n = 10; MXE: n = 8 per group). *P < 0.05, significantly different from corresponding VEH‐treated groups; two‐way ANOVA for repeated measures followed by Bonferroni post hoc test for multiple comparisons.
Marble burying
In the marble burying test, the total number of marbles buried by animals pretreated with MXE (0.5 and 1 mg·kg−1) was significantly higher than in control group (Figure 3). In particular, a significant drug effect was evident when considering the number of marbles fully covered by wood chip bedding rather than that of partly buried marbles. Indeed, no statistical differences were found in the mean number of marbles partly buried by VEH‐ and MEX‐treated groups. One‐way ANOVA confirmed that rats injected with MXE (0.5 and 1 mg·kg−1) exhibited a higher level of burying activity than control rats (significant main effect of treatment: F (11,96) = 6.153, P < 0.05). Conversely, the higher dose of MXE (2.5 mg·kg−1) did not significantly alter the number of buried marbles compared with the VEH‐treated group.
Figure 3.
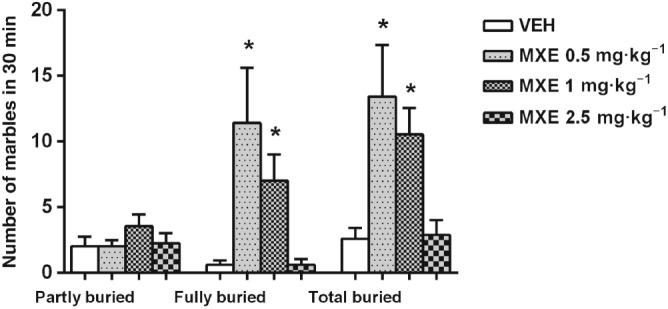
Number of marbles partly (≥67%) or entirely (>95%) covered with bedding by VEH‐ and MXE‐treated rats during the 30 min marble burying test (VEH: n = 10; MXE 0.5 mg·kg−1: n = 7; MXE 1 mg·kg−1: n = 11; MXE 2.5 mg·kg−1: n = 8 rats). Total buried stands for the sum of partly and fully buried marbles. Data are shown as means (±SEM). *P < 0.05, significantly different from corresponding VEH‐treated groups; one‐way ANOVA followed by Dunnett's multiple comparison test.
Self‐grooming behaviour
At the doses tested, MXE did not alter self‐grooming behaviour in rats, as the cumulative time (s) spent by MXE‐treated animals in face/head washing, body/genital/tail grooming, scratching and paw licking did not significantly differ from control group at any time interval, cumulative mean values being as follows: VEH: 42.5 s; MXE 0.5 mg·kg−1: 39.7 s; MXE 1 mg·kg−1: 45.2 s; MXE 2.5 mg·kg−1: 44.7 s; MXE 5 mg·kg−1; 40.3 s.
Elevated plus maze
As shown in Figure 4, the percentage of time spent in open arms was not significantly different between VEH‐ and MXE (0.5, 1 and 5 mg·kg−1)‐treated rats (panel A). Similarly, MXE (0.5 and 1 mg·kg−1) did not alter the percentage of time spent in closed arms (Figure 4A). However, the percentage of time spent in closed arms was increased in MXE 5 mg·kg−1‐treated rats with respect to the control group (one‐way ANOVA significant main effect of treatment: F (3,26) = 9.577, P < 0.05). Conversely, the percentage of entries in both arms (Figure 4B) was not significantly different between VEH‐ and MXE‐treated rats, while total arms entries were reduced in rats treated with MXE (5 mg·kg−1 ;Figure 4C) (significant main effect of treatment: F (3,26) = 11.51, P < 0.05).
Figure 4.

Effect of MXE (0.5, 1 and 5 mg·kg−1) in the elevated plus maze test (VEH and MXE 0.5 and 1 mg·kg−1: n = 8 per group; MXE 5 mg·kg−1: n = 6). Results are expressed as percentage of total time spent in open and closed arms (A), number of entries in open and closed arms (B) and number of total arms entries (C). Data are shown as means (±SEM). *P < 0.05, significantly different from corresponding VEH‐treated groups; one‐way ANOVA followed by Dunnett's multiple comparison test.
Social interaction
In the social interaction test, one‐way ANOVA detected a significant main effect of treatment. Specifically, as shown in Figure 5A, a significant decrease in the time spent in social interaction was observed in rats pretreated with the dose of 2.5 mg·kg−1 of MXE, compared with VEH‐treated rats (F (3,20) = 10.39, P < 0.05). Conversely, a positive (not statistically significant) trend was observed in rats treated with MXE(0.5 and 1 mg·kg−1). No significant effect was found at any dose of MXE in the number of social interactions (Figure 5B) although, when compared with VEH‐treated rats, a positive trend was found in animals pretreated with MXE (0.5 and 1 mg·kg−1) and a negative trend in those treated with MXE (2.5 mg·kg−1).
Figure 5.
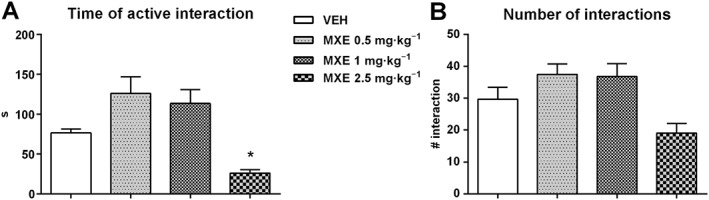
Effect of MXE (0.5, 1 and 2.5 mg·kg−1, n = 6 per group) in the social interactions test. Results are expressed as time (s) spent in active interaction (A) and number of interactions (B). Data are expressed as means (±SEM) during 10 min test. *P < 0.05, significantly different from VEH‐treated groups; one‐way ANOVA followed by Dunnett's multiple comparison test.
Forced swim test
In the forced swim test, one‐way ANOVA revealed a significant main effect of treatment (F (11,63) = 53.31, P < 0.05). Specifically, the highest dose of MXE tested (5 mg·kg−1) significantly reduced immobility and increased the time spent in swimming as compared with VEH group, which suggests an anti‐depressive effect (Figure 6). Accordingly, MXE (5 mg·kg−1) also induced a not significant, decreasing , trend in the time spent in climbing, compared with the control group.
Figure 6.
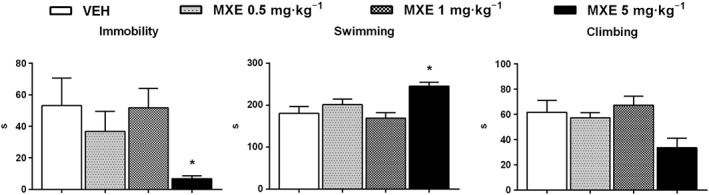
Effects of MXE (0.5, 1 and 5 mg·kg−1) in the forced swim test (VEH and MXE 0.5 and 1 mg·kg−1: n = 6 per group; MXE 5 mg·kg−1: n = 7). Data are shown as means (±SEM) time spent in immobility, swimming and climbing behaviour. *P < 0.05, significantly different from corresponding VEH‐treated groups; one‐way ANOVA followed by Dunnett's multiple comparison test.
Immunohistochemical assessment of MXE‐induced brain rpS6 phosphorylation
One‐way ANOVA showed a significant MXE main effect in PrL (F (2,15) = 18.53, P < 0.05), IL (F (2,15) = 27.72, P < 0.05), hippocampal areas CA1 (F (2,15) = 21.22, P < 0.05), CA2 (F (2,15) = 13.03, P < 0.05) and CA3 (F (2,15) = 14.38, P < 0.05), but not in accumbal nuclei (NAcC and NAcS) and BLA. As shown in Figure 7 (lower panel), MXE induced a significant increase in the mean number per mm2 (±SEM) of neurons expressing phosphorylated rpS6 in PrL at 1 and 5 mg·kg−1 doses, with the latter dose showing an increase lower than MXE (1 mg·kg−1), suggesting a plateau of effect, similar to the results from CA2. On the other hand,the effects of MXE in IL was dose‐related. Similar MXE dose‐related patterns of increased rpS6 phosphorylation was shown in CA1 and CA3.
Figure 7.
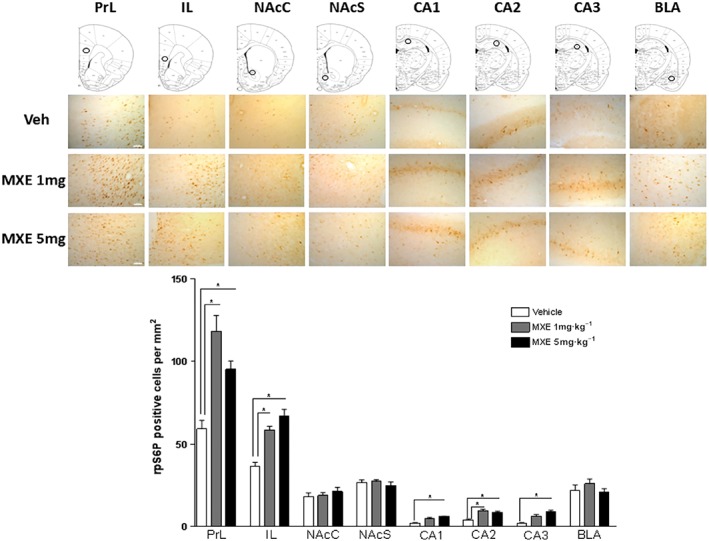
Immunohistochemical assessment of MXE‐induced rpS6P phosphorylation in drug addiction and reward‐related brain areas. Upper panel: schematic diagram of brain frontal sections (top row) showing the medium magnification microscopic field (circle) and representative high magnification images (0.3 mm2 sections) of region‐of‐interest within PrL and IL cortex; NAcC and NAcS; hippocampus CA1, CA2 and CA3; and BLA of rats treated with vehicle (Sal), MXE 1 or MXE 5 mg·kg−1. Scale bar: 100 μm. Lower panel: number of phosphorylated rpS6 positive neurons·mm−2 in PrL, IL, NAcC, NAcS, CA1, CA2, CA3 and BLA of rats treated with vehicle (Sal), MXE (1 mg·kg−1 or 5 mg·kg−1). Data are mean counts from three adjacent sections, from both hemispheres, for six rats per group. *P < 0.05, significantly different from corresponding VEH‐treated groups; one‐way ANOVA followed by Dunnett's post hoc test.
Discussion
This study characterizes the behavioural effects induced by an acute exposure to MXE in rats tested in a battery of behavioural assays. The range of MXE doses ≤5 mg·kg−1 used in our experiments was selected based on a pilot toxicity text showing acute toxicity and severe motor impairments at higher doses. MXE doses selected for behavioural testing were first evaluated at motor level. Our locomotor activity data, showing transient hypermotility and hypomotility at low and high doses of MXE, respectively, slightly differ from those reported by Botanas et al. (2015) and are in apparent contrast with those reported by Halberstadt et al. (2016) and Horsley et al. (2016). In Botanas et al. (2015) study, MXE (1.25, 2.5 and 5 mg·kg−1, i.p.) did not produce any significant change in the distance travelled and movement duration. However, the cumulative locomotor activity was measured during the three sessions of the conditioning phase of the place preference test, which makes it difficult to detect immediate and transient increments or decrements in motor activity. On the other hand, Halberstadt et al. (2016) showed that s.c. injections of a very high dose of MXE (10 mg·kg−1) induced locomotor hyperactivity, while a low dose (1 mg·kg−1) transiently reduced motor activity. Similarly, Horsley et al. (2016) reported that MXE (10 and 40 mg·kg−1, given s.c.) increased locomotor activity. While we did not test MXE doses higher than 5 mg·kg−1, which we excluded after initial toxicological screening, we observed opposite motor effects induced by MXE (1 and 5 mg·kg−1). A number of different experimental conditions might explain such discrepancies, including the different route of administration used in our studies and the other two studies, that is, i.p. or s.c. respectively. Further differences are the phases of the diurnal cycle when the experiments were carried out (Halberstadt et al., 2016), the rat strain used (Horsley et al., 2016) and motor parameters analysed. In particular, we evaluated horizontal, vertical activity and distance travelled in standard motor activity cages, while in the other two studies, motor activity was quantified as number of crossings between any of the eight sections within the behavioural pattern monitor and by a descriptive statistic (spatial d) that calculated the spatial structure of locomotor paths (Halberstadt et al., 2016), or by trajectory length in cm (corrected for deviations less than 3 cm) over 5 min time blocks during 30 min sessions (Horsley et al., 2016). Remarkably, irrespective of the specific motor parameter affected by MXE and of the direction of its effects, all of the three studies indicated a biphasic effect of MXE on locomotor activity, as previously reported for ketamine and MK‐801 (Castagné et al., 2012).
Given the recent use of MXE as self‐medication to alleviate chronic foot pain (Maskell et al., 2016), we evaluated the anti‐nociceptive effects of MXE in two different but complementary tests for analgesia, the tail‐flick and hot‐plate test. The highest dose of MXE tested (5 mg·kg−1) increased the latency time of reaction, indicating an analgesic effect, which was brief and did not reach statistical significance in the tail‐flick test but was long‐lasting and significantly different from VEH‐treated group in the hot‐plate test. This effect is unlikely to be related to the MXE‐induced motor effects, the depressant motor effect of MXE (5 mg·kg−1) is no longer more evident at the time of the test, that is, at 30 min after administration. As MXE shows a submicromolar affinity for the NMDA receptor (Roth et al., 2013), It is more likely that NMDA receptor antagonism accounts for the analgesic effect of high doses of MXE. Interestingly, in the tail‐flick test, i.p. administration of ketamine at doses up to 5 mg·kg−1 did not affect the basal latency in rats, that is, the nociceptive threshold (Huang et al., 2005), while significant analgesic effects have been reported following administration of higher (50 and 160 mg·kg−1) doses of ketamine (Baumeister and Advokat, 1991). Similarly, in the hot‐plate test, rats treated i.p. with ketamine 10–20 mg·kg−1 did not change their latency of reaction (Getova and Doncheva, 2011) and a significant analgesic effect of ketamine occurred only following administration of higher doses of ketamine (25 mg·kg−1) (Shikanai et al., 2014). That MXE induces its antinociceptive effects at doses lower than those classically reported for ketamine could be due to the presence of the methoxy group, which is likely to increase μ‐opioid receptor affinity, and to that of the N‐ ethylamino group that could increase potency of action. Accordingly, the insertion of the 2‐methoxy group in the structure of phencyclidine increases tail‐flick latency and reduces the time to onset of analgesia, compared with ketamine (Ahmadi and Mahmoudi, 2006). However, MXE‐induced antinociceptive effects might involve different sites in the brain, including non‐NMDA glutamate receptors, and other (not glutamatergic) neurotransmission systems (Forman, 1999). Indeed, MXE displayed affinity for the 5‐HT transporter, the dopamine transporter and the noradrenaline transporter but not for σ receptors (Roth et al., 2013; Hondebrink et al., 2017), was found to significantly activate the mesolimbic dopaminergic system (Mutti et al., 2016) and to inhibit monoamine transporters more potently than ketamine (Hondebrink et al., 2017).
In the marble burying test, contrary to anxiolytic (Archer et al., 1987), antidepressant (Nicolas et al., 2006) and antipsychotic drugs (Bruins Slot et al., 2008), MXE increased burying behaviour at low and intermediate doses, with the maximal effect observed at 0.5 mg·kg−1, while not inducing significant effects at higher doses. The effect of MXE we observed in this test is the opposite of that reported after acute exposure to nicotine (Anderson and Brunzell, 2012), morphine (Umathe et al., 2012; Kitanaka et al., 2015) and cannabinoids (Casarotto et al., 2010; Deiana et al., 2012) in mice. Quite unexpectedly, it is also dissimilar from that of NMDA receptor antagonists, which typically reduce marble‐burying behaviour (Egashira et al., 2008; Iijima et al., 2010). Yet it closely resembles the effects of the non‐selective serotonergic agonist meta‐chlorophenylpiperazine, which acutely increases the number of buried marbles at low but not high doses (Nardo et al., 2014), thus suggesting that MXE‐induced increased burying behaviour may involve the 5‐HT system rather than (or besides) the glutamatergic one. However, we cannot exclude, in this test, an influence of the stimulant motor effect of MXE (0.5 mg·kg−1), as hypermotility was still evident 30 min after drug administration, although it gradually diminished over time and was no longer significant 40 min after administration, that is, after the first 10 min of the 30 min test. To confirm the occurrence of repetitive behaviour induced by low doses of MXE and suggested by marble burying test data, self‐grooming behaviour was also examined. Rodent grooming behaviour consists of specific stereotyped patterns of sequential movements that can be affected by experimental manipulation, including administration of dopaminergic drugs (Kalueff et al., 2016). Given the reported stimulation by MXE of mesolimbic dopamine transmission (Mutti et al., 2016), we expected the time spent in self‐grooming to be increased following acute MXE administration. Thus, finding that MXE did not alter such a behaviour, even at a low dose previously showed to increase marble burying behaviour, was quite surprising. Notably, a lack of association between self‐grooming and marble burying behaviour in both rat and mouse models of autism has been reported (Reimer et al., 2015; Kratsman et al., 2016). Accordingly, these two types of repetitive behaviours have also been reported to go in the opposite direction in genetic mouse model of autism spectrum disorder (Sungur et al., 2014), thus suggesting that drug effects on marble burying and self‐grooming behaviour may diverge depending on the specific drug or animal model used.
To characterize the anxiety profile of rats acutely treated with MXE, we used two different animal tasks each measuring a specific type of anxiety, namely, the elevated plus maze and the social interaction test to evaluate spatial and social anxiety respectively. The elevated plus maze test gives information about the spatial anxiety of an animal, as the innate fear of rodents towards open spaces reduces the exploration of open arms. Non‐competitive NMDA receptor antagonists are typically considered potential anxiolytic substances (Wiley et al., 1995). However, studies investigating the anxiety‐related effects of ketamine in animal models are contradictory, reporting anxiogenic, anxiolytic or null effects (Babar et al., 2001; Becker et al., 2003). In our study, low to intermediate doses of MXE did not alter spatial anxiety in the elevated plus maze test. However, the highest dose tested (5 mg·kg−1) increased the time spent in closed arms suggesting that MXE may be anxiogenic at high doses. That MXE might exert different effects on anxiety depending on the unit dose administered is supported by data obtained in the social interaction test, where MXE (0.5 and 1 mg·kg−1) caused a positive, but not significant, trend in the time spent socializing and in the number of interactions following administration, while MXE (2.5 mg·kg−1) significantly reduced the time spent in interactions. The effect of MXE on anxiety is unrelated to motor effects, as the immediate and transient depressant effect of MXE (5 mg·kg−1) is no longer evident at the time of the test, that is, after 30 min from administration, and the dose of 2.5 mg·kg−1 has no significant motor effect. In keeping with our findings, a clinical study reported anxiolytic effects of ketamine at low doses and anxiogenic effects at higher doses (Krystal et al., 1994). Accordingly, preclinical studies have showed increased social interactions following administration of low doses of other non‐competitive NMDA receptor antagonists (Morales et al., 2013), and social isolation after exposure to high doses of the drug (Sams‐Dodd, 1996). Anxiolytic effects after low doses of serotonergic agents and anxiogenic‐like effects at higher doses have also been reported (Kolcsar et al., 2014; Gray and Hughes, 2015). Notably, among the reasons for using MXE, as reported in web discussion fora, are the increased social relationships experienced by users with low doses of MXE (Corazza et al., 2013; Zawilska, 2014).
Another intriguing effect reported by MXE users is its antidepressant‐like properties, in line with the capacity of non‐competitive NMDA receptor antagonists to display antidepressant effects in animal models of depression (Skolnick, 1999). Here, we found that acute administration of a high dose of MXE (5 mg·kg−1) significantly increased swimming time and decreased immobility time in rats, in line with the acute antidepressant effect showed by other NMDA receptor antagonists in the same test (Li et al., 2015). The observed MXE‐induced antidepressant‐like effect is not due to alterations in locomotor activity because the same dose of MXE did not enhance locomotor activity. Notably, we showed that a single injection of MXE induces anxiogenic and antidepressant effects, a pharmacological feature that this drug shares with ketamine (da Silva et al., 2010) and several selective 5‐HT reuptake inhibitors (SSRIs) (Griebel et al., 1994; Kurt et al., 2000; Pettersson et al., 2015). As such, we cannot exclude the possibility that, as reported for SSRIs and other agents that augment 5‐HT signalling, high doses of MXE may elicit an anxiety‐like state during the first few days (or weeks) of use, before anxiolytic effects emerge (Ravinder et al., 2011; Jonassen et al., 2015). Further studies will be necessary to confirm whether the anxiogenic effect induced by acute MXE administration reverses with continued drug administration.
Finally, we showed that MXE induced an increased phosphorylation of rpS6 in medial prefrontal (PrL and IL) and hippocampal areas in a dose‐related manner (with the exception of a bell‐shaped curve in PrL), whereas accumbal and BLA areas were not affected. This pattern of effects on rpS6 partly resembles those induced by ketamine, as a dose‐related effect of ketamine on rpS6 expression levels in PrL and IL, but not in the NAcS, has been recently described by Tedesco et al. (2013). In the same study, however, ketamine induced rpS6 phosphorylation in the NAcC and BLA, but not in the hippocampus. Blockade of NMDA receptors on inhibitory GABAergic neurons in prefrontal regions leads to an increase of glutamate and dopamine release in the prefrontal cortex (Lorrain et al., 2003). Enhanced glutamatergic transmission through glutamate AMPA and NMDA receptors affects Akt and ERK1/2 activities (Gong et al., 2006): Akt activates mTOR and downstream rpS6 (Ferrari et al., 1991; Ruvinsky and Meyuhas, 2006), whereas the ERK1/2 pathway induces rpS6 phosphorylation via p70S6K (Bessard et al., 2007) and, concomitantly, via p90 ribosomal s6 kinase (Roux et al., 2007). This broad regulation of rpS6 phosphorylation makes it a ‘convergent’ marker of increased neuronal activity (see Biever et al., 2015). We could, therefore, speculate that the effects of MXE on rpS6 in the prefrontal cortex and hippocampus are correlated with the antidepressant effects observed in the behavioural test, further supporting NMDA receptor blockade‐mediated, mood‐improving properties. Importantly, due to the lack of pharmacological characterization of MXE metabolites, we do not know whether MXE behavioural effects are (fully or partly) due to AMPA agonism, as recently shown for ketamine metabolites (Zanos et al., 2016). Further studies should investigate this potential mechanism for MXE and other ketamine‐like compounds.
In conclusion, our knowledge of the clinical effects of MXE originate from case reports of intoxication or self‐medication and self‐reported experiences described by users in Web discussion fora. We recently showed that MXE exerted discriminative stimulus effects similar to ketamine (Chiamulera et al., 2016) and induced clear rewarding effects in rats by stimulating mesolimbic dopaminergic transmission (Mutti et al., 2016). The present study adds further insight into the pharmacological effects of this new psychoactive substance by showing its ability to enhance repetitive/perseverative behaviour at low doses and to induce transient analgesia, social anxiety and antidepressant effects at high doses. Immunohistochemical analysis provided a ‘molecular snapshot’ of the neuroadaptive molecular effects of MXE at behaviourally active doses and confirmed that MXE shares with ketamine the ability to affect a marker of potential long‐term effects, such as the increased phosphorylation of rpS6 in the emotional and reward brain areas. Molecular findings support the idea that, similarly to ketamine, the rapid antidepressant effect induced by MXE might be mediated by rapid changes in neuroadaptive mechanisms.
Author contributions
M.T.Z. and S.A. carried out the behavioural experiments, analysis and discussion of data and contribute to the final version of the manuscript. P.F. and W.F. were involved in the design of the study, in the analysis and discussion of the data and participated in revising the article critically for important intellectual content. M.D.C. conducted the molecular experiments and analysis of data. C.C. carried out the biochemical experiments and analysis of data and contributed to the final version of the manuscript. L.F. was involved in the design of the research proposal, conduct and supervision of the behavioural experiments, analysis and discussion of data and contributed to the final version of the manuscript. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by funds from ‘Joint Project 2012’ from University of Verona, in collaboration with C.N.R. Institute of Neuroscience, Cagliari, and University of Cagliari, and from Fondazione Banco di Sardegna 2014.
Zanda, M. T. , Fadda, P. , Antinori, S. , Di Chio, M. , Fratta, W. , Chiamulera, C. , and Fattore, L. (2017) Methoxetamine affects brain processing involved in emotional response in rats. British Journal of Pharmacology, 174: 3333–3345. doi: 10.1111/bph.13952.
References
- Abe E, Ricard F, Darrouzain F, Alvarez JC (2013). An automated method for measurement of methoxetamine in human plasma by use of turbulent flow on‐ line extraction coupled with liquid chromatography and mass spectrometric detection. Anal Bioanal Chem 405: 239–245. [DOI] [PubMed] [Google Scholar]
- Adamowicz P, Zuba D (2015). Fatal intoxication with methoxetamine. J Forensic Sci 60: S264–S268. [DOI] [PubMed] [Google Scholar]
- Ahmadi A, Mahmoudi A (2006). Synthesis with improved yield and study on the analgesic effect of 2‐methoxyphencyclidine. Arzneimittelforschung 56: 346–350. [DOI] [PubMed] [Google Scholar]
- Albelda N, Joel D (2012). Animal models of obsessive‐compulsive disorder: exploring pharmacology and neural substrates. Neurosci Biobehav Rev 36: 47–63. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SM, Brunzell DH (2012). Low dose nicotine and antagonism of β2 subunit containing nicotinic acetylcholine receptors have similar effects on affective behavior in mice. PLoS One 7: e48665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer T, Fredriksson A, Lewander T, Söderberg U (1987). Marble burying and spontaneous motor activity in mice: interactions over days and the effect of diazepam. Scand J Psychol 28: 242–249. [DOI] [PubMed] [Google Scholar]
- Babar E, Ozgünen T, Melik E, Polat S, Akman H (2001). Effects of ketamine on different types of anxiety/fear and related memory in rats with lesions of the median raphe nucleus. Eur J Pharmacol 431: 315–320. [DOI] [PubMed] [Google Scholar]
- Baumeister A, Advokat C (1991). Evidence for a supraspinal mechanism in the opioid‐mediated antinociceptive effect of ketamine. Brain Res 566: 351–353. [DOI] [PubMed] [Google Scholar]
- Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G (2003). Ketamine‐induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 27: 687–700. [DOI] [PubMed] [Google Scholar]
- Bessard A, Frémin C, Ezan F, Coutant A, Baffet G (2007). MEK/ERK‐dependent uPAR expression is required for motility via phosphorylation of P70S6K in human hepatocarcinoma cells. J Cell Physiol 212: 526–536. [DOI] [PubMed] [Google Scholar]
- Biever A, Valjent E, Puighermanal E (2015). Ribosomal protein S6 phosphorylation in the nervous system: from regulation to function. Front Mol Neurosci 8: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botanas CJ, de la Peña JB, de la Pena IJ, Tampus R, Yoon R, Kim HJ et al (2015). Methoxetamine, a ketamine derivative, produced conditioned place preference and was self‐administered by rats: evidence of its abuse potential. Pharmacol Biochem Behav 133: 31–36. [DOI] [PubMed] [Google Scholar]
- Bruins Slot LA, Bardin L, Auclair AL, Depoortere R, Newman‐Tancredi A (2008). Effects of antipsychotics and reference monoaminergic ligands on marble burying behavior in mice. Behav Pharmacol 19: 145–152. [DOI] [PubMed] [Google Scholar]
- Cannon CZ, Kissling GE, Hoenerhoff MJ, King‐Herbert AP, Blankenship‐Paris T (2010). Evaluation of dosages and routes of administration of tramadol analgesia in rats using hot‐plate and tail‐flick tests. Lab Anim (NY) 39: 342–351. [DOI] [PubMed] [Google Scholar]
- Casarotto PC, Gomes FV, Resstel LB, Guimarães FS (2010). Cannabidiol inhibitory effect on marble‐burying behaviour: involvement of CB1 receptors. Behav Pharmacol 21: 353–358. [DOI] [PubMed] [Google Scholar]
- Castagné V, Wolinsky T, Quinn L, Virley D (2012). Differential behavioral profiling of stimulant substances in the rat using the LABORASTM system. Pharmacol Biochem Behav 101: 553–563. [DOI] [PubMed] [Google Scholar]
- Castelli MP, Fadda P, Casu A, Spanu MS, Casti A, Fratta W et al (2014). Male and female rats differ in brain cannabinoid CB1 receptor density and function and in behavioural traits predisposing to drug addiction: effect of ovarian hormones. Curr Pharm Des 20: 2100–2113. [DOI] [PubMed] [Google Scholar]
- Chiamulera C, Armani F, Mutti A, Fattore L (2016). The ketamine analogue methoxetamine generalizes to ketamine discriminative stimulus in rats. Behav Pharmacol 27: 204–210. [DOI] [PubMed] [Google Scholar]
- Coppola M, Mondola R (2012). Methoxetamine: from drug of abuse to rapid‐acting antidepressant. Med Hypotheses 79: 504–507. [DOI] [PubMed] [Google Scholar]
- Corazza O, Assi S, Schifano F (2013). From “special K” to “special M”: the evolution of the recreational use of ketamine and methoxetamine. CNS Neurosci Ther 19: 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Watanabe A, Yamasaki Y, Amada N, Arthur M, Fleming S et al (2012). Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9‐tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive‐compulsive behaviour. Psychopharmacology (Berl) 219: 859–873. [DOI] [PubMed] [Google Scholar]
- Egashira N, Okuno R, Harada S, Matsushita M, Mishima K, Iwasaki K et al (2008). Effects of glutamate‐related drugs on marble‐burying behavior in mice: implications for obsessive‐compulsive disorder. Eur J Pharmacol 586: 164–170. [DOI] [PubMed] [Google Scholar]
- Elian AA, Hackett J (2014). A polydrug intoxication involving methoxetamine in a drugs and driving case. J Forensic Sci 59: 854–858. [DOI] [PubMed] [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction (2014). Report on the risk assessment of 2‐(3‐methoxyphenyl)‐2‐(ethylamino)cyclohexanone (methoxetamine) in the framework of the council decision on new psychoactive substances, risk assessments, Publications Office of the European Union, Luxembourg.
- Fassette T, Martinez A (2016). An impaired driver found to be under the influence of methoxetamine. J Anal Toxicol 40: 700–702. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G (1991). Mitogen‐activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J Biol Chem 266: 22770–22775. [PubMed] [Google Scholar]
- Forman LJ (1999). NMDA receptor antagonism produces antinociception which is partially mediated by brain opioids and dopamine. Life Sci 64: 1877–1887. [DOI] [PubMed] [Google Scholar]
- Gagliese L, Melzack R (2000). Age differences in nociception and pain behaviours in the rat. Neurosci Biobehav Rev 24: 843–854. [DOI] [PubMed] [Google Scholar]
- Getova DP, Doncheva ND (2011). Effects of ketamine on memory and nociception in rats. Folia Med (Plovdiv) 53: 53–59. [DOI] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ (2006). Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity‐dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem 281: 18802–18815. [DOI] [PubMed] [Google Scholar]
- Gray VC, Hughes RN (2015). Drug‐, dose‐ and sex‐dependent effects of chronic fluoxetine, reboxetine and venlafaxine on open‐field behavior and spatial memory in rats. Behav Brain Res 281: 43–54. [DOI] [PubMed] [Google Scholar]
- Griebel G, Moreau JL, Jenck F, Misslin R, Martin JR (1994). Acute and chronic treatment with 5‐HT reuptake inhibitors differentially modulate emotional responses in anxiety models in rodents. Psychopharmacology (Berl) 113: 463–470. [DOI] [PubMed] [Google Scholar]
- Hajkova K, Jurasek B, Sykora D, Palenicek T, Miksatkova P, Kuchar M (2016). Salting‐out‐assisted liquid‐liquid extraction as a suitable approach for determination of methoxetamine in large sets of tissue samples. Anal Bioanal Chem 408: 1171–1181. [DOI] [PubMed] [Google Scholar]
- Halberstadt AL, Slepak N, Hyun J, Buell MR, Powell SB (2016). The novel ketamine analog methoxetamine produces dissociative‐like behavioral effects in rodents. Psychopharmacology (Berl) 233: 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer KE, Grager B, Müller DM, Rauber‐Lüthy C, Kupferschmidt H, Rentsch KM et al (2012). Ketamine‐like effects after recreational use of methoxetamine. Ann Emerg Med 60: 97–99. [DOI] [PubMed] [Google Scholar]
- Hondebrink L, Kasteel EEJ, Tukker AM, Wijnolts FMJ, Verboven AHA, Westerink RHS (2017). Neuropharmacological characterization of the new psychoactive substance methoxetamine. Neuropharmacology https://doi.org/10.1016/j.neuropharm.2017.04.035. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Hondebrink L, Verboven AH, Drega WS, Schmeink S, de Groot MW, van Kleef RG et al (2016). Neurotoxicity screening of (illicit) drugs using novel methods for analysis of microelectrode array (MEA) recordings. Neurotoxicology 55: 1–9. [DOI] [PubMed] [Google Scholar]
- Horsley RR, Lhotkova E, Hajkova K, Jurasek B, Kuchar M, Palenicek T (2016). Detailed pharmacological evaluation of methoxetamine (MXE), a novel psychoactive ketamine analogue‐behavioural, pharmacokinetic and metabolic studies in the Wistar rat. Brain Res Bull 126: 102–110. [DOI] [PubMed] [Google Scholar]
- Huang C, Long H, Shi YS, Han JS, Wan Y (2005). Ketamine enhances the efficacy to and delays the development of tolerance to electroacupuncture‐induced antinociception in rats. Neurosci Lett 375: 138–142. [DOI] [PubMed] [Google Scholar]
- Iijima M, Kurosu S, Chaki S (2010). Effects of agents targeting glutamatergic systems on marble‐burying behavior. Neurosci Lett 471: 63–65. [DOI] [PubMed] [Google Scholar]
- Imbert L, Boucher A, Delhome G, Cueto T, Boudinaud M, Maublanc J et al (2014). Analytical findings of an acute intoxication after inhalation of methoxetamine. J Anal Toxicol 38: 410–415. [DOI] [PubMed] [Google Scholar]
- Jonassen R, Chelnokova O, Harmer C, Leknes S, Landrø NI (2015). A single dose of antidepressant alters eye‐gaze patterns across face stimuli in healthy women. Psychopharmacology (Berl) 232: 953–958. [DOI] [PubMed] [Google Scholar]
- Kalueff AV, Stewart AM, Song C, Berridge KC, Graybiel AM, Fentress JC (2016). Neurobiology of rodent self‐grooming and its value for translational neuroscience. Nat Rev Neurosci 17: 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka J, Kitanaka N, Hall FS, Fujii M, Goto A, Kanda Y et al (2015). Memory impairment and reduced exploratory behavior in mice after administration of systemic morphine. J Exp Neurosci 9: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolcsar M, Gáll Z, Dogaru MT (2014). Dose dependent effects of serotonergic agents on anxiety. Acta Physiol Hung 101: 479–487. [DOI] [PubMed] [Google Scholar]
- Kratsman N, Getselter D, Elliott E (2016). Sodium butyrate attenuates social behavior deficits and odifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 102: 136–145. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD et al (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214. [DOI] [PubMed] [Google Scholar]
- Kurt M, Arik AC, Celik S (2000). The effects of sertraline and fluoxetine on anxiety in the elevated plus‐maze test in mice. J Basic Clin Physiol Pharmacol 11: 173–180. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu ZR, Ou BC, Wang YQ, Tan ZB, Deng CM et al (2015). Dopamine D2/D3 but not dopamine D1 receptors are involved in the rapid antidepressant‐like effects of ketamine in the forced swim test. Behav Brain Res 279: 100–105. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA (2003). Effects of ketamine and N‐methyl‐D‐aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117: 697–706. [DOI] [PubMed] [Google Scholar]
- Łukasik‐Głebocka M, Sommerfeld K, Tezyk A, Zielinska‐Psuja B, Druzdz A (2013). Acute methoxetamine intoxication – a case report with serum and urine concentrations. Przegl Lek 70: 671–673. [PubMed] [Google Scholar]
- Maskell KF, Bailey ML, Rose SR (2016). Self‐medication with methoxetamine as an analgesic resulting in significant toxicity. Pain Med 17: 1773–1775. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Varlinskaya EI, Spear LP (2013). Low doses of the NMDA receptor antagonists, MK‐801, PEAQX, and ifenprodil, induces social facilitation in adolescent male rats. Behav Brain Res 250: 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris H, Wallach J (2014). From PCP to MXE: a comprehensive review of the non‐medical use of dissociative drugs. Drug Test Anal 6: 214–232. [DOI] [PubMed] [Google Scholar]
- Mutti A, Aroni S, Fadda P, Padovani L, Mancini L, Collu R et al (2016). The ketamine‐like compound methoxetamine substitutes for ketamine in the self‐administration paradigm and enhances mesolimbic dopaminergic transmission. Psychopharmacology (Berl) 233: 2241–2251. [DOI] [PubMed] [Google Scholar]
- Nardo M, Casarotto PC, Gomes FV, Guimarães FS (2014). Cannabidiol reverses the mCPP‐induced increase in marble‐burying behavior. Fund Clin Pharmacol 28: 544–550. [DOI] [PubMed] [Google Scholar]
- Nicolas LB, Kolb Y, Prinssen EP (2006). A combined marble burying‐locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur J Pharmacol 547: 106–115. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2008). The Rat Brain in Stereotaxic Coordinates, Compact Sixth edn. Elsevier Academic Press: London, 2008. [Google Scholar]
- Pettersson R, Näslund J, Nilsson S, Eriksson E, Hagsäter SM (2015). Acute escitalopram but not contextual conditioning exerts a stronger “anxiogenic” effect in rats with high baseline “anxiety” in the acoustic startle paradigm. Psychopharmacology (Berl) 232: 1461–1469. [DOI] [PubMed] [Google Scholar]
- Ravinder S, Pillai AG, Chattarji S (2011). Cellular correlates of enhanced anxiety caused by acute treatment with the selective serotonin reuptake inhibitor fluoxetine in rats. Front Behav Neurosci 5: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer AE, de Oliveira AR, Diniz JB, Hoexter MQ, Chiavegatto S, Brandão ML (2015). Rats with differential self‐grooming expression in the elevated plus‐maze do not differ in anxiety‐related behaviors. Behav Brain Res 292: 370–380. [DOI] [PubMed] [Google Scholar]
- Roth BL, Gibbons S, Arunotayanun W, Huang XP, Setola V, Treble R et al (2013). The ketamine analogue methoxetamine and 3‐ and 4‐methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PLoS One 8: e59334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J et al (2007). RAS/ERK signaling promotes site‐specific ribosomal protein S6 phosphorylation via RSK and stimulates cap‐dependent translation. J Biol Chem 282: 14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O (2006). Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31: 342–348. [DOI] [PubMed] [Google Scholar]
- Sams‐Dodd F (1996). Phencyclidine‐induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol 7: 3–23. [PubMed] [Google Scholar]
- Satta V, Scherma M, Giunti E, Collu R, Fattore L, Fratta W et al (2016). Emotional profile of female rats showing binge eating behavior. Physiol Behav 163: 136–143. [DOI] [PubMed] [Google Scholar]
- Scherma M, Dessì C, Muntoni AL, Lecca S, Satta V, Luchicchi A et al (2015). Adolescent Δ(9)‐tetrahydrocannabinol exposure alters WIN55,212‐2 self‐administration in adult rats. Neuropsychopharmacology 41: 1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields JE, Dargan PI, Wood DM, Puchnarewicz M, Davies S, Waring WS (2012). Methoxetamine associated reversible cerebellar toxicity: three cases with analytical confirmation. Clin Toxicol 50: 438–440. [DOI] [PubMed] [Google Scholar]
- Shikanai H, Hiraide S, Kamiyama H, Kiya T, Oda K, Goto Y et al (2014). Subanalgesic ketamine enhances morphine‐induced antinociceptive activity without cortical dysfunction in rats. J Anesth 28: 390–398. [DOI] [PubMed] [Google Scholar]
- da Silva FC, do Carmo de Oliveira Cito M, da Silva MI, Moura BA, de Aquino Neto MR, Feitosa ML et al (2010). Behavioral alterations and pro‐oxidant effect of a single ketamine administration to mice. Brain Res Bull 83: 9–15. [DOI] [PubMed] [Google Scholar]
- Skolnick P (1999). Antidepressants for new millennium. Eur J Pharmacol 375: 31–40. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano MS, Fadda P, Frau R, Fattore L, Fratta W (2010). Cannabinoid self‐administration attenuates PCP‐induced schizophrenia‐like symptoms in adult rats. Eur Neuropsychopharmacol 20: 25–36. [DOI] [PubMed] [Google Scholar]
- Striebel JM, Nelson EE, Kalapatapu RK (2017). “Being with a Buddha”: a case report of methoxetamine use in a United States veteran with PTSD. Case Rep Psychiatry 2017: 2319094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungur AÖ, Vörckel KJ, Schwarting RK, Wöhr M (2014). Repetitive behaviors in the Shank1 knockout mouse model for autism spectrum disorder: developmental aspects and effects of social context. J Neurosci Methods 234: 92–100. [DOI] [PubMed] [Google Scholar]
- Tedesco V, Ravagnani C, Bertoglio D, Chiamulera C (2013). Acute ketamine‐induced neuroplasticity: ribosomal protein S6 phosphorylation expression in drug addiction‐related rat brain areas. Neuroreport 24: 388–393. [DOI] [PubMed] [Google Scholar]
- Umathe SN, Mundhada YR, Bhutada PS (2012). Differential effects of acute morphine, and chronic morphine‐withdrawal on obsessive‐compulsive behavior: inhibitory influence of CRF receptor antagonists on chronic morphine‐withdrawal. Neuropeptides 46: 217–221. [DOI] [PubMed] [Google Scholar]
- Ward J, Rhyee S, Plansky J, Boyer E (2011). Methoxetamine: a novel ketamine analog and growing health‐care concern. Clin Toxicol 49: 874–875. [DOI] [PubMed] [Google Scholar]
- Wikström M, Thelander G, Dahlgren M, Kronstrand R (2013). An accidental fatal intoxication with methoxetamine. J Anal Toxicol 37: 43–46. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Cristello AF, Balster RL (1995). Effects of site‐selective NMDA receptor antagonists in an elevated plus‐maze model of anxiety in mice. Eur J Pharmacol 294: 101–107. [DOI] [PubMed] [Google Scholar]
- Wood DM, Davies S, Puchnarewicz M, Johnston A, Dargan PI (2012). Acute toxicity associated with the recreational use of the ketamine derivative methoxetamine. Eur J Clin Pharmacol 68: 853–856. [DOI] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI et al (2016). NMDAR inhibition‐independent antidepressant actions of ketamine metabolites. Nature 533: 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawilska JB (2014). Methoxetamine – a novel recreational drug with potent hallucinogenic Properties. Toxicol Lett 230: 402–407. [DOI] [PubMed] [Google Scholar]