

Abstract
We review recent evidence concerning the significance of inhibitory GABA transmission and of neural disinhibition, that is, deficient GABA transmission, within the prefrontal cortex and the hippocampus, for clinically relevant cognitive functions. Both regions support important cognitive functions, including attention and memory, and their dysfunction has been implicated in cognitive deficits characterizing neuropsychiatric disorders. GABAergic inhibition shapes cortico‐hippocampal neural activity, and, recently, prefrontal and hippocampal neural disinhibition has emerged as a pathophysiological feature of major neuropsychiatric disorders, especially schizophrenia and age‐related cognitive decline. Regional neural disinhibition, disrupting spatio‐temporal control of neural activity and causing aberrant drive of projections, may disrupt processing within the disinhibited region and efferent regions. Recent studies in rats showed that prefrontal and hippocampal neural disinhibition (by local GABA antagonist microinfusion) dysregulates burst firing, which has been associated with important aspects of neural information processing. Using translational tests of clinically relevant cognitive functions, these studies showed that prefrontal and hippocampal neural disinhibition disrupts regional cognitive functions (including prefrontal attention and hippocampal memory function). Moreover, hippocampal neural disinhibition disrupted attentional performance, which does not require the hippocampus but requires prefrontal‐striatal circuits modulated by the hippocampus. However, some prefrontal and hippocampal functions (including inhibitory response control) are spared by regional disinhibition. We consider conceptual implications of these findings, regarding the distinct relationships of distinct cognitive functions to prefrontal and hippocampal GABA tone and neural activity. Moreover, the findings support the proposition that prefrontal and hippocampal neural disinhibition contributes to clinically relevant cognitive deficits, and we consider pharmacological strategies for ameliorating cognitive deficits by rebalancing disinhibition‐induced aberrant neural activity.
Linked Articles
This article is part of a themed section on Pharmacology of Cognition: a Panacea for Neuropsychiatric Disease? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.19/issuetoc
Abbreviations
- 5CSRT
five‐choice‐serial‐reaction‐time
- DMTP
delayed‐matching‐to‐place
- mGluR
metabotropic glutamate receptor
- PAM
positive allosteric modulator
Introduction
Cognitive deficits, including attentional and memory deficits, are a cross‐diagnostic symptom of many neuropsychiatric disorders that causes substantial functional disability and is a major treatment challenge. A prominent idea is that dysfunction within the prefrontal cortex and hippocampus causes such cognitive deficits (Millan et al., 2012). In this review, we will begin by highlighting evidence that hippocampal and prefrontal neural disinhibition, that is, deficient inhibitory GABA function, is a key neuropathological feature of important neuropsychiatric disorders characterized by cognitive deficits, including schizophrenia and age‐related cognitive decline (Table 1). Although inhibitory GABA neurotransmission has emerged as a key factor in shaping neural activity in the neocortex, including the prefrontal cortex, and hippocampus (Buzsaki and Wang, 2012; Isaacson and Scanziani, 2011; Mann and Paulsen, 2007), direct evidence for a causal contribution of prefrontal and hippocampal neural disinhibition to clinically relevant cognitive deficits has been lacking until recently. We will highlight that neural disinhibition within the prefrontal cortex or hippocampus has the potential to disrupt cognitive processing mediated by these regions and efferent sites, because regional inhibitory GABA transmission may be important to maintain balanced levels of neural activity within these respective regions, as well as in their projection sites. The review will, then, focus on recent studies examining the neural and cognitive impact of prefrontal and hippocampal reductions in inhibitory GABA tone by acute intra‐cerebral microinfusions of GABA antagonists in rats, using in vivo electrophysiology and translational behavioural assays of clinically relevant cognitive functions. These studies revealed that prefrontal and hippocampal neural disinhibition caused aberrant neural activity within the disinhibited regions and impaired key cognitive functions, including attention and memory, in a way that was consistent with regional neural disinhibition disrupting cognitive processing of the disinhibited regions and their projection sites. We will consider the theoretical implications of these findings, concerning the distinct relationships of regional inhibitory GABA tone and neural activity to distinct cognitive functions, their clinical implications, as well as potential pharmacological strategies to rebalance disinhibition‐induced aberrant neural activity in order to ameliorate cognitive impairments.
Table 1.
Some important neuropsychiatric disorders that have been associated with cortico‐hippocampal GABA dysfunction. All of these disorders show cognitive impairments (Millan et al., 2012).
| Disorder | References supporting GABA dysfunction |
|---|---|
| Schizophrenia | Benes and Berretta, 2001; Guidotti et al., 2005; Lewis and Moghaddam, 2006; Lisman et al., 2008; Fung et al., 2010; O'Donnell, 2011; Heckers and Konradi, 2015; Ruzicka et al., 2015 |
| Cognitive ageing and Alzheimer's disease | Huang and Mucke, 2012; Stanley et al., 2012; Spiegel et al., 2013; Nava‐Mesa et al., 2014; Busche and Konnerth, 2016; Thomé et al., 2016 |
| Autism | Rubenstein and Merzenich, 2003; Han et al., 2014 |
| Depression | Rajkowska et al., 2007; Luscher et al., 2011 |
| Bipolar disorder | Konradi et al., 2011 |
Prefrontal and hippocampal neural disinhibition in neuropsychiatric disorders characterized by cognitive deficits
The prefrontal cortex and the hippocampus play key roles in important cognitive functions, including attention and everyday memory (Bast, 2007; Bast, 2011; Chudasama and Robbins, 2006). Over the past 20 years, neural disinhibition, that is, reduced inhibitory GABA function, within these regions has emerged as a common feature of major neuropsychiatric disorders characterized by impairments in these important cognitive functions (Table 1).
The most compelling evidence comes from studies on the neurobiological mechanisms underlying schizophrenia. Alterations in GABA‐related, post mortem markers, consistent with reduced function of prefrontal and hippocampal inhibitory GABA neurons are a key neuropathological feature of schizophrenia, and many rodent models of schizophrenia show similar evidence for such neural disinhibition (Benes and Berretta, 2001; Lewis and Moghaddam, 2006; Lisman et al., 2008; O'Donnell, 2011; Heckers and Konradi, 2015; Ruzicka et al., 2015). The well‐characterized schizophrenia‐related cognitive and behavioural changes caused by NMDA receptor antagonists in humans and animal models have also been linked to neural disinhibition, with reduced GABAergic inhibition in the hippocampus and neocortex being a key effect of NMDA receptor antagonists, possibly reflecting a blockade of NMDA receptor‐mediated excitation of inhibitory GABA neurons (Lisman et al., 2008; Anticevic et al., 2012; Moghaddam and Krystal, 2012). Given that inhibitory GABA neurons play a key role in shaping hippocampal and neocortical neural oscillations (Mann and Paulsen, 2007; Buzsaki and Wang, 2012), neural disinhibition may also, at least partly, explain the aberrant oscillatory brain activity revealed by EEG and MEG measures in patients with schizophrenia (Lisman et al., 2008; Uhlhaas and Singer, 2012). Finally, neural disinhibition may underlie regional brain hyperactivity reported by clinical imaging studies of early‐stage schizophrenia (Schobel et al., 2009, 2013; Anticevic et al., 2015; Heckers and Konradi, 2015). More recently, consistent evidence for cortico‐hippocampal neural hyperactivity due to neural disinhibition has also emerged in cognitive ageing and early stages of Alzheimer's disease, with clinical and animal model studies showing a compromised inhibitory GABA system and aberrant increases in neural activity (Wilson et al., 2006; Sperling et al., 2010; Bakker et al., 2012; Huang and Mucke, 2012; Sanchez et al., 2012; Stanley et al., 2012; Verret et al., 2012; Schwab et al., 2013; Spiegel et al., 2013; Robitsek et al., 2015; Busche and Konnerth, 2016; Thomé et al., 2016; Haberman et al., 2017). Increased hippocampal neural activity may, at first glance, appear at odds with another hippocampal biomarker of cognitive ageing: in vitro electrophysiological recordings from hippocampal slices of aged rabbits and rats consistently reveal an enhanced post‐burst afterhyperpolarization in pyramidal neurons of the hippocampal CA1 subregion, which results in hypoexcitability of these neurons (Oh et al., 2010). However, contrasting with such hypoexcitability in CA1, the CA3 subregion of the aged rat hippocampus shows hyperexcitability in vitro (Simkin et al., 2015). These regional differences reported in vitro agree with some in vivo studies indicating that CA3 is the main locus of hippocampal neural hyperactivity associated with cognitive ageing in humans and rats (Wilson et al., 2006; Bakker et al., 2012). In light of the emerging evidence of hippocampal neural hyperactivity in ageing, it has been suggested that the hypoexcitability in CA1 neurons reflected by an enhanced post‐burst afterhyperpolarization in vitro may be a compensatory response to increased activity at CA3‐CA1 synapses due to CA3 hyperexcitability (Simkin et al., 2015).
In the long term, aberrant neural activity due to GABA dysfunction may lead to compensatory adjustments and excitotoxicity that could underlie the regional brain hypoactivity and atrophy characterizing later stages of schizophrenia and age‐related cognitive disorders (Huang and Mucke, 2012; Anticevic et al., 2015; Heckers and Konradi, 2015; Krystal and Anticevic, 2015). Direct evidence for this possibility comes from longitudinal functional and structural imaging studies in schizophrenia patients and in a rodent model of schizophrenia, in which hippocampal metabolic overactivity in the prodrome or at early stages of the mouse model predicted hippocampal atrophy in patients who had progressed to schizophrenia or at later stages of the mouse model respectively (Schobel et al., 2013). However, as described later, independent of the induction of atrophy, aberrant prefrontal and hippocampal neural activity caused by reduced GABA function may cause clinically relevant cognitive deficits, and recent studies in rodent models support this possibility.
Regional neural disinhibition may disrupt processing within the disinhibited region and in efferent sites
Inhibitory neurotransmission by GABA, which balances and controls excitatory neurotransmission, is important for shaping neural activity in the hippocampus and neocortex, including the prefrontal cortex (Mann and Paulsen, 2007; Isaacson and Scanziani, 2011; Buzsaki and Wang, 2012). Tightly controlled, spatio‐temporally specific neuronal disinhibition, that is, temporary reductions in the GABAergic inhibition of specific synaptic pathways, may open windows for enhanced processing of relevant stimuli, thereby facilitating learning and memory and, potentially, other cognitive processes (Letzkus et al., 2015).
However, tonic neural disinhibition within a brain region that is not restricted to specific synapses may disrupt both regional function and distal function in efferent brain sites (Figure 1). (Note: Tonic neural disinhibition, that is, long‐lasting neural disinhibition, such as resulting from GABA neuron dysfunction in neuropsychiatric disorders or from intra‐cerebral microinfusions of GABA‐A antagonists in rat models, would be expected to interfere with both phasic inhibition, which is caused by transient increases in synaptic GABA following firing of GABA neurons, and with tonic inhibition, which is caused by ambient GABA (Farrant and Nusser, 2005).) First, by interfering with spatio‐temporal control of regional neural activity, neural disinhibition within a brain region is likely to disrupt normal regional function. For example, single‐neuron recording studies in animal models show that stimulus‐selective, task‐appropriate tuning of neurons in the sensory cortices (Isaacson and Scanziani, 2011) and in the prefrontal cortex (Rao et al., 2000) requires intact GABA transmission. Second, by causing aberrant drive of projections, regional disinhibition may disrupt normal neural activity in efferent sites, thereby disrupting the normal function of these efferent sites. These two general hypotheses support two specific hypotheses concerning the cognitive effects of prefrontal and hippocampal neural disinhibition. First, given the importance of the prefrontal cortex for attention (Chudasama and Robbins, 2006) and of the hippocampus for everyday types of memory, such as episodic and place memory (Bast, 2007; Bast, 2011), prefrontal and hippocampal disinhibition may impair attention and everyday memory tasks, respectively. Second, considering strong hippocampo‐prefrontal projections, hippocampal disinhibition may disrupt prefrontal‐dependent cognitive function, such as attention, by disrupting prefrontal processing (Bast, 2011).
Figure 1.
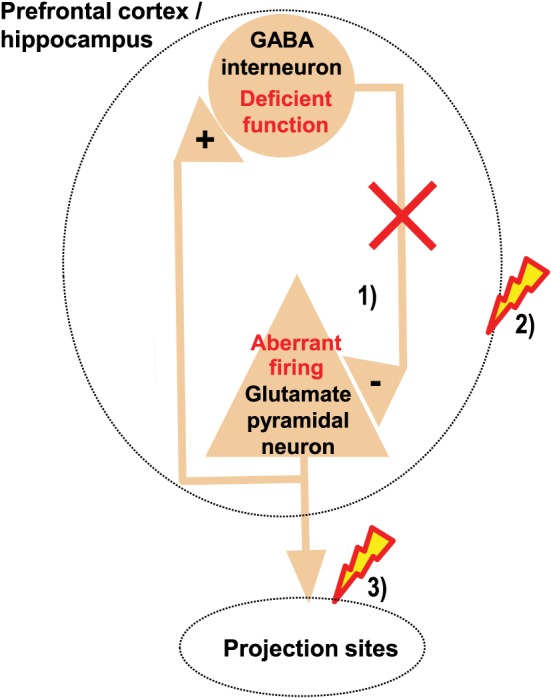
Regional GABA dysfunction may disrupt regional function and the function of projection sites. Deficient function of inhibitory GABA interneurons within the prefrontal cortex or hippocampus disrupts the spatio‐temporal control of excitatory glutamatergic neurons within these regions, causing aberrant firing of these neurons (1), which may disrupt the cognitive functions normally mediated by these regions (2). In addition, such aberrant firing may cause aberrant drive of neurons in projection sites, which may disrupt the functions normally mediated by these projection sites (3).
To test these hypotheses, we began investigating the neuro‐cognitive impact of prefrontal and hippocampal neural disinhibition in rats. We induced temporary prefrontal and hippocampal neural disinhibition, using acute microinfusion of subconvulsive doses of the selective GABAA receptor antagonist picrotoxin into the medial prefrontal cortex (Pezze et al., 2014) or hippocampus (McGarrity et al., 2016) and examined the effects on translational behavioural tests of attention and everyday‐type place learning. The hippocampal infusions targeted the temporal (also referred to as ventral) to intermediate hippocampus, because this part of the hippocampus features strong hippocampo‐prefrontal connectivity and corresponds to human anterior hippocampus, dysfunction of which has been implicated in schizophrenia (Bast, 2011). To link any cognitive effects to specific neural changes within prefrontal cortex or hippocampus, we measured how picrotoxin infusions affected neural activity in the vicinity of the infusion site, using in vivo electrophysiological recordings in anaesthetized rats.
Neural disinhibition in the prefrontal cortex and hippocampus enhances burst firing of neurons within these regions
In both the medial prefrontal cortex (Pezze et al., 2014) and the hippocampus (McGarrity et al., 2016), disinhibition by picrotoxin markedly enhanced firing of neurons in bursts, particularly increasing within‐burst firing rate and prevalence of burst firing (e.g. as reflected by increased percentage of spikes in burst) within the prefrontal cortex and increasing prevalence of burst firing, accompanied by a moderate increase in overall firing rate, in the hippocampus (Figure 2). Bursts are periods of relatively high firing that are separated by periods of comparatively little firing (Lisman, 1997). Our findings converge with the enhanced hippocampal burst firing reported following pharmaco‐ or optogenetic silencing of hippocampal inhibitory GABA interneurons in vitro (Lovett‐Barron et al., 2012) and in awake mice (Royer et al., 2012) and suggest a key role of GABAergic inhibition in the regulation of prefrontal and hippocampal burst firing.
Figure 2.
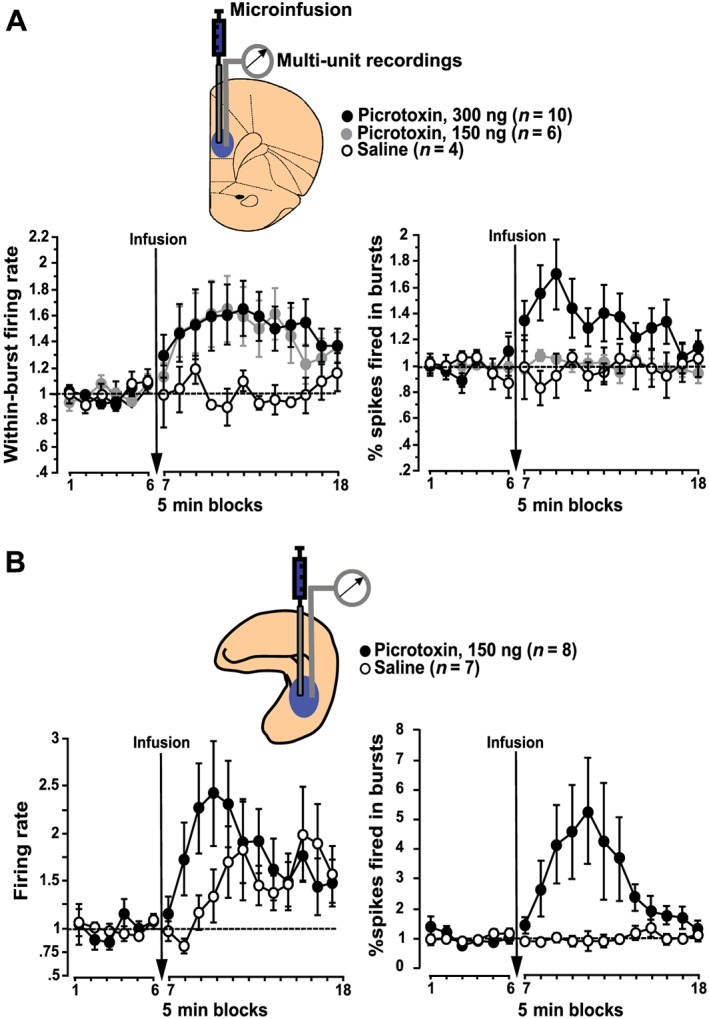
Neural disinhibition in prefrontal cortex and hippocampus enhances burst firing of neurons within these regions. Following local microinfusions of the GABA antagonist picrotoxin into the medial prefrontal cortex (A) or hippocampus (B) of anaesthetized rats, enhanced neuronal burst firing is the most marked effect revealed by in vivo electrophysiological recordings of multi‐unit activity in the vicinity of the infusion site. (A) In the medial prefrontal cortex, regional neural disinhibition by picrotoxin primarily increases within‐burst firing rate and, at the higher picrotoxin dose, also increases the prevalence of bursts, as reflected by an increased percentage of spikes fired in bursts. (B) In the hippocampus, local picrotoxin infusion markedly increases the prevalence of bursts, as reflected, for example, by an increased percentage of spikes fired in bursts, accompanied by a comparatively moderate increase in overall firing rate. All values show multi‐unit recording data normalized to baseline (average across the six 5 min blocks before infusion) and show mean ± SEM; prefrontal data graphs are adapted from Pezze et al. (2014) and hippocampal data from McGarrity et al. (2016).
Aberrant burst firing is likely to be detrimental to the cognitive functions of both the disinhibited region and their projection sites. Bursts have been suggested to be key units of neural information processing, increasing the reliability and selectivity of neural communication, and burst firing is particularly effective in driving post‐synaptic targets (Lisman, 1997; Cooper, 2002; Izhikevich et al., 2003; Larkum, 2013). In the visual cortex, short periods of vigorous firing in response to task‐relevant stimuli, separated by relatively quiescent periods, have recently been associated with task‐appropriate behavioural responding on a visual attention test, and it was proposed that such fine tuning of neural responses is widespread within cortical areas and important for a wide range of cognitive functions (Engel et al., 2016). Moreover, in the hippocampus, burst firing has been implicated in the encoding and readout of hippocampal memory (Takahashi and Magee, 2009; Xu et al., 2012). Our electrophysiological findings, in conjunction with the behavioural findings we outline below, support the idea that dysregulation of burst firing within the prefrontal cortex and hippocampus, by disruption of local GABA function, can disrupt both regional cognitive function, as well as the cognitive function of projection sites.
Neural disinhibition within the prefrontal cortex or hippocampus causes clinically relevant cognitive deficits
Disruption of prefrontal‐mediated cognitive function by prefrontal or hippocampal neural disinhibition
To test for attentional deficits, we used the five‐choice‐serial‐reaction‐time (5CSRT) task. This task requires rats to sustain and divide attention across a row of five apertures to detect brief light flashes occurring randomly in one of the apertures and to respond to these flashes to receive food reward. The 5CSRT task resembles human continuous performance tests, and its validity for assessing prefrontal‐mediated attentional mechanisms as impaired in schizophrenia and early age‐related cognitive decline has been well established (Chudasama and Robbins, 2006; Lustig et al., 2013; Romberg et al., 2013). Acute prefrontal (Pezze et al., 2014) or hippocampal (McGarrity et al., 2016) neural disinhibition by picrotoxin both caused attentional deficits on the 5CSRT test (Figure 3A, B).
Figure 3.
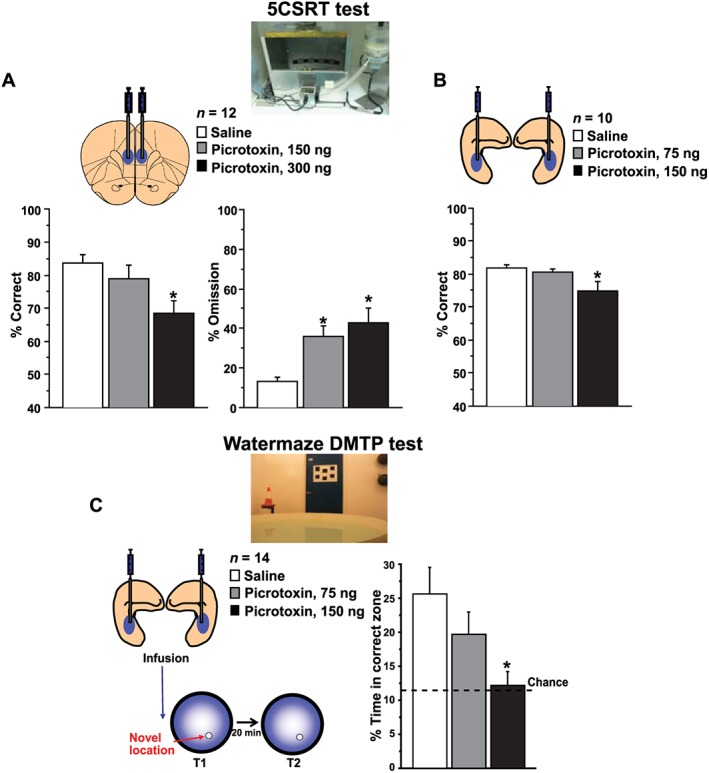
Neural disinhibition within the medial prefrontal cortex and hippocampus causes clinically relevant attentional and memory deficits. (A, B) Attentional deficits: neural disinhibition in the medial prefrontal cortex or hippocampus disrupts attentional performance on the 5CSRT task. The 5CSRT task requires rats to sustain and divide attention across a row of five apertures to detect brief light flashes occurring randomly in one of the apertures and to respond to these flashes to receive food reward. Following local microinfusions of the GABA antagonist picrotoxin into the medial prefrontal cortex (A) or hippocampus (B), rats show reduced attentional performance, as reflected by a reduced percentage of correct responses and, in case of the prefrontal disinhibition, also an increase in omitted trials. (C) Memory deficits: neural disinhibition in the hippocampus disrupts rapid place learning performance on the watermaze DMTP test. This highly hippocampus‐dependent test requires rats to learn, within one trial, the daily changing place of a hidden platform that offers escape from water, resembling the everyday task of remembering new places and routes. Following microinfusion of picrotoxin into the hippocampus, rats show reduced one‐trial place learning performance, as reflected by a marked reduction in search preference for the correct location. Prefrontal data graphs are adapted from Pezze et al. (2014) and hippocampal data graphs from McGarrity et al. (2016).
Attention as measured on the 5CSRT test requires neural activity within the medial prefrontal cortex, with both neurotoxic lesions (Chudasama and Muir, 2001; Passetti et al., 2002; Pezze et al., 2009; Chudasama et al., 2012) and functional inhibition of this region by the GABAA agonist muscimol (Pezze et al., 2014) markedly impairing attentional performance. Therefore, the attentional deficits caused by prefrontal picrotoxin, which are consistent with attentional impairments reported by others with medial prefrontal infusions of GABAA receptor antagonists in rats (Paine et al., 2011; Pehrson et al., 2013) and, recently, with optogenetic inactivation of parvalbumin‐positive prefrontal GABA neurons in mice (Kim et al., 2016), suggest that sustained attention depends on appropriately tuned prefrontal neural activity, with both too little and too much causing attentional impairments (Pezze et al., 2014). Similar to attention, aspects of cognitive flexibility, such as extra‐dimensional response shifts, are disrupted by neurotoxic lesions of the medial prefrontal cortex (Birrell and Brown, 2000) or functional inactivation by the sodium channel blocker bupivacaine (Floresco et al., 2008), as well as by neural disinhibition of the medial prefrontal cortex (Gruber et al., 2010; Enomoto et al., 2011), also suggesting a requirement for appropriately tuned medial prefrontal neural activity. However, not all cognitive functions requiring the prefrontal cortex are disrupted by prefrontal neural disinhibition. Response inhibition, as measured by the ability to withhold premature responses on the 5CSRT task, is impaired by prefrontal functional inhibition (Paine et al., 2011; Murphy et al., 2012; Pezze et al., 2014) or lesions (Chudasama and Muir, 2001; Passetti et al., 2002; Pezze et al., 2009; Chudasama et al., 2012), whereas prefrontal neural disinhibition does not change (Paine et al., 2011; Pezze et al., 2014) or, if more ventral portions of the medial prefrontal cortex are affected, may even improve response control (Murphy et al., 2012). This suggests that response inhibition requires prefrontal neural activity, but not the appropriate tuning of such activity.
The selective attentional deficit on the 5CSRT test following hippocampal neural disinhibition (Figure 3B), indicated by reduced accuracy without changes in any other performance measures, probably reflects disruption of extrahippocampal processing, most likely in the prefrontal cortex or ventral striatum by way of strong hippocampal functional connectivity to these sites (Bast, 2011). Previous lesion studies suggest that the hippocampus itself plays, if at all, only a minor role in mediating sustained attention on the 5CSRT task and related tests (see discussion in McGarrity et al., 2016). In contrast, as discussed in the preceding paragraph, sustained attention requires balanced prefrontal neural activity. Sustained attention on the 5CSRT test also requires an optimal level of prefrontal (Granon et al., 2000) and ventral striatal (Pezze et al., 2007) dopamine receptor stimulation, which may be disrupted by hippocampal neural disinhibition, given that hippocampal stimulation activates the meso‐prefrontal‐ventral striatal dopamine system (Mitchell et al., 2000; Floresco et al., 2001; Peleg‐Raibstein et al., 2005; Bast, 2007, 2011; Lodge and Grace, 2011). The attentional deficits following hippocampal neural disinhibition highlight that regional neural disinhibition can affect cognitive functions beyond those normally depending on the disinhibited region and result in impairments of cognitive functions mediated by projection sites of the disinhibited region.
The attentional deficits following hippocampal neural disinhibition, reflected by decreased response accuracy without changes in omissions on the 5CSRT test, are less pronounced than the attentional deficits following lesions (Chudasama and Muir, 2001; Passetti et al., 2002; Pezze et al., 2009; Chudasama et al., 2012) or functional inhibition and disinhibition (Paine et al., 2011; Pezze et al., 2014) of the medial prefrontal cortex, which all manifest as decreases in accuracy alongside increases in omissions (additionally, lesions and functional inhibition reduce inhibitory response control, as reflected by increased premature responses). However, other experimental manipulations primarily targeting the afferent modulation of the prefrontal cortex cause selective reductions in accuracy without increasing omissions, including selective manipulations of the cholinergic (McGaughy et al., 2002) or dopaminergic (Granon et al., 2000) modulation of the prefrontal cortex. Selective reductions in accuracy, without increases in omissions, have also been reported in the triple transgenic mouse model of Alzheimer's disease (Romberg et al., 2011) and the pilocarpine rat model of temporal lobe epilepsy (Faure et al., 2014), where the primary pathology is not within the prefrontal cortex but in medial temporal lobe regions, including the hippocampus. Interestingly, both the pilocarpine rat model (Kumar and Buckmaster, 2006) and the triple transgenic mouse model of Alzheimer's disease (Davis et al., 2014) show hippocampal hyperexcitability. The finding that hippocampal neural disinhibition causes attentional impairments suggests that hippocampal hyperexcitability may contribute to the attentional deficits in these rodent models.
Disruption of hippocampus‐mediated memory function by hippocampal neural disinhibition
To test for deficits in everyday‐type memory, we used the watermaze delayed‐matching‐to‐place (DMTP) task. This highly hippocampus‐dependent test requires rats to learn within one trial the daily changing place of a hidden platform that offers escape from water (Steele and Morris, 1999; Bast et al., 2009; Pezze and Bast, 2012), resembling the everyday task of remembering new places and routes. Similar place memory tests can be run in humans using virtual or real‐space analogues of the watermaze, and such tests have revealed marked deficits in schizophrenia and age‐related cognitive decline (Hort et al., 2007; Fajnerova et al., 2014). Hippocampal neural disinhibition by picrotoxin markedly disrupted rapid place learning performance on the watermaze DMTP test, as reflected by a marked reduction of search preference for the new location, which rats had to learn within the first trial of the day (McGarrity et al., 2016) (Figure 3C). Neural disinhibition within the medial prefrontal cortex did not impair such place learning performance, although it modulated behaviour on the DMTP task, biasing rats towards focused searching; experiments using functional inhibition of the medial prefrontal cortex by muscimol indicated that the prefrontal cortex is not required for this task (McGarrity et al., 2014). Previous studies suggest that performance on the watermaze DMTP test requires the hippocampus for the rapid encoding of new places and for the translation of such rapid place learning into behavioural performance. DMTP performance is disrupted by pharmacological manipulations targeting synaptic plasticity mechanisms mediated by NMDA (Steele and Morris, 1999) and dopamine receptors (Pezze and Bast, 2012) and by partial hippocampal lesions, including lesions restricted to temporal and intermediate hippocampus (Bast et al., 2009), which were targeted by the infusions in the study involving hippocampal neural disinhibition (McGarrity et al., 2016). Functional inhibition by the GABA agonist muscimol, targeting the intermediate hippocampus, also disrupts task performance (McGarrity et al., 2014). The requirement of temporal to intermediate hippocampus probably reflects that these regions feature functional connectivity to frontal and subcortical sites necessary to translate hippocampal learning into performance, although the specific relevant brain sites remain to be determined (Bast, 2007; Bast et al., 2009; Bast, 2011; McGarrity et al., 2014). Overall, neural disinhibition, causing aberrant neuronal bursting, may disrupt everyday‐type memory performance, as assessed on the watermaze DMTP task, by interfering with hippocampal encoding or readout of relevant place information or with passing on such information to hippocampal projection sites.
The finding that hippocampal neural disinhibition disrupts hippocampus‐dependent performance on the watermaze DMTP task (McGarrity et al., 2016) is consistent with other recent rodent studies reporting a learning‐related increase of hippocampal inhibitory synapses (Ruediger et al., 2012) and impaired memory performance following disruption of hippocampal GABA neuron function by molecular‐, opto‐ or pharmacogenetic approaches (Murray et al., 2011; Andrews‐Zwilling et al., 2012; Caputi et al., 2012; Donato et al., 2013; Gilani et al., 2014; Lovett‐Barron et al., 2014; Engin et al., 2015; Lee et al., 2016). Moreover, our findings support recent studies in humans and rodent models linking hippocampal overactivity and hyperexcitability to age‐related memory deficits (Koh et al., 2010; Bakker et al., 2012; Davis et al., 2014) and are consistent with the correlation of hippocampal overactivity with memory deficits in schizophrenia (Tregellas et al., 2014). However, hippocampal neural disinhibition may facilitate hippocampal synaptic plasticity (Wigstrom and Gustafsson, 1983; Martin et al., 2010). Such facilitation of hippocampal synaptic plasticity by neural disinhibition may under some circumstances be beneficial for memory, for example, if the neural disinhibition is spatio‐temporally regulated by endogenous plasticity (Donato et al., 2013) or if there is a pre‐existing deficit due to increased neural inhibition (Fernandez et al., 2007). Moreover, systemic injection of a selective inverse agonist to negatively modulate GABAA receptors containing the α5 subunit, which are predominantly expressed in hippocampus and constitute about 20% of hippocampal GABAA receptors, has been reported to facilitate hippocampal plasticity and performance on the watermaze DMTP test (Dawson et al., 2006), although other studies reported that transgenic reduction of hippocampal α5 subunit‐containing GABAA receptor expression disrupts aspects of hippocampus‐dependent memory (Prut et al., 2010; Engin et al., 2015). Interestingly, the selective inverse agonist at α5 subunit‐containing GABA receptors that was reported to enhance watermaze DMTP performance enhanced induction of long‐term potentiation at hippocampal (Schaffer collateral) synapses in vitro, without affecting the number of stimulation‐evoked population spikes, which may indicate that stimulation‐evoked neural burst firing was unchanged (Dawson et al., 2006). In contrast, hippocampal neural disinhibition caused by picrotoxin, which disrupted hippocampus‐dependent DMTP performance, altered the temporal organization of hippocampal neural activity, markedly enhancing burst‐pattern firing (McGarrity et al., 2016). Overall, hippocampus‐dependent memory performance appears to require hippocampal neural activity that is appropriately balanced by GABAergic inhibition, resembling the requirement of appropriately tuned prefrontal activity for prefrontal‐dependent cognitive functions (Pezze et al., 2014).
Significance of prefrontal and hippocampal inhibitory GABA transmission for distinct cognitive functions: distinct causal relationships linking regional neural activity to distinct cognitive functions
The recent studies discussed above show that inhibitory GABA transmission within the prefrontal cortex and hippocampus is required to regulate neuronal firing, especially burst‐pattern firing, and to sustain aspects of the cognitive functions supported by these regions and projection sites. Prefrontal neural disinhibition disrupts aspects of prefrontal‐dependent attentional performance and of cognitive flexibility, similar to prefrontal lesions and functional inhibition/inactivation (Gruber et al., 2010; Enomoto et al., 2011; Pezze et al., 2014), and hippocampal neural disinhibition disrupts hippocampus‐dependent memory performance, similar to hippocampal lesions and functional inhibition (McGarrity et al., 2016). (Note: permanent lesions and temporary functional inhibition or inactivation do not necessarily have the same cognitive and behavioural outcomes. Depending on the cognitive or behavioural function, lesions and temporary inhibition or inactivation may even have opposite effects, probably reflecting compensatory responses to permanent neural damage or lesion‐induced changes going beyond the target region of the lesion (Wang et al., 2015).). This suggests that neural activity within the prefrontal cortex or hippocampus, respectively, relates to these cognitive functions via an inverted U‐shaped function, with both too much and too little neural activity causing disruptions (Figure 4A). Moreover, the finding that hippocampal neural disinhibition impairs attentional performance, which does not require hippocampal neural activity, but is mediated by prefrontal‐striatal circuits, shows that the regulation of neural activity by GABAergic inhibition within a particular brain region can also be important for aspects of cognitive function that require balanced levels of neural activity within this region's projection sites (McGarrity et al., 2016). In other words, attention is largely insensitive to reductions of hippocampal neuron activity but is dirupted by aberrant tonic increases in hippocampal neural activity (Figure 4B).
Figure 4.
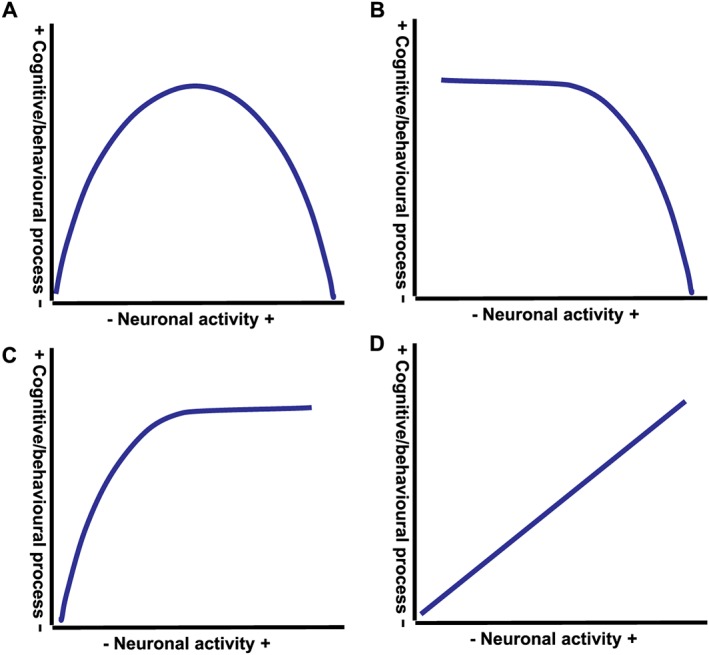
Distinct causal relationships link neural activity within medial prefrontal cortex and hippocampus to distinct cognitive and behavioural processes. Studies examining the cognitive and behavioural impact of bidirectional changes of neural activity within the medial prefrontal cortex or hippocampus, including by local infusions of GABA agonists and antagonists, show that neural activity within these regions can be linked to distinct cognitive and behavioural processes by distinct causal relationships. (A) Inverted U‐shaped relationship, with cognitive or behavioural process requiring a balanced level of neural activity and both too little and too much neural activity causing impairments; for example, the relationship between prefrontal neural activity and attentional performance and between hippocampal neural activity and rapid place learning performance. (B) Cognitive or behavioural process is not affected by reductions in neural activity but impaired by increases in neural activity; for example, the relationship between hippocampal neural activity and attentional performance. (C) Cognitive or behavioural process can be sustained as long as neural activity is above a minimal level; for example, the relationship between hippocampal and prefrontal neural activity and response control. (D) Monotonic positive relationship between neural activity and cognitive or behavioural process, with decreases in neural activity reducing the process and increases in neural activity increasing the process; for example, the relationship between hippocampal and prefrontal neural activity and locomotor activity.
However, other hippocampal and prefrontal functions may be less dependent on the regulation of regional neural firing by inhibitory GABA transmission. In contrast to prefrontal and hippocampal lesions or functional inhibition, neither medial prefrontal nor hippocampal neural disinhibition disrupt inhibitory response control on the 5CSRT test (as reflected by premature or perseverative responses; Pezze et al., 2014; McGarrity et al., 2016). This suggests that such response control requires neural activity within the hippocampo‐prefrontal circuit, but not the appropriate tuning of such activity. In other words, response control can be sustained as long as neural activity within the hippocampo‐prefrontal circuit is above a minimal level (Figure 4C).
Finally, there are also behavioural processes that are supported by prefrontal or hippocampal neural activity that may be enhanced by tonic reductions of local GABA transmission. Neural disinhibition of the (temporal) hippocampus (similar to direct chemical or electrical stimulation; Bast and Feldon, 2003) enhance locomotor activity (Bast et al., 2001a; McGarrity et al., 2016), which depends on neural activity within the temporal hippocampus and is reduced by temporary functional inhibition or inactivation of this region by muscimol or the sodium channel blocker tetrodotoxin (Bast et al., 2001b). Similarly, prefrontal neural disinhibition increases, whereas prefrontal functional inhibition decreases locomotor activity (Pezze et al., 2014). These findings suggest that hippocampal and prefrontal neural activity drives locomotor activity, with a monotonic positive relation between regional neural activity and locomotion (Figure 4D). These locomotor effects may reflect a positive modulation of ventral striatal dopamine transmission by neural activity within the hippocampo‐prefrontal circuit (Karreman and Moghaddam, 1996; Mitchell et al., 2000; Floresco et al., 2001; Bast and Feldon, 2003; Lodge and Grace, 2011).
In summary, regulation of prefrontal and hippocampal neural activty by GABAergic inhibition is important for cognitive and behavioural functions supported by these regions and by distal brain sites functionally connected to these regions. Neural disinhibition in the prefrontal cortex and hippocampus may have distinct effects on distinct cognitive or behavioural functions supported by these regions, reflecting distinct relationships of inhibitory GABA tone and neural activity to distinct functions: some functions require an intact inhibitory GABA tone and appropriately balanced neural activity levels, wheras other functions do not require the tuning of neural acitivy by GABAergic inhibition (as long as neural activity remains subconvulsive).
Prefrontal and hippocampal GABA dysfunction in neuropsychiatric disorders may account for important cognitive deficits characterizing these disorders
Prefrontal and hippocampal GABA dysfunction has been implicated in schizophrenia and age‐related cognitive decline, including early Alzheimer's disease (Benes and Berretta, 2001; Huang and Mucke, 2012; Stanley et al., 2012; Nava‐Mesa et al., 2014; Heckers and Konradi, 2015; Ruzicka et al., 2015; Busche and Konnerth, 2016). The neuro‐cognitive effects of acute pharmacological prefrontal and hippocampal neural disinhibition discussed above are mainly relevant to early stages of these disorders, before chronic disinhibition‐induced neural hyperactivity may lead to compensatory or excitotoxic effects, resulting in regional brain hypofunction and atrophy characteristic of later disease stages (Huang and Mucke, 2012; Schobel et al., 2013; Heckers and Konradi, 2015; Krystal and Anticevic, 2015). Given the close link between neural activity and metabolic activation (Sokoloff, 1981), the enhanced hippocampal neural activity, especially of burst‐pattern firing, caused by acute hippocampal GABA antagonism (McGarrity et al., 2016) is consistent with the hippocampal metabolic overactivity at rest reported by functional imaging studies in early stages of both schizophrenia and age‐related cognitive decline (Schobel et al., 2009; Sperling et al., 2010; Bakker et al., 2012; Schobel et al., 2013).
The rodent 5CSRT and watermaze DMTP tests have high validity to measure deficits in attention and memory relevant to clinical disorders, with related human paradigms – continuous performance tests and place learning tests in virtual and real‐space analogues of the watermaze, respectively – revealing marked deficits in schizophrenia and age‐related cognitive decline (Chudasama and Robbins, 2006; Hort et al., 2007; Lustig et al., 2013; Romberg et al., 2013; Fajnerova et al., 2014). Therefore, the findings that prefrontal and hippocampal neural disinhibition in rats causes impairments on these tests supports that prefrontal and hippocampal GABA dysfunction contributes to clinically relevant attentional and memory deficits. The memory and attentional deficits caused by hippocampal neural disinhibition (McGarrity et al., 2016) also support that causal relationships underly the recently reported correlations of hippocampal overactivity with both memory and attentional deficits in schizophrenia patients (Tregellas et al., 2014) and the association of hippocampal overactivity with memory deficits in amnestic mild cognitive impairment (Bakker et al., 2012).
Implications for pharmacological strategies to treat cognitive deficits
Drugs targeting neural network disruptions resulting from prefrontal and hippocampal neural disinhibition may offer much‐needed new treatment opportunities for cognitive deficits. As discussed above, recent findings show that key cognitive functions, including attention and memory, depend on balanced neural activity within the prefrontal cortex or hippocampus, with both too little and too much activity being detrimental. This has important implications for drug treatments targeting the adverse neuro‐cognitive effects of GABA dysfunction: drugs simply suppressing neural activity will be of limited suitability to treat cognitive deficits. Instead, it is critical to rebalance aberrant neural activity – that is, to curb excessive excitation and burst firing, without suppressing normal firing, as required for cognitive function.
Several candidate drugs are available that may achieve such rebalancing of aberrant neural activity induced by neural disinhibition. Of particular interest are second‐generation antiepileptics, including levetiracetam and lamotrigine, and positive allosteric modulators (PAMs) acting at the metabotropic glutamate receptor mGluR2. The second‐generation antiepileptics lamotrigine and levetiracetam block excessive hippocampal burst firing in vitro, while leaving basal neural activity largely unaffected, an effect suggested to reflect effects on state‐dependent cation currents that mainly contribute to high‐frequency firing (Kuo and Lu, 1997; De Smedt et al., 2007). Levetiracetam has been shown to ameliorate aberrant cortico‐hippocampal neural activity and improve cognitive deficits in rodent models of age‐related cognitive decline (Koh et al., 2010; Sanchez et al., 2012; Robitsek et al., 2015; Haberman et al., 2017) and in human cognitive ageing (Bakker et al., 2012). Lamotrigine has been under consideration for repurposing to ameliorate schizophrenia‐related cognitive deficits associated with cortico‐hippocampal neural disinhibition and has shown some promise in preclinical schizophrenia models, with limited benefits in clinical trials (Large et al., 2011). The failure in clinical trials may reflect that the proposed rebalancing mechanism of lamotrigine would mainly benefit early‐stage schizophrenia patients, characterized by aberrant regional brain hyperactivity, but be of limited benefit in long‐standing patients, as typically included in clinical trials, who show regional brain hypoactivity (Anticevic et al., 2015; Krystal and Anticevic, 2015). PAMs at the mGluR2 may rebalance activity by selectively curbing excessive glutamate release through activity‐dependent stimulation of mGluR2, a presynaptic autoreceptor controlling glutamate release at forebrain terminals (Fell et al., 2012). The mGluR2 has received much interest as a schizophrenia drug target, fuelled by findings that an orthosteric agonist improved cognitive deficits and psychosis‐related changes in rat models of schizophrenia, as well as positive and negative symptoms in an initial phase 2 clinical trial, although subsequent larger and longer clinical trials failed to support effectiveness against schizophrenia symptoms (Curley, 2012) and, more recently, an mGluR2 PAM also failed a phase 2 clinical trial (Litman et al., 2016). Importantly, as considered above with respect to lamotrigine, the proposed rebalancing mechanism of mGluR2 stimulation would mainly benefit early‐stage schizophrenia patients, characterized by aberrant regional brain hyperactivity, but be of limited benefit in long‐standing patients, as typically included in clinical trials, who show regional brain hypoactivity (Anticevic et al., 2015; Krystal and Anticevic, 2015). Consistent with this possibility, a recent re‐analysis of the clinical trials with the mGluR2 orthosteric agonist indicated beneficial effects in early‐stage, but not chronic, schizophrenia patients (Krystal and Anticevic, 2015).
What about drugs directly targeting GABA receptor function? Interestingly, substantial drug discovery efforts to improve cognitive function by targeting GABA receptors have focused on a negative modulation of GABA receptors, such as by inverse agonists selective for α5‐containing GABA receptors (Dawson et al., 2006; Ballard et al., 2009). However, the more recent findings reviewed here, highlighting the requirement of appropriate GABA tone in prefrontal cortex and hippocampus for important cognitive functions, including attention and memory, as well as the emergence of neural disinhibition as a feature of many disorders characterized by cognitive deficits, suggest a different approach. Drugs positively modulating GABA receptor function may have potential for ameliorating the neuro‐cognitive effects in disorders associated with neural disinhibition, as long as overstimulation of inhibitory GABA receptor function can be avoided, as this would interfere with cognitive function. GABA receptor modulators selective for receptor subunits with preferentially prefrontal and hippocampal expression, such as α2 and 5, have received particular interest as they may minimize sedation (Vinkers et al., 2010; Rudolph and Knoflach, 2011). Recent studies in rats showed that a PAM selective for α5‐containing GABA receptors reduces hippocampal hyperexcitability in the methylazoxymethanol acetate developmental model of schizophrenia (although the drug also reduced baseline hippocampal excitability in control rats, which may interfere with some hippocampus‐dependent functions) (Gill and Grace, 2011) and ameliorates age‐related memory impairments linked to hippocampal GABA dysfunction and hyperexcitability (Koh et al., 2013). However, an α2 subunit‐selective partial agonist failed to improve cognition in schizophrenia patients, with potential reasons discussed by the authors including insufficient receptor activity and sedative side effects due to insufficient subunit selectivity (Buchanan et al., 2011). As discussed above for the other drug candidates, long‐standing, chronic patients, as typically included in clinical trials, who show regional brain hypoactivity may show little benefit from any treatment targeting aberrant neural activity (Anticevic et al., 2015; Krystal and Anticevic, 2015). Moreover, the effectiveness of GABAergic approaches in treating prefrontal and hippocampal disinhibition in schizophrenia may be limited because the GABA system may be compromised beyond repair – indeed, GABAA receptors are already up‐regulated in schizophrenia patients, presumably as a compensatory response to presynaptic dysfunction (Benes and Berretta, 2001; Guidotti et al., 2005; Lewis and Moghaddam, 2006; Heckers and Konradi, 2015).
Conclusions and future directions
Prefrontal and hippocampal neural disinhibition causes aberrant regional neuron firing, characterized by enhanced bursting, and disrupts some clinically relevant cognitive functions of these regions, including attention and memory, whereas some cognitive and behavioural functions supported by these regions (e.g. inhibitory response) are spared. This highlights the finding that distinct cognitive and behavioural functions supported by a brain region can show distinct dependencies on regional inhibitory GABA transmission, reflecting distinct relationships to regional neural activity: some functions require an appropriate inhibitory GABA tone and balanced levels of neural activity, whereas other functions may not (as long as the disinhibition remains subconvulsive). To characterize the distinct causal relationships of prefrontal and hippocampal GABA tone and neural activity to specific cognitive functions, future studies will have to compare systematically the effects of both functional inhibition and functional disinhibition on tests of such cognitive functions. Acknowledging the diversity of the various receptor and neuron types making up the inhibitory GABA system (Ascoli et al., 2008; Rudolph and Knoflach, 2011), studies using intra‐cerebral microinfusions of broadly acting GABA agonists and antagonists may be complemented by studies using more selective ligands, to target specific GABA receptors subtypes (Rudolph and Knoflach, 2011), and opto‐ and pharmacogenetic methods, to modulate GABA transmission presynaptically with some specificity for different interneuron types (Royer et al., 2012; Lovett‐Barron et al., 2014; Nguyen et al., 2014).
Consistent with the idea that regional neural disinhibition can disrupt cognitive processing in distal sites, hippocampal neural disinhibition disrupts attentional performance that does not require the hippocampus but is mediated by prefrontal‐striatal mechanisms (McGarrity et al., 2016). This supports that hippocampal dysfunction can be partly manifested through deficits in prefrontal function, consistent with strong hippocampo‐prefrontal functional connectivity (Meyer‐Lindenberg et al., 2005; Bast, 2011). Further studies combining regional neural disinhibition with additional behavioural tests will be required to characterize further the ‘distal cognitive impact’, that is, the significance for cognitive functions mediated by efferent sites, of regional GABA tone and neural disinhibition (e.g. does hippocampal neural disinhibition also disrupt other prefrontal cognitive processes, such as aspects of cognitive flexibility?). Moreover, neurophysiological measurements in projection sites will be required to characterize the brain‐wide effects of regional neural disinhibition. Using translational imaging methods (e.g. metabolic radiological imaging or EEG measurements) for such measurements would make it possible to examine the role of hippocampo‐prefrontal neural disinhibition in generating important clinical biomarkers (e.g. regional metabolic hyperactivity and aberrant EEG oscillations) of some of the disorders associated with neural disinhibition (see section on ‘Prefrontal and hippocampal neural disinhibition in neuropsychiatric disorders characterized by cognitive deficits’).
The reviewed findings suggest that prefrontal and hippocampal neural disinhibition contributes to important cognitive deficits, including attentional and memory deficits, in neuropsychiatric disorders characterized by such neural disinhibition, including schizophrenia and age‐related cognitive decline (Table 1). Therefore, aberrant neural activity caused by neural disinhibition is a promising target for treatments aimed at restoring important cognitive functions in these disorders, especially at early disease stages, when disinhibition‐induced prefrontal and hippocampal hyperactivity is prevalent, rather than the hypoactivity and atrophy that characterize later disease stages (Huang and Mucke, 2012; Anticevic et al., 2015; Krystal and Anticevic, 2015). Suitable pharmacological treatments will have to rebalance the disinhibition‐induced aberrant neural activity by curbing excessive excitation and burst firing, while leaving intact normal firing, as required for cognitive function. Candidate drugs include second‐generation antiepileptics, including lamotrigine and levetiracetam, and mGluR2 PAMs. Preclinical studies in rodent models of prefrontal and hippocampal neural disinhibition can provide proof‐of‐concept for the suggested rebalancing actions of these candidate drugs, using a two‐step approach, involving (i) electrophysiological studies to determine if a candidate drug rebalances aberrant neural activity and (ii) translational behavioural assays to determine if the drug ameliorates impairments of clinically relevant cognitive functions. Such studies could pave the way for clinical trials, where the challenge will be to include patients at early disease stages who are most likely to benefit from treatments rebalancing aberrant neural activity.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
T.B. currently holds a BBSRC iCASE Award in partnership with Boehringer Ingelheim.
Acknowledgements
Our original research was supported by the University of Nottingham, the Royal Society (T.B.) and the Leverhulme Trust (M.P.). Our current research in this area is supported by the BBSRC DTP at the University of Nottingham and a BBSRC iCASE Award (in partnership with Boerhinger Ingelheim).
Bast, T. , Pezze, M. , and McGarrity, S. (2017) Cognitive deficits caused by prefrontal cortical and hippocampal neural disinhibition. British Journal of Pharmacology, 174: 3211–3225. doi: 10.1111/bph.13850.
References
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews‐Zwilling Y, Gillespie AK, Kravitz AV, Nelson AB, Devidze N, Lo I et al. (2012). Hilar GABAergic interneuron activity controls spatial learning and memory retrieval. PLoS One 7: e40555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevic A, Corlett PR, Cole MW, Savic A, Gancsos M, Tang Y et al. (2015). N‐methyl‐D‐aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biol Psychiatry 77: 569–580. [DOI] [PubMed] [Google Scholar]
- Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ et al. (2012). NMDA receptor function in large‐scale anticorrelated neural systems with implications for cognition and schizophrenia. Proc Nat Acad Sci 109: 16720–16725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso‐Nanclares L, Anderson SA, Barrionuevo G, Benavides‐Piccione R, Burkhalter A et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9: 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74: 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard TM, Knoflach F, Prinssen E, Borroni E, Vivian JA, Basile J et al. (2009). RO4938581, a novel cognitive enhancer acting at GABAA alpha5 subunit‐containing receptors. Psychopharmacology (Berl) 202: 207–223. [DOI] [PubMed] [Google Scholar]
- Bast T (2007). Toward an integrative perspective on hippocampal function: from the rapid encoding of experience to adaptive behavior. Rev Neurosci 18: 253–281. [DOI] [PubMed] [Google Scholar]
- Bast T (2011). The hippocampal learning‐behavior translation and the functional significance of hippocampal dysfunction in schizophrenia. Curr Opin Neurobiol 21: 492–501. [DOI] [PubMed] [Google Scholar]
- Bast T, Feldon J (2003). Hippocampal modulation of sensorimotor processes. Prog Neurobiol 70: 319–345. [DOI] [PubMed] [Google Scholar]
- Bast T, Wilson IA, Witter MP, Morris RG (2009). From rapid place learning to behavioral performance: a key role for the intermediate hippocampus. PLoS Biol 7: e1000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast T, Zhang WN, Feldon J (2001a). Hyperactivity, decreased startle reactivity, and disrupted prepulse inhibition following disinhibition of the rat ventral hippocampus by the GABA(A) receptor antagonist picrotoxin. Psychopharmacology (Berl) 156: 225–233. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang WN, Feldon J (2001b). The ventral hippocampus and fear conditioning in rats. Different anterograde amnesias of fear after tetrodotoxin inactivation and infusion of the GABA(A) agonist muscimol. Exp Brain Res 139: 39–52. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S (2001). GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25: 1–27. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci 20: 4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan RW, Keefe RS, Lieberman JA, Barch DM, Csernansky JG, Goff DC et al. (2011). A randomized clinical trial of MK‐0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry 69: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Konnerth A (2016). Impairments of neural circuit function in Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Wang XJ (2012). Mechanisms of gamma oscillations. Annu Rev Neurosci 35: 203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi A, Fuchs EC, Allen K, Le Magueresse C, Monyer H (2012). Selective reduction of AMPA currents onto hippocampal interneurons impairs network oscillatory activity. PLoS One 7: e37318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Doobay VM, Liu Y (2012). Hippocampal‐prefrontal cortical circuit mediates inhibitory response control in the rat. J Neurosci 32: 10915–10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Muir JL (2001). Visual attention in the rat: a role for the prelimbic cortex and thalamic nuclei? Behav Neurosci 115: 417–428. [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW (2006). Functions of frontostriatal systems in cognition: comparative neuropsychopharmacological studies in rats, monkeys and humans. Biol Psychol 73: 19–38. [DOI] [PubMed] [Google Scholar]
- Cooper DC (2002). The significance of action potential bursting in the brain reward circuit. Neurochem Int 41: 333–340. [DOI] [PubMed] [Google Scholar]
- Curley AA (2012). Opinions mixed on future for Lilly's mGluR2/3 agonist for schizophrenia [Online]. Available at: http://www.schizophreniaforum.org/news/opinions‐mixed‐future‐lilly%E2%80%99s‐mglur23‐agonist‐schizophrenia (accessed: 19 January 2017).
- Davis KE, Fox S, Gigg J (2014). Increased hippocampal excitability in the 3xTgAD mouse model for Alzheimer's disease in vivo. PLoS One 9: e91203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM et al. (2006). An inverse agonist selective for alpha5 subunit‐containing GABAA receptors enhances cognition. J Pharmacol Exp Ther 316: 1335–1345. [DOI] [PubMed] [Google Scholar]
- De Smedt T, Raedt R, Vonck K, Boon P (2007). Levetiracetam: the profile of a novel anticonvulsant drug‐part I: preclinical data. CNS Drug Rev 13: 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P (2013). Parvalbumin‐expressing basket‐cell network plasticity induced by experience regulates adult learning. Nature 504: 272–276. [DOI] [PubMed] [Google Scholar]
- Engel TA, Steinmetz NA, Gieselmann MA, Thiele A, Moore T, Boahen K (2016). Selective modulation of cortical state during spatial attention. Science 354: 1140–1144. [DOI] [PubMed] [Google Scholar]
- Engin E, Zarnowska ED, Benke D, Tsvetkov E, Sigal M, Keist R et al. (2015). Tonic Inhibitory control of dentate gyrus granule cells by alpha5‐containing GABAA receptors reduces memory interference. J Neurosci 35: 13698–13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T, Tse MT, Floresco SB (2011). Reducing prefrontal gamma‐aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol Psychiatry 69: 432–441. [DOI] [PubMed] [Google Scholar]
- Fajnerova I, Rodriguez M, Levcik D, Konradova L, Mikolas P, Brom C et al. (2014). A virtual reality task based on animal research – spatial learning and memory in patients after the first episode of schizophrenia. Front Behav Neurosci 8: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6: 215–229. [DOI] [PubMed] [Google Scholar]
- Faure JB, Marques‐Carneiro JE, Akimana G, Cosquer B, Ferrandon A, Herbeaux K et al. (2014). Attention and executive functions in a rat model of chronic epilepsy. Epilepsia 55: 644–653. [DOI] [PubMed] [Google Scholar]
- Fell MJ, McKinzie DL, Monn JA, Svensson KA (2012). Group II metabotropic glutamate receptor agonists and positive allosteric modulators as novel treatments for schizophrenia. Neuropharmacology 62: 1473–1483. [DOI] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci 10: 411–413. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Block AE, Tse MT (2008). Inactivation of the medial prefrontal cortex of the rat impairs strategy set‐shifting, but not reversal learning, using a novel, automated procedure. Behav Brain Res 190: 85–96. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Todd CL, Grace AA (2001). Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci 21: 4915–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry 167: 1479–1488. [DOI] [PubMed] [Google Scholar]
- Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S et al. (2014). Interneuron precursor transplants in adult hippocampus reverse psychosis‐relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A 111: 7450–7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KM, Grace AA (2011). Heterogeneous processing of amygdala and hippocampal inputs in the rostral and caudal subregions of the nucleus accumbens. Int J Neuropsychopharmacol 14: 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW (2000). Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci 20: 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AJ, Calhoon GG, Shusterman I, Schoenbaum G, Roesch MR, O'Donnell P (2010). More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J Neurosci 30: 17102–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Dong E, Grayson DR, Veldic M et al. (2005). GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology (Berl) 180: 191–205. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Koh MT, Gallagher M (2017). Heightened cortical excitability in aged rodents with memory impairment. Neurobiol Aging 54: 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Tai C, Jones CJ, Scheuer T, Catterall WA (2014). Enhancement of inhibitory neurotransmission by GABAA receptors having alpha2,3‐subunits ameliorates behavioral deficits in a mouse model of autism. Neuron 81: 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C (2015). GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res 167: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hort J, Laczo J, Vyhnalek M, Bojar M, Bures J, Vlcek K (2007). Spatial navigation deficit in amnestic mild cognitive impairment. Proc Natl Acad Sci U S A 104: 4042–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148: 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M (2011). How inhibition shapes cortical activity. Neuron 72: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhikevich EM, Desai NS, Walcott EC, Hoppensteadt FC (2003). Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci 26: 161–167. [DOI] [PubMed] [Google Scholar]
- Karreman M, Moghaddam B (1996). The prefrontal cortex regulates the basal release of dopamine in the limbic striatum: an effect mediated by ventral tegmental area. J Neurochem 66: 589–598. [DOI] [PubMed] [Google Scholar]
- Kim H, Ahrlund‐Richter S, Wang X, Deisseroth K, Carlen M (2016). Prefrontal parvalbumin neurons in control of attention. Cell 164: 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M (2010). Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology 35: 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Rosenzweig‐Lipson S, Gallagher M (2013). Selective GABA(A) alpha5 positive allosteric modulators improve cognitive function in aged rats with memory impairment. Neuropharmacology 64: 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradi C, Zimmerman EI, Yang CK, Lohmann KM, Gresch P, Pantazopoulos H et al. (2011). Hippocampal interneurons in bipolar disorder. Arch Gen Psychiatry 68: 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Anticevic A (2015). Toward illness phase‐specific pharmacotherapy for schizophrenia. Biol Psychiatry 78: 738–740. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS (2006). Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci 26: 4613–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Lu L (1997). Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol 121: 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large CH, Bison S, Sartori I, Read KD, Gozzi A, Quarta D et al. (2011). The efficacy of sodium channel blockers to prevent phencyclidine‐induced cognitive dysfunction in the rat: potential for novel treatments for schizophrenia. J Pharmacol Exp Ther 338: 100–113. [DOI] [PubMed] [Google Scholar]
- Larkum M (2013). A cellular mechanism for cortical associations: an organizing principle for the cerebral cortex. Trends Neurosci 36: 141–151. [DOI] [PubMed] [Google Scholar]
- Lee V, MacKenzie G, Hooper A, Maguire J (2016). Reduced tonic inhibition in the dentate gyrus contributes to chronic stress‐induced impairments in learning and memory. Hippocampus 26: 1276–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Luthi A (2015). Disinhibition, a circuit mechanism for associative learning and memory. Neuron 88: 264–276. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B (2006). Cognitive dysfunction in schizophrenia: convergence of gamma‐aminobutyric acid and glutamate alterations. Arch Neurol 63: 1372–1376. [DOI] [PubMed] [Google Scholar]
- Lisman JE (1997). Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20: 38–43. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S et al. (2008). Circuit‐based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman RE, Smith MA, Doherty JJ, Cross A, Raines S, Gertsik L et al. (2016). AZD8529, a positive allosteric modulator at the mGluR2 receptor, does not improve symptoms in schizophrenia: a proof of principle study. Schizophr Res 172: 152–157. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA (2011). Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci 32: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett‐Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR et al. (2014). Dendritic inhibition in the hippocampus supports fear learning. Science 343: 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett‐Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH et al. (2012). Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci 15: 423–430 S421–423. [DOI] [PubMed] [Google Scholar]
- Luscher B, Shen Q, Sahir N (2011). The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry 16: 383–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig C, Kozak R, Sarter M, Young JW, Robbins TW (2013). CNTRICS final animal model task selection: control of attention. Neurosci Biobehav Rev 37: 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EO, Paulsen O (2007). Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci 30: 343–349. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, Orser BA (2010). Alpha5GABAA receptor activity sets the threshold for long‐term potentiation and constrains hippocampus‐dependent memory. J Neurosci 30: 5269–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarrity S, Mason R, Fone KC, Pezze M, Bast T (2016). Hippocampal neural disinhibition causes attentional and memory deficits. Cereb Cortex Advance Article: 1–16. [DOI] [PubMed] [Google Scholar]
- McGarrity S, Somerled S, Eaton C, Bast T, Pezze M (2014). Medial prefrontal cortex is not required, but can modulate, hippocampus‐dependent behaviour on the watermaze delayed‐matching‐to‐place test. FENS Abstr 7: 1403. [Google Scholar]
- McGaughy J, Dalley JW, Morrison CH, Everitt BJ, Robbins TW (2002). Selective behavioral and neurochemical effects of cholinergic lesions produced by intrabasalis infusions of 192 IgG‐saporin on attentional performance in a five‐choice serial reaction time task. J Neurosci 22: 1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Lindenberg AS, Olsen RK, Kohn PD, Brown T, Egan MF, Weinberger DR et al. (2005). Regionally specific disturbance of dorsolateral prefrontal‐hippocampal functional connectivity in schizophrenia. Arch Gen Psychiatry 62: 379–386. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Agid Y, Brune M, Bullmore ET, Carter CS, Clayton NS et al. (2012). Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11: 141–168. [DOI] [PubMed] [Google Scholar]
- Mitchell SN, Yee BK, Feldon J, Gray JA, Rawlins JN (2000). Activation of the retrohippocampal region in the rat causes dopamine release in the nucleus accumbens: disruption by fornix section. Eur J Pharmacol 407: 131–138. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Krystal JH (2012). Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull 38: 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ER, Fernando AB, Urcelay GP, Robinson ES, Mar AC, Theobald DE et al. (2012). Impulsive behaviour induced by both NMDA receptor antagonism and GABAA receptor activation in rat ventromedial prefrontal cortex. Psychopharmacology (Berl) 219: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L et al. (2011). Parvalbumin‐positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci 14: 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava‐Mesa MO, Jimenez‐Diaz L, Yajeya J, Navarro‐Lopez JD (2014). GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer's disease. Front Cell Neurosci 8: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen R, Morrissey MD, Mahadevan V, Cajanding JD, Woodin MA, Yeomans JS et al. (2014). Parvalbumin and GAD65 interneuron inhibition in the ventral hippocampus induces distinct behavioral deficits relevant to schizophrenia. J Neurosci 34: 14948–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P (2011). Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull 37: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, Disterhoft JF (2010). Learning and aging related changes in intrinsic neuronal excitability. Front Aging Neurosci 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine TA, Slipp LE, Carlezon WA Jr (2011). Schizophrenia‐like attentional deficits following blockade of prefrontal cortex GABAA receptors. Neuropsychopharmacology 36: 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passetti F, Chudasama Y, Robbins TW (2002). The frontal cortex of the rat and visual attentional performance: dissociable functions of distinct medial prefrontal subregions. Cereb Cortex 12: 1254–1268. [DOI] [PubMed] [Google Scholar]
- Pehrson AL, Bondi CO, Totah NK, Moghaddam B (2013). The influence of NMDA and GABA(A) receptors and glutamic acid decarboxylase (GAD) activity on attention. Psychopharmacology (Berl) 225: 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg‐Raibstein D, Pezze MA, Ferger B, Zhang WN, Murphy CA, Feldon J et al. (2005). Activation of dopaminergic neurotransmission in the medial prefrontal cortex by N‐methyl‐d‐aspartate stimulation of the ventral hippocampus in rats. Neuroscience 132: 219–232. [DOI] [PubMed] [Google Scholar]
- Pezze M, Bast T (2012). Dopaminergic modulation of hippocampus‐dependent learning: blockade of hippocampal D1‐class receptors during learning impairs 1‐trial place memory at a 30‐min retention delay. Neuropharmacology 63: 710–718. [DOI] [PubMed] [Google Scholar]
- Pezze M, McGarrity S, Mason R, Fone KC, Bast T (2014). Too little and too much: hypoactivation and disinhibition of medial prefrontal cortex cause attentional deficits. J Neurosci 34: 7931–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze MA, Dalley JW, Robbins TW (2007). Differential roles of dopamine D1 and D2 receptors in the nucleus accumbens in attentional performance on the five‐choice serial reaction time task. Neuropsychopharmacology 32: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze MA, Dalley JW, Robbins TW (2009). Remediation of attentional dysfunction in rats with lesions of the medial prefrontal cortex by intra‐accumbens administration of the dopamine D(2/3) receptor antagonist sulpiride. Psychopharmacology (Berl) 202: 307–313. [DOI] [PubMed] [Google Scholar]
- Prut L, Prenosil G, Willadt S, Vogt K, Fritschy JM, Crestani F (2010). A reduction in hippocampal GABAA receptor alpha5 subunits disrupts the memory for location of objects in mice. Genes Brain Behav 9: 478–488. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel‐Hidalgo JJ (2007). GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman‐Rakic PS (2000). Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci 20: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitsek J, Ratner MH, Stewart T, Eichenbaum H, Farb DH (2015). Combined administration of levetiracetam and valproic acid attenuates age‐related hyperactivity of CA3 place cells, reduces place field area, and increases spatial information content in aged rat hippocampus. Hippocampus 25: 1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg C, Bussey TJ, Saksida LM (2013). Paying more attention to attention: towards more comprehensive cognitive translation using mouse models of Alzheimer's disease. Brain Res Bull 92: 49–55. [DOI] [PubMed] [Google Scholar]
- Romberg C, Mattson MP, Mughal MR, Bussey TJ, Saksida LM (2011). Impaired attention in the 3xTgAD mouse model of Alzheimer's disease: rescue by donepezil (Aricept). J Neurosci 31: 3500–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC et al. (2012). Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci 15: 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2: 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov 10: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger S, Spirig D, Donato F, Caroni P (2012). Goal‐oriented searching mediated by ventral hippocampus early in trial‐and‐error learning. Nat Neurosci 15: 1563–1571. [DOI] [PubMed] [Google Scholar]
- Ruzicka WB, Subburaju S, Benes FM (2015). Circuit‐ and diagnosis‐specific DNA methylation changes at gamma‐aminobutyric acid‐related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiat 72: 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A 109: E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I et al. (2013). Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 78: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Kelly MA, Corcoran CM, Van Heertum K, Seckinger R, Goetz R et al. (2009). Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophr Res 114: 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Yu S, Wong W, McGeer EG, McGeer PL (2013). GAD65, GAD67, and GABAT immunostaining in human brain and apparent GAD65 loss in Alzheimer's disease. J Alzheimers Dis 33: 1073–1088. [DOI] [PubMed] [Google Scholar]
- Simkin D, Hattori S, Ybarra N, Musial TF, Buss EW, Richter H et al. (2015). Aging‐related hyperexcitability in CA3 pyramidal neurons is mediated by enhanced A‐type K+ channel function and expression. J Neurosci 35: 13206–13218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L (1981). Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J Cereb Blood Flow Metab 1: 7–36. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV et al. (2010). Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med 12: 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel AM, Koh MT, Vogt NM, Rapp PR, Gallagher M (2013). Hilar interneuron vulnerability distinguishes aged rats with memory impairment. J Comp Neurol 521: 3508–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EM, Fadel JR, Mott DD (2012). Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiol Aging 33: 431 e431–431 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele RJ, Morris RG (1999). Delay‐dependent impairment of a matching‐to‐place task with chronic and intrahippocampal infusion of the NMDA‐antagonist D‐AP5. Hippocampus 9: 118–136. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Magee JC (2009). Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62: 102–111. [DOI] [PubMed] [Google Scholar]
- Thomé A, Gray DT, Erickson CA, Lipa P, Barnes CA (2016). Memory impairment in aged primates is associated with region‐specific network dysfunction. Mol Psychiatry 21: 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E et al. (2014). Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry 171: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W (2012). Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large‐scale networks. Neuron 75: 963–980. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149: 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkers CH, Mirza NR, Olivier B, Kahn RS (2010). The inhibitory GABA system as a therapeutic target for cognitive symptoms in schizophrenia: investigational agents in the pipeline. Expert Opin Investig Drugs 19: 1217–1233. [DOI] [PubMed] [Google Scholar]
- Wang J, Bast T, Wang YC, Zhang WN (2015). Hippocampus and two‐way active avoidance conditioning: contrasting effects of cytotoxic lesion and temporary inactivation. Hippocampus 25: 1517–1531. [DOI] [PubMed] [Google Scholar]
- Wigstrom H, Gustafsson B (1983). Facilitated induction of hippocampal long‐lasting potentiation during blockade of inhibition. Nature 301: 603–604. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Gallagher M, Eichenbaum H, Tanila H (2006). Neurocognitive aging: prior memories hinder new hippocampal encoding. Trends Neurosci 29: 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Sudhof TC (2012). Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 73: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]