

Abstract
Background and Purpose
Cysteinyl leukotrienes (CysLTs) are pro‐inflammatory lipid mediators that exacerbate disease state in several asthma phenotypes including asthma induced by allergen, virus and exercise. However, the role of CysLTs in irritant‐induced airway disease is not well characterized. The purpose of the current study was to investigate the effect of montelukast, a CysLT1 receptor antagonist, on parameters of irritant‐induced asthma induced by inhalation of chlorine in the mouse.
Experimental Approach
BALB/c mice were exposed to chlorine in air (100 ppm, for 5 min). Montelukast (3 mg·kg−1) or the vehicle (1% methylcellulose) was administered 24 and 1 h prior to chlorine exposure and 1 h prior to outcome measurements. Twenty‐four hours after exposure, responses to inhaled aerosolized methacholine, cell composition and an array of cytokines/chemokines in bronchoalveolar lavage (BAL) fluid were measured. Neutralizing antibodies against IL‐6 and VEGF were administered prior to exposures.
Key Results
Montelukast reduced chlorine ‐induced airway hyperresponsiveness (AHR) to methacholine in the peripheral lung compartment as estimated from dynamic elastance, but not in large conducting airways. Montelukast treatment attenuated chlorine‐induced macrophage influx, neutrophilia and eosinophilia in BAL fluid. Chlorine exposure increased VEGF, IL‐6, the chemokines KC and CCL3 in BAL fluid. Montelukast treatment prevented chlorine‐induced increases in VEGF and IL‐6. Anti‐IL‐6 antibody inhibited chlorine‐induced neutrophilia and reduced AHR.
Conclusions and Implications
Pre‐treatment with montelukast attenuated chlorine‐induced neutrophilia and AHR in mice. These effects are mediated, in part, via IL‐6.
Abbreviations
- AHR
airway hyperresponsiveness
- BAL
bronchoalveolar lavage
- CysLTs
cysteinyl leukotrienes
- IIA
irritant‐induced asthma
Introduction
Irritant‐induced asthma (IIA) is a subtype of asthma that occurs following inhalation of noxious substances, with a short latency and does not require prior sensitization (Brooks and Bernstein, 2011; Tarlo and Lemiere, 2014). One of the most commonly reported toxic exposures that results in the development of IIA is inhalation of chlorine gas and chlorine‐related compounds (see White and Martin, 2010). Chlorine is a highly reactive substance that mediates its inhalational toxicity through oxidative stress (Martin et al., 2003), resulting in a predominantly neutrophilic airway inflammation and airway hyperresponsiveness (AHR) to inhaled aerosolized methacholine (MCh). Chlorine‐induced neutrophilia is responsible for further oxidative damage and the consequent airway dysfunction and is sufficient to engage the Nrf2‐mediated antioxidant response (McGovern et al., 2015).
Cysteinyl‐leukotrienes (CysLTs) are lipid mediators which have been extensively studied in the context of allergen‐driven airway dysfunction but have received less attention in the context of responses to irritants. Montelukast (MK) is a highly selective antagonist of CysLT1 receptors (Jones et al.,1995), the predominant CysLT receptor expressed in the bronchial tree (Lynch et al., 1999). While CysLTs have not been usually associated with oxidative stress‐induced, neutrophil‐driven pulmonary disease, there is evidence that montelukast can affect processes associated with neutrophilic inflammation. Treatment with montelukast reduced neutrophilic airway inflammation and prevented alterations in static compliance following the induction of experimental emphysema with porcine pancreatic elastase in mice (Ikeda et al., 2014). In addition, montelukast may be protective against oxidative damage, as treatment with this drug prevented increases in glutathione in models of acute injury including haemorrhagic shock and unilateral ureteral obstruction in rats (Al‐Amran et al., 2013; Otunctemur et al., 2015). Although montelukast has been used extensively in the treatment of allergen, viral and exercise‐induced asthma, the efficacy of montelukast treatment following oxidative‐stress induced IIA has not been thoroughly evaluated. Therefore, we sought to examine the effect of montelukast treatment on chlorine‐induced airway dysfunction in mice exposed to inhaled chlorine. For this, BALB/C mice were treated with montelukast prior to chlorine exposure. Following exposure, we assessed measured airway function in response to inhaled MCh, collected bronchoalveolar lavage (BAL) fluid for analysis inflammatory cell populations and quantified levels of several cytokine and chemokines. We evaluated the endogenous antioxidant response by quantifying Nrf2 nuclear translocation in bronchial epithelial cells in vivo and assessed the capacity of CysLTs to directly induce Nrf2 nuclear translocation in vitro in human bronchial epithelial cells. Montelukast was effective at preventing chlorine‐induced increases in IL‐6 and VEGF, and therefore, we assessed the roles of these cytokines using an IL‐6 neutralizing antibody or an anti‐VEGF antibody prior to chlorine exposure and evaluated the inflammatory response and pulmonary mechanics.
Our results showed that montelukast was effective at preventing chlorine‐induced AHR in the peripheral lung compartment, pulmonary neutrophilia and eosinophilia and increases in IL‐6 and VEGF. Treatment with montelukast appeared to enhance the endogenous antioxidant response by increasing Nrf2 nuclear translocation in bronchial epithelial cells, although this was likely to be mediated via indirect mechanisms.
Methods
Animals and protocol
All animal care and experimental procedures complied with the guidelines of the Canadian Council for Animal Care and protocols were approved by the Animal Care Committee of McGill University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male BALB/c mice (18–22 g) were purchased from Charles River (Wilmington, MA, USA). Housing conditions were as follows. All rodents were provided with corn cob bedding + Enviro dry and a Nestlet for enrichment. Humidity was maintained between 30 and 70%. Light cycles are based on a 12 h cycle (7:00 h to 19:00 h of light). Room temperatures are maintained between 18 and 24°C. All the mouse cages are on automated watering system using reverse osmosis water. Mice were fed irradiated rodent chow provided by Teklad. For all experimental procedures, mice were provided with water and food ad libitum throughout the experiment.
Four groups were studied: (1) methylcellulose (MC)/air exposed; (2) montelukast (MK)/air exposed; (3) MC/Cl2 exposed; and (4) MK/Cl2 exposed. A dose of 3 mg·kg−1 of montelukast (Cayman Chemical, Ann Arbor, MI, USA) was prepared in a 1% MC or 1% MC alone was administered in a volume of 200 μL using a 22 gauge gavage needle (Kent Scientific Corporation, CI, USA). Mice were treated three times; 24 h prior to chlorine exposure, 1 h prior to chlorine exposure and 1 h prior to evaluation of AHR (Figure 1). The dose of montelukast was based on prior studies of its efficacy against late allergic inflammation and bronchoconstriction in rats (Ihaku et al., 1999).
Figure 1.
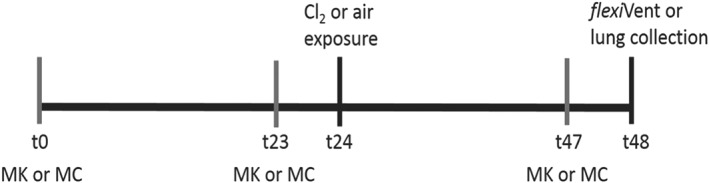
Study design. Mice were treated with 3 mg·kg−1 of montelukast (MK) prepared in a 1% MC solution. Control mice received 1% MC alone. montelukast or MC was administered in a volume of 200 μL using a 22 gauge gavage needle. Mice were treated three times: 24 h prior to chlorine (Cl2) exposure, 1 h prior to chlorine exposure and 1 h prior to evaluation of AHR.
Exposure to chlorine
Twenty‐four hours following treatment with montelukast or MC, mice were exposed to chlorine at a concentration of 100 ppm for 5 min using a nose‐only exposure device as previously described (McGovern et al., 2010). Chlorine gas was mixed with room air using a Dynacalibrator (Model 230‐28A, VICI Metronics, WA, USA).
Evaluation of respiratory system mechanics and methacholine (MCh) challenge
Twenty‐four hours following chlorine exposure, respiratory system mechanics were measured in response to aerosolized MCh (McGovern et al., 2013). Prior to measurements, mice were sedated with xylazine hydrochloride (8 mg·kg−1 i.p.) and anaesthetized with sodium pentobarbital (30 mg·kg−1 i.p). Following anaesthesia, the trachea was cannulated with an 18 gauge cannula. Animals were connected to a computer‐controlled small animal ventilator (flexiVent, Scireq, Montreal, PQ, Canada) and paralysed using pancuronium chloride (0.2 mg·kg−1 i.p.). Mice were ventilated in a quasi‐sinusoidal fashion with a tidal volume of 0.18 mL and 150 breaths per minute. A positive end‐expiratory pressure of 3.0 cm H2O was used. The mice received an initial baseline challenge of saline followed by increasing doses of nebulized MCh; 6.25, 12.5, 25, 50 mg·mL−1. The response of large airways (Newtonian resistance, RN parameter), small airways and lung tissue resistance (tissue damping, G parameter) and lung parenchymal stiffness (elastance, H parameter) were assessed using the constant‐phase model (Bates, 2009).
Bronchoalveolar lavage
Following lung function measurements, mice were killed with an overdose of sodium pentobarbital (80 mg·kg−1, i.p.) and BAL was performed. The lungs were lavaged with 1 mL of sterile PBS and placed in a 1.5 mL Eppendorf tube on ice. BAL fluid was centrifuged at 850 × g for 5 min at 4°C, and the supernatant was retained for cytokine analysis. The cell pellet was re‐suspended in sterile PBS, and total live and dead cells were counted using Trypan Blue exclusion. Differential cell counts were performed following preparation of cells on slides using a cytocentrifuge (Cytospin™ 4, Thermo Fisher Scientific, Inc., Waltham, MA, USA) and a commercial stain (Diff‐Quik® method, Medical Diagnostics, Düdingen, Germany). Differential cell counts, including epithelial cells, were determined on a count of 300 cells.
Antibody treatments
Mice were treated with 100 μg of anti‐IL‐6R monoclonal antibody (R&D Systems, Minneapolis, MN) by i.p. injection, 4 h before chlorine exposure. Another set of mice were similarly treated with 100 μg of anti‐VEGF monoclonal antibody (Biolegend, San Diego, CA) and isotype control. Purified rat IgG (R & D Systems) was administered as control.
mRNA analysis of central lung tissues
Twenty‐four hours after exposure to chlorine, the right lung was removed, followed by isolation of the parahilar region of the lung (termed central portion), immersed in RNAlater, snap‐frozen in liquid nitrogen and then stored at −80°C. After thawing, the tissue was homogenized and RNA was extracted using the RNeasy Minikit (Qiagen, Toronto, ON, Canada). Reverse transcription was performed using Superscript II enzyme (Invitrogen, Life Technologies, Burlington, ON, Canada). The following primer sequences were used: ho‐1 forward: GAATGAACACTCTGGAGATGACAC, ho‐1 reverse: TGTGAGGGACTCTGGTCTTTG; gpx‐2 forward: GTTCTCGGCTTCCCTTGC, gpx‐2 reverse: TTCAGGACTCCTCGTTCTGA. nqo‐1 forward: AGCCAATCAGCGTTCGGTA, nqo‐1 reverse: GAATGGGCCAGTACAATCAGG. sod‐1 forward: CAGGACCTCATTTTAATCCTCAC, sod‐1 reverse: TGCCCAGGTCTCCAACAT. S9 was used as an endogenous control.
Nrf2 nuclear translocation analysis in vivo
Following BAL, the left lung was isolated and instilled intra‐tracheally with 10% buffered formalin for fixation. Tissue was embedded in paraffin, and 5‐μm‐thick sections were prepared prior to staining. For Nrf2 immunofluorescence staining, sections were first de‐paraffinized and rehydrated. The slides were placed in boiling water for 8 min using an antigen unmasking solution (Vector Labs, Burlingame, CA, USA) for antigen retrieval. Sections were incubated in 2% Triton X‐100 with TBS for membrane permeabilization, protein blocking was performed and slides were placed in a humidified chamber overnight at 4°C. Sections were incubated overnight in a humidified chamber at 4°C with rabbit polyclonal IgG antibody against Nrf2 (#F0112; Santa Cruz Biochemistry, CA, USA) at a 1:100 dilution in Antibody Diluent with Background Reducing Components (Dako, Carpinteria, CA, USA). Following incubation, FITC Goat Anti‐Rabbit IgG (#554020; Becton Dickinson, CA, USA) at a 1:400 dilution in double‐ distilled H2O was applied for 45 min at room temperature. Nuclear staining was done using Hoechst (1:4000 in double‐ distilled H2O; Bio‐Rad, CA, USA). The sections were mounted using Fluoroshield and were visualized using an Olympus BX51 microscope. Images were taken using QImaging Retina 2000r camera. Nrf2 nuclear translocation was quantified using commercial software (Image‐Pro Plus, Media Cybernetics, Inc., MD, USA) as previously described (Manders, 1993; Smallcombe and McMillan, 2002; McGovern et al., 2015).
Nrf2 translocation assay in vitro
In order to assess whether CysLTs had a direct effect on Nrf2 nuclear translocation, we utilized a human bronchial epithelial cell line (BEAS‐2B, ATCC) cells containing a Nrf2‐luciferase reporter. BEAS‐2B cells were stably transfected with a pGL4.28 plasmid (Promega, Madison, WI, USA) containing an antioxidant response element (ARE) (G TAC CGC AGT CAC AGT GAC TCA GCA GAA TCG CTA G) upstream of a hygromycin B resistance gene and a firefly luciferase construct.
The cells were incubated for 24 h in DMEM containing 0.1% FBS with montelukast (10 uM) (Jones et al., 1995) or vehicle. Following incubation, sodium hypochlorite (3 μM) was added. Four hours later, culture medium was removed and cells were lysed using 40 μL of Reporter Lysis buffer (Promega, Madison, WI, USA) that was added. Whole cell lysates were collected and centrifuged at 15 800 × g for 3 min; 10 μL of supernatant was transferred to a 96‐well plate for reading in the Tecan iControl luciferase system.
Measurement of cytokine levels in BAL
To assess the production of the cytokines IL‐6, IL‐10, IL‐17, TNF‐α, the chemokines KC, CCL3, CXCL2, CCL5, and VEGF in BAL, we used MILLIPLEX MAP Mouse Cytokine/Chemokine kit (Millipore Corporation, Billerica, MA, USA) according to the manufacturer's instructions and using a Luminex xMAP.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Airway responses to MCh were analysed in GraphPad Prism version 6 (GraphPad software, San Diego, CA, USA) by two‐way ANOVA followed by Tukey's multiple comparison test comparing all experimental groups with each other. One‐way ANOVA and Newman–Keuls multiple comparison test were used for all other analyses involving three or more groups. The Kruskal–Wallis test was used for the analysis of immunofluorescence with Dunn's multiple comparison post hoc test because the data were not normally distributed. A P value of <0.05 was considered significant.
Materials
The compounds used in these experiments were supplied as follows: chlorine by VICI Metronics (Poulsbo, Washington); methacholine, pancuronium bromide, sodium hypochlorite by Sigma‐Aldrich (St Louis, MO) and montelukast by Cayman Chemicals (Ann Arbor, Michigan);
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Montelukast treatment reduces airway hyperresponsiveness
Evaluation of airway mechanics was performed in montelukast‐ or MC‐treated mice following either chlorine or air exposure. Montelukast treatment did not significantly reduce Newtonian resistance (RN) (Figure 2A), indicating that large AHR was not inhibited, but it did prevent chlorine‐induced increases in tissue damping (G) and tissue elastance (H) (Figure 2B, C), indicating an effect on the peripheral airways. MC treatment alone had no effect on airway responses to MCh in air‐exposed groups for any parameter.
Figure 2.
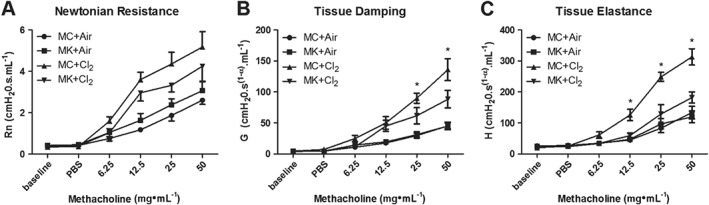
Montelukast reduces chlorine‐induced airway hyperresponsiveness. Twenty‐four hours following chlorine (Cl2) exposure, respiratory system mechanics were evaluated in response to inhaled MCh. (A) Montelukast (MK; 3 mg·kg−1) treatment did not prevent increases in Newtonian resistance. (B) Montelukast prevented chlorine‐induced increases in tissue damping at the two highest concentrations of MCh. (C) Montelukast prevented chlorine‐induced increases in tissue elastance at the three highest concentrations of MCh. (A–C) Montelukast treatment alone had no effect on AHR as no difference was observed between montelukast + Air and MC + Air groups for any parameter. MC + Air, MK + Air n = 7 per group; MC + Cl2, MK + Cl2 n = 9 per group. *P < 0.05, significant difference between (MC + Cl2) and (MK + Cl2) groups.
Montelukast reduces inflammation in BAL
Chlorine‐induced airway inflammation and epithelial cell shedding were assessed in BAL fluid. Total cell counts in MK‐treated groups were lower compared with MC‐treated mice following chlorine (Figure 3A). Montelukast treatment prevented chlorine‐induced increase in macrophages (Figure 3B) and also reduced the neutrophil influx, which was significantly lower in the montelukast groups compared with MC mice following chlorine (Figure 3C). Montelukast also prevented the chlorine‐induced increases in eosinophils (Figure 3E). Lymphocytes were increased following chlorine exposure but were unaffected by montelukast pretreatment (data not shown). Epithelial cell numbers were unaltered by chlorine exposure and by montelukast (data not shown). Montelukast did not affect cell populations in air‐exposed mice compared with air‐MC treatment (Figure 3A–D).
Figure 3.
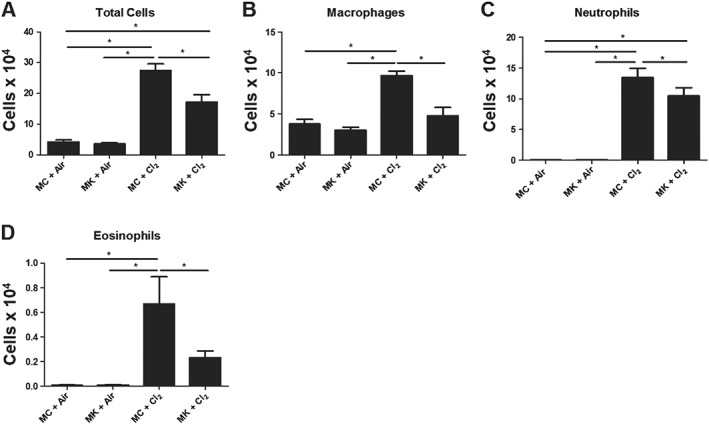
Montelukast prevents pulmonary inflammation following chlorine exposure. (A) Montelukast (MK) treatment reduced total number of inflammatory cells in chlorine (Cl2)‐exposed mice. (B) Montelukast reduced macrophages compared with (MC + Cl2) ‐exposed mice. (C) Montelukast prevented neutrophil influx in chlorine‐treated groups. (D) Montelukast prevented chlorine‐induced increase in eosinophil numbers. (A–D) There was no difference in inflammatory cell profiles between MC + Air and MK + Air‐treated groups. n = 10 per group; *P < 0.05, significantly different as indicated.
Nrf2 nuclear translocation by immunofluorescence analysis
To address whether montelukast treatment could affect the endogenous antioxidant response, Nrf2 nuclear translocation was evaluated. Figure 4A shows illustrative examples of airways. Hoechst stain was used to mark nuclei (Figure 4A, panels i–iv) and Nrf2 staining is visualized in green (Figure 4A, panels v–viii). Figure 4A, panels ix–xii shows the images merged. Following chlorine exposure in the presence of montelukast, there was evidence of translocation of Nrf2 to nuclei in the epithelium. Exposure to chlorine increased the Nrf2 nuclear immunofluorescence (Figure 4B). The nuclear localization of Nrf2 was further increased by montelukast treatment both in air‐exposed mice and mice exposed to chlorine (Figure 4B).
Figure 4.
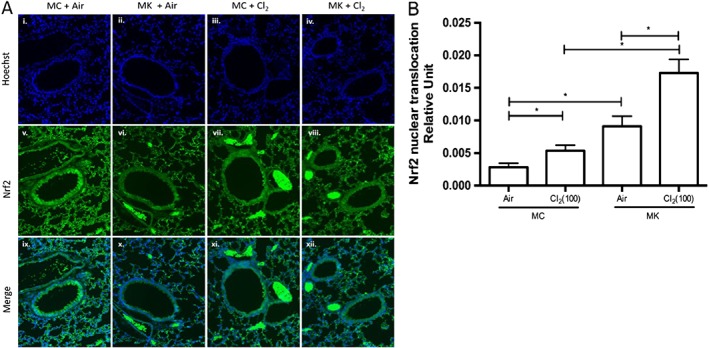
Montelukast increases Nrf2 nuclear translocation. Immunofluorescent staining for Nrf2 was performed 24 h following chlorine (Cl2) exposure. (A, panels i–iv) Nuclei were stained with Hoechst. (A, panels v–viii) Illustrative images showing airways stained for Nrf2. (A, panels ix–xii) Merge of nuclear and Nrf2 staining. (B) Quantification of Nrf2 nuclear translocation using Image ProPlus software. Nrf2 translocation occurred after chlorine exposure, and this translocation was enhanced by montelukast treatment. MC + Air, MK + Air n = 6 per group; MC + Cl2, MK + Cl2 n = 10 per group; 10–20 airways analysed per mouse. *P < 0.05, significantly different as indicated.
Nrf2‐dependent antioxidant gene expression
To evaluate whether montelukast affected Nrf2‐dependent antioxidant enzyme gene expression, qPCR was performed on lung tissue isolated from the parahilar region of the lung. We previously reported that only the parahilar region and not the peripheral lung tissue showed increases in Nrf2 mRNA 24 h after chlorine exposure (McGovern et al., 2015). mRNA levels of gpx‐2, nqo‐1 and ho‐1 increased after chlorine exposure (Figure 5A–C). Montelukast treatment prevented increases in HO‐1 coding for haem oxygenase ‐1 (Figure 5C). However, Sod‐1 expression was not affected by chlorine exposure or by montelukast (Figure 5D). Montelukast treatment also did not significantly affect the transcript levels in the lung tissue of air‐exposed animals.
Figure 5.
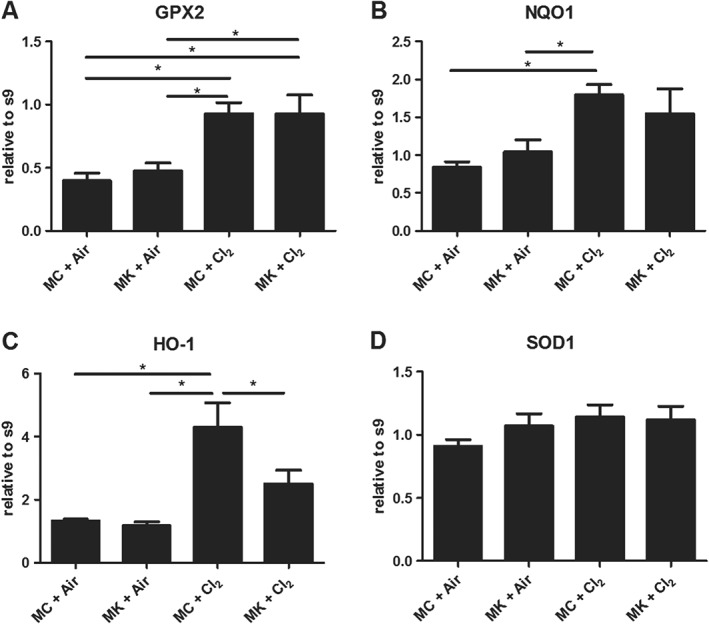
Montelukast (MK) effects on antioxidant gene expression. Nrf2 second phase enzyme expression increased at 24 h after chlorine (Cl2) exposure (A–C). (A) glutathione peroxidase 2 (GPX2). (B) Haem oxygenase‐1 (HO‐1). (C) NAD(P)H dehydrogenease, quinione 1 (NQO1). (D) The Nrf2‐independent enzyme SOD 1 (SOD‐1) did not change significantly. MC + Air, MK + Air n = 6 per group; MC + Cl2, MK + Cl2 n = 10 per group. *P < 0.05, significantly different as indicated. s9, ribosomal protein S9.
Nrf2 nuclear translocation luciferase assay in BEAS‐2B cells
Previously, we reported that sodium hypochlorite induced Nrf2 nuclear translocation in BEAS‐2B cells (McGovern et al., 2015). Therefore, we sought to determine whether treatment with the CysLT LTD4 would stimulate Nrf2 nuclear translocation directly and whether montelukast affected this response. Incubation of BEAS‐2B cells with sodium hypochlorite markedly increased Nrf2 nuclear translocation and the translocation was unaffected by treatment with montelukast (Figure 6A). Treatment of cells with LTD4 or montelukast in the absence of sodium hypochlorite did not induce Nrf2 translocation (Figure 6B). We also observed that addition of LTD4, in the presence of sodium hypochlorite did not further increase Nrf2 nuclear translocation, regardless of montelukast treatment (Figure 6C).
Figure 6.
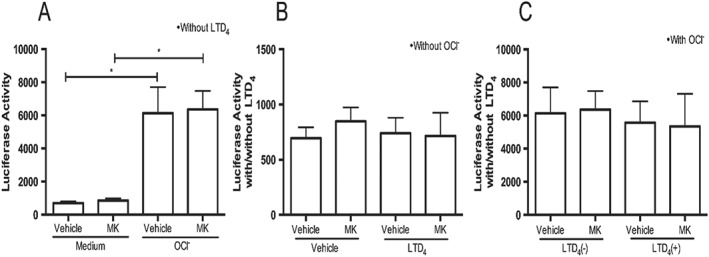
Montelukast (MK) did not affect Nrf2 nuclear translocation. (A) Hypochlorite (OCl‐) induces Nrf2 nuclear translocation (ARE activity) in BEAS‐2B cells. (B) LTD4 did not affect Nrf2 nuclear translocation (ARE activity) in BEAS‐2B cells in the absence of hypochlorite. (C) LTD4 treatment alone did not influence Nrf2 nuclear translocation. n = 3 independent experiments. *P < 0.05, significantly different as indicated.
Chemokine and cytokine levels in BAL fluid
To address a potential mechanism by which montelukast could prevent chlorine‐induced AHR and neutrophilia, we evaluated several pro‐inflammatory cytokines and chemokines. Exposure to chlorine resulted in an increase in IL‐6, VEGF, CCL3 and KC levels in BAL. (Figure 7A–E). Montelukast treatment prevented the increases in IL‐6 and VEGF but did not affect significantly CCL3 and KC (Figure 7 A–D). Other mediators measured were unaffected by chlorine exposure.
Figure 7.

Montelukast prevents chlorine‐induced increases in IL‐6 and VEGF. Cytokine and chemokine levels in the BAL were measured 24 h following chlorine (Cl2) exposure. (A) Chlorine induced an increase in IL‐6 release compared with air‐exposed mice, an effect prevented by montelukast (MK) treatment. (B) Montelukast treatment prevented increases in VEGF release following chlorine exposure. (C) Chlorine did not affect levels of CXCL2. (D) Chlorine increased CCL3 levels, but montelukast had did not prevent this increase. (E) KC was increased following chlorine, but montelukast did not prevent this increase. MC + Air, MK + Air n = 8 per group, MC + Cl2, MK + Cl2, n = 10 per group. *P < 0.05, significantly different as indicated.
Neutralization of IL‐6 prevents chlorine‐induced neutrophilia
Given that montelukast prevented chlorine‐induced AHR, neutrophil influx and IL‐6 levels in BAL, we used an IL‐6 neutralizing antibody to explore whether or not CysLT1 receptor‐mediated effects of chlorine exposure were potentially mediated by IL‐6. We found that chlorine‐induced neutrophilia was attenuated following neutralization of IL‐6 (Figure 8C). While macrophage and eosinophil numbers were fewer in BAL from chlorine exposed mice treated with anti‐IL‐6 antibody, compared with IgG‐chlorine, we did not observe statistically significant changes (Figure 8B, D). Neutralization of IL‐6 had no effect on numbers of BAL lymphocytes or epithelial cells (data not shown).
Figure 8.
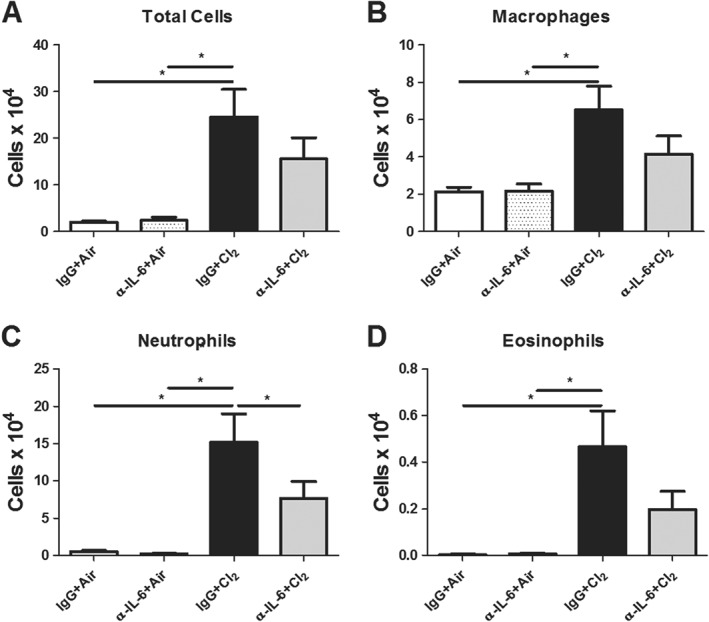
Treatment with the antibody against IL‐6 prevents chlorine‐induced neutrophilia. Mice were treated with the IL‐6 antibody, prior to chlorine (Cl2) exposure. (A) Chlorine induced an increase in total inflammatory cell numbers compared with air‐exposed mice and the IL‐6 antibody prevented this increase. (B) Chlorine induced an increase in macrophage numbers. (C) The IL‐6 antibody prevented neutrophil influx in chlorine‐exposed groups. (D) Eosinophils were increased in chlorine‐exposed groups treated with IgG, but not following the IL‐6 antibody compared with air‐exposed mice. (A–D) The IL‐6 antibody did not influence inflammatory cell populations in air‐exposed groups compared with IgG‐treated control mice. IgG + Air, α‐IL‐6 + Air, n = 6 per group, IgG + Cl2, n = 11. α‐IL‐6 + Cl2 n = 10. *P < 0.05, significantly different as indicated.
Neutralization of IL‐6 partly attenuates chlorine‐induced AHR
Neutralization of IL‐6 did not prevent increases in large airway responsiveness as reflected in RN following chlorine exposure (Figure 9A, C). However, treatment with anti‐IL‐6 did have a small but significant effect on chlorine‐induced increases in G and H at the highest concentration of MCh, corresponding to effects on peripheral airways (Figure 9B, D, E).
Figure 9.
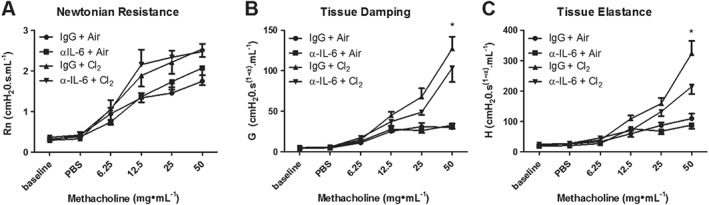
The IL‐6 antibody partly attenuates chlorine‐induced AHR. (A) The IL‐6 antibody did not prevent changes in Newtonian resistance. (B) The IL‐6 antibody prevented increased tissue damping at the highest dose of MCh. (C) The IL‐6 antibody effectively prevented increased tissue elastance at the highest dose of MCh. (A–C) The IL‐6 antibody had no effect on AHR in air‐exposed groups compared with IgG‐treated control mice. IgG + Air, α‐IL‐6 + air n = 6 per group, IgG + Cl2, α‐IL‐6 + Cl2 n = 7 per group. *P < 0.05, significant effect of the IL‐6 antibody.
Anti‐VEGF antibody partly reduced chlorine‐induced inflammation and AHR
Mice were treated with the anti‐VEGF antibody 1 h prior to chlorine exposure and demonstrated a significant reduction in total cell numbers compared with IgG‐treated control mice (Figure 10A). The change in total cell counts was attributable to a reduction in eosinophils and in epithelial cell shedding (Figure 10D, F). Anti‐VEGF antibody did not prevent the neutrophil influx in chlorine‐exposed groups compared with IgG‐treated control mice (Figure 10C). Likewise, lymphocytes and epithelial cells were not significantly affected by antibody treatment (data not shown). Anti‐VEGF treatment did not affect RN, G or H parameters of respiratory mechanics (Figure 10G–I).
Figure 10.
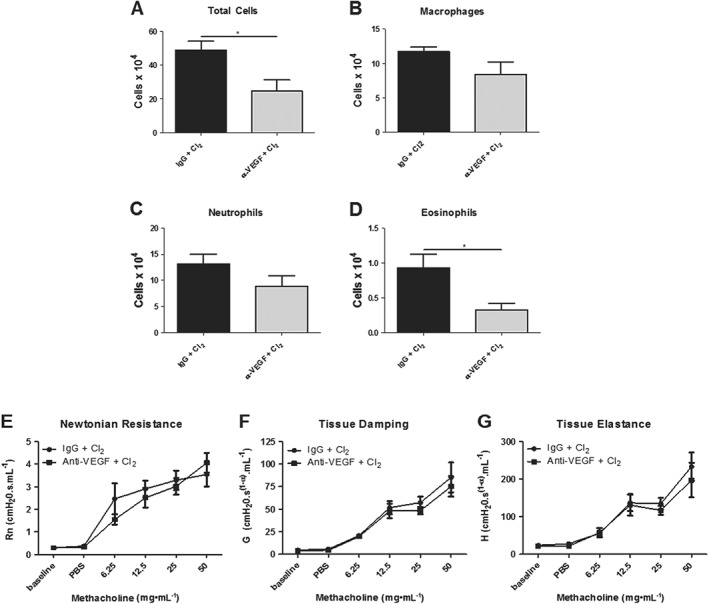
Effects of a VEGF antibody on chlorine‐induced inflammation and AHR. (A) The VEGF antibody decreased total cell numbers compared with IgG‐treated control mice. (B, C) Macrophage and neutrophils numbers were not affected by the VEGF antibody. (D) Eosinophils were decreased in mice treated with the VEGF antibody compared with IgG‐treated control mice. (E–G) The VEGF antibody had no effect on chlorine‐induced Newtonian resistance, tissue damping or tissue elastance. n = 8 per group. *P < 0.05, significant effect of the VEGF antibody.
Discussion
In the present study, we examined the effect of acute antagonism of the CysLT1 receptor in a well characterized model of IIA induced by chlorine exposure. Exposure of mice to inhaled chlorine increased AHR and pulmonary inflammation. These increases were prevented by pretreatment with the selective CysLT1 receptor antagonist montelukast. Several cytokines and chemokines increased in BAL fluid after chlorine exposure, and treatment with montelukast was effective in preventing the increases in VEGF and IL‐6. IL‐6 has been implicated in neutrophil recruitment, and therefore, we utilized an IL‐6 neutralization antibody to determine whether CysLT1 receptor‐dependent changes in neutrophilia and AHR were related to IL‐6. Neutralization of IL‐6 attenuated chlorine‐induced pulmonary neutrophilia and AHR. Anti‐VEGF treatment reduced BAL fluid eosinophilia but failed to prevent chlorine‐induced AHR. These results suggested that CysLT1 receptor‐driven airway dysfunction and neutrophilic inflammation was mediated, in part, via an IL‐6 dependent mechanism.
Montelukast inhibited the development of AHR after chlorine exposure implicating CysLTs in its development. We have demonstrated that these mediators are synthesized following chlorine exposure with a slow time course, reaching a peak by 24 h after exposure (McGovern et al., 2016). Hypochlorite activates TRPA1 channels which leads to neuropeptide release from airway sensory nerves (Hox et al., 2013), which in the context of hyperpnea‐induced bronchoconstriction is upstream of CysLT release (Yang et al., 1997). However, the time course of the synthesis of CysLTs is dissimilar from that triggered by activation of sensory nerves unless the neural effects of chlorine are slow in onset. Thus, currently, the cells of origin of CysLTs and the precise triggers for their synthesis in our model of IIA remain uncertain.
We have previously shown that AHR induced by chlorine exposure in mice is neutrophil‐dependent (McGovern et al., 2015). The effects of montelukast on AHR are possibly explained by the observed reduction in neutrophils in BAL fluid, which although significant was not complete. In a recent study, Ikeda et al. (2014) reported that montelukast inhibited the development of AHR and both eosinophilic and neutrophilic airway inflammation in studies of experimental emphysema and asthma. Montelukast treatment significantly reduced the number of lymphocytes and neutrophils in the BAL fluid induced by the administration of elastase. In our current study, the inhibition of AHR was largely observed in lung compartments which reflect responses of the peripheral lung. The changes in peripheral lung mechanics are a reflection of small airway narrowing and/or closure leading also to an apparent increase in stiffness of the parenchyma through loss of functioning units. Perhaps if a more complete reduction in BAL neutrophilia had been achieved, effects on AHR located to the large airways might also have been observed. However, it is possible that the mechanisms leading to AHR in the peripheral lung may not be identical to those affecting the large airways.
Chlorine is a potent oxidant that leads to airway dysfunction and inflammation in association with oxidative stress. In turn, oxidative stress triggers Nrf2 translocation and the induction of second‐phase enzymes that may limit the effects of oxidative damage. Consistent with previous published results (McGovern et al., 2016), Chlorine caused translocation of Nrf2 to the nucleus in the airway epithelium. Although we found evidence of enhancement of this translocation in bronchial epithelial cells in vivo by montelukast treatment, mRNA expression of Nrf2‐dependent enzymes in the lung parenchyma was not significantly further up‐regulated by montelukast. The reason for this discrepancy is not clear, but it suggests that the mitigation of the airway dysfunction by montelukast was unrelated to oxidant defences. Although the Nrf2‐regulated genes in the lungs include almost all the relevant antioxidants, such as HO‐1 and several members of the glutathione S‐transferase family, perhaps their effects would be evident only at time points beyond 24 h. Indeed, we have recently shown that Nrf2 deficiency in chlorine‐exposed mice does not affect outcomes at 24 h post‐exposure (Ano et al., 2017). The effects of montelukast on Nrf2 translocation in the airway epithelium in vivo appear to have been mediated indirectly. BEAS‐2B cells exposed to hypochlorite demonstrated translocation of Nrf2, but this was not reproduced by stimulation of the CysLT1 receptor by LTD4 and was not inhibited by montelukast.
To address the underlying mechanisms of montelukast treatment in chlorine‐induced airway inflammation and AHR, BAL cytokine levels were assayed. While precise mechanisms by which montelukast may prevent inflammation and pulmonary dysfunction after injury remain unclear, montelukast treatment did reduce the levels of pro‐inflammatory cytokines associated with oxidative stress, as well as neutrophilia in vivo and in vitro including VEGF and IL‐6 (Lee et al., 2004; Maeba et al., 2005; Mullol et al., 2010). In the current study, exposure to chlorine caused an increase in the neutrophil chemoattractant chemokine KC. This result is consistent with a previous report that found KC to be increased by 6 h after exposure to chlorine (Tian et al., 2008) and may account in part for the rapid influx of neutrophils (White and Martin, 2010). Although montelukast treatment did not prevent changes in KC, it did prevent chlorine‐induced increases in IL‐6 and VEGF, both of which have been implicated in neutrophilia. Several previous publications have demonstrated the anti‐inflammatory effects of montelukast. This compound inhibits lipopolysaccharide‐induced production of IL‐1β, IL‐6, TNF‐α and MCP‐1 from peripheral blood mononuclear cells of subjects with asthma (Maeba et al., 2005) and protects against lung injury induced by haemorrhagic shock in rats, reducing serum levels of TNF‐α and IL‐6 (Al‐Amran et al., 2013). Perhaps of greater relevance, montelukast reduced GM‐CSF, IL‐6 and IL‐8 production by human nasal epithelial cells, stimulated with fetal bovine serum (Mullol et al., 2010). Taken together, our results suggest that montelukast treatment may prevent neutrophilic inflammation and subsequent AHR by inhibiting the synthesis and release of chemokines such as IL‐6. Further exploration of the mechanisms of action of montelukast on inflammatory mediator release and AHR might be usefully pursued using precision cut lung slices.
VEGF has been previously reported to increase over time after chlorine‐inducedairway injury (Mo et al., 2015). It is a potent angiogenic factor and contributes to angiogenesis in the lung (Carmeliet et al., 1996) and enhanced microvascular permeability (Leung et al., 1989; Dvorak et al., 1995; Medford and Millar, 2006). In colon cancer cells, montelukast was shown to inhibit growth of colon cancer xenografts primarily by reducing VEGF expression (Savari et al., 2013). Furthermore, montelukast has been shown to reduce vascular permeability by regulating VEGF expression in the lungs of mice with allergen‐induced asthma (Lee et al., 2004). Our results show montelukast effectively prevents increased levels of VEGF in BAL following chlorine exposure. Although neutralization of VEGF led to inhibition of eosinophil numbers, it did not significantly inhibit neutrophilia after chlorine. Anti‐VEGF also had no effect on AHR, suggesting that eosinophils may not contribute to chlorine‐induced AHR.
Chlorine‐induced airway and lung dysfunction are well‐documented consequences of acute exposure to high concentrations of the gas (Van Sickle et al., 2009). Other accidental exposures occur in the domestic setting through inappropriate mixing of cleaning products (Gorguner et al., 2004). Exposure to bleach alone in concentrations below currently defined acceptable limits may lead to decline in lung function in asthmatic subjects (Sastre et al., 2011). Perhaps of greater public health concern is the effect of attendance in early life at chlorinated swimming pools, on the development of allergy and asthma (Bernard et al., 2015). Our results suggest that preventive treatment with a leukotriene modifier may be a useful adjunct to the treatment of respiratory symptoms caused by chlorine exposure, but appropriate trials in human subjects are required to confirm these conclusions.
In conclusion, the administration of montelukast reduced AHR and neutrophilic airway inflammation following exposure to inhaled chlorine and montelukast also increased Nrf2 nuclear translocation. However, these effects on endogenous antioxidant pathways appear to be unrelated to changes in AHR and inflammation. Decreasing neutrophilia following montelukast treatment was associated with reductions in levels of IL‐6 and VEGF in BAL fluid. Neutralization of IL‐6, but not VEGF‐A, attenuated chlorine‐induced neutrophilia and AHR in the peripheral lung. Our data suggest that montelukast prevents chlorine‐induced AHR and inflammation partly through an IL‐6‐dependent mechanisms.
Author contributions
T.M. and J.G.M. were involved in the conception and design of the study; analysis, acquisition of data and interpretation were performed by Y.H., S.A., B.A., M.S., T.M., J.G.M.; Y.H., T.M. and J.G.M. drafted and/or revised critically for intellectual content; Y.H., B.A., T.M., S.A., M.S. and J.G.M. finally approved the version to be published.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by an operating grant from the Canadian Institutes of Health Research (MOP‐126131).
Hamamoto, Y. , Ano, S. , Allard, B. , O'Sullivan, M. , McGovern, T. K. , and Martin, J. G. (2017) Montelukast reduces inhaled chlorine triggered airway hyperresponsiveness and airway inflammation in the mouse. British Journal of Pharmacology, 174: 3346–3358. doi: 10.1111/bph.13953.
References
- Al‐Amran FG, Hadi NR, Hashim AM (2013). Cysteinyl leukotriene receptor antagonist montelukast ameliorates acute lung injury following haemorrhagic shock in rats. Eur J Cardiothorac Surg 43: 421–427. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano S, Panariti A, Allard B, O'Sullivan M, McGovern TK, Hamamoto Y et al (2017). Inflammation and airway hyperresponsiveness after chlorine exposure are prolonged by Nrf2 deficiency in mice. Free Radic Biol Med 102: 1–15. [DOI] [PubMed] [Google Scholar]
- Bates JH (2009). Pulmonary mechanics: a system identification perspective. Conf Proc IEEE Eng Med Biol Soc 2009: 170–172. [DOI] [PubMed] [Google Scholar]
- Bernard A, Nickmilder M, Dumont X (2015). Chlorinated pool attendance, airway epithelium defects and the risks of allergic diseases in adolescents: interrelationships revealed by circulating biomarkers. Environ Res 140: 119–126. [DOI] [PubMed] [Google Scholar]
- Brooks SM, Bernstein IL (2011). Irritant‐induced airway disorders. Immunol Allergy Clin North Am 31: 747–768 vi. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M et al (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435–439. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF, Brown LF, Detmar M, Dvorak AM (1995). Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 146: 1029–1039. [PMC free article] [PubMed] [Google Scholar]
- Gorguner M, Aslan S, Inandi T, Cakir Z (2004). Reactive airways dysfunction syndrome in housewives due to a bleach‐hydrochloric acid mixture. Inhal Toxicol 16: 87–91. [DOI] [PubMed] [Google Scholar]
- Hox V, Alpizar YA, Voedisch S, Callebaut I, Bobic S, Sharify A et al (2013). Crucial role of transient receptor potential ankyrin 1 and mast cells in induction of non‐allergic airway hyperreactivity in mice. Am J Respir Crit Care Med 187: 486–493. [DOI] [PubMed] [Google Scholar]
- Ihaku D, Cameron L, Suzuki M, Molet S, Martin J, Hamid Q (1999). Montelukast, a leukotriene receptor antagonist, inhibits the late airway response to antigen, airway eosinophilia, and IL‐5‐ expressing cells in Brown Norway rats. J Allergy Clin Immunol 104: 1147–1154. [DOI] [PubMed] [Google Scholar]
- Ikeda G, Miyahara N, Koga H, Fuchimoto Y, Waseda K, Kurimoto E et al (2014). Effect of a cysteinyl leukotriene receptor antagonist on experimental emphysema and asthma combined with emphysema. Am J Respir Cell Mol Biol 50: 18–29. [DOI] [PubMed] [Google Scholar]
- Jones TR, Labelle M, Belley M, Champion E, Charette L, Evans J et al (1995). Pharmacology of montelukast sodium (Singulair), a potent and selective leukotriene D4 receptor antagonist. Can J Physiol Pharmacol 73: 191–201. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Kim SR, Park HS, Jin GY, Lee YC (2004). Cysteinyl leukotriene receptor antagonist regulates vascular permeability by reducing vascular endothelial growth factor expression. J Allergy Clin Immunol 114: 1093–1099. [DOI] [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246: 1306–1309. [DOI] [PubMed] [Google Scholar]
- Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM et al (1999). Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399: 789–793. [DOI] [PubMed] [Google Scholar]
- Maeba S, Ichiyama T, Ueno Y, Makata H, Matsubara T, Furukawa S (2005). Effect of montelukast on nuclear factor kappaB activation and proinflammatory molecules. Ann Allergy Asthma Immunol 94: 670–674. [DOI] [PubMed] [Google Scholar]
- Manders EMM (1993). Measurement of co‐localization of objects in dualcolor confocal images. J Microsc 169: 375. [DOI] [PubMed] [Google Scholar]
- Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH et al (2003). Chlorine‐induced injury to the airways in mice. Am J Respir Crit Care Med 168: 568–574. [DOI] [PubMed] [Google Scholar]
- McGovern T, Goldberger M, Chen M, Allard B, Hamamoto Y, Kanaoka Y et al (2016). CysLT1 receptor is protective against oxidative stress in a model of irritant‐induced asthma. J Immunol 197: 266–277. [DOI] [PubMed] [Google Scholar]
- McGovern TK, Goldberger M, Allard B, Farahnak S, Hamamoto Y, O'Sullivan M et al (2015). Neutrophils mediate airway hyperresponsiveness following chlorine‐induced airway injury in the mouse. Am J Respir Cell Mol Biol 52: 513–522. [DOI] [PubMed] [Google Scholar]
- McGovern TK, Powell WS, Day BJ, White CW, Govindaraju K, Karmouty‐Quintana H et al (2010). Dimethylthiourea protects against chlorine induced changes in airway function in a murine model of irritant induced asthma. Respir Res 11: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, Martin JG (2013). Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J Vis Exp : e50772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medford AR, Millar AB (2006). Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): paradox or paradigm? Thorax 61: 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Chen J, Humphrey DM Jr, Fodah RA, Warawa JM, Hoyle GW (2015). Abnormal epithelial structure and chronic lung inflammation after repair of chlorine‐induced airway injury. Am J Physiol Lung Cell Mol Physiol 308: L168–L178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullol J, Callejas FB, Mendez‐Arancibia E, Fuentes M, Alobid I, Martinez‐Anton A et al (2010). Montelukast reduces eosinophilic inflammation by inhibiting both epithelial cell cytokine secretion (GM‐CSF, IL‐6, IL‐8) and eosinophil survival. J Biol Regul Homeost Agents 24: 403–411. [PubMed] [Google Scholar]
- Otunctemur A, Ozbek E, Cakir SS, Dursun M, Cekmen M, Polat EC et al (2015). Beneficial effects montelukast, cysteinyl‐leukotriene receptor antagonist, on renal damage after unilateral ureteral obstruction in rats. Int Braz J Urol 41: 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre J, Madero MF, Fernandez‐Nieto M, Sastre B, del P, V, Potro MG, et al (2011). Airway response to chlorine inhalation (bleach) among cleaning workers with and without bronchial hyperresponsiveness. Am J Ind Med 54: 293–299. [DOI] [PubMed] [Google Scholar]
- Savari S, Liu M, Zhang Y, Sime W, Sjolander A (2013). CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One 8: e73466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallcombe A, McMillan D (2002). Co‐localization; how it is determined, and how it is analysed with the Bio‐Rad LaserPix image analysis software? [Online] Available at: https://www.microscopyu.com/pdfs/Casavan_Media_Cybernetics_Application_Note_1.pdf) (accessed September 25, 2015).
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlo SM, Lemiere C (2014). Occupational asthma. N Engl J Med 370: 640–649. [DOI] [PubMed] [Google Scholar]
- Tian X, Tao H, Brisolara J, Chen J, Rando RJ, Hoyle GW (2008). Acute lung injury induced by chlorine inhalation in C57BL/6 and FVB/N mice. Inhal Toxicol 20: 783–793. [DOI] [PubMed] [Google Scholar]
- Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F et al (2009). Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med 27: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CW, Martin JG (2010). Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc Am Thorac Soc 7: 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XX, Powell WH, Hojo M, Martin JG (1997). Hyperpnea‐induced bronchoconstriction is dependent on tachykinin‐induced cysteinyl leukotriene synthesis. J Appl Physiol 82: 538–544. [DOI] [PubMed] [Google Scholar]