

Abstract
Background and Purpose
To further the development of new agents for the treatment of adrenocortical carcinoma (ACC), we characterized the molecular and cellular mechanisms of cytotoxicity by the adrenalytic compound ATR‐101 (PD132301‐02).
Experimental Approach
We compared the effects of ATR‐101, PD129337, and ABC transporter inhibitors on cholesterol accumulation and efflux, on cortisol secretion, on ATP levels, and on caspase activation in ACC‐derived cell lines. We examined the effects of these compounds in combination with methyl‐β‐cyclodextrin or exogenous cholesterol to determine the roles of altered cholesterol levels in the effects of these compounds.
Key Results
ATR‐101 caused cholesterol accumulation, ATP depletion, and caspase activation within 30 minutes after addition to ACC‐derived cells, whereas PD129337 did not. Suppression of cholesterol accumulation by methyl‐β‐cyclodextrin or exogenous cholesterol, prevented ATP depletion and caspase activation by ATR‐101. ATR‐101 blocked cholesterol efflux and cortisol secretion, suggesting that it inhibited ABCA1, ABCG1, and MDR1 transporters. Combinations of ABCA1, ABCG1, and MDR1 inhibitors were also cytotoxic. Combinations of ATR‐101 with inhibitors of ABCG1, MDR1, or mitochondrial functions had increased cytotoxicity. Inhibitors of steroidogenesis reduced ATP depletion by ATR‐101, whereas U18666A enhanced cholesterol accumulation and ATP depletion together with ATR‐101. ATR‐101 repressed ABCA1, ABCG1, and IDOL transcription by mechanisms that were distinct from the mechanisms that caused cholesterol accumulation.
Conclusions and Implications
Inhibition of multiple ABC transporters and the consequent accumulation of cholesterol mediated the cytotoxicity of ATR‐101. Compounds that replicate these effects in tumours are likely to be useful in the treatment of ACC.
Abbreviations
- ABCA1
ATP‐binding cassette transporter A1, ABC1, CERP
- ABCG1
ATP‐binding cassette transporter G1, ABC8
- ACAT
acyl‐coenzyme A: cholesterol acyltransferase
sterol O‐acyltransferase, SOAT
- ACC
adrenocortical carcinoma
- ATR‐101
1‐[[1‐[4‐(dimethylamino)phenyl]cyclopentyl]methyl]‐3‐[2,6‐di(propan‐2‐yl)phenyl]urea;hydrochloride, PD132301‐02, CI‐984
- MDR1
multiple drug resistance protein 1
- MβCD
methyl‐β‐cyclodextrin
- NBD‐cholesterol
22‐(N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)amino)‐23,24‐bisnor‐5‐cholen‐3β‐ol
- PD129337
1‐[2,6‐di(propan‐2‐yl)phenyl]‐3‐[(1‐phenylcyclopentyl)methyl]urea
- U18666A
3β‐(2‐diethylaminoethoxy)‐5‐androsten‐17‐one, (3S,8R,9S,10R,13S,14S)‐3‐[2‐(diethylamino)ethoxy]‐10,13‐dimethyl‐1,2,3,4,7,8,9,11,12,14,15,16‐dodecahydrocyclopenta[a]phenanthren‐17‐one hydrochloride
Introduction
Control of the levels of cholesterol (The term “cholesterol” is used to denote unesterified cholesterol in this paper) is essential for cell functions and viability (see Maxfield and Van Meer, 2010). The cholesterol levels in adrenocortical cells are affected by many pathways, some of which are unique to the adrenal cortex. Studies of anti‐atherosclerosis agents have identified compounds that cause selective degeneration of the adrenal cortex (adrenalytic activity) in several species (Dominick et al., 1993; Reindel et al., 1994; Matsuo et al., 1996; Sliskovic et al., 1998; Tanaka et al., 1998). Here we have investigated the adrenalytic compound ATR‐101, also known as PD132301‐02, as a prospective agent for the treatment of adrenocortical carcinoma (ACC). We focused on ATR‐101 because of its cytotoxicity in ACC‐derived cells and its anti‐xenograft and adrenalytic activities (Cheng et al., 2016).
ACC is a rare cancer that has few treatment options. The adrenalytic compound mitotane is a first‐line drug for ACC treatment despite its poor efficacy, unfavourable pharmacokinetics, severe side effects and potential drug interactions (Maiter et al., 2016). Clinical trials of molecularly targeted agents have not demonstrated therapeutic benefit for ACC patients (see Creemers et al., 2016). The differences in the genetic and epigenetic changes among different ACC tumours suggest that different molecular mechanisms underlie the malignancy of different ACC tumours (Assie et al., 2014; Zheng et al., 2016). Adrenalytic compounds can potentially be used for the treatment of ACCs that have different determinants of malignancy.
ATR‐101 inhibits the establishment and impedes the growth of ACC cell xenografts in nude mice (Cheng et al., 2016). The inhibition of xenograft growth in animals that are treated with ATR‐101 correlates with increased apoptosis of xenograft cells. ATR‐101 causes mitochondrial dysfunction in ACC‐derived cells and toxicity due to reactive oxygen species (ROS) in cultured cells and in the zona fasciculata layer of the guinea pig adrenal cortex (Cheng et al., 2016).
The tissue‐specific toxicity of adrenalytic compounds correlates with cholesterol accumulation. The cholesterol level of guinea pig adrenal glands rises within an hour after ATR‐101 administration and increases threefold in 24 h (Wolfgang et al., 1995). ATR‐101 toxicity in cynomologous monkeys is limited to cholesterol‐rich tissues, including the adrenal cortex, the corpus luteum and sebaceous glands (Reindel et al., 1994). Low‐density lipoprotein‐deficient rabbits are resistant to the adrenalytic effect of FR145237 (Matsuo et al., 1996). However, the mechanisms whereby ATR‐101 causes cholesterol accumulation and their roles in ATR‐101 cytotoxicity were unknown.
The adrenal cortex has high rates of cholesterol uptake, synthesis, trafficking, metabolism, and efflux that must be balanced to support steroidogenesis and to prevent the accumulation of toxic levels of cholesterol (see Miller and Bose, 2011). Cholesterol is imported from low‐density lipoprotein particles at a steady‐state rate that depends on the level of low‐density lipoprotein receptors (see Goldstein and Brown, 2009). Cholesterol from high‐density lipoprotein particles enters steroidogenic cells through diffusion that is facilitated by the SR‐BI receptor (Reaven et al., 2001). Adrenal glands and testes have a unique cholesterol biosynthetic pathway to ensure cholesterol availability for steroidogenesis (Mitsche et al., 2015).
Adrenocortical cells must respond rapidly to stress and to other signals that induce steroidogenesis within minutes after a stimulus (Bose et al., 2002; Fallahsharoudi et al., 2015). Many inhibitors of steroidogenesis cause a rapid increase in the cholesterol content of the adrenal glands or of cultured adrenocortical cells (Dibartolomeis et al., 1986; Lehoux and Lefebvre, 1991; Sbiera et al., 2015). Some compounds increase both cholesterol and cholesteryl ester levels, indicating that that the increases in cholesterol levels do not require the inhibition of cholesterol esterification (Brecher and Hyun, 1978; Lehoux and Lefebvre, 1991; Pandey and Rudraiah, 2015). Consequently, changes in the amount of cholesterol consumed by steroidogenesis are not buffered by changes in cholesterol ester storage.
Cholesterol trafficking is required both for the maintenance of appropriate cholesterol levels of different cell membranes and for cholesterol efflux (reviewed in Ikonen, 2008). ATP‐binding cassette (ABC) transporters [ABCA1, ABCG1, multiple drug resistance protein 1 (MDR1)] regulate the vesicular trafficking of cholesterol between different membranes (Debry et al., 1997; Luker et al., 1999; Tarling and Edwards, 2011; Yamauchi et al., 2015). Disruptions to cholesterol trafficking contribute to cholesterol accumulation and toxicity in several human diseases (see Vanier, 2010; Porto, 2014; Sahakitrungruang, 2015).
Cholesterol efflux is controlled by the active transport of cholesterol to extracellular acceptors by ABC transporters. The ABCA1 and ABCG1 transporters are thought to be the principal conduits for cholesterol export from macrophages (Wang et al., 2007; Out et al., 2008). ABCA1 and ABCG1 are enriched in the adrenal cortex, yet no adrenocortical dysfunction was reported in Abca1/Abcg1 double knockout mice (Out et al., 2008). Different phenotypes have been reported for Abca1 single knockout mice (Christiansen‐Weber et al., 2000; Mcneish et al., 2000; Orso et al., 2000). It is possible that compensatory effects of other transporters or differences in genetic backgrounds or mouse husbandry affect the phenotypes that are produced by Abca1 and/or Abcg1 deletion.
The MDR1/P‐glycoprotein, also known as ABCB1, can influence both cholesterol levels and steroid secretion. Cells that overexpress MDR1 have higher levels of cholesterol uptake and cholesterol ester storage (Luker et al., 1999; Tessner and Stenson, 2000). Ectopically expressed MDR1 does not increase cholesterol efflux in HEK293, HeLa or 77.1 cells (Le Goff et al., 2006; Morita et al., 2007). MDR1 is required to maintain normal circulating corticosterone levels in mice and for steroid secretion by mouse adrenocortical cells (Altuvia et al., 1993; Muller et al., 2003). It is not known if the effects of MDR1 on cholesterol levels and on steroid secretion are direct and independent or each other or if these effects are indirect consequences of a single molecular function of MDR1.
Acyl‐coenzyme A: cholesterol acyltransferase (ACAT) inhibition was proposed to cause cholesterol accumulation and cytotoxicity in ACC cell lines (Sbiera et al., 2015; Lapensee et al., 2016). Most ACAT inhibitors do not cause cholesterol accumulation or cytotoxicity under normal cell culture conditions, and differences in the ACAT inhibitory activities of different compounds do not correlate with differences in cytotoxicity (Junquero et al., 2001; Rodriguez and Usher, 2002; An et al., 2008; Pokhrel et al., 2012). Only a small proportion of ACAT inhibitors have adrenalytic activity, even though many of them reduce serum cholesterol levels in animals (see Sliskovic et al., 2002). No adverse events related to adrenocortical damage were reported in phase II or III clinical trials of ACAT inhibitors (Tardif et al., 2004; Meuwese et al., 2009). Ablation of the gene encoding ACAT1 in mice eliminates cholesterol esterification in the adrenal cortex, but it does not cause adrenocortical damage, altered corticosteroid levels or cholesterol accumulation in the adrenals (Meiner et al., 1996). It is therefore unlikely that ACAT inhibition is sufficient to cause cytotoxicity or adrenalytic activity.
The roles of cholesterol uptake, synthesis, trafficking, storage, metabolism and efflux in the control of the cholesterol levels in ACC cells are not well understood. We investigated the effects of ATR‐101 and of non‐adrenalytic ACAT inhibitors on several pathways that can affect the cholesterol levels of adrenocortical cells. We found that the inhibition of cholesterol efflux and of steroidogenesis were the principal mechanisms that correlated with cytotoxic cholesterol accumulation in ACC‐derived cells cultured with ATR‐101. The combined cytotoxic and anti‐steroidogenic effects of ATR‐101 in ACC cells have the potential to provide dual benefits in the treatment of ACC.
Methods
Cell culture conditions
H295R cells were obtained from ATCC (Manassas, VA) and were validated by analysis of their corticosteroid profile. BD140C cells were obtained from Dr Kimberly Bussey (Tgen, Phoenix, Arizona). H295R and BD140C cells were cultured in DMEM without glucose (Gibco, Gaithersburg, MD) supplemented with 10 mM galactose, 5% FBS, 0.1 mg·mL−1 penicillin–streptomycin, 2 mM L‐glutamine, 5 mM sodium HEPES, 1 mM sodium pyruvate and the compounds indicated in each experiment. In the experiments that examined the effects of compounds in the absence of cholesterol in the medium, the medium of cells that were cultured under standard conditions was replaced with serum‐free medium containing the indicated compounds. The cells were cultured in serum‐free medium for the indicated time and were analysed using the same protocols that were used for cells that were cultured in serum‐containing medium.
Visualization of cholesterol levels and of cholesterol esterification
The cells were seeded in 96‐well ibiTreat μ‐plates and allowed to adhere for 48 h prior to the start of each experiment. The compounds indicated in each experiment were added, and the cells were cultured for the time indicated. After fixation, cholesterol was detected by incubating the cells with 100 μg·mL−1 filipin III at 37 C for 2 h. The bound filipin III was imaged by fluorescence microscopy using 377 ± 11 nm excitation and 447 ± 60 nm emission wavelengths. Images were captured using either a 4× or a 60× objective. Cholesterol esterification was visualized by culturing the cells with the indicated compounds followed by incubation with 1 μM NBD‐cholesterol for 2 h. NBD‐cholesterol esters were imaged by fluorescence microscopy using 485 ± 20 nm excitation wavelengths. Images were captured using either a 20× or a 60× objective.
ATP and caspase 3/7 assays
The ATP levels of cells were measured by lysis in CellTiter‐Glo reagent (Promega, Madison, WI) and measurement of the luminescence. The caspase 3/7 activities of cells were measured by lysis in Apo‐ONE reagent (Promega) and measurement of the fluorescence.
Extracellular cholesterol, cholesterol efflux, cortisol secretion and doxorubicin clearance
To quantify total extracellular cholesterol that was associated with cells, we measured the total amount of cholesterol that was dislodged from cells during a brief wash. After culturing cells with the indicated compounds, the culture medium was removed. Medium lacking cholesterol was added to the cells, and the wash medium was collected after 30 s. To establish if the cholesterol that was released from the cells was exported by ABCA1, the wash step was performed in parallel using media with and without 50 μM glibenclamide, which inhibits ABCA1 activity. The total amount of cell‐associated cholesterol that was released into the wash medium was measured using a fluorometric enzyme‐linked assay (Cayman Chemical, Ann Arbor, MI). Cholesterol esters were hydrolyzed using cholesterol esterase. Cholesterol was oxidized by cholesterol oxidase, producing hydrogen peroxide. The hydrogen peroxide was reacted with 10‐acetyl‐3,7‐dihydroxyphenoxazine in the presence of horseradish peroxidase to produce resorufin. Resorufin fluorescence was measured using 555 nm exitation and 590 nm emission wavelengths in a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Cholesterol efflux was measured by replacing the culture medium with serum‐free medium supplemented with 5 μg·mL−1 apoA1 (Sigma, St. Louis, MO) and the indicated compounds. After the indicated time, the serum‐free medium was collected, and the total cholesterol concentration was measured using the fluorometric enzyme‐linked assay (Cayman Chemical).
Cortisol secretion was measured by replacing the culture medium with fresh medium containing the indicated compounds. The medium was collected at the indicated times. The cortisol concentration in the medium was measured using indirect elisa (Arbor Assays, Ann Arbor, MI).
Doxorubicin was imaged by fluorescence microscopy in live cells using 485·20 nm−1 excitation wavelengths and either a 10× or 20× objective.
Transcript measurement
mRNA was isolated (Qiagen) and was reverse transcribed using the Transcriptor First Strand cDNA synthesis kit (Roche). The relative amounts of cDNAs corresponding to the indicated transcripts were quantified using qPCR assays with specific primers (Supporting Information Table S1).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The data were analysed using GraphPad Prism v7.00 software. Data are shown as means ±2 SD. The number of samples included in each analysis is indicated in each figure legend and refers to separate cultures of cells from the number of experiments indicated in the legend. Groups that were used for statistical analysis included at least five samples. Technical replicates were performed to ensure the reliability of the measurements and were not included in the analysis of variance. A P value <0.05 was interpreted to indicate statistical significance. The results of all statistical tests are shown in the Supplementary Information.
Materials
ATR‐101 was synthesized and purified as described (Trivedi et al., 1994; Cheng et al., 2016). Benzamil, ketoconazole, PD129337, U18666A, and zosuquidar were supplied by Sigma; glibenclamide by Abcam (Cambridge, MA); cyclosporin A, rhodamine 123, doxorubicin, abiraterone acetate, metyrapone, trilostane and anastrozole were supplied by Cayman Chemical; olesoxime by Tocris (Minneapolis, MN). These compounds were dissolved in DMSO at concentrations ranging from 50 to 250 mM. The final concentration of DMSO for all samples within each experiment was the same and ranged from 0.1 to 0.4% for all experiments. NBD‐cholesterol from Molecular Probes (Eugene, OR), cholesterol, cholesterol linoleate and α‐tocopherol from Sigma were dissolved in ethanol at concentrations ranging from 20 to 240 mM. The final concentration of ethanol for all samples within each experiment was the same and ranged from 0.02 to 0.2% for all experiments. Verapamil, methyl‐β‐cyclodextrin and cholesterol:methyl‐β‐cyclodextrin (from Sigma) were dissolved in the cell culture media.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Roles of cholesterol accumulation versus the inhibition of cholesterol esterification in ATR‐101 cytotoxicity
As ATR‐101 causes cholesterol accumulation and ATP depletion in the adrenal glands of guinea pigs, we examined the effects of ATR‐101 on cholesterol and ATP levels, as well as on caspase 3/7 activities, in ACC‐derived cells. Cholesterol accumulation was detected 15 min after ATR‐101 addition to H295R cells (Figure 1A; Supporting Information Fig. S1A). ATP depletion and caspase activation were detected 30 and 15 min after ATR‐101 addition respectively (Figure 1B). The rapid accumulation of cholesterol, ATP depletion and caspase activation after ATR‐101 addition to cells indicates that these effects of ATR‐101 were causes rather than consequences of ATR‐101 cytotoxicity.
Figure 1.

Effects of ATR‐101 versus PD129337 on cholesterol accumulation, cholesterol esterification, ATP levels and caspase 3/7 activities in ACC‐derived cells. (A) Time‐dependence of the effect of ATR‐101 on cholesterol levels in H295R cells. The cells were cultured with DMSO vehicle or with 60 μM ATR‐101 for the times indicated above the images. The images show filipin III binding to cholesterol in fields containing 5500–7800 cells. The scale bars denote 100 μm. The fluorescence intensities of the cell populations and the statistical significance of the differences are shown in Supporting Information Fig. S1A. (B) Time‐dependence of the effects of ATR‐101 on the ATP levels and on the caspase 3/7 activities in H295R cells. The cells were cultured with DMSO vehicle or with 100 μM ATR‐101 for the indicated times. The ATP levels (left graphs) and the caspase 3/7 activities (right graphs) were measured in cells that were grown in parallel. The graphs show the means ± 2SD of five samples from three experiments. *P < 0.05, significantly different from DMSO control; two‐way ANOVA with Sidak's post hoc tests. (C) Effects of ATR‐101 versus PD132997 on the cholesterol levels in H295R and BD140C cells. H295R (upper images) and BD140C (lower images) cells were cultured with DMSO vehicle, ATR‐101 or PD129337 at the indicated concentrations for 4 h. The images show filipin III binding to cholesterol and are representative of images collected in two separate experiments for each cell line. The scale bars denote 10 μm. The full fields from which the images were cropped are shown in Supporting Information Fig. S1D. (D) Effects of different concentrations of ATR‐101 versus PD132997 on the ATP levels and caspase activities in H295R and BD140C cells. H295R (upper panels) and BD140C (lower panels) were cultured with the indicated concentrations of ATR‐101 or PD129337 for 4 h. The ATP levels (left graphs) and the caspase 3/7 activities (right graphs) were measured in cells that were grown in parallel. The graphs show the means ± 2SD of eight samples from four experiments and five samples from three experiments in H295R and BD140C cells respectively. *P < 0.05, significantly different from PD129337; two‐way ANOVA with Sidak's post hoc tests. The ATP levels and the caspase activities of H295R and BD140C cells that were cultured with ATR‐101 or PD132997 for 24 h are shown in Supporting Information Fig. S1B. (E) Effects of ATR‐101 or PD129337 on cholesterol esterification in H295R and BD140C cells. H295R (upper images) and BD140C (lower images) cells were cultured with DMSO vehicle or with the indicated concentrations of ATR‐101 or PD129337 for 2 h, followed by an additional 2 h after the addition of 1 μg·mL−1 NBD‐cholesterol. The images show NBD‐cholesterol ester (green) and Hoechst (blue) fluorescence and are representative of images collected in five separate experiments for each cell line. The scale bars denote 30 μm. The effects of different concentrations of ATR‐101 and of PD129337 on cholesterol esterification are shown in Supporting Information Fig. S1C. The full fields from which the images were cropped are shown in Supporting Information Fig. S1E.
To investigate the specificity of the effect of ATR‐101 on cholesterol accumulation, we compared the effects of ATR‐101 and PD129337 in ACC‐derived cell lines. ATR‐101 and PD129337 have closely related molecular structures, and both of them inhibit cholesterol esterification (ACAT activity), but only ATR‐101 has adrenalytic activity (Trivedi et al., 1993; Trivedi et al., 1994). ATR‐101 caused cholesterol accumulation in or near the plasma membrane of both H295R and BD140C cells (Figure 1C). PD129337 did not cause cholesterol accumulation, even when it was added at a fivefold higher concentration than ATR‐101 (Figure 1C).
To evaluate the specificity of the effects of ATR‐101 on ATP depletion and caspase activation, we compared the effects of ATR‐101 and PD129337 in ACC‐derived cell lines. ATR‐101 reduced the ATP level and increased the caspase 3/7 activity of H295R and BD140C cells (Figure 1D). The concentrations of ATR‐101 that caused ATP depletion and caspase activation were similar to those that caused cholesterol accumulation both in H295R and in BD140C cells. PD129337 did not reduce the ATP level or increase the caspase 3/7 activity, even when it was added at a fivefold higher concentration than ATR‐101 for up to 24 h (Figure 1D; Supporting Information Fig. S1B). ATR‐101 therefore caused cholesterol accumulation, ATP depletion and caspase activation by specific mechanisms that required structural determinants that are not present in PD129337.
To evaluate the potential roles of cholesterol accumulation and of ACAT inhibition in ATR‐101 cytotoxicity, we compared the effects of ATR‐101 and PD129337 on cholesterol esterification, the ATP level and caspase 3/7 activity in ACC‐derived cell lines. Both ATR‐101 and PD129337 inhibited NBD‐cholesterol esterification and accumulation in lipid droplets in H295R and BD140C cells (Figure 1E). PD129337 inhibited cholesterol esterification more efficiently than ATR‐101 did, consistent with its lower ACAT inhibitory coefficient (Supporting Information Fig. S1C) (Trivedi et al., 1993; Trivedi et al., 1994). However, PD129337 did not cause ATP depletion or caspase activation at any concentration tested (Figure 1D). The concentration of ATR‐101 that was required for ATP depletion and caspase activation was more than two orders of magnitude higher than the concentration that inhibited cholesterol esterification (Figure 1D). ACAT inhibition alone therefore did not cause cytotoxicity in these ACC‐derived cells.
Effects of cholesterol sequestration and of exogenous cholesterol on ATR‐101 cytotoxicity
To determine if cholesterol accumulation was required for ATR101 cytotoxicity, we evaluated the effects of cholesterol sequestration by methyl‐β‐cyclodextrin (MβCD) on ATP depletion and caspase activation by ATR‐101. Addition of MβCD together with ATR‐101 prevented cholesterol accumulation in H295R cells (Figure 2A; Supporting Information Fig. S2A). MβCD also prevented ATP depletion and caspase activation both at 4 and 24 h after ATR‐101 addition to H295R cells (Figure 2B; Supporting Information Fig. S2B). Cells that were cultured with ATR‐101 alone were small and rounded, whereas cells that were cultured with ATR‐101 in the presence of MβCD remained flat and adherent for at least 30 h (Supporting Information Fig. S2E). MβCD alone had little effect on the ATP level, the caspase 3/7 activity or the morphology of H295R cells (Figure 2B; Supporting Information Fig. S2B, E). MβCD did not eliminate all effects of ATR‐101, indicating that it did not prevent ATR‐101 entry into cells or effects that were independent of cholesterol accumulation (Supporting Information Fig. S2F; Figure 4D).
Figure 2.
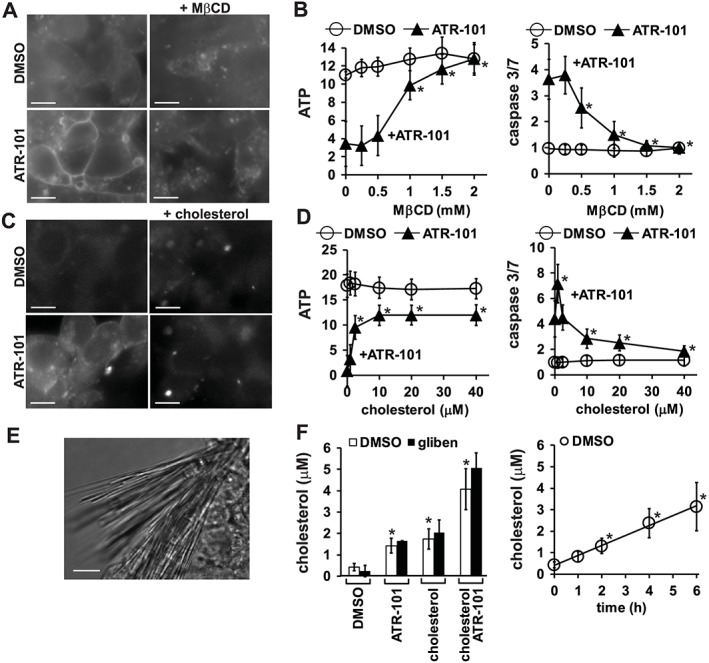
Effects of ATR‐101 in combination with MβCD or with exogenous cholesterol on cholesterol accumulation and on cytotoxicity. (A) Effects of ATR‐101 and MβCD separately and in combination on the cholesterol levels in H295R cells. The cells were cultured with DMSO vehicle (upper images) or with 100 μM ATR101 (lower images), alone (left images) or together with of 2 mM MβCD (right images) for 4 h. The images show filipin III binding to cholesterol and are representative of images from four separate experiments. The cholesterol levels of H295R cells that were cultured with ATR‐101 and MβCD for 24 h are shown in Supporting Information Fig. S2A. The full fields from which the images were taken are shown in Supporting Information Fig. S2J. Representative phase contrast images of fields of cells that were cultured with ATR‐101 and MβCD separately and in combination are shown in Supporting Information Fig. S2E. The scale bars denote 10 μm. (B) Effects of MβCD on ATP depletion and caspase 3/7 activation by ATR‐101 in H295R cells. The cells were cultured with DMSO vehicle or 50 μM ATR‐101 together with the indicated concentrations of MβCD for 4 h. The ATP levels (left graphs) and the caspase 3/7 activities (right graphs) were measured in cells that were grown in parallel. The graphs show the means ± 2SD of six samples from three experiments. *P < 0.05, significant effect of MβCD; one‐way ANOVA with Dunnett's post hoc tests. The ATP levels and the caspase 3/7 activities of cells that were cultured with ATR‐101 and MβCD for 24 h are shown in Supporting Information Fig. S2B. (C) Effects of ATR‐101 and exogenous cholesterol separately and in combination on the intracellular cholesterol levels in H295R cells. The cells were cultured with DMSO vehicle (upper images) or with 50 μM ATR‐101 (lower images) together with ethanol control (left images) or 10 μM exogenous cholesterol (right images) for 4 h. The images show filipin III binding to cholesterol and are representative of two separate experiments. The scale bars denote 10 μm. The levels of intracellular cholesterol in H295R cells that were cultured with ATR‐101 and exogenous cholesterol for 24 h are shown in Supporting Information Fig. S2A. The full fields from which the images were cropped are shown in Supporting Information Fig. S2K. Representative phase contrast images of cells that were cultured with ATR‐101 and exogenous cholesterol separately and in combination are shown in Supporting Information Fig. S2E. (D) Effects of exogenous cholesterol on ATP depletion and caspase 3/7 activation by ATR‐101 in H295R cells. The cells were cultured with DMSO vehicle or 50 μM ATR‐101 together with the indicated concentrations of exogenous cholesterol for 24 h. The ATP levels (left graph) and the caspase 3/7 activities (right graph) were measured in cells that were grown in parallel. The graphs show the means ± 2SD of five samples from three experiments. *P < 0.05, significant effect of exogenous cholesterol; one‐way ANOVA with Dunnett's post hoc tests. The ATP levels and the caspase 3/7 activities of H295R cells that were cultured with ATR‐101 and exogenous cholesterol for 4 h are shown in Supporting Information Fig. S2C. (E) Cholesterol crystallization at the plasma membrane of cells cultured with ATR‐101. H295R cells were cultured with 40 μM ATR‐101 for 24 h and were imaged using phase contrast microscopy. The scale bar denotes 10 μm. (F) Comparison of the amounts of extracellular cholesterol that were associated with H295R cells that were cultured with ATR‐101 and exogenous cholesterol separately and in combination (left graph), and the amount of cholesterol effluxed from control cells (right graph). The cells were cultured for 4 h in serum‐containing medium containing 100 μM ATR‐101 or 40 μM cholesterol separately and in combination. The culture medium was removed, and the extracellular cholesterol that was associated with the cells was recovered by washing the cells using an equal volume of serum‐free medium with or without 50 μM glibenclamide. The amounts of total cholesterol in the wash media were measured (left graph). The amount of cholesterol effluxed from cells was measured by removing the medium and adding an equal volume of serum‐free medium to control cells. The serum‐free medium was removed at the times indicated, and the amounts of total cholesterol were measured (right graph). *P < 0.05, significantly different from DMSO controls (left graph); significantly different from time =0 (right graph); one‐way ANOVA with Dunnett's post hoc tests.
We examined the effects of adding exogenous cholesterol together with ATR‐101 to H295R cells. Unexpectedly, cells that were cultured with ATR‐101 together with low concentrations of exogenous cholesterol had lower intracellular cholesterol levels than cells that were cultured with ATR‐101 alone for 4 or 24 h (Figure 2C; Supporting Information Fig. S2A). Consistent with the decrease in intracellular cholesterol, the addition of exogenous cholesterol partially restored the ATP level of cells that were cultured with ATR‐101 (Figure 2D; Supporting Information Fig. S2C). Exogenous cholesterol also reduced caspase activation in cells that were cultured with ATR‐101 for 24 (Figure 2D). Cells that were cultured with ATR‐101 in the presence of exogenous cholesterol remained flat and adherent for at least 30 h, whereas cells that were cultured with ATR‐101 alone were small and rounded (Supporting Information Fig. S2E). Exogenous cholesterol alone had no detectable effect on the cholesterol level, the ATP level, the caspase 3/7 activity or morphology of H295R cells (Figure 2C, D; Supporting Information Fig. S2A, C, E). The reduction in the intracellular cholesterol level of cells that were cultured with ATR‐101 upon addition of exogenous cholesterol correlated with an increase in total cell‐associated extracellular cholesterol (see Figure 2E, F below). In contrast to exogenous cholesterol, exogenous cholesterol linoleate did not prevent the accumulation of cholesterol or ATP depletion caused by ATR‐101 (Supporting Information Fig. S2A, D). Moderate cholesterol : MβCD concentrations also reduced ATP depletion by ATR‐101, whereas high cholesterol : MβCD concentrations increased the cholesterol level and caused ATP depletion both alone and in combination with ATR‐101 (Supporting Information Fig. S2H, I). The suppression of ATR‐101‐dependent cholesterol accumulation, ATP depletion and caspase activation by exogenous cholesterol corroborated the essential role of cholesterol accumulation in ATR‐101 cytotoxicity.
To establish if exogenous cholesterol affected ATR‐101 entry into the cells or cholesterol esterification, we tested the effects of exogenous cholesterol on NBD‐cholesterol esterification, with or without ATR‐101. There was no significant difference in NBD‐cholesterol esterification, or in its inhibition by ATR‐101, between cells that were cultured with or without exogenous cholesterol (Supporting Information Fig. S2G).
The consistent relationship between cholesterol accumulation and ATP depletion in cells that were cultured with ATR‐101 alone and the suppression of cholesterol accumulation and the prevention of ATP depletion by MβCD as well as by exogenous cholesterol support the hypothesis that cholesterol accumulation is necessary for ATR‐101 cytotoxicity.
Cholesterol crystallization at the plasma membrane of cells that are cultured with ATR‐101
We observed that H295R cells that were cultured with ATR‐101 were associated with crystals that emanated from the cell membrane (Figure 2E). These needle‐shaped crystals were similar to cholesterol crystals that are associated with cholesterol‐loaded macrophages as well as other cells with high cholesterol levels (Kellner‐Weibel et al., 1999). H295R cells that were grown with ATR‐101 in combination with MβCD did not produce crystals, consistent with sequestration of the cholesterol by MβCD (Supporting Information Fig. S2A).
H295R cells that were cultured with ATR‐101 in combination with exogenous cholesterol produced more extracellular crystals than cells that were cultured with ATR‐101 or exogenous cholesterol separately (Supporting Information Fig. S2G). We quantified the total extracellular cholesterol that was associated with cells that were cultured with ATR‐101 and exogenous cholesterol separately and in combination. ATR‐101 and extracellular cholesterol increased the total extracellular cholesterol that was associated with H295R cells (Figure 2F). The concurrent increase in cholesterol crystals and decrease in intracellular cholesterol caused by exogenous cholesterol in the presence of ATR‐101 suggests that exogenous cholesterol reduced intracellular cholesterol accumulation by facilitating cholesterol crystallization at the plasma membrane. The total amount of extracellular cholesterol that was associated with cells after 4 h culture with ATR‐101 and exogenous cholesterol was larger than the total amount of cholesterol that was exported from control cells in the same time (Figure 2F). The passive discharge of cholesterol was therefore sufficient to prevent intracellular cholesterol accumulation in the presence, but not in the absence of exogenous cholesterol.
ATR‐101 effects on cholesterol accumulation and on the ATP levels of cells that are cultured without serum cholesterol
We examined the effects of ATR‐101 on cholesterol accumulation, ATP depletion and caspase activation in H295R cells that were cultured in serum‐free medium to determine the influence of cholesterol in the medium on ATR‐101 cytotoxicity. The medium was replaced with serum‐free medium with or without ATR‐101, and the cells were cultured for 4 h. Cells that were cultured in with ATR‐101 in serum‐free medium had higher cholesterol levels than cells that were cultured in serum‐free medium lacking ATR‐101 (Figure 3A). ATR‐101 caused ATP depletion and caspase activation with the same potency in cells that were cultured in serum‐free medium as in cells cultured under standard conditions (Figure 3B). MβCD suppressed ATP depletion and caspase activation by ATR‐101 in serum‐free medium. Cholesterol accumulation was therefore required for ATP depletion and for caspase activation by ATR‐101 also in cells that were cultured in serum‐free medium. Excess cellular cholesterol can therefore mediate ATR‐101 cytotoxicity even when the cells are cultured with ATR‐101 in the absence of serum cholesterol.
Figure 3.

Effects of ATR‐101 on cholesterol trafficking and efflux. (A) Effects of ATR‐101 and MβCD separately and in combination on the cholesterol levels in H295R cells that were cultured for 4 h in serum‐free medium. The medium of cells that were cultured under standard conditions was replaced with serum‐free media containing apoA‐I with either DMSO vehicle (upper images) or 100 μM ATR‐101 (lower images), alone (left images) or in combination with of 4 mM MβCD (right images). The images show filipin III binding to cholesterol and are representative of two separate experiments. The full fields from which the images were cropped are shown in Supporting Information Fig. S3B. The scale bars denote 10 μm. (B) Effects of MβCD on ATP depletion and caspase 3/7 activation by ATR‐101 in H295R cells that were cultured in serum‐free medium. The cells were cultured as described in part A, and the ATP levels and caspase 3/7 activities were measured. The graphs show the means ± 2SD of eight samples from four experiments and six samples from three experiments for the ATP and caspase 3/7 assays respectively. *P < 0.05, significant effect of MβCD; unpaired two‐tailed Student's t‐tests. (C) Effects of ATR‐101 on the rates of cholesterol efflux and on the ATP levels of H295R cells during culture in serum‐free medium. The medium of cells that were cultured under standard conditions was replaced with serum‐free medium containing apoA‐I with DMSO vehicle or 100 μM ATR‐101. At the indicated times, the cholesterol level in the medium (upper graph) and the ATP level in the cells (lower graph) were measured. The graphs show the means ± 2SD of six samples from two experiments. *P < 0.05, significantly different from DMSO control; two‐way ANOVA with Sidak's post hoc tests. (D) Effects of ATR‐101 and of ABC transporter inhibitors on cholesterol efflux and on the ATP levels of H295R cells during culture in serum‐free medium. The levels of cholesterol in the medium (upper graph) and of cellular ATP (lower graph) were measured in the same cultures 4 h after replacing the standard culture medium with serum‐free media containing apoA‐I and the indicated concentrations of ATR‐101, PD129337, glibenclamide, benzamil or verapamil. The graphs show the means ± 2SD of six samples from three experiments for cells cultured with ATR‐101, PD129337 or glibenclamide and two samples from one experiment for cells cultured with benzamil or verapamil. *P < 0.05, significantly different from DMSO controls (0 μM compound); one‐way ANOVA with Dunnett's post hoc tests. (E) Effects of ATR‐101 in combination with olesoxime on cholesterol efflux and on the ATP levels in H295R cells. The cells were cultured with the indicated concentrations of ATR‐101 alone or ATR‐101 in combination with 40 μM olesoxime for 4 h. The media were replaced with serum‐free medium containing apoA‐I, and the levels of cholesterol in the medium (upper graph) and of cellular ATP (lower graph) were measured after incubation for an additional 4 h. The graphs show the means ± 2SD of six samples from two experiments and nine samples from three experiments for the cholesterol efflux and ATP assays respectively. *P < 0.05, significantly different from control without olesoxime; two‐way ANOVA with Sidak's post hoc tests.
ATR‐101 effects on cholesterol efflux
To identify potential causes of cholesterol accumulation in cells that were cultured with ATR‐101, we measured the rates of cholesterol efflux from H295R cells with or without ATR‐101. H295R cells that were cultured without ATR‐101 produced a linear increase in the total cholesterol concentration of the culture medium over 4 h (Figure 3C). ATR‐101 inhibited cholesterol efflux, and there was no detectable increase in the cholesterol concentration of the medium of cells that were cultured with ATR‐101 (Figure 3C). The concentration of ATR‐101 that inhibited cholesterol efflux was similar to the concentrations that caused cholesterol accumulation, ATP depletion and caspase activation (Figure 3D). In contrast, PD129337 had no detectable effect on cholesterol efflux. The concurrent decrease in total soluble cholesterol that was exported to the culture medium and the increase in cell‐associated cholesterol indicate that ATR‐101 redirected the pathways for cholesterol discharge from cells. ATR‐101 inhibited cholesterol efflux to soluble lipoprotein particles, which requires active export by ABC transporters, and caused an increase in passive extrusion of crystalline and cell‐associated cholesterol (Figures 3D and 2F).
We compared the effect of ATR‐101 on cholesterol efflux with the effects of known ABC transporter inhibitors. Glibenclamide blocked cholesterol efflux in H295R cells at a concentration that inhibits ABCA1 (50 μM; Figure 3D) (Nieland et al., 2004). Benzamil did not reduce cholesterol efflux from H295R cells at concentrations that inhibit ABCG1 and that caused cholesterol accumulation in H295R cells (50 μM; Figure 3D; Supporting Information Fig. S5C) (Cserepes et al., 2004). Verapamil did not reduce cholesterol efflux at a concentration that inhibits MDR1 and that caused doxorubicin accumulation in H295R cells (50 μM; Figure 3D; Supporting Information Fig. S4C) (Bentz et al., 2013). More than 10‐fold higher concentrations of verapamil and benzamil were required to inhibit cholesterol efflux and to cause ATP depletion than are required to inhibit MDR1 and ABCG1 respectively.
We compared the effects of individual ABC transporter inhibitors on cholesterol accumulation and on the ATP level of H295R cells. Glibenclamide did not cause cholesterol accumulation and did not reduce the ATP level of cells (Figure 3D, Supporting Information Fig. S3A). Verapamil and benzamil caused cholesterol accumulation but did not reduce the ATP levels of H295R cells at the concentrations that inhibit ABCG1 and MDR1 activity respectively (Supporting Information Figs S3A, S5C and S4C; Figure 3D) (Cserepes et al., 2004; Bentz et al., 2013). The inhibition of cholesterol efflux was neither sufficient nor necessary to cause cholesterol accumulation, and cholesterol accumulation was not sufficient to cause ATP depletion in ACC‐derived cells.
We tested if the inhibition of cholesterol efflux by ATR‐101 required ATP depletion. Olesoxime reduced ATP depletion by ATR‐101, but it had no detectable effect on the inhibition of cholesterol efflux by ATR‐101 from the same cells, indicating that the restoration of a nearly normal ATP level did not restore cholesterol efflux (Figure 3E). Glucose and α‐tocopherol also restored the ATP level and did not restore cholesterol efflux (Supporting Information Fig. S3C).
ATR‐101 effects on cortisol secretion and on doxorubicin clearance
We measured cortisol secretion from cells that were cultured with ATR‐101 to determine the effect of ATR‐101 on ABC transporters that export cholesterol metabolites. H295R cells that were cultured without ATR‐101 produced a linear increase in the cortisol concentration of the medium over 8 h (Figure 4A). ATR‐101 inhibited cortisol secretion at the earliest time when cortisol secretion was detected 4 h after ATR‐101 addition to the cells. ATR‐101 blocked both basal and forskolin‐stimulated cortisol secretion as efficiently as the MDR1 inhibitor verapamil (Figure 4A; Supporting Information Fig. S4A). The same concentration of ATR‐101 inhibited cortisol secretion as was required to cause cholesterol accumulation, caspase activation and ATP depletion in H295R cells (Figure 4B). PD129337 did not reduce, but rather increased, cortisol secretion at concentrations that were up to fivefold higher than the concentration of ATR‐101 that inhibited cortisol export. ACAT inhibition alone did not reduce cortisol secretion. ATR‐101 therefore inhibited cortisol secretion by mechanisms that required specific functional groups that are not present in PD129337.
Figure 4.

Effects of ATR‐101 on cortisol efflux and on doxorubicin accumulation. (A) Effects of ATR‐101 on the rate of cortisol secretion from H295R cells cultured with or without forskolin. The medium of the cells was replaced with media containing DMSO vehicle or 100 μM ATR‐101 alone, or together with 100 μM forskolin. The cortisol levels in the media were measured at the times indicated. The graph shows the means ± 2SD of five samples from two experiments. *P < 0.05, significantly different from corresponding samples containing ATR‐101; two‐way ANOVA with Sidak's post hoc tests. (B) Effects of ATR‐101 and of PD129337 on cortisol secretion and on ATP levels. The medium of H295R cells was replaced with media containing the indicated concentrations of ATR‐101 or PD129337, and the levels of cortisol in the media (upper graph) and of ATP in the cells (lower graph) were measured in the same cultures after 4 h. The graphs show the means ± 2SD of six samples from two experiments and five samples from two experiments for the cortisol secretion and ATP assays respectively. *P < 0.05, significantly different from DMSO controls; two‐way ANOVA with Sidak's post hoc tests. (C) Effects of ATR‐101 in combination with exogenous cholesterol or α‐tocopherol on cortisol secretion and ATP depletion. The medium of H295R cells was replaced with media containing the indicated concentrations of ATR‐101 together with vehicle, 40 μM exogenous cholesterol or 40 μM α‐tocopherol. The levels of cortisol in the media (upper graph) and of ATP in the cells (lower graph) were measured in the same cultures after 4 h. The graphs show the means ± 2SD of six samples from two experiments. *P < 0.05, significant effect of ATR‐101; one‐way ANOVA with Dunnett's post hoc tests. (D) Effects of ATR‐101 and MβCD separately and in combination on doxorubicin accumulation in H295R cells. The cells were cultured in the presence of 25 μM doxorubicin together with DMSO vehicle (upper images) or 100 μM ATR‐101 (lower images) alone (left images) or in combination with 4 mM MβCD (right images). The levels of doxorubicin in the cells were imaged by fluorescence microscopy after 2 h. The images show doxorubicin fluorescence and are representative of images from two separate experiments. The scale bars denote 200 μm.
We investigated if the inhibition of cortisol secretion by ATR‐101 required ATP depletion. Exogenous cholesterol as well as α‐tocopherol restored normal or nearly normal ATP levels, but neither compound prevented the inhibition of cortisol secretion by ATR‐101 in the same cells (Figure 4C).
To determine if ATR‐101 inhibits MDR1 activity using an independent assay, we measured the accumulation of doxorubicin in H295R cells that were cultured in medium containing doxorubicin with or without ATR‐101. The level of doxorubicin fluorescence was fivefold higher in H295R cells that were cultured with doxorubicin in the presence of ATR‐101 for 2 h than in control cells (Figure 4D). ATR‐101 caused doxorubicin accumulation in the presence of MβCD, indicating that ATR‐101 inhibited doxorubicin clearance independently of cholesterol accumulation or ATP depletion.
Cytotoxicity and cholesterol accumulation by combinations of ABC transporter inhibitors
Because ATR‐101 inhibited both cholesterol, cortisol and doxorubicin export, and because inhibition of individual ABC transporters did not cause ATP depletion, we hypothesized that the simultaneous inhibition of several ABC transporters was required for ATR‐101 cytotoxicity. We tested the effects of different combinations of ABC transporter inhibitors on the ATP levels and on the caspase 3/7 activities of H295R cells. Glibenclamide, benzamil and zosuquidar in combination reduced the ATP level and increased the caspase 3/7 activity of the cells (Figure 5A). When any one of the ABC transporter inhibitors was omitted, no decrease in the ATP level was detected, and the caspase 3/7 activity was reduced. Similarly, glibenclamide, benzamil and verapamil in combination caused ATP depletion in H295R cells, but the pairwise combinations had only a partial effect (Supporting Information Fig. S5A). Thus, the simultaneous inhibition of ABCA1, ABCG1, MDR1 and potentially other targets of verapamil, zosuquidar, benzamil and glibenclamide was required to mimic the effects of ATR‐101 on the ATP level and on the caspase 3/7 activity of H295R cells.
Figure 5.
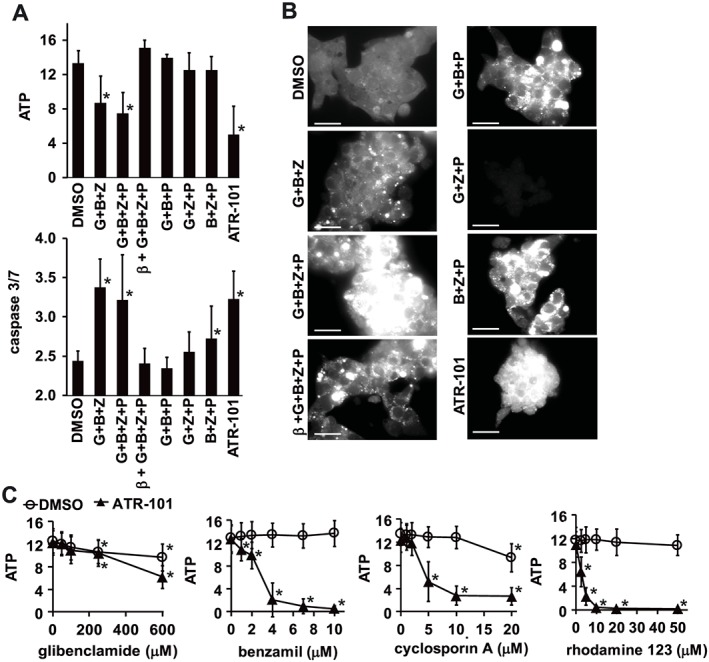
Combined effects of ABC transporter inhibitors, substrates, and of ATR‐101 on ATP levels, caspase 3/7 activities and cholesterol levels in H295R cells. (A) Effects of different combinations of ABC transporter inhibitors on the ATP level and on the caspase 3/7 activity of H295R cells. The cells were cultured in serum‐free media containing 50 μM each of the indicated combinations of glibenclamide (G), benzamil (B) and zosuquidar (Z), 10 μM PD129337 (P) and 4 mM MβCD (β) for 4 h. The ATP levels (upper graph) and caspase 3/7 activities (lower graph) were measured in parallel cultures. The graphs show the means ± 2SD of six samples from three experiments. *P < 0.05, significantly different from DMSO controls; one‐way ANOVA with Dunnett's post hoc tests. (B) The cholesterol levels of H295R cells that were cultured as described in panel A were visualized by filipin III binding. The scale bars denote 30 μm. (C) Effects of ATR‐101 in combination with glibenclamide, benzamil, cyclosporin A or rhodamine 123 on the ATP levels of H295R cells. H295R cells were cultured with DMSO vehicle or 20 μM ATR‐101 together with the indicated concentrations of glibenclamide, benzamil, cyclosporin A or rhodamine 123. The ATP levels were measured 4 h after addition of each compound and were obtained in separate experiments. The graphs show the means ± 2SD of six samples from three experiments. *P < 0.05, significant effect of ABC inhibitors and substrates alone and in combination with ATR‐101; one‐way ANOVA with Dunnett's post hoc tests.
Glibenclamide, benzamil and zosuquidar increased the cholesterol level of H295R cells (Figure 5B). PD129337 further increased the cholesterol level together with these ABC inhibitors, but it did not enhance ATP depletion or caspase 3/7 activation (Figure 5A). MβCD reduced the cholesterol level and suppressed the effects on the ATP level and on the caspase 3/7 activity (Figure 5A, B). Several other combinations of ABC transporter inhibitors and PD129337 increased the cholesterol level, but had little or no effect on the ATP level or on the caspase 3/7 activity (Figure 5A, B). Cholesterol accumulation was necessary for ATP depletion and for caspase activation by these ABC inhibitors, but it was not sufficient for cytotoxicity. It is possible that the accumulation of other steroids or cholesterol metabolites requires inhibition of all three ABC transporters and is necessary for their cytotoxicity.
Cytotoxicity of ATR‐101 in combination with ABC transporter inhibitors and substrates
We investigated if the potency of ATR‐101 was enhanced when it was applied in combination with compounds that targeted individual ABC transporters. We tested ATR‐101 in combination with individual ABC transporter inhibitors and substrates at concentrations that were not cytotoxic when tested separately. ATR‐101 in combination with glibenclamide did not increase the efficiency of ATP depletion (Figure 5C; Supporting Information Fig. S5B). In contrast, ATR‐101 in combination with each of benzamil, cyclosporin A, verapamil and rhodamine 123 caused ATP depletion at concentrations that did not deplete ATP individually (Figure 5C; Supporting Information Fig. S5B). ATR‐101 and the MDR1 substrate rhodamine‐123 in combination reduced the ATP level of H295R cells at concentrations that were 10‐fold and fivefold lower than the ATR‐101 and rhodamine‐123 concentrations that were required to reduce the ATP level separately (Figure 5C; Supporting Information Fig. S5B). MβCD suppressed ATP depletion by ATR‐101 in combination with these ABC transporter inhibitors and substrates, suggesting that ATP depletion by these combinations of inhibitors required cholesterol accumulation (Supporting Information Fig. S5B). The potency of ATR‐101 can therefore be enhanced by combining it with individual ABC inhibitors or substrates.
Roles of steroids and steroidogenesis in ATR‐101 cytotoxicity
Inhibition of MDR1 by ATR‐101 was predicted to cause the accumulation of steroids and other products of cholesterol metabolism. We examined the effects of several structurally dissimilar inhibitors of steroidogenic enzymes on ATP depletion by ATR‐101. Most of these inhibitors reduced ATP depletion by ATR‐101, suggesting that steroid accumulation contributed to ATR‐101 cytotoxicity (Figure 6A). The concentrations of the inhibitors that reduced ATP depletion by ATR‐101 were consistent with their inhibitory coefficients for specific steroidogenic enzymes (Takahashi et al., 1990; Johansson et al., 1998; Garrido et al., 2014). The reduction of ATP depletion by ATR‐101 when it was combined with any one of several different inhibitors of steroidogenesis is consistent with the hypothesis that steroid accumulation contributes to ATR‐101 cytotoxicity.
Figure 6.
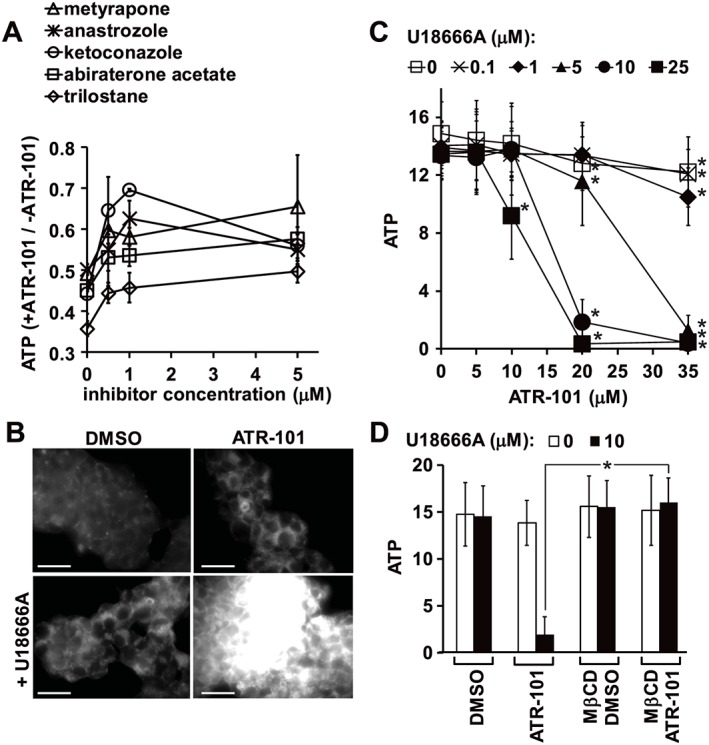
Effects of steroids and inhibitors of steroidogenesis on ATR‐101 cytotoxicity. (A) Effects of inhibitors of steroidogenic enzymes on ATP depletion by ATR‐101 in H295R cells. The cells were cultured with 35 μM ATR‐101 or DMSO vehicle together with different concentrations the indicated inhibitors of steroidogenesis. The ATP levels of the cells were measured after 4 h. The graph shows the ratio between the ATP levels of cells that were cultured with each concentration of each inhibitor together with ATR‐101 versus cells that were cultured with the same concentration of each inhibitor alone. Each data point represents the mean ± 2SD of two pairs of cell cultures grown with and without ATR‐101. The data prior to calculation of the ratios are shown in Supporting Information Fig. S6A. (B) Effects of ATR‐101 and U18666A on the cholesterol levels in H295R cells. The cells were cultured with 20 μM ATR‐101 and 8 μM U18666A, separately and in combination in serum‐free medium. The images show filipin III binding to cholesterol and are representative of two separate experiments. The scale bars denote 30 μm. (C) Combined effects of ATR‐101 and U18666A on the ATP level of H295R cells. The cells were cultured with DMSO vehicle or the indicated concentrations of U18666A and ATR‐101 in serum‐free medium for 4 h and the ATP levels were measured. The graph shows the means ± 2SD of six samples from three experiments. *P < 0.05, significant effect of ATR‐101 in the presence of the indicated concentration of U18666A; two‐way ANOVA with Dunnett's post hoc tests. (D). Effects of MβCD on ATP depletion by the combined effects of ATR‐101 together with U18666A. The cells were cultured with 20 μM ATR‐101 and 10 μM U18666A, separately and in combination, with or without 2 mM MβCD in serum‐free medium. The graph shows the means ± 2SD of six samples from three experiments. *P < 0.05, significant effect of MβCD; unpaired two‐tailed Student's t‐tests.
To investigate potential mechanisms whereby steroid accumulation could contribute to ATR‐101 cytotoxicity, we tested the effects of ATR‐101 in combination with 3β‐(2‐diethylaminoethoxy)‐5‐androsten‐17‐one (U18666A). ATR‐101 and U18666A in combination caused a greater than additive increase in cholesterol accumulation (Figure 6B). ATR‐101 and U18666A in combination also caused ATP depletion at concentrations that had no detectable effect on the ATP level separately (Figure 6C). MβCD suppressed ATP depletion by ATR‐101 in combination with U18666A, suggesting that their combined effect on the ATP level required cholesterol accumulation (Figure 6D). U18666A and endogenous steroids can inhibit cholesterol trafficking, suggesting that steroid accumulation could contribute to ATR‐101 cytotoxicity by inhibiting cholesterol trafficking.
Effects of ATR‐101 on ABC transporter and steroidogenic gene transcription
We investigated the effects of ATR‐101 on transcription of genes whose products modulate cholesterol levels and steroidogenesis. ATR‐101 reduced the levels of ABCA1, ABCG1 and IDOL transcripts within an hour after addition to H295R cells (Figure 7A). Transcription of these genes was repressed by lower ATR‐101 concentrations than were required for the cytotoxic effects of ATR‐101 or for the activation of CHOP transcription, indicating that their repression was not due to general cell stress. Similar changes in transcript levels were observed 4 and 8 h after ATR‐101 addition. The levels of several steroid biosynthetic gene transcripts, including SULT2A1, HSD3B2 and CYP17A1 were reduced in cells cultured with ATR‐101 (Figure 7B).
Figure 7.
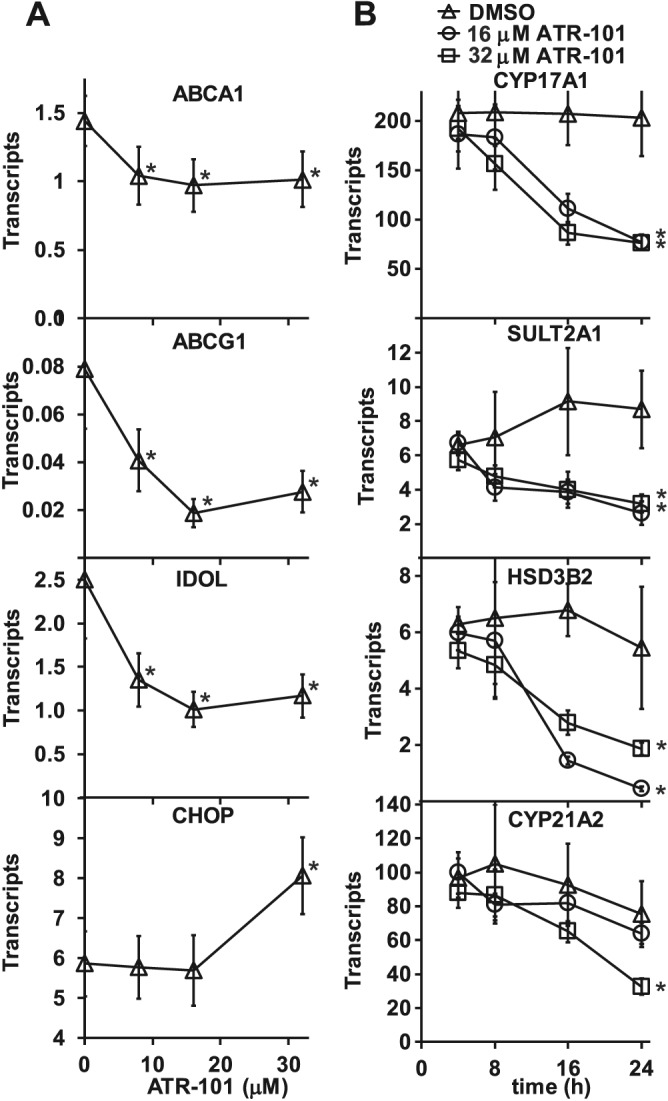
ATR‐101 effects on the transcription of genes that affect cholesterol and steroid levels. (A) Effects of ATR‐101 on ABCA1, ABCG1, IDOL and CHOP transcript levels. The levels of the transcripts indicated in each graph were measured in cells that were cultured with the concentrations of ATR‐101 indicated at the bottom of the figure for 1 h. The transcript levels were normalized by the RPL9 transcript levels. The graphs show the means ± 2SD of five samples from four experiments. *P < 0.05, significant effect of ATR‐101; one‐way ANOVA with Dunnett's post hoc tests. (B). Effects of ATR‐101 on steroidogenic gene transcription. The levels of the transcripts indicated in each graph were measured in cells that were cultured for the times indicated at the bottom of the figure with the concentrations of ATR‐101 indicated by the symbols shown at the top of the figure. The transcript levels were normalized by the RPL9 transcript levels. The graphs show the means ± 2SD of two samples from two experiments. *P < 0.05, significantly different from DMSO controls; one‐way ANOVA with Dunnett's post hoc tests; all samples cultured for different times with ATR‐101 were compared with all samples cultured for different times without ATR‐101, n = 8.
PD129337 and ATR‐101 had equivalent effects on the levels of ABCA1 as well as ABCG1 transcripts (Supporting Information Fig. S7A). They repressed transcription of these genes by mechanisms that were distinct from the selective inhibition of cholesterol and cortisol export by ATR‐101. The transcription of ABCA1, ABCG1 and IDOL genes is activated by liver X receptor complexes in response to hydroxysterol binding (see Wollam and Antebi, 2011). Molecular dynamics simulations predicted that ATR‐101 and PD129337 can bind to the ligand binding pocket of liver X receptor α (Supporting Information Fig. S7B). ATR‐101 and PD129337 binding to liver X receptor α could displace hydroxysterol ligands in a manner similar to that which has been described for unsaturated fatty acids (Ou et al., 2001).
Discussion
ACC has a poor prognosis in most cases because no existing drugs can halt tumour growth indefinitely and because the high levels of circulating steroids that are produced by many tumours suppress immune responses and disrupt other physiological functions. Current combination treatments that are used to counteract tumour growth and to suppress excess steroid production are plagued by adverse effects, treatment resistance and drug interactions that can compromise efficacy. Compounds that counteract both tumour growth and steroid production would provide new treatment options for ACC patients. The cytotoxic cholesterol accumulation and the inhibition of cortisol secretion that are caused by ATR‐101 demonstrate that a single agent can inhibit ACC cell viability and steroid production.
ATR‐101 inhibited cholesterol efflux and cortisol secretion with rapid kinetics at the same concentration that caused ATP depletion and caspase activation. The suppression of ATR‐101 cytotoxicity through the sequestration of cholesterol by MβCD and through the enhancement of cholesterol crystallization by exogenous cholesterol indicates that cholesterol accumulation is necessary for ATR‐101 cytotoxicity.
The increased effects of ATR‐101 and U18666A in combination on the cholesterol and ATP levels were consistent with a role of cholesterol trafficking in ATR‐101 cytotoxicity. Both the intracellular trafficking of cholesterol between different cell membranes as well as cholesterol efflux to lipoprotein particles are regulated by ABC transporters (Debry et al., 1997; Luker et al., 1999; Wang et al., 2007; Out et al., 2008; Tarling and Edwards, 2011; Yamauchi et al., 2015). Because of the close relationship between cholesterol trafficking and efflux, our data do not distinguish whether ATR‐101 inhibited these processes independently, or if the inhibition of cholesterol trafficking and efflux were interdependent consequences of ABC transporter inhibition.
The rapid inhibition of cortisol secretion and doxorubicin clearance by ATR‐101 suggest that ATR‐101 inhibited MDR1 activity. ATR‐101 administration reduces the levels of circulating steroids in mice with ACC cell xenografts and in dogs stimulated with ACTH (Cheng et al., 2016; Lapensee et al., 2016). Some MDR1 inhibitors reduced circulating corticosteroid levels in clinical trials, suggesting that MDR1 inhibition can reduce steroid secretion in man (Guthrie et al., 1983; Shapiro et al., 1990; Kobayashi et al., 1996). The simultaneous accumulation of cholesterol and inhibition of steroid secretion caused by ATR‐101 can potentially provide dual benefits to ACC patients. To our knowledge, ATR‐101 is the first compound that has been found to inhibit both cholesterol and cortisol export.
The similar cytotoxicity of ATR‐101 and of combinations of ABC transporter inhibitors that inhibit ABCA1, ABCG1 and MDR1 is consistent with the hypothesis that ATR‐101 cytotoxicity requires the simultaneous inhibition of several pathways that can facilitate cholesterol efflux or metabolism (Figure 8). Cholesterol accumulation was necessary for ATP depletion both by ATR‐101 and by combinations of ABC transporter inhibitors. However, cholesterol accumulation was not sufficient for ATP depletion. The differences in cytotoxicity among different combinations of ABC transporter inhibitors could be due to the distinct distributions of cholesterol or to effects that are independent of cholesterol accumulation. The many molecular and cellular mechanisms that mediate ATR‐101 cytotoxicity can account for the tissue and cell type specificity of its adrenalytic activity.
Figure 8.
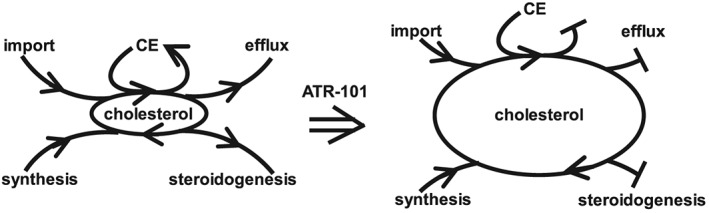
Model for the mechanisms whereby ATR‐101 causes cytotoxic cholesterol accumulation in ACC cells. The ovals at the centre of the diagrams represent cholesterol trafficking (perimeter) and cholesterol accumulation (area) in cells in the absence (left) and in the presence (right) of ATR‐101. CE: cholesterol esters.
ATR‐101 inhibits mitochondrial oxidative phosphorylation and causes the release of ROS in ACC‐derived cells (Cheng et al., 2016). Both cholesterol accumulation and mitochondrial dysfunctions are necessary for ATR‐101 cytotoxicity, as compounds that prevent either of these effects suppress ATR‐101 cytotoxicity. Previous studies have shown that disruptions to cholesterol trafficking can affect mitochondrial membrane potential, F1F0‐ATPase activity and ATP levels in mouse brain and neurons (Yu et al., 2005). Conversely, perturbations to mitochondrial functions can affect steroidogenesis and cholesterol efflux (Midzak et al., 2011; Graham, 2015). It is possible that ATR‐101 causes cholesterol accumulation and mitochondrial dysfunctions by independent mechanisms or that the effects of ATR‐101 on these processes are interrelated.
Disruptions to cholesterol trafficking and metabolism underlie several pathological conditions that are characterized by cholesterol accumulation. StAR mutations cause congenital lipoid adrenal hyperplasias that are associated with a marked reduction in steroidogenesis and elevated cholesterol levels in steroidogenic cells (Sahakitrungruang, 2015). Mutations in Niemann–Pick type C (NPC) intracellular cholesterol transporters or lysosomal acid lipase cause NPC, Wolman and cholesterol ester storage diseases that are associated with impaired cholesterol trafficking and increased cholesterol accumulation in many tissues (Vanier, 2010; Porto, 2014). A combination of genetic and dietary factors cause atherosclerosis that is associated with elevated serum cholesterol and cholesterol accumulation in macrophage foam cells and vascular smooth muscle cells (Buja et al., 1979). CYP46A1 depletion in the striatum of mice mimics Huntington's disease phenotypes and increases the cholesterol level in the brain (Boussicault et al., 2016). Cholesterol accumulation is therefore toxic to adrenocortical as well as other cells under physiological conditions.
ATR‐101 has relatively low potency in assays of cytotoxicity in ACC cells, xenograft suppression in nude mice and adrenalytic activity in guinea pigs (Cheng et al., 2016). The increased cytotoxicity of ATR‐101 in combination with inhibitors of MDR1 and ABCG1 suggests that the low efficiencies of MDR1 and ABCG1 inhibition by ATR‐101 limit its potency. The enhancement of ATR‐101 potency by compounds with mechanisms of action that overlap those that were required for ATR‐101 cytotoxicity suggests that higher potency and potentially improved therapeutic efficacy can be achieved by combining compounds with complementary mechanisms of cytotoxicity and adrenalytic activity.
Author contributions
V.E.B. designed and performed all of the experiments, analyzed the data, produced the figures and drafted the manuscript. T.K.K. established the overall objectives of the study, interpreted the implications of the results and wrote the final paper.
Conflict of interest
T.K.K. is named as an inventor in patent WO 2013142214 entitled ‘Compounds and methods for treating aberrant adrenocortical cell disorders’. The children of T.K.K. own shares in Millendo Inc., which has licensed the patent.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of ATR‐101 compared with PD129337 on the cholesterol levels, ATP levels, caspase 3/7 activities, and cholesterol esterification, at different times in H295R cells.
Figure S2 Effects of ATR‐101 in combination with MβCD and of or with exogenous cholesterol in on cholesterol levels, ATP levels, caspase 3/7 activities, and cholesterol crystallization.
Figure S3 Effects of ATR‐101 and of ABC transporter inhibitors on the cholesterol levels of H295R cells that were cultured in serum‐free medium for 4 h.
Figure S4 Effects of ATR‐101 and of MDR1 inhibitors on cortisol secretion and doxorubicin accumulation.
Figure S5 Combined effects of ABC transporter inhibitors with each other and with ATR‐101 on the ATP levels of H295R cells.
Figure S6 Effects of ATR‐101 in combination with inhibitors of steroidogenesis on the ATP levels of H295R cells.
Figure S7 Effects of ATR‐101 compared with PD129337 on transcript levels in H295R cells.
Table S1 Primer sequences used for qPCR.
Acknowledgements
We thank Dr. Yunhui Cheng and other members of the Kerppola laboratory for critical discussions. We thank Arthur Wolin and Anna Chiarella for their experiments investigating the combined effects of ATR‐101 and U18666A on ACC‐derived cells. The research was supported by the National Institute on Drug Abuse (DA030339).
Burns, V. E. , and Kerppola, T. K. (2017) ATR‐101 inhibits cholesterol efflux and cortisol secretion by ATP‐binding cassette transporters, causing cytotoxic cholesterol accumulation in adrenocortical carcinoma cells. British Journal of Pharmacology, 174: 3315–3332. doi: 10.1111/bph.13951.
References
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuvia S, Stein WD, Goldenberg S, Kane SE, Pastan I, Gottesman MM (1993). Targeted disruption of the mouse mdr1b gene reveals that steroid hormones enhance MDR gene expression. J Biol Chem 268: 27127–27132. [PubMed] [Google Scholar]
- An S, Jang YS, Park JS, Kwon BM, Paik YK, Jeong TS (2008). Inhibition of acyl‐coenzyme A:cholesterol acyltransferase stimulates cholesterol efflux from macrophages and stimulates farnesoid X receptor in hepatocytes. Exp Mol Med 40: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O et al (2014). Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46: 607–612. [DOI] [PubMed] [Google Scholar]
- Bentz J, O'connor MP, Bednarczyk D, Coleman J, Lee C, Palm J et al (2013). Variability in P‐glycoprotein inhibitory potency (IC(5)(0)) using various in vitro experimental systems: implications for universal digoxin drug‐drug interaction risk assessment decision criteria. Drug Metab Dispos 41: 1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose HS, Lingappa VR, Miller WL (2002). Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 417: 87–91. [DOI] [PubMed] [Google Scholar]
- Boussicault L, Alves S, Lamaziere A, Planques A, Heck N, Moumne L et al (2016). CYP46A1, the rate‐limiting enzyme for cholesterol degradation, is neuroprotective in Huntington's disease. Brain 139: 953–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecher PI, Hyun Y (1978). Effect of 4‐aminopyrazolopyrimidine and aminoglutethimide on cholesteryl metabolism and steroidogenesis in the rat adrenal. Endocrinology 102: 1404–1413. [DOI] [PubMed] [Google Scholar]
- Buja LM, Kovanen PT, Bilheimer DW (1979). Cellular pathology of homozygous familial hypercholesterolemia. Am J Pathol 97: 327–357. [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Kerppola RE, Kerppola TK (2016). ATR‐101 disrupts mitochondrial functions in adrenocortical carcinoma cells and in vivo. Endocr Relat Cancer 23: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen‐Weber TA, Voland JR, Wu Y, Ngo K, Roland BL, Nguyen S et al (2000). Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high‐density lipoprotein cholesterol deficiency. Am J Pathol 157: 1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers SG, Hofland LJ, Korpershoek E, Franssen GJ, Van Kemenade FJ, De Herder WW et al (2016). Future directions in the diagnosis and medical treatment of adrenocortical carcinoma. Endocr Relat Cancer 23: R43–R69. [DOI] [PubMed] [Google Scholar]
- Cserepes J, Szentpetery Z, Seres L, Ozvegy‐Laczka C, Langmann T, Schmitz G et al (2004). Functional expression and characterization of the human ABCG1 and ABCG4 proteins: indications for heterodimerization. Biochem Biophys Res Commun 320: 860–867. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debry P, Nash EA, Neklason DW, Metherall JE (1997). Role of multidrug resistance P‐glycoproteins in cholesterol esterification. J Biol Chem 272: 1026–1031. [DOI] [PubMed] [Google Scholar]
- Dibartolomeis MJ, Williams C, Jefcoate CR (1986). Inhibition of ACTH action on cultured bovine adrenal cortical cells by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin through a redistribution of cholesterol. J Biol Chem 261: 4432–4437. [PubMed] [Google Scholar]
- Dominick MA, Mcguire EJ, Reindel JF, Bobrowski WF, Bocan TM, Gough AW (1993). Subacute toxicity of a novel inhibitor of acyl‐CoA: cholesterol acyltransferase in beagle dogs. Fundam Appl Toxicol 20: 217–224. [DOI] [PubMed] [Google Scholar]
- Fallahsharoudi A, De Kock N, Johnsson M, Ubhayasekera SJ, Bergquist J, Wright D et al (2015). Domestication effects on stress induced steroid secretion and adrenal gene expression in chickens. Sci Rep 5: 15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido M, Peng HM, Yoshimoto FK, Upadhyay SK, Bratoeff E, Auchus RJ (2014). A‐ring modified steroidal azoles retaining similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J Steroid Biochem Mol Biol 143: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS (2009). The LDL receptor. Arterioscler Thromb Vasc Biol 29: 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A (2015). Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic Biol Med 89: 982–992. [DOI] [PubMed] [Google Scholar]
- Guthrie GP Jr, Mcallister RG Jr, Kotchen TA (1983). Effects of intravenous and oral verapamil upon pressor and adrenal steroidogenic responses in normal man. J Clin Endocrinol Metab 57: 339–343. [DOI] [PubMed] [Google Scholar]
- Ikonen E (2008). Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9: 125–138. [DOI] [PubMed] [Google Scholar]
- Johansson M, Larsson C, Bergman A, Lund BO (1998). Structure‐activity relationship for inhibition of CYP11B1‐dependent glucocorticoid synthesis in Y1 cells by aryl methyl sulfones. Pharmacol Toxicol 83: 225–230. [DOI] [PubMed] [Google Scholar]
- Junquero D, Pilon A, Carilla‐Durand E, Patoiseau JF, Tarayre JP, Torpier G et al (2001). Lack of toxic effects of F 12511, a novel potent inhibitor of acyl‐coenzyme A: cholesterol O‐acyltransferase, on human adrenocortical cells in culture. Biochem Pharmacol 61: 387–398. [DOI] [PubMed] [Google Scholar]
- Kellner‐Weibel G, Yancey PG, Jerome WG, Walser T, Mason RP, Phillips MC et al (1999). Crystallization of free cholesterol in model macrophage foam cells. Arterioscler Thromb Vasc Biol 19: 1891–1898. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Weiss RE, Vogelzang NJ, Vokes EE, Janisch L, Ratain MJ (1996). Mineralocorticoid insufficiency due to suramin therapy. Cancer 78: 2411–2420. [DOI] [PubMed] [Google Scholar]
- Lapensee CR, Mann JE, Rainey WE, Crudo V, Hunt SW 3rd, Hammer GD (2016). ATR‐101, a selective and potent inhibitor of Acyl‐CoA Acyltransferase 1, induces apoptosis in H295R adrenocortical cells and in the adrenal cortex of dogs. Endocrinology 157: 1775–1788. [DOI] [PubMed] [Google Scholar]
- Le Goff W, Settle M, Greene DJ, Morton RE, Smith JD (2006). Reevaluation of the role of the multidrug‐resistant P‐glycoprotein in cellular cholesterol homeostasis. J Lipid Res 47: 51–58. [DOI] [PubMed] [Google Scholar]
- Lehoux JG, Lefebvre A (1991). Short‐term effects of ACTH on the low‐density lipoprotein receptor mRNA level in rat and hamster adrenals. J Mol Endocrinol 6: 223–230. [DOI] [PubMed] [Google Scholar]
- Luker GD, Nilsson KR, Covey DF, Piwnica‐Worms D (1999). Multidrug resistance (MDR1) P‐glycoprotein enhances esterification of plasma membrane cholesterol. J Biol Chem 274: 6979–6991. [DOI] [PubMed] [Google Scholar]
- Maiter D, Bex M, Vroonen L, T'sjoen G, Gil T, Banh C et al (2016). Efficacy and safety of mitotane in the treatment of adrenocortical carcinoma: a retrospective study in 34 Belgian patients. Ann Endocrinol (Paris) 77: 578–585. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Hashimoto M, Suzuki J, Iwanami K, Tomoi M, Shimomura K (1996). Difference between normal and WHHL rabbits in susceptibility to the adrenal toxicity of an acyl‐CoA:cholesterol acyltransferase inhibitor, FR145237. Toxicol Appl Pharmacol 140: 387–392. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, Van Meer G (2010). Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol 22: 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcneish J, Aiello RJ, Guyot D, Turi T, Gabel C, Aldinger C et al (2000). High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP‐binding cassette transporter‐1. Proc Natl Acad Sci U S A 97: 4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M et al (1996). Disruption of the acyl‐CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc Natl Acad Sci U S A 93: 14041–14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwese MC, De Groot E, Duivenvoorden R, Trip MD, Ose L, Maritz FJ et al (2009). ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: the CAPTIVATE randomized trial. JAMA 301: 1131–1139. [DOI] [PubMed] [Google Scholar]
- Midzak AS, Chen H, Aon MA, Papadopoulos V, Zirkin BR (2011). ATP synthesis, mitochondrial function, and steroid biosynthesis in rodent primary and tumor Leydig cells. Biol Reprod 84: 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WL, Bose HS (2011). Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 52: 2111–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsche MA, Mcdonald JG, Hobbs HH, Cohen JC (2015). Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell‐type specific pathways. Elife 4: e07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita SY, Kobayashi A, Takanezawa Y, Kioka N, Handa T, Arai H et al (2007). Bile salt‐dependent efflux of cellular phospholipids mediated by ATP binding cassette protein B4. Hepatology 46: 188–199. [DOI] [PubMed] [Google Scholar]
- Muller MB, Keck ME, Binder EB, Kresse AE, Hagemeyer TP, Landgraf R et al (2003). ABCB1 (MDR1)‐type P‐glycoproteins at the blood‐brain barrier modulate the activity of the hypothalamic‐pituitary‐adrenocortical system: implications for affective disorder. Neuropsychopharmacology 28: 1991–1999. [DOI] [PubMed] [Google Scholar]
- Nieland TJ, Chroni A, Fitzgerald ML, Maliga Z, Zannis VI, Kirchhausen T et al (2004). Cross‐inhibition of SR‐BI‐ and ABCA1‐mediated cholesterol transport by the small molecules BLT‐4 and glyburide. J Lipid Res 45: 1256–1265. [DOI] [PubMed] [Google Scholar]
- Orso E, Broccardo C, Kaminski WE, Bottcher A, Liebisch G, Drobnik W et al (2000). Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1‐deficient mice. Nat Genet 24: 192–196. [DOI] [PubMed] [Google Scholar]
- Ou J, Tu H, Shan B, Luk A, Debose‐Boyd RA, Bashmakov Y et al (2001). Unsaturated fatty acids inhibit transcription of the sterol regulatory element‐binding protein‐1c (SREBP‐1c) gene by antagonizing ligand‐dependent activation of the LXR. Proc Natl Acad Sci U S A 98: 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Out R, Jessup W, Le Goff W, Hoekstra M, Gelissen IC, Zhao Y et al (2008). Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circ Res 102: 113–120. [DOI] [PubMed] [Google Scholar]
- Pandey A, Rudraiah M (2015). Analysis of endocrine disruption effect of Roundup® in adrenal gland of male rats. Toxicol Rep 2: 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhrel L, Maezawa I, Nguyen TD, Chang KO, Jin LW, Hua DH (2012). Inhibition of Acyl‐CoA: cholesterol acyltransferase (ACAT), overexpression of cholesterol transporter gene, and protection of amyloid beta (Abeta) oligomers‐induced neuronal cell death by tricyclic pyrone molecules. J Med Chem 55: 8969–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto AF (2014). Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases. Pediatr Endocrinol Rev 12 (Suppl 1): 125–132. [PubMed] [Google Scholar]
- Reaven E, Leers‐Sucheta S, Nomoto A, Azhar S (2001). Expression of scavenger receptor class B type 1 (SR‐BI) promotes microvillar channel formation and selective cholesteryl ester transport in a heterologous reconstituted system. Proc Natl Acad Sci U S A 98: 1613–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reindel JF, Dominick MA, Bocan TM, Gough AW, Mcguire EJ (1994). Toxicologic effects of a novel acyl‐CoA:cholesterol acyltransferase inhibitor in cynomolgus monkeys. Toxicol Pathol 22: 510–518. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Usher DC (2002). Anti‐atherogenic effects of the acyl‐CoA:cholesterol acyltransferase inhibitor, avasimibe (CI‐1011), in cultured primary human macrophages. Atherosclerosis 161: 45–54. [DOI] [PubMed] [Google Scholar]
- Sahakitrungruang T (2015). Clinical and molecular review of atypical congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab 20: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbiera S, Leich E, Liebisch G, Sbiera I, Schirbel A, Wiemer L et al (2015). Mitotane Inhibits sterol‐O‐acyl transferase 1 triggering lipid‐mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells. Endocrinology 156: 3895–3908. [DOI] [PubMed] [Google Scholar]
- Shapiro R, Carroll PB, Tzakis AG, Cemaj S, Lopatin WB, Nakazato P (1990). Adrenal reserve in renal transplant recipients with cyclosporine, azathioprine, and prednisone immunosuppression. Transplantation 49: 1011–1013. [DOI] [PubMed] [Google Scholar]
- Sliskovic DR, Picard JA, O'brien PM, Liao P, Roark WH, Roth BD et al (1998). Alpha‐substituted malonester amides: tools to define the relationship between ACAT inhibition and adrenal toxicity. J Med Chem 41: 682–690. [DOI] [PubMed] [Google Scholar]
- Sliskovic DR, Picard JA, Krause BR (2002). ACAT inhibitors: the search for a novel and effective treatment of hypercholesterolemia and atherosclerosis. Prog Med Chem 39: 121–171. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Luu‐The V, Labrie F (1990). Inhibitory effect of synthetic progestins, 4‐MA and cyanoketone on human placental 3 beta‐hydroxysteroid dehydrogenase/5‐4‐ene‐isomerase activity. J Steroid Biochem Mol Biol 37: 231–236. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Terasawa T, Hagihara H, Ishibe N, Sawada M, Sakuma Y et al (1998). Inhibitors of acyl‐CoA:cholesterol O‐acyltransferase. 3. Discovery of a novel series of N‐alkyl‐N‐[(fluorophenoxy)benzyl]‐N′‐arylureas with weak toxicological effects on adrenal glands. J Med Chem 41: 4408–4420. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Gregoire J, L'allier PL, Anderson TJ, Bertrand O, Reeves F et al (2004). Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation 110: 3372–3377. [DOI] [PubMed] [Google Scholar]
- Tarling EJ, Edwards PA (2011). ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci U S A 108: 19719–19724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessner TG, Stenson WF (2000). Overexpression of MDR1 in an intestinal cell line results in increased cholesterol uptake from micelles. Biochem Biophys Res Commun 267: 565–571. [DOI] [PubMed] [Google Scholar]
- Trivedi BK, Holmes A, Stoeber TL, Blankley CJ, Roark WH, Picard JA et al (1993). Inhibitors of acyl‐Coa:cholesterol acyltransferase. 4. A novel series of urea ACAT inhibitors as potential hypocholesterolemic agents. J Med Chem 36: 3300–3307. [DOI] [PubMed] [Google Scholar]
- Trivedi BK, Purchase TS, Holmes A, Augelli‐Szafran CE, Essenburg AD, Hamelehle KL et al (1994). Inhibitors of acyl‐CoA:cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N‐phenyl‐N'‐[(1‐phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity. J Med Chem 37: 1652–1659. [DOI] [PubMed] [Google Scholar]
- Vanier MT (2010). Niemann‐Pick disease type C. Orphanet J Rare Dis 5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH et al (2007). Macrophage ABCA1 and ABCG1, but not SR‐BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117: 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang GH, Macdonald JR, Vernetti LA, Pegg DG, Robertson DG (1995). Biochemical alterations in guinea pig adrenal cortex following administration of PD 132301‐2, an inhibitor of acyl‐CoA:cholesterol acyltransferase. Life Sci 56: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Wollam J, Antebi A (2011). Sterol regulation of metabolism, homeostasis, and development. Annu Rev Biochem 80: 885–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Iwamoto N, Rogers MA, Abe‐Dohmae S, Fujimoto T, Chang CC et al (2015). Deficiency in the lipid exporter ABCA1 impairs retrograde sterol movement and disrupts sterol sensing at the endoplasmic reticulum. J Biol Chem 290: 23464–23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M (2005). Altered cholesterol metabolism in Niemann–Pick type C1 mouse brains affects mitochondrial function. J Biol Chem 280: 11731–11739. [DOI] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA et al (2016). Comprehensive Pan‐Genomic characterization of adrenocortical carcinoma. Cancer Cell 29: 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of ATR‐101 compared with PD129337 on the cholesterol levels, ATP levels, caspase 3/7 activities, and cholesterol esterification, at different times in H295R cells.
Figure S2 Effects of ATR‐101 in combination with MβCD and of or with exogenous cholesterol in on cholesterol levels, ATP levels, caspase 3/7 activities, and cholesterol crystallization.
Figure S3 Effects of ATR‐101 and of ABC transporter inhibitors on the cholesterol levels of H295R cells that were cultured in serum‐free medium for 4 h.
Figure S4 Effects of ATR‐101 and of MDR1 inhibitors on cortisol secretion and doxorubicin accumulation.
Figure S5 Combined effects of ABC transporter inhibitors with each other and with ATR‐101 on the ATP levels of H295R cells.
Figure S6 Effects of ATR‐101 in combination with inhibitors of steroidogenesis on the ATP levels of H295R cells.
Figure S7 Effects of ATR‐101 compared with PD129337 on transcript levels in H295R cells.
Table S1 Primer sequences used for qPCR.