

Abstract
Dianxianning (DXN) is a traditional Chinese formula, and has been approved in China for treating epilepsy since 1996. Here anti-Alzheimer’s disease activity of DXN has been reported. DXN improved AD-like symptoms of paralysis and 5-HT sensitivity of transgenic Aβ1-42 C. elegans. In worms, DXN significantly increased Aβ monomers and decreased the toxic Aβ oligomers, thus reducing Aβ toxicity. DXN significantly suppressed the expression of hsp-16.2 induced by juglone, and up-regulated sod-3 expression. These results indicated that DXN increased stress resistance and protected C. elegans against oxidative stress. Furthermore, DXN could significantly promote DAF-16 nuclear translocation, but it did not activate SKN-1. The inhibitory effect of DXN on the Aβ toxicity was significantly reverted by daf-16 RNAi, rather than skn-1 RNAi or hsf-1 RNAi. These results indicated that DAF-16 is at least partially required for the anti-AD effect of DXN. In conclusion, DXN improved Aβ-induced pathological characteristics partially through DAF-2/DAF-16 insulin like pathway in transgenic worms. Together with our data obtained by Morris water maze test, the results showed that DXN markedly ameliorated cognitive performance impairment induced by scopolamine in mice. All the results support that DXN is a potential drug candidate to treat Alzheimer’s diseases.
Introduction
Alzheimer’s disease is a progressive neurodegenerative disorder which is becoming more prevalent in ageing populations worldwide. It is neuropathologically characterized by extensive neuronal loss and the presence of neurofibrillary tangles and senile plaques1. Nowadays over 46 million people suffer from AD in the world, and it is estimated to increase to 131.5 million by 20502. So far, there are only five FDA-approved anti-AD drugs on the market. Unfortunately, these drugs can only delay the onset of dementia, and none of them can halt or reverse the disease. Those medications for treating AD have never met the medical need, it is urgent to find effective and safe drugs against AD.
A lot of evidence have shown that the pathological hallmarks of AD are included Aβ aggregation and deposition with senile plaque appearance, tau hyper phosphorylation with tangle formation, which destroy synapses, induce brain inflammation, eventually lead neuronal death and severe brain shrinkage3. Among them, the Aβ is generated in brain from the amyloid precursor protein (APP) after sequential proteolytic cleavage by β-secretase and γ-secretase enzymes4. Under normal conditions, the produced Aβ is quickly removed from the brain. However, the mutations that increase Aβ production and the factors that decrease Aβ clearance or enhance Aβ aggregation will lead to Aβ self-aggregates into assemblies ranging from oligomers to protofibrils, fibrils and amyloid plaques5. Aβ cascade hypothesis of AD is widely accepted6. Accordingly, the anti-AD therapies primarily aim to lower the level of Aβ generation, aggregation or accumulation in the brain of AD patients. However, many of such drug candidates were demonstrated to manifest only limited benefit in AD patients. The reason is mainly explained by that other complex pathogenic cascades of related Aβ induced toxicity, are not affected, thereby the overall effects of those therapies being negligible4, 7. Obviously, more factors of the pathogenic cascades need to be considered in the process of anti-AD drug discovery.
Previous studies have indicated that AD patients have an increased risk of developing epileptic seizures, and the risk of epileptic activity is highest in those patients with early-onset dementia who over-express amyloid precursor protein (APP) and Aβ6. Epidemiological data indicates that patients with AD and seizure disorders have greater cognitive impairment, faster progression of symptoms, and more severe neuronal loss at autopsy than those without seizures8. There is a close relationship between epilepsy and AD, and Aβ has been identified to be the link between these two disorders9. In fact, antiepileptic drugs have been involved in preclinical studies or clinical trials for treating AD. Levetiracetam can suppress neuronal network dysfunction and reverse AD damage and cognitive impairment in mice6. Lamotrigine can attenuate deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice and effectively promote their learning memory behavior10. Valproic acid can inhibit Aβ generation, neuritic plaque production, ameliorate cognitive performance in AD mice, but it is unlikely to affect patient cognitive function11, 12. Carbamazepine and phenytoin can even impair patient cognitive function11. Anyhow, repurposing antiepileptic drugs is still available to discover potential anti-AD drug candidates.
The Dianxian Ning (DXN) has been approved in China Food and Drug Administration as an antiepileptic drug since 1996. DXN consists of eight Chinese medicine herbs, namely Valeriana jatamansi Rhizoma et Radix, Rhizoma Acori tatarinowii, Ramulus Uncariae cum Uncis, Semen Pharbitidis, Semen Euphorbiae, Radix et Rhizoma Valeriana officinalis, Rhizoma et Radix Nardostachys, and menthol crystal13. According to the theory of traditional Chinese medicine (TCM), DXN can clinically eliminate phlegm for resuscitation, stop wind, and calm the spirit13. In fact, TCM similarly function on a multi-targeted manner to treat the symptoms. Previous work reported that Valeriana jatamansi Rhizoma et Radix possesses anti-inflammatory, anti-oxidant activities14, 15. Rhizoma Acori Tatarinowii can exert neuroprotective action to battle against AD1. Ramulus Uncariae cum Uncis can inhibit Aβ formation, and compounds extracted from this herb have neuroprotective activity16, 17. However, how those eight herb components in one formula of DXN work together, and whether DXN can act as a complete formula or to be reduced, or as a resource of any effective compound for treating AD remains unclear.
Caenorhabditis elegans is an inexpensive tool to be widely used to evaluate anti-AD drug candidates based on Aβ hypothesis18. In this study, we investigated the effect of DXN on Aβ-induced injury using transgenic C. elegans model which exhibits several pathological behaviors due to Aβ toxicity. We also examined whether DXN can improve cognitive performance impairments induced by scopolamine in mice by Morris water maze test. Finally, we explored the underlying mechanism that DXN exerted its possible anti-AD action. Our results provide evidences for that DXN as a traditional Chinese anti-epileptic medicine is likely to be a potential drug candidate to fight against AD. Also, our results provide clues to introduce anti-epileptic TCM as potential drug candidate discovery resources for treating AD.
Results
DXN ameliorated AD-like symptoms induced by Aβ1-42 expression
To determine whether the DXN can protect against the toxicity induced by Aβ1-42 expression, transgenic C. elegans smg-1 (cc546)I temperature sensitive Aβ strain CL4176 was used, and human Aβ1-42 peptide has been transferred into worms and controlled by the promoter of myo-3, thus the over expression of Aβ1-42 in the muscle cells can induce AD-like symptom of Aβ-dependent paralysis. We found that DXN significantly delayed worm paralysis in a dose-dependent manner (Fig. 1A).
Figure 1.

The effects of DXN on AD-like symptoms in Aβ transgenic C. elegans. (A) DXN delayed AD worm paralysis induced by Aβ over-expression in muscle tissues in smg-1 (cc546)I temperature sensitive Aβ strain CL4176. Data are the average of three replicates with about 180 worms in each group. ***Indicated that there was significant difference between treatment group and control group at P < 0.001. (B) DXN ameliorated 5-HT hypersensitivity induced by Aβ over-expression in nerve cells in snb-1/Aβ1-42 strain CL2355. Worm strain CL2122 without Aβ expression in nerve cells was used as a transgenic control. Data are the average of three replicates with about 90 worms in each group. There is significant difference among these groups when symbols are different (P < 0.05).
Serotonin (5-HT) is an important neurotransmitter that regulates worm behaviors of locomotion, egg-laying, olfactory learning and mating19–21. Exogenous 5-HT can inhibit C. elegans locomotion, leading active worm into paralysis. C. elegans snb-1/Aβ1-42 strain CL2355 shows serotonin hypersensitivity when Aβ are over-expressed in nerve system22, 23. Here we investigated whether DXN treatment could reverse serotonin hypersensitivity induced by the toxicity of Aβ to nerve cells in the transgenic snb-1/Aβ1-42 worms. Our results showed that snb-1/Aβ1-42 worms were hypersensitive to exogenous 5-HT, but CL2122 without Aβ expression in nerve cells were not. DXN significantly alleviated the paralysis symptom of hypersensitive response to 5-HT in a dose-dependent manner (Fig. 1B).
Scopolamine is an anti-cholinergic agent to induce aspects of the memory loss and cognitive impairment, which resemble the hallmark symptoms observed in AD associated with cholinergic dysfunction at least on some degree. The scopolamine model has been widely used for screening possible AD symptom therapeutic agents to improve cognitive performances24. In present work, this model was used to evaluate the effects of DXN on learning and memory impairment by the Morris water maze test. Compared with the normal control group, the memory training results indicated that the mean escape latencies were significantly lengthened in scopolamine treated mice on the 5th day of the experiment. DXN improved learning and memory performance of mice as piracetam (Table 1). In probe test, the swimming time in the target quadrant of DXN treated animals significantly lengthened in contrast to the model group (Fig. 2A). The platform frequency remarkably increased in DXN and piracetam treated groups (Fig. 2B). Compared to model group, the length of swimming path between drug treatment groups and normal control group were not significant in probe test (Fig. 2C). These results suggested that DXN indeed promoted learning and memory performance in scopolamine treated mice.
Table 1.
Comparison of average escape latency time for each group in the hidden platform trial.
| Treatment | Escape latency time/second | ||||
|---|---|---|---|---|---|
| 1st day | 2nd day | 3rd day | 4th day | 5th day | |
| Control | 38.37 ± 3.25 | 33.60 ± 4.45 | 29.76 ± 2.96 | 21.36 ± 4.19 | 16.54 ± 2.72# |
| Model | 45.06 ± 1.90 | 42.64 ± 1.87 | 32.68 ± 3.93 | 33.28 ± 2.72 | 34.18 ± 2.68^ |
| 0.5 g/kg Piracetam | 41.01 ± 2.62 | 33.02 ± 3.04 | 29.83 ± 2.79 | 28.06 ± 3.33 | 21.07 ± 2.61#& |
| 0.39 g/kg-DXN | 35.77 ± 3.65 | 31.08 ± 3.63 | 33.11 ± 3.51 | 27.61 ± 3.63 | 25.75 ± 3.22& |
| 2.35 g/kg-DXN | 43.86 ± 3.29 | 35.43 ± 3.08 | 34.29 ± 3.66 | 27.87 ± 3.05 | 20.71 ± 3.14#& |
Data are the average of 14 animals in each group. There is significant difference among these groups when symbols are different (P < 0.05).
Figure 2.

The effects of DXN on cognitive impairment in mice induced by scopolamine in Morris water maze test (MWM). (A) The mean time in probe trial of MWM. (B) The mean platform frequency in probe trial of MWM. (C) The swimming path length in probe trial of MWM. Data are the average of 14 animals in each group. There is significant difference among these groups when symbols are different (P < 0.05).
DXN exerted anti-AD like action by its principal component Valeriana jatamansi Rhizoma et Radix
DXN is a kind of traditional Chinese medicine, and it is composed of eight herbs. Previous works have suggested that three herbs of Valerianae jatamansi Rhizoma et Radix (Vj), Rhizoma Acori tatarinowii (Ra) and Ramulus Uncariae cum Uncis (Ru) in DXN have anti-AD activity1, 14–17. In present work, Vj, Ra and Ru at an equivalent dose in DXN were firstly used to tested whether they can ameliorate AD-like symptom of delaying paralysis in worms. We further tried to investigate that whether DXN can be reduced to one or more herbs which can function as the principal component, and finally some effective compounds based anti-Aβ therapy for treating AD can be extracted from the right principal herbs.
The results showed that Vj delayed worm paralysis as DXN complete formula to a similar degree. Ra and Ru significantly alleviated the AD-like symptom in worms to a much lesser degree (Fig. 3A). These results supported that Vj was the principal component of DXN. It is deserved to notice that Vj alone also significantly shortened worm body length, but DXN did not (Fig. 3B). In contrast to drug treatment from eggs, worms were administrated with DXN or Vj when incubation temperature was upshifted to induce Aβ expression, and at that time animals were on the same development stage. The results showed that DXN and Vj did delay worm paralysis, but to a less degree (Fig. 3C). Of course, shorter dosing time and less DXN and Vj entry into worms led the decline in anti-AD activity of DXN and Vj for delaying paralysis. Anyway, the effect of Vj largely decreased than that of DXN did (Fig. 3A,C). Taken together, those results supported that complete DXN was a rational formula as it is and it should not be reduced. Additionally, transgenic worm smg-1[myo-3::GFP] was further used to test whether DXN treatment non-specifically affected gene expressions, the results showed that neither DXN nor Vj treatment altered exogenous gfp gene expression (Fig. 3D,E), suggesting that DXN and Vj treatment possible inhibiting non-specific protein expression could be rule out.
Figure 3.

DXN and its principal component Valeriana jatamansi (Vj) delayed worm paralysis similarly, but it could not be reduced due to its safety. (A) Paralysis assays for the temperature sensitive Aβ strain CL4176 treated with DXN or other herbs of Ra, Ru and Vj. Data are the average of three replicates with about 180 worms in each group. ***Indicated that there was significant difference between treatment group and control group at P < 0.001. (B) The effects of DXN and Vj on the body length of AD worms tested. The concentration of herb of Ra, Ru and Vj was equivalent to each of them containing in DXN. There is significant difference among these groups when symbols are different (P < 0.05). (C) Paralysis assays for the temperature sensitive Aβ strain CL4176 treated with DXN from the onset of paralysis. Data are the average of three replicates with about 180 worms in each group. ***Indicated that there was significant difference between treatment group and control group at P < 0.001. (D) The effect of DXN on Vj on GFP expression in smg-1[myo-3p::GFP] worms. (E) The relative mean fluorescence intensity of worms treated with or without drugs. Data are the average of three replicates with about 90 worms in each group. The scale bar was 40 μm.
Iridoids and sesquiterpenoid derived from Vj has been reported to have neuroprotective activity25. Here, we further evaluated compound 1–15 extracted from Vj (Fig. 4), and treated worms with each compound at relative final concentration due to their different maximum saturated solubility in DMSO. In contrast to Vj, almost all of them delayed worm paralysis induced by over-expression of Aβ to a much lesser degree, except that 100 μM of compound 15 did not have any anti-AD activity, 250 μM of compound 11 significantly promoted worm paralysis (Fig. 5). It is deserved to notice that compound 6 is baldrinal, which is regarded as the marker substance of Vj and DXN, and its content is 0.930 mg per gram of DXN or Vj coarse powder. In Fig. 5, 160 µM baldrinal was at a dosage higher by 2.6-fold than that contained in Vj used. Previous work showed that an iridoid, 10-O-trans-p-coumaroylcatalpol at similar dose or less significantly inhibits α-synuclein aggregation in worms26. So that, it cannot be excluded that more active compound will be isolated from Vj in the future work. Because relative large amount of baldrinal is contained in Vj, its anti-AD activity is still dissecting in our lab. At present, it is reasonable to believe that DXN needs complete Vj.
Figure 4.

Compounds of 1–15 extracted from Valeriana jatamansi 1: 8-hydroxyl-pathcouli alcohol; 2: 8-acetoxyl-pathchouli alcohol; 3: Ursolic acid; 4: 3β- Hydroxy-5α,8α-epidioxyergosta-6,22-diene; 5: stigmast-5-ene-3β-ylformate; 6: baldrinal; 7: (-)-bornyl ferulate; 8: vanillin; 9: (3S, 4S, 5S, -7S, 8S, 9S)-3,8-ethoxy-7-dihydroxy-4,8- dimethylperhydro cyclopenta - [c] pyran; 10: valerol A; 11: salicylic acid ;12: (4β,8β)-8-ethoxy-3-methyl -10- methylen-2,9-dioxatricyclo[4.3.1.03,7]decan-4-ol; 13: Valeriananoids D; 14: 5- (hydroxymethyl) - 2-furfuraldehyde; 15: 8-methylvalepotriate.
Figure 5.

The effects of compound 1–15 extracted from DXN on AD-like symptoms in the temperature sensitive Aβ worms. Data are the average of three replicates with about 180 worms in each group. ***Indicated that there was significant difference between treatment group and control group at P < 0.001.
DXN reduced Aβ oligomers rather than amyloid deposits in transgenic C. elegans
To explore whether the DXN affects the aggregation of the large Aβ species, unc-54/human Aβ1-42 worm strain CL2006 that constituently over expresses human Aβ1-42 in muscle cells was used. Although memantine is clinically used as a selected NMDA receptor antagonist, evidence recently indicates that it lowers the Aβ level and decreases Aβ plaques to support memantine may be a disease-modifying drug for treating AD27–30. Therefore, memantine has been used as a positive control. We found the DXN had no remarkable effect on the deposits of the Aβ1-42 peptide, suggesting that the DXN could not decrease the aggregation of the Aβ species significantly (Fig. 6A,B). As expected, memantine significantly lowered Aβ deposits (Fig. 6A,B). Considering the current evidence shows that soluble Aβ oligomers, rather than senile plaques (SPs) or Aβ fibrils, are the toxic species to link to neurodegeneration and cognitive decline31, 32, we performed the Western blotting analyses to test whether the DXN can reduce the toxic Aβ oligomers.
Figure 6.

The effect of DXN on Aβ deposits or Aβ oligomers in AD worms. (A) ThS staining images of Aβ deposits in unc-54/human Aβ1-42 worms after treatment with or without 15 mg/mL DXN, and 10 mM memantine (MEM) was used as positive control. Wild type N2 was used as ThS Aβ-specific staining control. The scale bar was 20 μm. Data are the average of three replicates with about 24 worms in each group. (B) Quantification of ThS-positive particles in head region of unc-54/human Aβ1-42 worms. There is significant difference among these groups when symbols are different (P < 0.05). (C) Western blot of Aβ species in the temperature sensitive Aβ strain CL4176 treated with or without 15 mg/mL DXN. (D) Band intensity of 4 KD Aβ monomers or 20 KD oligomers in C were analyzed by Image J software. Data are the average of three replicates with about 400–500 worms in each group.
Primary antibody of 6E10 against Aβ was used in western blotting analysis, and the relative densities of Aβ monomer of 4 kDa band and higher molecular weight oligomer of 20 kDa band were quantified22. The result showed that DXN significantly reduced the level of Aβ oligomers and increased the level of Aβ monomers (Fig. 6C,D). It indicated that DXN could significantly change the Aβ oligomers with high neurotoxicity into Aβ monomers, which is much less toxic than Aβ oligomers.
DXN delaying paralysis of AD worms was partially mediated by DAF-16 activation
DAF-16 is a crucial component of DAF-2 insulin like pathway to regulate many gene expressions for defending against various stresses including oxidative stress and proteotoxicity33, its activation has been proved to be benefit for delaying paralysis induced by human Aβ excessive expression in the muscle in C. elegans 34. To examine whether DAF-16 is required for DXN protecting worms from severe stress of aberrant Aβ generation and accumulation, we knocked down expression of the transcription factor DAF-16 by RNAi, and found that the inhibitory effect of DXN on worm paralysis significantly decreased (Fig. 7A). We further constructed a daf-16 mutant strain under genetic background of the temperature sensitive Aβ strain CL4176 by standard genetic crosses. Treating the AD worms with daf-16 mutation, a similar result has been obtained (Fig. 7B). These results indicated that DXN reduced Aβ toxicity at least partially through a DAF-16-based mechanism. After DXN treatment, DAF-16 was indeed activated and displayed nuclear translocation from the cytosol to the nucleus (Fig. 7C,D). Further, its typical downstream target SOD-3 was markedly up-regulated (Fig. 8A,B). According to our knowledge, DAF-16 activation increases stress resistance and then leads to lengthen lifespan. Expectedly, the result of lifespan assay on unc-54/human Aβ1-42 worm strain CL2006 treated with DXN showed that DXN did significantly extend worm lifespan (Fig. 3S).
Figure 7.

DXN inhibiting Aβ toxicity was at least partially mediated by DAF-16. (A) DXN inhibiting worm paralysis induced by Aβ over-expression was partially reverted by daf-16 RNAi. (B) The effect of DXN was similar partially reverted by daf-16 deletion. (C) Pdaf-16::gfp reporter was induced in worm strain TJ356 after treatment with 15 mg/mL DXN, and heat shock treatment at 37 °C for 15 min as a positive control. The scale bar was 80 μm. (D) Quantification of DAF-16 nuclear localization. In A and B, data are the average of three replicates with about 120-180 worms in each group. In C and D, animals were about 90 individuals. ***Indicated that there was significant difference between treatment group and control group at P < 0.001. There is significant difference among these groups when symbols are different (P < 0.05).
Figure 8.

SOD-3 was required for DXN inhibiting Aβ toxicity. (A) sod-3 expression was up-regulated after 15 mg/mL DXN treatment, and worms were treated with 20 mM juglone as positive control. The scale bar was 40 μm. (B) Quantification of sod-3 expression. (C) 15 mg/mL DXN inhibited the expression of Phsp-16.2::GFP induced by 40 μM juglone, and worms treated with 20 mM juglone were used as positive control. The scale bar was 25 μm. (D) Quantification of the expression of hsp-16.2. In (A–D), data are the average of three replicates with about 90 worms in each group. There is significant difference among these groups when symbols are different (P < 0.05). (E) The effects of sod-3 RNAi and hsp16.2 RNAi on DXN inhibiting worm paralysis induced by Aβ1-42 over-expression. Data are the average of three replicates with about 120–180 worms in each group. ***Indicated that there was significant difference between treatment groups and control group at P < 0.001.
As a chaperon, HSP-16.2 can be induced by the abnormal Aβ proteins to increase their sequestration, degradation, and refolding35. In our experiment, the expression of hsp-16.2 was examined after DXN pretreatment by using TJ375. The transgenic worm strain TJ375 has a GFP reporter gene controlled by the promoter of hsp-16.2, it exhibits an excessive GFP expression in the pharynx after exposure to oxidative stress such as juglone. The results showed that DXN alone did not significantly affect the hsp-16.2::gfp expression. However, when C. elegans were pretreated with DXN and further administrated with juglone, the fluorescence density in the pharynx of worms under juglone-induced oxidative stress significantly decreased compared to the DXN untreated group (Fig. 8C,D). We further treated AD worms with RNAi for sod-3 and hsp-16.2 (Fig. 8E). The results showed that sod-3 RNAi significantly promoted worm paralysis suggesting that SOD-3 is required for battling against Aβ toxicity. It is seemed to find that DXN delayed AD-like symptoms to the same degree as DXN treatment alone without sod-3 RNAi, but it did not indicate that SOD-3 was not required for DXN-mediated neuroprotection. Possibly, the compensatory upregulation of other sod genes or related detoxifying signaling pathway counteracted the effect of sod-3 RNAi on DXN-mediated delaying worm paralysis36. As for hsp16.2 RNAi, no obvious effect was observed. It might be explained by that hsp-16.2 is only expressed under stress37.
Transcription factor SKN-1 modulates genes that fight against oxidative and xenobiotic stress and genes that regulate protein homeostasis33. After skn-1 RNAi, the Aβ-induced paralysis phenotype in the temperature sensitive Aβ strain CL4176 was still significantly delayed by DXN treatment (Fig. 1S-A), indicating that SKN-1 was not involved in the protective effect of DXN. It was further validated by SKN-1 did not appear nuclear translocation at all (Fig. 1S-B).
HSF-1 regulates many gene expressions in response to stress38, and has been recognized to control disaggregation and degradation of Aβ34. In present work, after hsf-1 RNAi, the Aβ-induced paralysis phenotype in the temperature sensitive Aβ strain CL4176 was still significantly delayed by DXN treatment (Fig. 1S-C), indicating that HSF-1 was not involved in the protective effect of DXN. In present work, RNAi for hsf-1 and skn-1 significantly promoted worm thermal stress sensitivity (Fig. 1S-D). Therefore, the lack of effect of RNAi on worm paralysis after DXN treatment could not result from that the RNAi treatment did not work.
Discussion
DXN has been clinically used to treat epilepsy in China since 1990s. In present study, it strongly ameliorated Aβ-induced paralysis and 5-HT hypersensitivity in the transgenic C. elegans in a dose dependent manner (Fig. 1). Furthermore, DXN significantly alleviated learning and memory impairment induced by scopolamine in mice (Fig. 2). Those results indicated that DXN was potential to serve as a drug candidate for treating AD. In fact, we also tested the anti-AD activity of other two traditional Chinese formulas of Dianxianping tablet (DXP) and Guishaozhenxian tablet (GS), these two formulas also have been approved by CFDA for treating epilepsy. Our results showed that DXP and GS are less effective in comparison to DXN at their concentration available (Fig. 2S), and both of them could not be used on a larger dosage due to that they have severely delayed worm growth and development at their concentration in present work. Therefore, although there is close link between epilepsy and AD, whether an anti-epileptic drug can be used for treating AD still needs to be carefully investigated in details.
In present work, Vj has been proved to exert its delaying paralysis of AD-like symptom as a principal component (Fig. 3A). Further, compounds of 1–15 isolated from Vj significantly delayed worm paralysis and have anti-AD activity on a certain degree, except that compound 15 did not affect worm paralysis and compound 11 even aggravated AD-like symptom. In comparison with the complete herb of Vj, they only exhibited moderate protective effect (Fig. 5). Similarly, coffee extract can protect worms from Aβ toxicity, whereas caffeine does only to a much lesser degree19. Previously, Vj has been demonstrated to have anti-inflammatory and anti-oxidant activity14, 15. Several other compounds of iridoids, sesquiterpenoids extracted from this species and other related species of genus Valeriana have been shown to have neuroprotective effects25, 39. It is reasonable to conclude that these compounds can exert additive or synergistic effect for anti-AD. Of course, it cannot be excluded that more active compound will be isolated from Vj in the future work.
DXN, as a kind of traditional Chinese medicine, is composed of eight herbs. Among the herbs, besides Vj, Ra and Ru have anti-AD activity1, 14–17. Our results show that Ra and Ru did not ameliorate AD-like symptom in worms additively or synergistically (Fig. 3A), but complete DXN did not delay nematode growth any more (Fig. 3B), indicating that DXN is less toxic than Vj alone. It is especially important for a potential anti-AD drug to act effectively to be used just at asymptomatic and preclinical stage, and the disease manifests its clinical symptoms after a decade or more, thus anti-AD therapy being a long term prevention40. The present evidence supported that DXN could not be reduced. Further work needs to investigate on the actions of these herbs through a feasible AD model in vivo or in vitro based on other respects of the pathogenesis of this devastating disease.
After DXN treatment, Aβ oligomers significantly reduced, but Aβ monomers increased in AD worms (Fig. 6C,D). It is well recognized that Aβ oligomers induce the death of neurons and are responsible for AD-related memory loss. Moreover, Aβ oligomers are closely related to severity of dementia in AD patients1, 39. Accumulating evidence suggests that Aβ oligomers but not Aβ monomers or Aβ deposits are correlated with Aβ toxicity41, 42. Our results supported that DXN strongly ameliorating Aβ toxicity in the transgenic C. elegans overexpressed human Aβ1-42 might be a consequence of its directly or indirectly promoting the shift from the toxic Aβ oligomer to less-toxic monomer form.
Small HSPs are Low-molecular-weight Heat Shock Proteins (12–43 kD) in response to a series of injuries including thermal stress and oxidative stress43. Moreover, small HSPs also participate in preventing the accumulation of several different types of toxic proteins such as Aβ and ployQ44. HSP16.2 can suppress Aβ toxicity by assisting abnormal protein sequestration, degradation, and refolding in AD C. elegans 35. Here, DXN treatment did not increase the expression of hsp-16.2 (Fig. 8C,D), indicating that toxic Aβ oligomer reduction after DXN treatment was not mediated by molecular chaperon HSP-16.2. Then whether DXN can directly bind to Aβ proteins to inhibit their aggregation and increase their less toxic monomers needs further investigated. However, DXN counteracted the up-regulation of hsp-16.2 induced by juglone (Fig. 8C,D). Since juglone is a reactive oxygen species generator45, it gave us a notion that DXN could reduce Aβ toxicity through anti-oxidant activity. It agrees with the previous work that components of DXN, Vj and Rhizoma Acori tatarinowii possess high antioxidant activity14, 46, 47.
SOD-3 protects C. elegans from oxidative stress and participates in suppressing the Aβ toxicity48, 49. In present work, our results showed that DXN indeed promoted the expression of sod-3 (Fig. 8A,B). It supported that DXN increased organism stress resistance and armed C. elegans to defend against oxidative stress induced by aberrant Aβ proteins50. SOD-3 locates downstream of DAF-16 in C. elegans 51, 52. By turns, DAF-16 is a FOXO transcription factor and acts as a crucial component in the DAF-2/DAF-16 insulin like signaling pathway34, 53. The activation of DAF-16 will lead to elevated expression of genes in responses to a wide range of stressors54, 55. Our results showed that DXN could significantly activate DAF-16 to translocate from cytosol to nucleus (Fig. 7C,D), and DAF-16 is required for the delaying paralysis effect of DXN (Fig. 7A,B). HSF-1 and SKN-1 are transcriptional factors in parallel to DAF-16, which also can promote the expression of stress responsive genes to inhibit the toxicity of Aβ33, 34, 38. In our experiment, hsf-1 and skn-1 RNAi did not affect the DXN delaying paralysis of anti-AD action (Fig. 1SA,B). Additionally, SKN-1 did not exhibit nuclear translocation after DXN treatment (Fig. 1SC). In conclusion, as shown in Fig. 9, DXN suppressing Aβ-induced pathological behaviors was at least partially mediated by DAF-16 activation in worms. DXN also directly or indirectly reduced Aβ oligomers and increased less toxic Aβ monomers.
Figure 9.
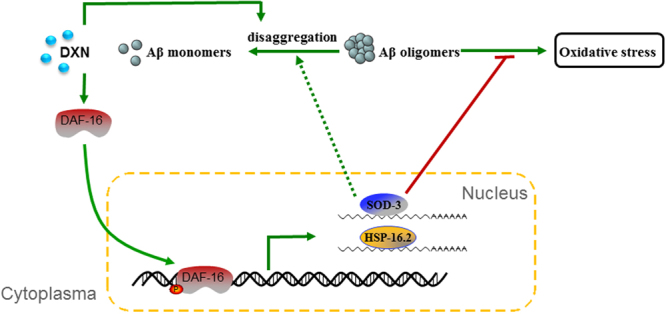
Schematic diagram of DXN action on suppressing Aβ toxicity. DXN directly or indirectly reduced Aβ oligomers and increased less toxic Aβ monomers, further, DXN induced DAF-16 activation to regulate its downstream stress responsive gene expressions, such as sod-3, hsp-16.2, at least partially to exert its anti-AD activity.
Materials and Methods
Preparation of DXN
DXN is composed of eight herb drugs at dose of 500 g Valerianae jatamansi Rhizoma et Radix (Vj), 200 g Semen Pharbitidis, 200 g Ramulus Uncariae cum Uncis (Ru), 500 g Rhizoma Acori tatarinowii (Ra), 200 g Rhizoma et Radix Nardostachyos, 15 g Semen Euphorbiae, 0.3 g Menthol, 0.62 g Radix et Rhizoma Valeriana officinalis. DXN was prepared according to WS3-B-2823-97 in Drug Standard issued by Ministry of public health, China (Fourteenth prescriptions of Chinese Medicine). Briefly, herb drugs were crushed into coarse powder and extracted by diacolation and decoction methods. Among them, 200 g Vj coarse powder without extraction and menthol were directly added into the mixtures of other herb drug extractions. Finally, the mixtures were crushed into 1000 tablets of DXN. Quality control of DXN was performed by HPLC56. Baldrinal was used as marker substance and its content was 0.930 mg/g DXN. Total iridoids and sesquiterpenoid were measured by ultraviolet spectrophotometry according to Li et al. (2016)57. Total iridoids and sesquiterpenoid were 22.7 mg/g DXN.
In present work, DXN was produced by Kunming Chinese Medicine Factory Co., Ltd. (License Number: H.M.L.N. Z53020771). It was pressed and grinded into powder with a pestle in a mortar, and then dissolved in sterile water, centrifuged at 10,000 rpm for 10 min, the resultant supernatant containing DXN at a concentration of 150 mg/mL as a stock solution was stored at 4 °C before use.
C. elegans strains and maintenance
The wild-type C. elegans N2, transgenic C. elegans CL2355 [snb-1/Aβ1-42/long 3′-UTR + mtl-2::GFP]; CL2122 dvIs15 [(pPD30.38) unc-54(vector) + (pCL26) mtl-2::GFP]; CL4176 smg-1(cc546)I; dvIs27[myo-3p::Aβ(1-42)::let-851 3′UTR) + rol-6(su1006)]X; CL2006, dvIs2 [pCL12 (unc-54/human Aβ peptide1-42 minigene) + pRF4]; CL2179, smg-1[myo-3::GFP]; TJ356 [zIs356 (Pdaf-16:: daf-16a/b::GFP + rol-6)]; TJ375[gpIs1(hsp-16.2::GFP)]; CF1553 [muIs84 ((pAD76) sod-3p::GFP + rol-6)]; LG333[geIs7(skn-1b::GFP)]; CF1038 [daf-16(mu86) I] were obtained from Caenorhabditis Genetics Center (CGC) (University of Minnesota, Minneapolis, MN). All worms were propagated at 20 °C except CL4176 at 16 °C on solid nematode growth medium (NGM) seeded with standard food resource of E. coli OP50.
Paralysis assay
Transgenic C. elegans of temperature sensitive Aβ strain (CL4176) maintained at 16 °C were egg-synchronized onto the NGM plates containing with 0, 1 mg/mL, 5 mg/mL, 15 mg/mL DXN. Other principal herbs of Vj, Ra and Ru were used at an equivalent dose as they were in DXN, and 3.2 mg/mL Vj, 3.7 mg/mL Ra and 0.6 mg/mL Ru was used, respectively. For compounds of 1–15 extracted from Vj, they were all diluted at indicated final concentrations in 0.1%DMSO according to their maximum saturated solubility. Compounds of 1–7 were relatively used at a final concentration of 50 μM, 100 μM, 20 μM, 20 μM, 20 μM, 160 μM, 120 μM, compounds of 8–11 were used at 250 μM and compounds of 12–15 were used at 100 μM. Worms were induced to express human Aβ1-42 till they were at L3 stage larvae by up-shifting the incubation temperature from 16 °C to 25 °C for 34 h. The transgenic worms were scored at 2 h intervals for paralysis till all animals in negative control group were paralyzed. Each worm was gently touched with a platinum loop to identify as the paralysis, nematodes were considered to be paralyzed if they did not move at all or only moved their heads58.
Fluorescence staining of Aβ deposits
Transgenic C. elegans with unc-54/human Aβ1-42 maintained at 20 °C were egg-synchronized onto NGM plates and grown until the L4 stage, worms were then moved to fresh NGM plates containing 0, 1 mg/mL, 5 mg/mL, 15 mg/mL DXN. Two days later, Thioflavine-S (ThS) staining was performed as described previously22. Briefly, worms were collected by washing with M9 and were fixed in 4% paraformaldehyde in PBS, pH7.4, at 4 °C for 24 h. The fixative solution was replaced by permeabilization solution (5% fresh β-mercaptoethanol, 1% Triton X-100, 125 mM Tris-HCl, pH7.4), and incubated in a 37 °C incubator for another 24 h. The animals were washed three times in PBS-T (PBS plus 0.1% Triton X-100), stained with 0.125% ThS (Sigma) in 50% ethanol for 2 min, and the samples were destained with 50% ethanol twice for 2 min. Stained samples were resuspended in 100 μL PBS, and finally mounted and observed by fluorescence microscopy (BX53; Olympus, Japan). Amyloid deposits in the anterior area of each animal were quantified by scoring the number of ThS reactive deposits. C. elegans N2 were used as amyloid deposit control.
Exogenous serotonin sensitivity assay
After egg-synchronized, the transgenic snb-1/Aβ1-42 worm were placed at 20 °C on NGM plates seeded with OP50 for 48 h. The worms were treated with 0, 1 mg/mL, 5 mg/mL, 15 mg/mL DXN for another 48 h, respectively. 30 worms in each group were washed with M9 buffer for three times and were transferred into 200 μL M9 buffer containing 1 mg serotonin (Sigma), paralyzed worms were scored after 5 min. Animals were considered to be paralyzed if they did not move at all within 5 sec. Worm strain CL2122 without Aβ expression in nerve cells was used as a transgenic control.
Western blotting
Human Aβ1-42 in the temperature sensitive Aβ strain (CL4176) was identified by immunoblotting on a Tris-Tricine gel by the standard Western blotting assay. Wild type N2 was used as negative control of the expression of Aβ. Worms were treated as paralysis assay with or without 15 mg/mL DXN. Nematodes were then washed three times in M9 to remove the bacteria after incubation temperature up-shifting for 40 h and finally harvested in distilled water with protease inhibitor cocktail (Sigma, P2714) added, quickly frozen in liquid nitrogen and stored at -80 °C. Worms were boiled in sample lysis buffer (1× protease inhibitor cocktail, 62 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 4% β-mercaptoethanol) for 10 min, then seated on ice to cool, centrifuged at 14,000 g for 5 min. Total protein content in the supernatant was measured by Lowry method. Proteins were boiled for 5 min in sample loading buffer (62 mM Tris-HCl, pH6.8, 2% SDS, 10% glycerol, 4% β-mercaptoethanol, 0.0005% bromophenol blue). Samples were performed electrophoresis on the Tris-Tricine gel with 70 μg total protein in each lane. After the gel was transferred to 0.22μm PVDF membrane, the membrane was boiled in PBS for 15 min before blocking with a solution of 5% milk in TBS-Tween (100 mM Tris-HCl, pH7.5, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature. Blotting was carried out overnight at 4 °C. Aβ proteins were detected by 6E10 (Biolegend, 803001) at 1:1000 dilution. Internal control of α-tubulin was probed by polyclonal antibody against it (Sigma, T6199) at 1:1000 dilution. Goat anti-mouse HRP-labelled IgG was used as a secondary antibody.
Nuclear localization of DAF-16
Worm daf-16::gfp (TJ356) expressing GFP as a reporter was used for detecting the intracellular distribution of DAF-1659. Synchronized L1 larvae worms were fed with or without 15 mg/mL DXN for 72 h. Subsequent to this treatment, worms were mounted on glass slides and their nuclear localization of DAF-16 were observed under a fluorescence microscope (BX53; Olympus, Japan). Worms are scored based on two categories of cytosolic and nuclear distribution with respect to the major localization of the DAF-16::GFP fusion protein. DAF-16 intracellular location was indicated by the ratio of nuclear translocation in the experimental population.
RNA interference (RNAi)
RNAi gene expression clones were constructed as described by Fraser et al.60. Briefly, PCR products were synthesized using Q5 High-Fidelity DNA Polymerase (New England Biolabs, USA) with genomic DNA as template by relative primers listed in Table 2, and then inserted into L4440 vector (Addgene plasmid 1654). Recombinant plasmids were subsequently transformed into the E. coli HT115 (DE3) bacterial strain using a standard method. The desired positive colonies were verified by sequencing. The sequences of target genes were available from the web site http://www.wormbase.org/#01-23-6.
Table 2.
List of primers for amplifying target genes.
| Gene | Primer name | Primer sequence | Restriction enzyme |
|---|---|---|---|
| daf-16 | daf-16-F | 5′ AAAACTGCAGAGTACAGCAATTCCCAAATGAAA 3′ | Pst I |
| daf-16-R | 5′ CCCAAGCTTAATTGGATTTCGAAGAAGTGGAT 3′ | Hind III | |
| skn-1 | skn-1-F | 5′ AAAACTGCAGGGGTACCCACTTGCCCTATT 3′ | Pst I |
| skn-1-R | 5′CCCAAGCTTTCATTTCAGCCACTCACTGC 3′ | Hind III | |
| hsf-1 | hsf-1-F | 5′AAAACTGCAGGAAAAAAAGTAGGAGCAAAAAAT3′ | Pst I |
| hsf-1-R | 5′CGGGGTACCAGTCAAAAAGCTGAAAAAATCGG3′ | Kpn I | |
| hsp16.2 | hsp16.2-F | 5′ CGGGGTACC ATTCAGCAGATTTCTCTTCGACGATT 3′ | Kpn I |
| hsp16.2-R | 5′ CCGCTCGAGTGTCACTTTACCACTATTTCCGTCC 3′ | Xho I | |
| sod-3 | sod-3-F | 5′ CGGGGTACCAGCTCCTTTTAAATTAAGACA 3′ | Kpn I |
| sod-3-R | 5′ CCGCTCGAGTATTCTTCCAGTTGGCAAT 3′ | Xho I |
HT115 (DE3) bacteria containing the target genes and expressing double stranded RNAs (dsRNA) that inactivate specified genes were used as previously described58, 61. Target gene expressions were knocked down by feeding the worms with dsRNA-containing E. coli HT115 strains from egg to adulthood at 16 °C. Except for skn-1, worms were exposed to RNAi bacteria from L3 stage larvae. The next generation of these worms were transferred to another RNAi plates and used for paralysis assay as above.
Quantification of Psod-3::gfp expression via fluorescence microscopy
The transgenic C. elegans strain CF1553 expressing GFP as a reporter for inducible sod-3 expression was used in our study. Age-synchronized L1 stage transgenic C. elegans were treated with or without 15 mg/mL DXN for 72 h, the expression of sod-3::gfp was evaluated through measuring the GFP fluorescence intensity of each whole worm. At least 25 randomly selected worms from each group were observed and photographed by fluorescence microscopy on a glass slide. To quantitatively measure the GFP fluorescence intensity in each whole worm, the Image J software (NIH, Bethesda, MD, USA) was used.
Quantification of Phsp-16.2::gfp expression via fluorescence microscopy
In the transgenic strain TJ375, GFP is fused to hsp-16.2 promoter for reporting the expression of HSP-16.2. Age-synchronized L1 stage Phsp-16.2::gfp worms were treated with or without 15 mg/mL DXN for 48 h, followed by exposure to 20 mM juglone for 24 h. The expression of hsp-16.2::gfp was evaluated through measuring the fluorescence intensity of the GFP reporter. At least 25 randomly selected worms from each group were measured by fluorescence microscopy on a glass slide. To quantitatively measure the GFP fluorescence intensity in the area anterior of the pharyngeal bulb in individual worms, Image J software (NIH, Bethesda, MD, USA) was used.
Morris water maze test
The Morris water maze test was performed as described by Morris (1981)62. The apparatus was a circular pool of 120 cm diameter filled with water, and maintained at 22.0 ± 0.5 °C by a calefaction stick. A transparent platform of 11 cm diameter 1 cm below the water surface was placed at a fixed point of one quadrant. 4-6 weeks old Kunming mice were provided by the Experiment Animal Center of Lanzhou University. Animals were treated with DXN at dose of 0.39 g/kg and 2.34 g/kg by i.g. for 21 days, mice in positive control group administrated piracetam at dose of 0.5 g/kg by i.g. Amnesia was induced by scopolamine at dose of 3 mg/kg by i.p. 30 min after DXN or piracetam administration. Animals in control group were only received normal saline by i.g.. One day prior to the experiment, animals were trained to swim for 60 sec in the absence of the platform. Mice were performed single trial per day for consecutive 5 days, and escape latency of the time mice taken to swim to the platform was recorded by an automated tracking system (Ethovision XT software). 24 h after the experiment, a probe trial was performed to test for spatial memory.
All experiments were approved by the Animal Care and Use Committee of Lanzhou University, and the methods were carried out in accordance to the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China.
Statistical analysis
Statistical analyses were performed by SPSS 17.0 software. The statistical significances of the results were analyzed by using one-way analysis of variance (ANOVA). Except for paralysis assays, log rank survival test was carried out to compare the significance level among treatments. The P value of 0.05 or less was considered to be significant statistically.
Electronic supplementary material
Acknowledgements
This work was supported by the Ministry of Science and Technology new drug project of MOST of China (2015ZX09501-004-003-008); National Natural Science Foundation of China (31560176; 81403145; 81560715); Fundamental Research Funds for the Central Universities of China (lzujbky-2017-206). Nematode strains used in this work were provided by CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Author Contributions
Dejuan Zhi designed the experiment and analysis the data, wrote the manuscript. Dong Wang prepared Figures and Table 1 except for Figures 3C-E, 4, 5, 6C-D, 8E, 1S-D, 3S. Wenqi Yang prepared Figures 3C-E, 6C-D, 8E, 1S-D, 3S. Hongyu Li, Shuqian Zhu, Xin Wang, Ningbo Wang, Juan Dong, Meizhu Wang corrected the manuscript. Ziyun Duan prepared Fig. 5. Hongyu Li put forward guideline opinions. Juan Dong prepared Figure 3S, Na Wang prepared RNAi clones of hsf-1, skn-1, daf-16, sod-3 and hsp-16.2. Dongqing Fei and Zhanxin Zhang prepared compounds 1–15 in Figure 4. All authors reviewed and considered the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11628-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hou YJ, et al. Smart soup, a traditional Chinese medicine formula, ameliorates amyloid pathology and related cognitive deficits. PloS one. 2014;9(11):e111215. doi: 10.1371/journal.pone.0111215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prince, M. et al. World Alzheimer Report 2015. The global impact of dementia. An analysis of prevalence, incidence, cost and trends. Alzheimer’s disease international, London (2015).
- 3.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 4.Mucke L. Neuroscience: Alzheimer’s disease. Nature. 2009;461:895–897. doi: 10.1038/461895a. [DOI] [PubMed] [Google Scholar]
- 5.Golde TE, Petrucelli L, Lewis J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010;223:252–266. doi: 10.1016/j.expneurol.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez PE, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bica L, Crouch PJ, Cappai R, White AR. Metallo-complex activation of neuroprotective signalling pathways as a therapeutic treatment for Alzheimer’s disease. Mol. BioSyst. 2009;5:134–142. doi: 10.1039/B816577G. [DOI] [PubMed] [Google Scholar]
- 8.Vossel KA, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop JJ, Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: two faces of the same coin? Neuromol. Med. 2010;12:48–55. doi: 10.1007/s12017-009-8097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang MY, et al. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging. 2014;35:2713–2725. doi: 10.1016/j.neurobiolaging.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Kwan P, Brodie MJ. Neuropsychological effects of epilepsy and antiepileptic drugs. Lancet. 2001;357:216–222. doi: 10.1016/S0140-6736(00)03600-X. [DOI] [PubMed] [Google Scholar]
- 12.Qing H, et al. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med. 2008;205:2781–2789. doi: 10.1084/jem.20081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He L, et al. Reevaluation of the effect of Dianxianning on seizure rate of refractory epilepsy as additive treatment in clinical practice. Front. Med. 2011;5:229–234. doi: 10.1007/s11684-011-0139-5. [DOI] [PubMed] [Google Scholar]
- 14.Jugran A, et al. Association of ISSR markers with some biochemical traits of Valeriana jatamansi Jones. Ind. Crop. Prod. 2013;44:671–676. doi: 10.1016/j.indcrop.2012.09.004. [DOI] [Google Scholar]
- 15.You JS, et al. Evaluation of anxiolytic activity of compound Valeriana jatamansi Jones in mice. BMC Complem. Altern. Med. 2012;12:1. doi: 10.1186/1472-6882-12-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujiwara H, et al. Uncaria rhynchophylla, a Chinese medicinal herb, has potent antiaggregation effects on Alzheimer’s beta-amyloid proteins. J. Neurosci. Res. 2006;84:427–433. doi: 10.1002/jnr.20891. [DOI] [PubMed] [Google Scholar]
- 17.Xian, Y. F. et al. Bioassay-guided isolation of neuroprotective compounds from Uncaria rhynchophylla against beta-amyloid-induced neurotoxicity. Evid-Based Compl. Alternat. Med. 2012 (2012). [DOI] [PMC free article] [PubMed]
- 18.Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown MK, Luo Y. Bilobalide modulates serotonin-controlled behaviors in the nematode Caenorhabditis elegans. BMC Neurosci. 2009;10:1. doi: 10.1186/1471-2202-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuttley WM, Atkinson-Leadbeater KP, Van Der Kooy D. Serotonin mediates food-odor associative learning in the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:12449–12454. doi: 10.1073/pnas.192101699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/S0896-6273(00)81199-X. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, et al. Amyloid-β-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 2006;26:13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arya U, Dwivedi H, Subramaniam JR. Reserpine ameliorates Aβ toxicity in the Alzheimer’s disease model in Caenorhabditis elegans. Exp. Gerontol. 2009;44:462–466. doi: 10.1016/j.exger.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Wesnes KA, et al. Cholinesterase inhibition in the scopolamine model of dementia. Ann. NY Acad. Sci. 1991;640(1):268–271. doi: 10.1111/j.1749-6632.1991.tb00231.x. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, et al. Iridoids from the roots of Valeriana jatamansi and their neuroprotective effects. Fitoterapia. 2011;82:1133–1136. doi: 10.1016/j.fitote.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 26.Shukla V, et al. Iridoid compound 10-O-trans-p-coumaroylcatalpol extends longevity and reduces alpha synuclein aggregation in Caenorhabditis elegans. CNS Neurol. Disord-DR. 2012;11(8):984–992. doi: 10.2174/1871527311211080007. [DOI] [PubMed] [Google Scholar]
- 27.Scholtzova H, Wadghiri YZ, Douadi M, et al. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer’s-disease-model transgenic mice shown as by micromagnetic resonance imaging. J. Neurosci. Res. 2008;86(12):2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alley GM, Bailey JA, Chen DM, et al. Memantine lowers amyloid-β peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J. Neurosci. Res. 2010;88(1):143–154. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Coria H, Green KN, Billings LM, et al. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am. J. Pathol. 2010;176(2):870–880. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu MY, Wang S, Yao WF, et al. Memantine improves spatial learning and memory impairments by regulating NGF signaling in APP/PS1 transgenic mice. Neurosci. 2014;273:141–151. doi: 10.1016/j.neuroscience.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Lacor PN, et al. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesné S, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 33.Murphy, C. T. & Hu, P. J. Insulin/insulin-like growth factor signaling in C. elegans, WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.164.1, http://www.wormbook.org (2013). [DOI] [PMC free article] [PubMed]
- 34.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 35.Fonte V, et al. Suppression of in vivo β-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J. Biol. Chem. 2008;283:784–791. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- 36.Back P, Matthijssens F, Vlaeminck C, Braeckman BP, Vanfleteren JR. Effects of sod gene overexpression and deletion mutation on the expression profiles of reporter genes of major detoxification pathways in Caenorhabditis elegans. Exp. Gerontol. 2010;45(7):603–610. doi: 10.1016/j.exger.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Leroux MR, Melki R, Gordon B, Batelier G, Candido EP. Structurefunction studies on small heat shock protein oligomeric assembly and interaction with unfolded polypeptides. J. Biol. Chem. 1997;272:24646–24656. doi: 10.1074/jbc.272.39.24646. [DOI] [PubMed] [Google Scholar]
- 38.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong F, et al. Iridoids and sesquiterpenoids of Valeriana stenoptera and their effects on NGF-induced neurite outgrowth in PC12 cells. Phytochemistry. 2015;118:51–60. doi: 10.1016/j.phytochem.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 40.Golde TE, Schneider LS, Koo EH. Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuevas ME, et al. Soluble Aβ1-40 Peptide Increases Excitatory Neurotransmission and Induces Epileptiform Activity in Hippocampal Neurons. J. Alzheimers Dis. 2011;23:673–687. doi: 10.3233/JAD-2011-091717. [DOI] [PubMed] [Google Scholar]
- 42.Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394:595–599. doi: 10.1038/29106. [DOI] [PubMed] [Google Scholar]
- 44.Edwards HV, Cameron RT, Baillie GS. The emerging role of HSP20 as a multifunctional protective agent. Cell. Signal. 2011;23:1447–1454. doi: 10.1016/j.cellsig.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Heidler T, Hartwig K, Daniel H, Wenzel U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11:183–195. doi: 10.1007/s10522-009-9239-x. [DOI] [PubMed] [Google Scholar]
- 46.Prasad R, et al. Valeriana jatamansi partially reverses liver cirrhosis and tissue hyperproliferative response in rat. Method. Find. Exp. Clin. 2010;32:713. doi: 10.1358/mf.2010.32.10.1522224. [DOI] [PubMed] [Google Scholar]
- 47.Zhang W, et al. Characterization of polysaccharides with antioxidant and immunological activities from Rhizoma Acori Tatarinowii. Carbohyd. Polym. 2015;133:154–162. doi: 10.1016/j.carbpol.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 48.Minniti AN, et al. The protein oxidation repair enzyme methionine sulfoxide reductase a modulates Aβ aggregation and toxicity in vivo. Antioxid. Redox Sig. 2015;22:48–62. doi: 10.1089/ars.2013.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science. 2000;290:147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- 50.Chauhan V, Chauhan A. Oxidative stress in Alzheimer’s disease. Pathophysiol. 2006;13:195–208. doi: 10.1016/j.pathophys.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Chávez V, Mohri-Shiomi A, Maadani A, Vega LA, Garsin DA. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics. 2007;176:1567–1577. doi: 10.1534/genetics.107.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McElwee J, Bubb K, Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell. 2003;2:111–121. doi: 10.1046/j.1474-9728.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- 53.Chen X, et al. Ethosuximide ameliorates neurodegenerative disease phenotypes by modulating DAF-16/FOXO target gene expression. Mol. Neurodegene. 2015;10:1–14. doi: 10.1186/1750-1326-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 55.Tullet JMA, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di H, et al. Determination of valtrate, acevaltrate, and their degradation product-baldrinal in Valeriana wallichii by HPLC analysis. Chinese Tradit. Herb. Drugs. 2007;12:046. [Google Scholar]
- 57.Li, P. et al. Content determination of total iridoid compounds and baldrinal and 11-ethoxyviburtinal from Valerianae Jatamansi Rhizoma et Radix. Chinese J. Inform. Tradit. Chinese Med. 88–91 (2016).
- 58.Dostal V, Roberts CM, Link CD. Genetic mechanisms of coffee extract protection in a Caenorhabditis elegans model of β-amyloid peptide toxicity. Genetics. 2010;186:857–866. doi: 10.1534/genetics.110.120436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henderson ST, Johnson TE. Daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001;11:1975–1980. doi: 10.1016/S0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 60.Fraser AG, et al. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- 61.Keowkase R, Aboukhatwa M, Luo Y. Fluoxetine protects against amyloid-beta toxicity, in part via daf-16 mediated cell signaling pathway, in Caenorhabditis elegans. Neuropharmacology. 2010;59:358–365. doi: 10.1016/j.neuropharm.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morris RGM. Spatial localization does not require the presence of local cues. Learn. Motiv. 1981;12:239–260. doi: 10.1016/0023-9690(81)90020-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.