

Abstract
Aim
Selective androgen receptor modulators (SARMs) induce anabolic effects on muscle without the adverse effects of androgenic steroids. In this first‐in‐human study, we report the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of the SARM GSK2881078.
Methods
In Part A, healthy young men (n = 10) received a single dose of study drug (0 mg, 0.05 mg, 0.1 mg, 0.2 mg GSK2881078 or matching‐placebo). In Part B, repeat‐dose cohorts in men (n = 65) were 0.05 mg, 0.2 mg then 0.08 mg, 0.24 mg, 0.48 mg, 0.75 mg, or placebo; in women (n = 24) they were 0.24 mg, 0.35 mg, or placebo (7 days for 0.5 mg, 14 days for other doses).
Results
PK analysis showed dose‐proportional increases in exposure and a long >100‐h half‐life. No significant effects on vital signs, electrocardiograms, cardiac telemetry or standard clinical laboratory studies were observed. A dose–response effect was observed on lowering both high‐density lipoprotein and sex hormone‐binding globulin. In females at 0.35 mg, differences from placebo were −0.518 (95% confidence interval: –0.703, −0.334) mmol l–1 and −39.1 (−48.5, −29.7) nmol l–1, respectively. Women showed greater sensitivity to these parameters at lower doses than men. Drug‐related adverse events (AEs) were mild. One woman developed a drug rash and was withdrawn. Two men had elevated creatine phosphokinase after physical exertion during follow‐up. A serious AE occurred in a subject on placebo.
Conclusions
These data demonstrate pharmacodynamic effects with acceptable tolerability and support further clinical evaluation of this SARM.
Keywords: biomarkers, drug development, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
Selective androgen receptor modulators (SARMs) can increase muscle strength without the side effects of testosterone, which can be a useful treatment for muscle‐wasting diseases.
Although some SARMs are in development, no SARM has been approved for clinical use by the United States Food and Drug Administration.
What this Study Adds
The novel SARM GSK2881078 is well tolerated, with expected pharmacodynamic effects (e.g. lowering of high‐density lipoprotein and of sex hormone‐binding globulin in men and women, and of testosterone in men).
The results support further development of this new compound.
Tables of Links
| TARGETS |
|---|
| Nuclear hormone receptors 2 |
| Androgen receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
One of the debilitating consequences of chronic illness is the impairment of physical function due to muscle wasting, which also often accompanies advanced age. Sarcopenia, defined as a marked loss of muscle mass and strength due to ageing, is associated with impaired physical function and higher rates of institutionalization, mortality and diminished quality of life 3, 4, 5, 6, 7. Similarly, cachexia describes the accelerated loss of body weight, which often includes loss of muscle mass, that occurs in persons with acute or chronic illness including cancer, heart failure, chronic obstructive pulmonary disease, kidney disease, arthritis, burn injury and acquired immune deficiency syndrome 8.
Physical therapy and exercise are the current standards of treatment for muscle wasting, and these have been shown to be effective for increasing muscle mass and improving physical function 9. However, to date there is no medication approved by either the United States Food and Drug Administration (FDA) or the United Kingdom Medicines and Healthcare Products Regulatory Agency that can be used with the standard of care to treat sarcopenia or cachexia of any aetiology.
Testosterone has shown therapeutic efficacy, improving muscle mass and physical function, especially in conjunction with exercise 10, 11. However, testosterone replacement therapy has been associated with significant side effects 12, 13, 14, 15, 16. These include cardiovascular events, prostatic stimulation and enlargement, a possible increased risk of prostate cancer and elevated haemoglobin levels. Selective androgen receptor modulators (SARMs) have the potential to act selectively on the androgen receptor to produce anabolic effects on muscle, without adverse effects (AEs) on the prostate in men or androgenic effects in women, including hirsutism 17, 18, 19.
SARMs bind to the androgen receptor and induce conformational changes that facilitate interactions with specific co‐activator and co‐repressor proteins 20, 21. These interactions result in downstream regulation of target genes and other receptor‐mediated pathways 11, 17, 18. Induction of anabolic effects on muscle and bone, overall body metabolism and physical function could be useful for many clinical conditions in which subjects experience muscle wasting and impaired physical function. However, the development of a SARM with the balance of beneficial effects on muscle without AEs has proven difficult. SARMs currently in development include enobosarm 22, 23, LGD‐4033 24 and GSK2881078. These SARM compounds, and MK‐0773, which was discontinued 25, have shown common pharmacodynamic (PD) changes associated with androgen receptor agonists: lowering of high‐density lipoprotein (HDL), thyroxine‐binding globulin (TBG) and sex hormone‐binding globulin (SHBG); increased muscle mass; and positive effects on selected measures of physical function 19, 22, 23, 24, 25. Only enobosarm was tested in a Phase III registration trial but it was not approved by the FDA.
The SARM GSK2881078 [(R)‐1‐(1‐(methylsulfonyl)propan‐2‐yl)‐4‐(trifluoromethyl)‐1H–indole‐5‐carbonitrile, Chemical Abstracts Service registry number 1539314–06‐1] shows greater than 100‐fold selectivity for the androgen receptor over glucocorticoid, mineralocorticoid and progesterone receptors. GSK2881078 at 0.3 mg kg–1 day–1 dosed orally once daily (QD) for 28 days restored the weight of the levator ani muscle in orchiectomized rats to that of sham‐operated rats but produced only a minor increase in prostate weight compared with vehicle‐treated orchiectomized rats (unpublished data on file, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, PA, USA 19406).
This was the first study of GSK2881078 in humans, evaluating the safety, tolerability, pharmacokinetics (PK), and PD of the drug after oral administration of single‐ascending doses and repeat‐ascending doses to healthy male and healthy postmenopausal female subjects. The purpose of this first‐in‐human study was to provide sufficient confidence in the human safety and PK characteristics of GSK2881078 to inform progression to further repeat‐dose and proof‐of‐concept studies. The dose range proposed in this study was based on a low starting dose, using less than the minimum anticipated biological effect level (MABEL; see Supporting Information) derived from preclinical studies, including 13‐week toxicology studies in rats. Subsequent dose escalation was done to show clear PD effects and explore supra‐therapeutic exposures within the no observable AE level, if supported by adequate safety and tolerability at previous doses.
Methods
Study design
This two‐part, randomized, double‐blind [investigators and subjects blinded to treatment; sponsor (GlaxoSmithKline) unblinded for subject safety purposes] first‐in‐human, dose‐escalation, placebo‐controlled study was designed to evaluate the safety, tolerability, PK and PD of the SARM GSK2881078 in single and repeat doses in healthy male subjects and healthy postmenopausal female subjects.
In Part A, a four‐way crossover design, each subject (males only) received a single dose (D) of GSK2881078 at weekly intervals over 4 weeks (D1, 0.1 mg; D2, 0.2 mg; D3, 0 mg; D4, 0.05 mg) or matching placebo (P) in varying sequences (D1‐D2‐D3‐P, D1‐D2‐P‐D4, D1‐P‐D3‐D4, or P‐D2‐D3‐D4). (‘P’ designates intact blind, while ‘0 mg’ dose denotes investigator unblinded. A 0‐mg dose was given to evaluate clearance during one investigator‐unblinded dosing period owing to measurable baseline GSK2881078 concentrations in subjects dosed in the previous dosing period. Subjects remained blinded.) On day −1, subjects reported for study assessments and a meal. Following an overnight fast (≥8 h), subjects were dosed on day 1, then assessed over a 24‐h period prior to discharge the next day to continue a 6‐day washout period before returning for the next dose. Dose selection for Part B was based, in part, on data from Part A.
In Part B, each subject received repeat administration of study drug over 7 days (placebo or 0.05 mg GSK2881078) or over 14 days. Males and females were included in Part B, with male dosing cohort data reviewed prior to females being dosed. In consideration of the long half‐life (t1/2) observed in Part A, a loading regimen [twice daily (BID) dosing] was used to approximate steady‐state exposures during 14 days of dosing (for males: placebo or GSK2881078 0.2 mg BID days 1–3, then 0.08 mg QD days 4–14, 0.24 mg BID days 1–3, then 0.24 mg QD days 4–14, 0.48 mg BID days 1–3, then 0.48 mg QD days 4–14, or 0.75 mg BID days 1–3, then 0.75 mg QD days 4–14; for females: placebo or GSK2881078 0.24 mg BID days 1–3, then 0.24 mg QD days 4–14; or 0.35 mg BID days 1–3, then 0.35 mg QD days 4–14). The BID loading dose was given on days 1–3 at 08:00 h and 18:00 h, with subsequent single doses given at 08:00 h. On day −1, subjects reported for study assessments and a meal, followed by an overnight fast and dosing on the morning of day 1. Postdose, subjects underwent safety and PK assessments. In the group of subjects that received 0.24 mg doses, dosing on day 12 was under fed conditions (subjects received a standard meal 30 min prior to dosing) to provide exposure comparison to fasted conditions (day 14). All subjects remained at the study sites for the duration of the study. The last follow‐up visit was at approximately day 42.
The investigators and sponsor complied with all regulatory requirements relating to safety reporting to regulatory authorities, institutional review boards (IRBs) and investigators. The study protocol was approved by independent IRBs at the respective study sites. [The IRBs included the contract research organization [CRO], Parexel International Baltimore EPCU and Aspire IRB, Santee, CA, USA; and the CRO Quintiles Phase One Services and Midlands IRB, Overland Park, KS, USA]. The study was conducted in accordance with Good Clinical Practice and all applicable regulatory requirements, as well as the guiding principles of the 2008 Declaration of Helsinki. All subjects gave written informed consent before enrolment, which included compliance with the requirements and restrictions listed in the consent form.
Population studied
For Part A, only men were recruited. For Part B, men were initially enrolled, followed by separate cohorts of postmenopausal women. Men aged 18–50 years and postmenopausal women aged 50–70 years were required to have body mass index within the range of 19–32 kg m–2 and be in generally good health, with absence of clinically significant chronic disease including diabetes and hypertension, no use of chronic medications and no evidence of muscle wasting as assessed by a physician. Acceptable laboratory values and electrocardiogram (ECG) data were also required. Reasons for exclusion from the study included a history of cardiovascular, pulmonary, gastrointestinal, hepatic, renal, endocrinological, haematological or immunological conditions or malignancy that was not in complete remission for at least 5 years (or 1 year for non‐melanoma skin carcinoma). Women could not have taken postmenopausal hormone replacement therapy within 6 months of the first dose of study drug (see Supporting Information for complete inclusion and exclusion criteria).
Assessments
For the primary objective of assessing safety and tolerability, clinical laboratory tests, vital signs, ECGs, physical examinations, echocardiograms, cardiac biomarkers and cardiac telemetry were conducted, and AEs were monitored.
For the secondary objective of examining the PK profile of GSK2881078, plasma samples were collected and assayed using a validated analytical method based on protein precipitation, followed by high‐performance liquid chromatography/mass spectrometry/mass spectrometry (HPLC‐MS/MS) analysis.
The concentrations of GSK2881078 in plasma samples were calculated from calibration plots of standards prepared at known concentrations of GSK2881078 in human plasma. A weighted 1/x2 linear regression was applied over the range 0.05–50 ng ml–1 for GSK2881078. Acetonitrile containing internal standards ([13C3 15N]‐GSK2881078) at concentrations of 1 ng ml–1 was added to plasma samples. After vortex mixing, the deproteinized samples were centrifuged for approximately 4 min at a minimum of ~1640 g. The sample reconstituted in water was analysed using a TurboIonSpray Interface and multiple reaction monitoring. Chromatography was performed using a 50 × 2.1 mm i.d. Waters, HSS T3 (1.8 μm) column and eluted at a flow rate of 0.7 ml min–1. The gradient mobile phase consisted of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) and was run from 40% to 85% of mobile phase B in 3 mins. A Sciex API‐5000 triple quadrupole mass spectrometer (Applied Biosciences, Concord, ON, Canada) operated in negative ion mode. The temperature of the probe was maintained at 500°C, with a curtain gas setting of 20 and collision gas setting of 4. GSK2881078 was monitored by multiple reactions monitoring of 375 to 209. Internal standard [13C3 15N]‐GSK2881078 was monitored by multiple reactions monitoring of 379 to 210.
Urine samples were analysed for GSK2881078 using an analytical method based on liquid–liquid extraction followed by ultra‐HPLC‐MS/MS. Acetonitrile containing internal standards ([13C3 15N]‐GSK2974872) at concentrations of 20 000 pg ml–1 were added to urine samples. The lower limit of quantification for GSK2881078 was 50 pg ml–1 with higher limits of quantification of 50 000 pg ml–1. Analytical computer systems included Analyst Version 1.6.1 and SMS2000 version 2.3 (GlaxoSmithKline, King of Prussia, PA, USA).
Quality control (QC) samples, prepared at three different analyte concentrations and stored with study samples, were analysed with each batch of samples against separately prepared calibration standards. For the analysis to be acceptable, no more than one‐third of the QC results were to deviate from the nominal concentration by more than 15%, and at least 50% of the results from each QC concentration were to be within 15% of nominal.
PK analyses of the concentration–time data for plasma GSK2881078 were conducted using noncompartmental Model 200 (for extravascular administration) Phoenix Build 6.3.0.395, WinNonlin 6.3 (Pharsight Corporation, St Louis, MO, USA). Actual elapsed time from dosing was used to estimate all individual plasma PK parameters for evaluable subjects. The maximum observed concentration (Cmax) and the time at which Cmax was observed (Tmax) were determined directly from the raw concentration–time data. The area under the plasma concentration–time curve over the dosing interval (AUC(0–τ)) and the area under the plasma concentration–time curve from time zero to the last quantifiable time point (AUC(0–t)) were calculated by a combination of linear and logarithmic trapezoidal methods. The linear trapezoidal method was used for all incremental trapezoids arising from increasing concentrations, and the logarithmic trapezoidal method was used for those arising from decreasing concentrations. In addition, the predose (trough) concentration (Cτ) was determined, where τ is the end of dosing interval.
Fasting blood samples for biomarker assessment were drawn in the Part B predose baseline and during treatment to detect levels of hormones [serum luteinizing hormone (LH), SHBG, follicle‐stimulating hormone (FSH), prolactin, inhibin B, oestradiol, progesterone, testosterone and dihydrotestosterone (DHT)] and for lipid panels [plasma total cholesterol, low‐density lipoprotein (LDL), very‐low‐density lipoprotein (VLDL), HDL, triglycerides and serum apolipoproteins A1 and B (ApoA1 and ApoB)]. An ad hoc calculation and summary of free testosterone 26 was also conducted for subjects in the 14‐day treatment regimens.
Fasting samples were drawn in Parts A and B to assess cardiovascular biomarkers [plasma brain natriuretic peptide (BNP) and troponin]. Biomarkers assessed only in Part B were adrenal [plasma adrenocorticotropic hormone (ACTH), serum cortisol and dehydroepiandrosterone sulphate] and metabolic [serum insulin‐like growth‐factor‐1, insulin‐like growth factor‐binding protein 3, thyroxine (T4), free T4 and thyroid‐stimulating hormone] and plasma fasting insulin and glucose.
Statistical analyses
The sample size for each cohort was based on feasibility; no formal power/sample size calculations were performed, and no formal statistical hypotheses were tested. Version 9.1 or higher of the SAS system (Cary, NC, USA) was used to analyse the data. For Part B 14‐day repeat‐dose cohorts, placebo subjects were combined across cohorts for summarization and statistical analysis. Part B results are reported by gender. Safety data were summarized using the population of subjects who received at least one dose of study drug. Biomarker analyses were conducted on subjects in the safety population who had biomarker assessments at baseline and at least one postbaseline assessment. PK data were analysed for the subjects in the safety population for whom a PK sample was obtained and analysed.
For Part B of the study, change from baseline biomarker, lipid and hormone values were analysed separately in male and female subjects using analysis of covariance, with baseline covariates. Dose proportionality of Cmax and AUC(0–τ) on day 14 in males following QD administration of GSK2881078 was analysed using appropriate power and analysis of variance (ANOVA) models. Achievement of steady‐state exposure was assessed visually and from the estimate of the slope from the linear regression of log Cτ vs. time on days 4–8. The effect of food on GSK2881078 PK parameters was assessed by comparing day 12 (fed) to day 14 (fasted) PK parameters (Cmax and AUC(0–τ)) following administration of 0.24 mg GSK2881078 QD to males, using ANOVA. PK parameters were natural log‐transformed prior to statistical analysis.
Results
Subjects
A total of 99 subjects were enrolled. Treatments and subject demographics for Parts A and B are summarized in Table 1. All subjects enrolled in Part A completed the study. Four of the 89 subjects in Part B withdrew from treatment, three of them because of AEs.
Table 1.
Subject demographics
| Part A | |
|---|---|
| N | 10 |
| Mean age, years (SD) | 34.4 (7.90) |
| Male, n (%) | 10 (100) |
| African American/African heritage (%) | 9 (90) |
| White (%) | 1 (10) |
| Part B | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Intervention | |||||||||
| Placebo 7‐day | GSK2881078 0.05 mg 14‐day | Placebo 14‐day | GSK2881078 | ||||||
| 0.2 mg BID, then 0.08 mg QD | 0.24 mg BID, then 0.24 mg QD | 0.24 mg BID, then 0.24 mg QD | 0.35 mg BID, then 0.35 mg QD | 0.48 mg BID, then 0.48 mg QD | 0.75 mg BID, then 0.75 mg QD | ||||
| Total | 4 | 12 | 18 | 9 | 9 | 12 | 6 | 12 | 7 |
| Male, n (%) | 4 (100) | 12 (100) | 12 (67) | 9 (100) | 9 (100) | 0 | 0 | 12 (100) | 7 (100) |
| Mean age, years (SD) | 38.5 (2.65) | 35.1 (10.41) | 47.6 (13.14) | 40.4 (8.09) | 35.1 (14.62) | 58.8 (5.08) | 61.7 (5.28) | 33.6 (14.43) | 36.3 (15.03) |
| Black, n (%) | 3 (75) | 8 (67) | 5 (28) | 6 (67) | 5 (56) | 3 (25) | 0 | 4 (33) | 1 (14) |
| White, n (%) | 1 (25) | 4 (33) | 12 (67) | 2 (22) | 3 (33) | 9 (75) | 6 (100) | 7 (58) | 4 (57) |
| Other, n (%) | 0 | 0 | 1 (6) | 1 (11) | 1 (11) | 0 | 0 | 1 (8) | 2 (28) |
BID, twice daily; QD, once daily; SD standard deviation
PK
Plasma concentrations of GSK2881078 over time exhibited an initial rapid absorption phase following single and repeated administration (Figure 1). Plasma PK parameter estimates for Part A are summarized in Table 2. A dose proportional increase was observed in geometric mean Cmax. Median tmax was comparable across doses. The geometric mean t1/2 was long (>100 h) and similar across all treatments. Here, the quantifiable concentrations after administration of placebo or 0 mg GSK2881078 were included in the analysis for the preceding treatment dose to better characterize GSK2881078 elimination.
Figure 1.
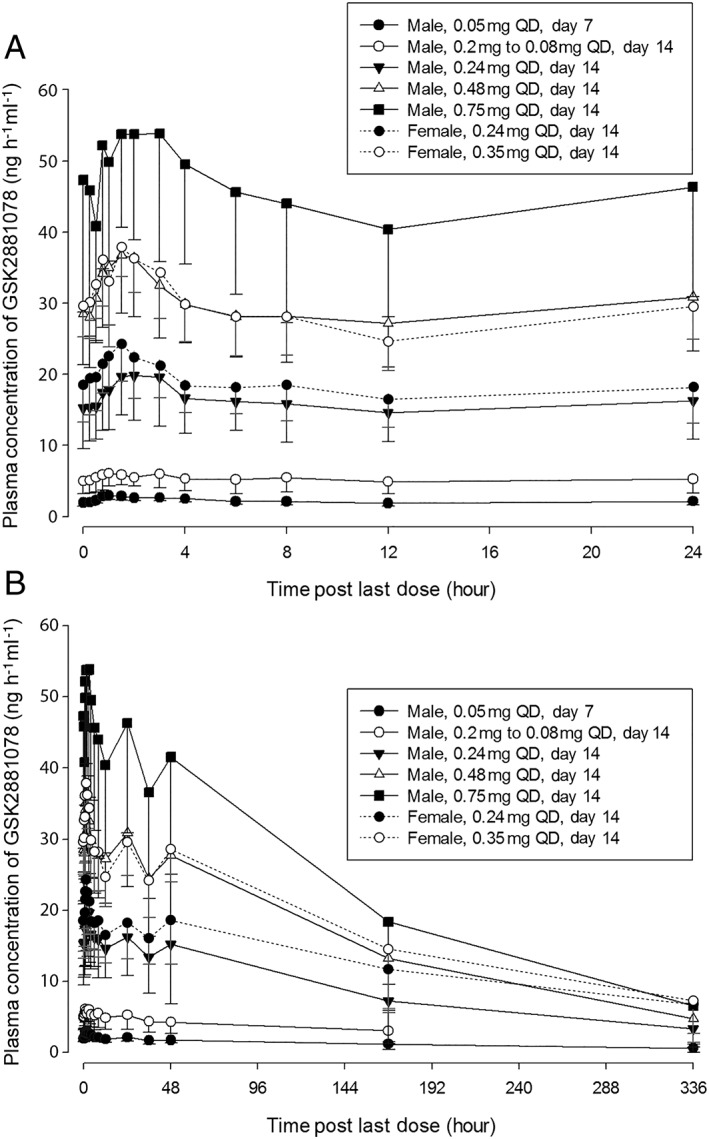
Plasma concentration over (A) the first 24‐h period following the last dose in Part B, and (B) the full 14‐day period following the final dose in Part B. QD, once daily
Table 2.
Summary of selected plasma GSK2881078 pharmacokinetic (PK) parameters for Part A
| PK parameter | Treatment | n | Geometric mean (CVb%) | 95% CI |
|---|---|---|---|---|
|
AUC
(0–t)
(ng∙h ml –1 ) |
0.05 mg | 7 | 91.9 (48) | (60.1, 140.5) |
| 0.1 mg | 5 | 69.3 (96) | (25.3, 189.6) | |
| 0.2 mg | 7 | 298.3 (43) | (203.8, 436.7) | |
| C max (ng ml –1 ) | 0.05 mg | 7 | 1.1 (32) | (0.8, 1.5) |
| 0.1 mg | 5 | 1.7 (24) | (1.2, 2.2) | |
| 0.2 mg | 7 | 3.5 (17) | (3.0, 4.1) | |
| t 1/2 (h) | 0.05 mg | 7 | 116.28 (30.3) | (88.4, 152.94) |
| 0.1 mg | 2 | 103.40 | – | |
| 0.2 mg | 6 | 127.40 (41.0) | (84.24, 192.66) | |
| Median (range) | ||||
| t max (h) | 0.05 mg | 7 | 1.00 (0.50, 1.00) | – |
| 0.1 mg | 5 | 1.00 (1.00, 1.50) | – | |
| 0.2 mg | 7 | 1.02 (0.75, 2.00) | – |
AUC(0–t), area under the curve from time zero to last quantifiable time point; CI, confidence interval; Cmax, maximum observed concentration; CVb, coefficient of variability; t1/2, apparent terminal elimination half‐life; tmax, time to maximum concentration
Plasma PK parameters for Part B are summarized in Table 3 for male and female subjects. The median tmax was similar across treatments and days, and no differences were observed between genders (males: 1.00–2.00 h; females: 1.26–1.50 h). Unscheduled plasma samples were taken following the last dose better to characterize GSK2881078 elimination. Consequently, the time of the last measurable concentration (tlast) after the last dose was not consistent (ranging from 48–822 h in males and 671–746 h in females), resulting in higher intersubject variability for AUC(0–t) both in males and females on days 7–9 and day 14 [coefficient of variability (CVb) ranging from 59–99% in males and 31–59% in females] compared with days 1 and 12 (CVb ranging from 18–33% in males and 11–22% in females).
Table 3.
Summary of plasma GSK2881078 pharmacokinetic parameters in Part B
| MALES | |||||
|---|---|---|---|---|---|
| PK parameter | Treatment | Day | n | Geometric mean (CVb%) | 95% CI |
| AUC (0–t) (ng.h ml –1 ) | 0.05 mg | 1 | 12 | 12.0 (18) | (10.7, 13.5) |
| 0.05 mg | 7–9 | 12 | 429.1 (59) | (302.7, 608.1) | |
| 0.2 mg to 0.08 mg | 1 | 9 | 23.7 (18) | (20.6, 27.2) | |
| 0.2 mg to 0.08 mg | 14 | 9 | 612.5 (99) | (324.4, 1156.6) | |
| 0.24 mg | 1 | 9 | 35.0 (18) | (30.5, 40.1) | |
| 0.24 mg | 12a | 9 | 339.6 (29) | (272.1, 423.9) | |
| 0.24 mg | 14 | 9 | 2662.8 (85) | (1507.9, 4702.2) | |
| 0.48 mg | 1 | 12 | 58.2 (21) | (50.9, 66.5) | |
| 0.48 mg | 14 | 12 | 5355.2 (58) | (3802.6, 7541.7) | |
| 0.75 mg | 1 | 7 | 88.0 (33) | (65.1, 118.9) | |
| 0.75 mg | 14 | 6 | 7562.9 (60) | (4216.6, 13564.7) | |
| AUC (0–τ) (ng.h ml –1 ) | 0.05 mg | 7–9 | 12 | 50.5 (15) | (45.8, 55.7) |
| 0.2 mg to 0.08 mg | 14 | 9 | 119.3 (34) | (92.4, 154.0) | |
| 0.24 mg | 12a | 9 | 339.6 (29) | (272.1, 423.9) | |
| 0.24 mg | 14 | 9 | 369.3 (30) | (295.0, 462.3) | |
| 0.48 mg | 14 | 12 | 688.7 (24) | (592.6, 800.4) | |
| 0.75 mg | 14 | 6 | 1037.8 (32) | (750.1, 1435.9) | |
| C max (ng ml –1 ) | 0.05 mg | 1 | 12 | 1.0 (29) | (0.8, 1.2) |
| 0.05 mg | 7–9 | 12 | 3.2 (18) | (2.9, 3.6) | |
| 0.2 mg to 0.08 mg | 1 | 9 | 3.4 (21) | (2.9, 4.0) | |
| 0.2 mg to 0.08 mg | 14 | 9 | 6.1 (33) | (4.8, 7.8) | |
| 0.24 mg | 1 | 9 | 5.1 (12) | (4.6, 5.6) | |
| 0.24 mg | 12a | 9 | 16.6 (26) | (13.7, 20.2) | |
| 0.24 mg | 14 | 9 | 20.9 (34) | (16.2, 27.0) | |
| 0.48 mg | 1 | 12 | 8.6 (25) | (7.3, 10.0) | |
| 0.48 mg | 14 | 12 | 36.6 (22) | (31.8, 42.1) | |
| 0.75 mg | 1 | 7 | 12.7 (31) | (9.6, 16.9) | |
| 0.75 mg | 14 | 6 | 56.6 (29) | (42.0, 76.2) | |
| t 1/2 (h) | 0.05 mg | 7–9 | 11 | 115.9 (40.5) | (89.18, 150.62) |
| 0.2 mg to 0.08 mg | 14 | 4 | 102.98 (62.7) | (41.18, 257.51) | |
| 0.24 mg | 14 | 9 | 108.12 (48.7) | (75.82, 154.18) | |
| 0.48 mg | 14 | 11 | 106.24 (31.0) | (86.67, 130.22) | |
| 0.75 mg | 14 | 6 | 103.74 (44.9) | (66.18, 162.64) | |
| Median (range) | |||||
| t max (h) | 0.05 mg | 1 | 12 | 1.00 (0.48, 3.02) | ‐ |
| 0.05 mg | 7–9 | 12 | 1.00 (0.72, 4.00) | ‐ | |
| 0.2 mg to 0.08 mg | 1 | 9 | 1.50 (0.50, 4.03) | ‐ | |
| 0.2 mg to 0.08 mg | 14 | 9 | 1.53 (0.52, 3.02) | ‐ | |
| 0.24 mg | 1 | 9 | 1.02 (0.57, 2.02) | ‐ | |
| 0.24 mg | 12a | 9 | 2.00 (0.00, 23.90) | ‐ | |
| 0.24 mg | 14 | 9 | 2.00 (0.75, 48.00) | ‐ | |
| 0.48 mg | 1 | 12 | 1.50 (0.75, 2.00) | ‐ | |
| 0.48 mg | 14 | 12 | 1.50 (0.75, 48.00) | ‐ | |
| 0.75 mg | 1 | 7 | 1.50 (1.03, 2.00) | ‐ | |
| 0.75 mg | 14 | 6 | 1.25 (0.25, 3.00) | ‐ |
| FEMALES | |||||
|---|---|---|---|---|---|
| PK parameter | Treatment | Day | n | Geometric mean (CVb%) | 95% CI |
| AUC (0–t) (ng.h ml –1 ) | 0.24 mg | 1 | 12 | 29.9 (22) | (26.0.1, 34.3) |
| 0.24 mg | 14 | 11 | 5301.1 (59) | (3669.3, 7658.5) | |
| 0.35 mg | 1 | 6 | 46.2 (11) | (41.0, 52.1) | |
| 0.35 mg | 14 | 5 | 7126.9 (31) | (4867.3, 10435.5) | |
| AUC (0–τ) (ng.h ml –1 ) | 0.24 mg | 14 | 11 | 430.7 (24) | (367.6, 504.6) |
| 0.35 mg | 14 | 5 | 674.2 (17) | (549.0, 828.1) | |
| C max (ng ml –1 ) | 0.24 mg | 1 | 12 | 5.2 (31) | (4.3, 6.3) |
| 0.24 mg | 14 | 11 | 24.6 (20) | (21.5, 28.1) | |
| 0.35 mg | 1 | 6 | 6.8 (22) | (5.4, 8.5) | |
| 0.35 mg | 14 | 5 | 38.5 (13) | (32.7, 45.3) | |
| t 1/2 (h) | 0.24 mg | 14 | 9 | 149.29 (41.7) | (109.71, 203.15) |
| 0.35 mg | 14 | 5 | 148.64 (26.5) | (107.54, 205.45) | |
| Median (range) | |||||
| t max (h) | 0.24 mg | 1 | 12 | 1.26 (0.75, 1.98) | – |
| 0.24 mg | 14 | 11 | 1.50 (0.75, 2.00) | – | |
| 0.35 mg | 1 | 6 | 1.29 (0.95, 1.50) | – | |
| 0.35 mg | 14 | 5 | 1.50 (1.00, 3.00) | – |
AUC(0–t), area under the curve from time zero to last quantifiable time point; AUC(0–τ), AUC from time 0 to the end of the dosing interval (τ); AUC(0–24), the AUC from time 0 to 24 h; BID, twice daily; CI, confidence interval; Cmax, maximum observed concentration; CVb, coefficient of variability; PK, pharmacokinetic; QD, once daily; tmax, time to maximum concentration; t1/2, apparent terminal elimination half‐life
Day 12 for analysis of food effect
In males and females, the geometric means for AUC(0–τ) and Cmax differed by less than 20% at a comparable dose (0.24 mg). Intersubject variability for AUC(0–τ) and Cmax was similar in males and females, and typically within the range of 12–34%. The t1/2s were long (geometric means >100 h) and similar on days 7–9 and day 14 across all treatments in males. In females, the geometric mean t1/2 at a comparable dose (0.24 mg) appeared 38% longer than in males. It is likely that this was attributable to the differences in tlast, which was 340 h for four of the nine males compared with >620 h in all other instances.
The urine PK parameters of renal clearance (CLR) and percentage dose excretion at 24 h (fe24) were comparable between males and females [geometric mean CLR (ml h–1): 11.08 in males, 10.04 in females; fe24 (%): 1.53 in males, 1.93 in females]. Statistical analysis of dose proportionality at day 14 in males showed that slope estimates for both AUC(0–τ) and Cmax were close to 1 (0.969 and 0.987, respectively) and the 90% confidence intervals (CIs) both included 1 [(0.871, 1.067) and (0.888, 1.086), respectively]. Assessment of the food effect suggested bioequivalence in AUC(0–τ). The fed:fasted ratio of the geometric least squares mean for AUC(0–τ) was 0.920 (90% CI 0.890, 0.951). For Cmax, the fed:fasted ratio was 0.794 (90% CI 0.727, 0.867).
PD and biomarkers
Analysis of reproductive hormones in male subjects showed reductions in testosterone, DHT, and SHBG relative to baseline and placebo at all active doses (Table 4). The reductions in testosterone at the two highest doses continued for 14 days after cessation of GSK2881078 treatment and were increasing toward baseline by follow‐up day 28 at the lower doses and in all doses by follow‐up day 42 (Figure 2). SHBG levels were stable or increasing by follow‐up day 28 for all doses and, with the exception of the lowest dose (0.2 mg BID then 0.08 mg QD), had not yet returned to baseline by follow‐up day 42 (Figure 3). Adjusted mean changes from baseline on day 14 showed no apparent dose response in DHT or testosterone but there appeared to be a dose‐response relationship for SHBG lowering (Table 4 and Figure 3). There was no apparent effect of treatment on calculated free testosterone (Figure 4). Reductions in FSH were also observed at all doses but with no apparent dose‐response relationship. There was no consistent reduction in LH with treatment. There were no clinically meaningful changes in oestradiol, inhibin, progesterone or prolactin in male subjects.
Table 4.
ANCOVA results for change from baseline in selected lipids, adrenal and metabolic hormone biomarkers in males
| Parameter | Day | Statistic a | Placebo (n = 12) | GSK2881078 | |||
|---|---|---|---|---|---|---|---|
| 0.08 mg (n = 9) | 0.24 mg (n = 9) | 0.48 mg (n = 12) | 0.75 mg (n = 6) | ||||
| Lipids | |||||||
| Apolipoprotein A1 (G/L) | 14 | N | 12 | 9 | 8 | 12 | 6 |
| Adjusted mean | −0.082 | −0.264 | −0.297 | −0.418 | −0.576 | ||
| difference from placebo (95% CI) | −0.182 (−0.307, −0.057) | −0.215 (−0.344, −0.087) | −0.336 (−0.452, −0.221) | −0.494 (−0.636, −0.352) | |||
| HDL (mmol l –1 ) | 14 | N | 10 | 9 | 9 | 12 | 0 |
| Adjusted mean | −0.082 | −0.248 | −0.348 | −0.439 | NA | ||
| difference from placebo (95% CI) | −0.165 (−0.317, −0.014) | −0.265 (−0.417, −0.114) | −0.357 (−0.499, −0.215) | NA | |||
| Triglycerides (mmol l –1 ) | 14 | N | 10 | 9 | 9 | 12 | 0 |
| Adjusted mean | 0.010 | −0.300 | −0.293 | −0.329 | NA | ||
| difference from placebo (95% CI) | −0.310 (−0.591, −0.028) | −0.303 (−0.585, −0.021) | −0.339 (−0.604, −0.074) | NA | |||
| VLDL (mmol l –1 ) | 14 | N | 10 | 9 | 9 | 12 | 0 |
| Adjusted mean | 0.022 | −0.120 | −0.116 | −0.129 | NA | ||
| difference from placebo (95% CI) | −0.142 (−0.259, −0.024) | −0.137 (−0.255, −0.020) | −0.150 (−0.265, −0.036) | NA | |||
| Adrenal biomarkers | |||||||
| ACTH (ng l –1 ) | 8 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | −3.06 | −9.94 | 2.80 | 20.37 | 0.75 | ||
| difference from placebo (95% CI) | −6.88 (−22.65, 8.88) | 5.86 (−9.25, 20.96) | 23.43 (9.43, 37.43) | 3.81 (−13.32, 20.95) | |||
| ACTH (ng l –1 ) | 14 | n | 11 | 8 | 9 | 12 | 6 |
| Adjusted mean | −6.15 | −7.91 | 2.08 | 4.79 | 5.62 | ||
| difference from placebo (95% CI) | −1.75 (−17.89, 14.38) | 8.23 (−7.00, 23.46) | 10.94 (−3.19, 25.08) | 11.77 (−5.47, 29.02) | |||
| Cortisol (nmol l –1 ) | 8 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | 4.37 | −38.55 | −6.42 | 95.73 | −65.69 | ||
| difference from placebo (95% CI) | −42.92 (−116.31, 30.47) | −10.79 (−84.20, 62.62) | 91.36 (23.41, 159.31) | −70.07 (−153.32, 13.18) | |||
| Cortisol (nmol l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | −20.59 | −26.54 | 0.79 | 5.24 | 14.09 | ||
| difference from placebo (95% CI) | −5.94 (−79.33, 67.44) | 21.39 (−52.02, 94.80) | 25.83 (−42.11, 93.78) | 34.68 (−48.57, 117.93) | |||
| Metabolic biomarkers | |||||||
| TBG (nmol l –1 ) | 14 | n | 12 | 8 | 9 | 12 | 6 |
| Adjusted mean | −6.6 | −54.9 | −56.2 | −63.4 | −65.4 | ||
| difference from placebo (95% CI) | −48.3 (−81.5, −15.0) | −49.6 (−81.9, −17.4) | −56.8 (−86.5, −27.0) | −58.8 (−95.2, −22.4) | |||
| IGF‐1 (μg l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | 11.4 | 18.4 | 17.9 | 32.2 | −15.9 | ||
| difference from placebo (95% CI) | 6.9 (−23.7, 37.5) | 6.4 (−24.1, 37.0) | 20.8 (−7.8, 49.4) | −27.3 (−62.9, 8.2) | |||
| IGF‐3 (nmol l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | 8.4 | 6.1 | 5.1 | 5.3 | −20.6 | ||
| difference from placebo (95% CI) | −2.3 (−23.6, 19.0) | −3.3 (−24.6, 17.9) | −3.1 (−22.9, 16.6) | −29.0 (−53.3, −4.6) | |||
| Free thyroxine (pmol l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | 0.26 | −1.16 | 0.79 | 3.00 | 1.83 | ||
| difference from placebo (95% CI) | −1.42 (−2.6, −0.2) | 0.53 (−0.7, 1.7) | 2.74 (1.6, 3.8) | 1.57 (0.3, 2.9) | |||
| Reproductive hormones | |||||||
| DHT (nmol l –1 ) | 14 | n | 12 | 9 | 8 | 12 | 5 |
| Adjusted mean | −0.13 | −0.78 | −0.84 | −0.92 | −0.71 | ||
| difference from placebo (95% CI) | −0.65 (−0.89, −0.40) | −0.71 (−0.96, −0.46) | −0.79 (−1.01, −0.56) | −0.57 (−0.87, −0.28) | |||
| SHBG (nmol l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | −3.24 | −15.95 | −19.50 | −21.11 | −22.84 | ||
| difference from placebo (95% CI) | −12.72 (−15.9, −9.5) | −16.27 (−19.4, −13.1) | −17.87 (−20.8, −15.0) | −19.61 (−23.2, −16.1) | |||
| Testosterone (nmol l –1 ) | 14 | n | 12 | 9 | 8 | 12 | 5 |
| Adjusted mean | −0.62 | −6.19 | −6.69 | −7.78 | −6.75 | ||
| difference from placebo (95% CI) | −5.58 (−8.15, −3.00) | −6.07 (−8.71, −3.43) | −7.16 (−9.53, −4.79) | −6.14 (−9.22, −3.06) | |||
| FSH (IU l –1 ) | 14 | n | 10 | 9 | 9 | 12 | 0 |
| Adjusted mean | −0.406 | −1.163 | −0.807 | −0.926 | NA | ||
| difference from placebo (95% CI) | −0.757 (−1.369, −0.145) | −0.402 (−1.001, 0.198) | −0.521 (−1.079, 0.037) | NA | |||
| LH (IU l –1 ) | 14 | n | 12 | 9 | 9 | 12 | 6 |
| Adjusted mean | 0.49 | −0.23 | −1.07 | 1.46 | −1.07 | ||
| difference from placebo (95% CI) | ‐0.72 (−2.18, 0.73) | −1.56 (−3.0, −0.12) | 0.97 (−0.36, 2.30) | −1.56 (−3.22, 0.10) | |||
ANCOVA, analysis of covariance; ACTH, adrenocorticotropic hormone; CI, confidence interval; DHT, dihydrotestosterone; FSH, follicle‐stimulating hormone; HDL, high‐density lipoprotein; IGF‐1, insulin‐like growth factor‐1; IGF‐3, insulin‐like growth factor‐3; NA, not applicable; SHBG, sex hormone‐binding globulin; TBG, thyroxine‐binding globulin; VLDL, very‐low‐density lipoprotein
For between treatment comparisons to be reported, the sample size in each treatment group involved in the comparison must have been at least three.
Figure 2.
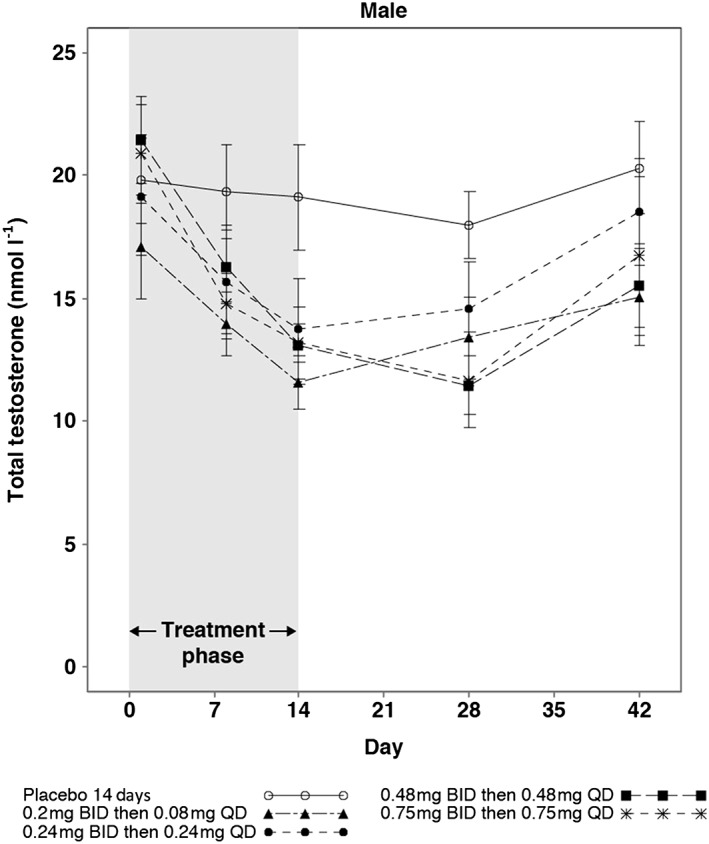
Mean (SE) total testosterone up to 42 days in male subjects with 14‐day treatment. BID, twice daily; QD, once daily; SE, standard error
Figure 3.
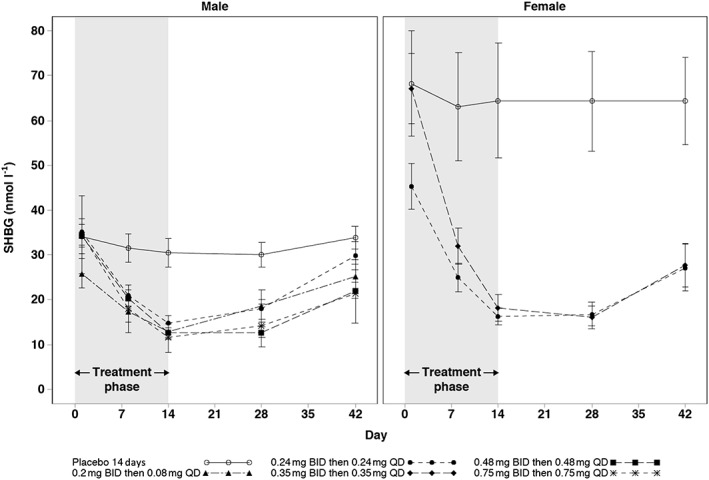
Mean (SE) SHBG up to 42 days in male and female subjects with 14‐day treatment. BID, twice daily; QD, once daily; SE, standard error; SHBG, sex hormone‐binding globulin
Figure 4.
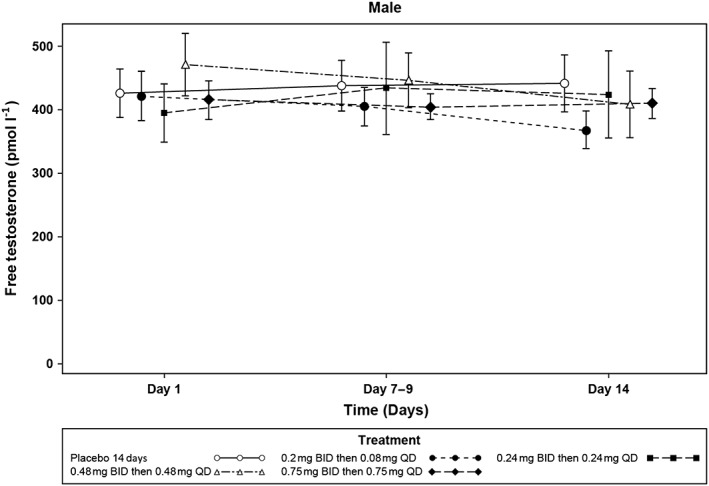
Mean (SE) free testosterone up to 14 days in male subjects with 14‐day treatment. Free testosterone was estimated by calculation 26. BID, twice daily; QD, once daily; SE, standard error
In female subjects, SHBG was reduced in a dose‐dependent manner (Table 5). SHBG levels remained suppressed on follow‐up day 28, and although levels started to increase by follow‐up day 42, they had still not returned to baseline (Figure 3). There were no other clinically meaningful changes in female reproductive hormones.
Table 5.
ANCOVA results for change from baseline in selected lipids, adrenal and metabolic hormones biomarkers in females
| Parameter | Day | Statistic | Placebo (n = 6) | GSK2881078 | |
|---|---|---|---|---|---|
| 0.24 mg (n = 12) | 0.35 mg (n = 6) | ||||
| Lipids | |||||
| Apolipoprotein A1 (G/L) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | 0.038 | −0.370 | −0.457 | ||
| difference from placebo | −0.408 | −0.495 | |||
| (95% CI) | (−0.539, −0.277) | (−0.650, −0.339) | |||
| HDL (mmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | −0.039 | −0.335 | −0.558 | ||
| difference from placebo | −0.295 | −0.518 | |||
| (95% CI) | (−0.450, −0.141) | (−0.703, −0.334) | |||
| Triglycerides (mmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | 0.042 | −0.348 | −0.502 | ||
| difference from placebo | −0.390 | −0.544 | |||
| (95% CI) | (−0.595, −0.184) | (−0.796, −0.292) | |||
| VLDL (mmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | 0.014 | −0.159 | −0.234 | ||
| difference from placebo | −0.173 | −0.249 | |||
| (95% CI) | (−0.267, −0.079) | (−0.364, −0.134) | |||
| Adrenal biomarkers | |||||
|
Day 8
ACTH (ng l –1 ) |
8 | n | 6 | 12 | 6 |
| Adjusted mean | −2.03 | 3.64 | 25.87 | ||
| difference from placebo | 5.66 | 27.89 | |||
| (95% CI) | (−13.15, 24.48) | (6.12, 49.67) | |||
|
Day 14
ACTH (ng l –1 ) |
14 | n | 5 | 12 | 5 |
| Adjusted mean | −8.77 | −0.59 | 1.44 | ||
| difference from placebo | 8.18 | 10.21 | |||
| (95% CI) | (−11.92, 28.28) | (−13.81, 34.23) | |||
| Cortisol (nmol l –1 ) | 8 | n | 6 | 12 | 6 |
| Adjusted mean | 21.56 | 50.30 | 60.93 | ||
| difference from placebo | 28.74 | 39.37 | |||
| (95% CI) | (−51.25, 108.72) | (−52.92, 131.66) | |||
| Cortisol (nmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | −50.29 | −42.92 | −4.38 | ||
| difference from placebo | 7.37 | 45.91 | |||
| (95% CI) | (−76.24, 90.98) | (−52.18, 144.00) | |||
| DHEAS (μmol l –1 ) | 8 | n | 6 | 12 | 6 |
| Adjusted mean | −0.23 | −0.11 | 0.29 | ||
| difference from placebo | 0.12 | 0.52 | |||
| (95% CI) | (−0.5, 0.7) | (−0.2, 1.2) | |||
| DHEAS (μmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | −0.46 | −0.27 | 0.43 | ||
| difference from placebo | 0.19 | 0.89 | |||
| (95% CI) | (−0.4, 0.8) | (0.2, 1.6) | |||
| Metabolic hormones | |||||
| TBG (nmol l –1 ) | n | 5 | 12 | 5 | |
| Adjusted mean | −6.7 | −83.5 | −90.5 | ||
| difference from placebo | −76.8 | −83.9 | |||
| (95% CI) | (−113.4, −40.3) | (−127.5, −40.3) | |||
| IGF‐1 (μg l –1 ) | n | 5 | 12 | 5 | |
| Adjusted mean | −18.0 | 48.0 | 41.8 | ||
| difference from placebo | 66.0 | 59.8 | |||
| (95% CI) | (12.9, 119.1) | (1.8, 117.9) | |||
| IGF‐3 (nmol l –1 ) | n | 5 | 12 | 5 | |
| Adjusted mean | −2.5 | −17.7 | −8.9 | ||
| difference from placebo | −15.2 | −6.4 | |||
| (95% CI) | (−31.1, 0.7) | (−25.1, 12.3) | |||
| Free thyroxine (pmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | −0.01 | 0.59 | 1.49 | ||
| difference from placebo | 0.60 | 1.50 | |||
| (95% CI) | (−0.3, 1.5) | (0.4, 2.6) | |||
| Reproductive hormones | |||||
| SHBG (nmol l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | −4.87 | −32.65 | −43.99 | ||
| difference from placebo | −27.78 | −39.12 | |||
| (95% CI) | (−36.7, −18.9) | (−48.5, −29.7) | |||
| Oestradiol (pmol l –1 ) | 14 | n | 4 | 12 | 5 |
| Adjusted mean | −22.59 | −33.04 | −26.90 | ||
| difference from placebo | −10.45 | −4.32 | |||
| (95% CI) | (−27.1, 6.2) | (−23.5, 14.9) | |||
| FSH (IU l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | 1.044 | 3.890 | −14.978 | ||
| difference from placebo | 2.846 | −16.022 | |||
| (95% CI) | (−7.204, 12.896) | (−28.892, −3.152) | |||
| LH (IU l –1 ) | 14 | n | 5 | 12 | 5 |
| Adjusted mean | 2.13 | 1.53 | −5.33 | ||
| difference from placebo | −0.61 | −7.46 | |||
| (95% CI) | (−6.64, 5.42) | (−14.61, −0.31) | |||
ACTH, adrenocorticotropic hormone; ANCOVA, analysis of covariance; DHEAS, dehydroepiandrosterone sulphate; FSH, follicle‐stimulating hormone; HDL, high‐density lipoprotein; IGF, insulin‐like growth factor; LH, luteinizing hormone; SHBG, sex hormone‐binding globulin; TBG, thyroxine‐binding globulin
HDL, ApoA1, triglycerides and VLDL were reduced with GSK2881078 treatment relative to baseline and placebo both in males and females (Tables 4 and 5). No changes were observed for total cholesterol, LDL or total ApoB. In both males and females, HDL levels were returning to baseline by the end of the study (Figure 5).
Figure 5.
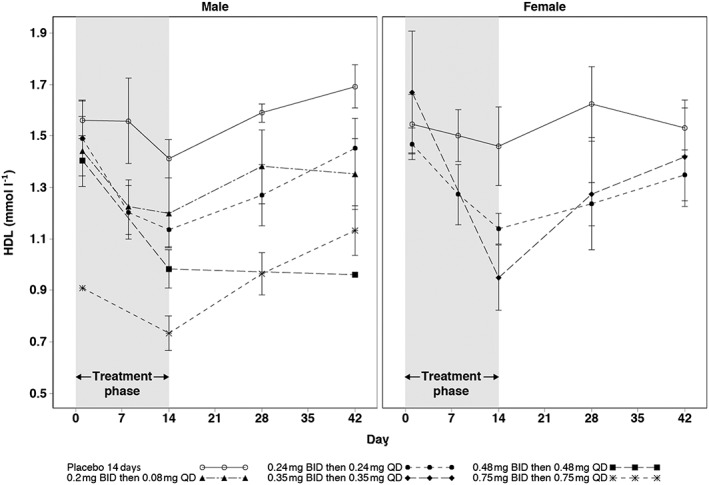
Mean (SE) HDL up to 42 days in male and female subjects with 14‐day treatment. BID, twice daily; HDL, high‐density lipoprotein; QD, once daily, SE, standard error
There was no consistent effect on adrenal biomarkers in male subjects. An increase in ACTH and cortisol was observed in the male 0.48‐mg group on day 8. However, this effect was not observed at a higher dose and did not persist on day 14 (Table 4). This isolated increase in men was not considered clinically meaningful because cortisol levels remained within the normal range, and a dose–response relationship was not observed. ACTH and cortisol elevations decreased with continued dosing beyond day 8 and this was not associated with other clinical or safety signals. No clinically meaningful changes in adrenal hormones were observed in women (Table 5). For metabolic biomarkers, TBG was reduced at all doses both in males and females. In addition, free T4 was increased at the high doses in males (0.48 mg and 0.75 mg) and females (0.35 mg) (Tables 4 and 5) without clinical effect. Specifically, there was no increase in heart rate and no tachycardia or other clinical symptoms. There were no clinically meaningful changes in other assessed metabolic biomarkers. Levels of the cardiac biomarker BNP were unaffected in male and female subjects over 14 days of dosing. All troponin levels were in the normal range, with the exception of one result on day 4 for a subject in the 0.48‐mg group; repeat samples for troponin were normal and ECGs were unremarkable.
Safety
Overall, treatment was well tolerated. In Part A, two subjects (20%) had a single AE, one with extremity pain and one with an upper respiratory tract infection, both of which were mild. In Part B, 48 subjects (54%) had at least one AE. The most commonly reported AEs overall were: headache and upper respiratory tract infection (eight subjects, 9%); medical device site reaction (five subjects, 6%); and constipation, dyspepsia, and palpitations (each reported in three subjects, 3% each). The most common AEs (occurring in two or more subjects) considered related to the study drug were constipation, dyspepsia and nausea.
In Part B, three subjects withdrew owing to AEs. A female subject on placebo developed chest pain on day 12. She was evaluated with an exercise stress test, which was abnormal (both events were serious AEs). She had emergency cardiac catheterization, which was unremarkable, and was followed without further dosing. A female subject on active treatment developed a maculopapular rash on day 10, and a skin biopsy was consistent with a drug reaction. She was discontinued from further dosing and treated with topical corticosteroids with resolution of the rash. She also had elevated alanine aminotransferase (ALT) to 2.9 × upper limit of normal (ULN) with no increase in bilirubin. In addition, a male on active treatment developed a shortened QT interval after his first dose and withdrew from the study. This ECG finding was not considered to be drug related. A female on active treatment developed atopic dermatitis, not considered drug related, toward the end of her dosing period, and also had an elevation of ALT to 2.3 × ULN, with no increase in bilirubin. Two men on active treatment experienced muscle soreness after demanding physical activity in the follow‐up period, 14 and 28 days, respectively, after the last dose. Both men showed marked elevation of creatine phosphokinase (CPK) (17 841 IU l–1 and 4590 IU l–1, respectively, normal CPK level is <294 IU l–1), and mild elevation of ALT (115 IU l–1 and 81 IU l–1, respectively, with ULN <50 IU l–1). The CPK and ALT values resolved over 3 weeks in both men. The elevated CPK values were considered severe but not drug related. No other subjects showed elevation of ALT ≥2 × ULN during treatment.
Discussion
Investigations into SARM compounds at various stages of preclinical and clinical development are ongoing. These agents could address deleterious muscle wasting phenomena that occur with advanced age and chronic disease 22, 23, 24, 25, 27. The current study was a randomized, double‐blind, placebo‐controlled investigation of the safety, tolerability, PK and PD of GSK2881078, a novel SARM compound, in single and repeat doses in healthy male and postmenopausal female subjects.
Overall, the dose range of GSK2881078 evaluated was well tolerated among a sample of healthy male and postmenopausal female subjects. AEs occurred in half the study population, with a similar distribution between active treatment and placebo groups. Although SARMs and oral androgens are associated with elevations in liver enzymes 25, no clinically significant hepatic signals were observed.
There was an approximate dose‐proportional increase in exposures of GSK2881078 in the dose range evaluated in this study. Food did not significantly alter AUC(0–τ) but was associated with a 21% decrease in Cmax. Less than 2% of the GSK2881078 dose was excreted in urine, and CLR was similar in males and females. While trough concentrations were used to assess steady‐state attainment in the study, with an observed t1/2 in humans that was much longer than anticipated, the collection of trough samples only on days 4–8 limited an adequate assessment of steady‐state attainment. With an estimated t1/2 >100 h, steady‐state is expected to be achieved much later than day 8. The long t1/2 probably also accounted for the measurable concentrations in subjects while receiving 0 mg GSK2881078 preceded by an active dosing regimen 5 days earlier in Part A. A washout period longer than 5 days is needed in future studies of this molecule. Given the long t1/2 of GSK2881078 and the objective of emulating steady‐state exposure profiles over the 14‐day course of the study, a loading regimen was devised based on the Part A data, which would elevate plasma exposures rapidly and maintain them over the study period. As a result, direct comparison of PK results to address accumulation of GSK2881078 from day 1 to day 14 is not possible using standard PK approaches but will be addressed in future analyses making use of population PK modelling.
Following repeat dosing, there was an apparent 38% difference in t1/2 between genders for the 0.24 mg BID then 0.24 mg QD treatment, which was probably attributable to tlast occurring much earlier for some of the male subjects. As the quantifiable concentrations with placebo or 0 mg GSK2881078 following single‐dose administration in Part A were included in the analysis for the preceding treatment dose to better characterize GSK2881078 elimination, tlast exceeded the last scheduled sampling time for the corresponding treatment, accounting for the high variability of AUC(0–t) (CVb ranging from 43% to 96%) and the apparent lack of dose proportionality in contrast to Part B, where additional samples permitted improved characterization of the long t1/2.
Consistent with other oral androgens and other SARMs under investigation, GSK2881078 (doses of 0.2 mg BID then 0.08 mg QD and higher) was associated with reductions in HDL, ApoA1, triglycerides and VLDL relative to baseline and placebo in male and female subjects 22, 24. This was an expected result because androgen receptor agonists are known to affect hepatic metabolism. There were no apparent changes in total cholesterol, LDL or ApoB. The clinical implications of androgen‐associated lipid alterations are not clear.
Reductions in testosterone, DHT, SHBG and FSH were observed relative to baseline and placebo in male subjects, and reductions in SHBG in female subjects receiving GSK2881078. The accompanying decrease in SHBG in both males and females is consistent with hepatic effects observed with other SARMs 22, 25. Free testosterone did not change in either males or females, consistent with the decrease in SHBG. No clinically meaningful changes were observed in other reproductive hormones. Androgen receptor agonists can act centrally on the hypothalamic–pituitary–gonadal axis, to suppress the signals for gonadal hormonal secretion, LH and FSH. In the present study, neither free testosterone nor LH levels declined, suggesting that the decrease in total testosterone was not due to hypothalamic inhibition. Thus, in men, total testosterone levels can fall, as observed. Effects in the postmenopausal women were minimal. In addition, androgen receptor agonists also suppress production of binding globulins such as SHBG in the liver, leading to lower levels of endogenous sex steroids (testosterone and oestradiol), typically without significant effect on free hormone levels. All hormonal levels were returning to baseline values by the end of the study.
There were no consistent, clinically meaningful changes in adrenal hormones in the male or female subjects. In male and female subjects, TBG was reduced relative to baseline and placebo. As noted, androgen receptor agonists act on the liver to reduce the levels of binding globulins, such as TBG and SHBG. Free hormone levels typically are unaffected, although here, men and women alike showed increases in free T4 at the higher doses. The clinical significance of this is unclear, although there was no evidence of a clinical response to this, such as an increased heart rate. There were no clinically meaningful changes in any other metabolic biomarkers. Monitoring of BNP and troponin showed no evidence of myocardial injury.
Comparison of our results with those from longer duration studies of other SARMs in development (enobosarm 22 and LGD‐4033 24) reveals modest differences. Our high‐dose male cohort (0.75 mg) yielded approximately similar levels of SHBG and total testosterone suppression as those seen in high‐dose cohorts of enobosarm and LGD‐4033, suggesting similar levels of androgen receptor agonism. Adverse effects were generally not different between SARMs. Of note were elevations in ALT seen with enobosarm, leading to study discontinuation in one subject receiving the highest dose examined, 3 mg QD. One possible differentiating feature of GSK2881078 is the long t1/2 of the compound. GSK2881078 has a terminal t1/2 of 7.5 days, considerably longer than enobosarm (22 h 28) or LGD (24–36 h 24), which may limit peak : trough compound excursion. While daily testosterone concentrations are diurnal 29, a more static exposure may offer more sustained anabolic stimulation of muscle.
Currently, there are no approved therapies for the prevention or treatment of deleterious muscle wasting, although there is a clinical need for safe anabolic compounds such as SARMs. GSK2881078 has demonstrated clear target engagement shown by significant reductions in SHBG, TBG and HDL. Good safety and tolerability were also demonstrated and are consistent with the broad safety margin shown in preclinical toxicology studies (unpublished data on file, GlaxoSmithKline, King of Prussia, PA, USA). These data, combined with the dose‐proportional plasma levels and predictable biomarker profiles of GSK2881078, provide a pharmacological rationale for further clinical study of this novel SARM for the treatment of muscle wasting.
Competing Interest
This study (NCT02045940) was funded by GlaxoSmithKline (GSK). R.V.C., P.T. and J.W. were employees of GSK at the time of the study and they hold stock in GSK; A.C.W., S.A. and M.H.M. are employees of and hold stock in GSK.
Funding for this study was provided by GSK. The authors would like to thank Denise Shortino for her contribution to the statistical design of the study; and Ronald V Goldwater, MD and Phil Leese, MD, for their contributions to the clinical conduct of the study. Editorial support and graphic services were provided by Elizabeth Rosenberg, PhD, and Susan Abulhawa, BS, of AOI Communications, L.P., and were funded by GSK.
Contributors
All authors meet the criteria for authorship set forth by the International Committee of Medical Journal Editors. R.V.C. and S.A. contributed to the concept and design of the study, analysed and interpreted the data and wrote sections of the first draft; A.C.W. analysed and interpreted the data and wrote sections of the first draft; P.T. and J.W. contributed to the concept and design of the study and analysed and interpreted the data; M.H.M. analysed and interpreted the data and wrote sections of the first draft. All authors commented critically throughout, approved the final draft for submission and agree to be accountable for the content.
Supporting information
Data S1 Minimum Anticipated Biological Effect Level (MABEL) explanation and Inclusion and Exclusion Criteria.
Clark, R. V. , Walker, A. C. , Andrews, S. , Turnbull, P. , Wald, J. A. , and Magee, M. H. (2017) Safety, pharmacokinetics and pharmacological effects of the selective androgen receptor modulator, GSK2881078, in healthy men and postmenopausal women. Br J Clin Pharmacol, 83: 2179–2194. doi: 10.1111/bcp.13316.
ClinicalTrials.org identification: NCT02045940
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 2015; 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gill TM, Gahbauer EA, Murphy TE, Han L, Allore HG. Risk factors and precipitants of long‐term disability in community mobility: a cohort study of older persons. Ann Intern Med 2012; 156: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European Consensus on Definition and Diagnosis: report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010; 39: 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guralnik JM, LaCroix AZ, Branch LG, Kasl SV, Wallace RB. Morbidity and disability in older persons in the years prior to death. Am J Public Health 1991; 81: 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jette AM, Branch LG. Impairment and disability in the aged. J Chronic Dis 1985; 38: 59–65. [DOI] [PubMed] [Google Scholar]
- 7. Batsis JA, Mackenzie TA, Lopez‐Jimenez F, Bartels SJ. Sarcopenia, sarcopenic obesity, and functional impairments in older adults: NHANES 1999–2004. Nutr Res 2015; 35: 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ali S, Garcia JM. Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options – a mini‐review. Gerontology 2014; 60: 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pahor M, Guralnik JM, Ambrosius WT, Blair S, Bonds DE, Church TS, et al. Effect of structured physical activity on prevention of major mobility disability in older adults: the LIFE study randomized clinical trial. JAMA 2014; 311: 2387–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casaburi R, Bhasin S, Cosentino L, Porszasz J, Somfay A, Lewis MI, et al. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 170: 870–878. [DOI] [PubMed] [Google Scholar]
- 11. Bhasin S, Jasuja R. Selective androgen receptor modulators as function promoting therapies. Curr Opin Clin Nutr Metab Care 2009; 68: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette AM, et al. Adverse events associated with testosterone administration. N Engl J Med 2010; 363: 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Corona G, Maseroli E, Rastrelli G, Isidori AM, Sforza A, Mannucci E, et al. Cardiovascular risk associated with testosterone‐boosting medications: a systematic review and meta‐analysis. Expert Opin Drug Saf 2014; 13: 1327–1351. [DOI] [PubMed] [Google Scholar]
- 14. Kloner RA, Carson C 3rd, Dobs A, Kopecky S, Mohler ER 3rd. Testosterone and cardiovascular disease. J Am Coll Cardiol 2016; 67: 545–557. [DOI] [PubMed] [Google Scholar]
- 15. Snyder PJ, Bhasin S, Cunningham GR, Matsumoto AM, Stephens‐Shields AJ, Cauley JA, et al. Effects of testosterone treatment in older men. N Engl J Med 2016; 374: 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Budoff MJ, Ellenberg SS, Lewis CE, Mohler ER 3rd, Wenger NK, Bhasin S, et al. Testosterone treatment and coronary artery plaque volume in older men with low testosterone. JAMA 2017; 317: 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Negro‐Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J Clin Endocrinol Metab 1999; 84: 3459–3462. [DOI] [PubMed] [Google Scholar]
- 18. Cadilla R, Turnbull P. Selective androgen receptor modulators in drug discovery: medicinal chemistry and therapeutic potential. Curr Top Med Chem 2006; 6: 245–270. [DOI] [PubMed] [Google Scholar]
- 19. Bhasin S. Selective androgen receptor modulators as function promoting therapies. J Frailty Aging 2015; 4: 121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Narayanan R, Coss CC, Yepuru M, Kearbey JD, Miller DD, Dalton JT. Steroidal androgens and nonsteroidal, tissue‐selective androgen receptor modulator, S‐22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol Endocrinol 2008; 22: 2448–2465. [DOI] [PubMed] [Google Scholar]
- 21. Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol 2009; 16: 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, et al. The selective androgen receptor modulator GTx‐024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double‐blind, placebo‐controlled phase II trial. J Cachexia Sarcopenia Muscle 2011; 2: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobs AS, Boccia RV, Croot CC, Gabrail NY, Dalton JT, Hancock ML, et al. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double‐blind, randomised controlled phase 2 trial. Lancet Oncol 2013; 14: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basaria S, Collins L, Dillon EL, Orwoll K, Storer TW, Miciek R, et al. The safety, pharmacokinetics, and effects of LGD‐4033, a novel nonsteroidal oral, selective androgen receptor modulator, in healthy young men. J Gerontol A Biol Sci Med Sci 2013; 68: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papanicolaou DA, Ather SN, Zhu H, Zhou Y, Lutkiewicz J, Scott BB, et al. A phase IIA randomized, placebo‐controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK‐0773 in female participants with sarcopenia. J Nutr Health Aging 2013; 17: 533–543. [DOI] [PubMed] [Google Scholar]
- 26. Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab 1999; 84: 3666–3672. [DOI] [PubMed] [Google Scholar]
- 27. Anker MS, von Haehling S, Springer J, Banach M, Anker SD. Highlights of the mechanistic and therapeutic cachexia and sarcopenia research 2010 to 2012 and their relevance for cardiology. Int J Cardiol 2013; 162: 73–76. [DOI] [PubMed] [Google Scholar]
- 28. Coss CC, Jones A, Dalton JT. Pharmacokinetic drug interactions of the selective androgen receptor modulator GTx‐024 (Enobosarm) with itraconazole, rifampin, probenecid, celecoxib and rosuvastatin. Invest New Drugs 2016; 34: 458–467. [DOI] [PubMed] [Google Scholar]
- 29. Brambilla DJ, Matsumoto AM, Araujo AB, McKinlay JB. The effect of diurnal variation on clinical measurement of serum testosterone and other sex hormone levels in men. J Clin Endocrinol Metab 2009; 94: 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Minimum Anticipated Biological Effect Level (MABEL) explanation and Inclusion and Exclusion Criteria.