

Abstract
Introduction
Optimal management of soft tissue sarcoma (STS) remains a challenge. A nationwide survey assessed the quality of STS care in the Netherlands, thereby aiming to identify potentialities for improvement through more centralized disease management.
Methods
From the Netherlands Cancer Registry (NCR), data were obtained on 3317 adult STS patients (excluding gastrointestinal stromal tumor, GIST) diagnosed in 2006–2011. Logistic regression models were employed to compare outcomes on selected clinical indicators reflecting prevailing STS guidelines between high-volume (≥10 resections annually) and low-volume (<10 resections) hospitals, between academic and general hospitals, and between sarcoma research centers and other hospitals, adjusted for case mix. Analyses were performed on imputed datasets (m = 50), generated through multiple imputations by chained equations.
Results
Overall, 89% of patients underwent surgical resection. Resection status remained unknown in 24% (excluding those with metastasized disease), and grade was not documented for one-third of tumors. Microscopic residual disease was detected in 20% with an increased risk for older patients, larger and deeply located tumors, and those located in the (retro)peritoneum or upper extremity. Almost half of patients with an R1 resection received adjuvant radiotherapy. Following adjustment for case mix factors, patients treated in high-volume hospitals less often had macroscopic residual disease (R2 resection; adjusted odds ratio: 0.54). A strongly skewed distribution of surgical volumes was observed.
Conclusions
These survey results indicate a potential for improving Dutch STS care. More centralized sarcoma management should improve definitive pathology reporting on tumor characteristics, adherence to treatment guidelines and overall disease outcome.
Electronic supplementary material
The online version of this article (doi:10.1245/s10434-017-6003-3) contains supplementary material, which is available to authorized users.
Soft tissue sarcomas (STS) form a heterogeneous group of tumors deriving from mesenchymal progenitor cells, often showing differentiation towards different mesenchymal cells (e.g., fibrous tissue, adipose tissue, smooth and striated muscle). The etiology remains unknown. They account for 1% of all malignant tumors; incidence rates increase with age and show a slight male predominance.1,2
STS are primarily defined by the local aggressive growth pattern and categorized in more than 50 histological subtypes.3,4 Tumor entities vary substantially in biological behavior and tumor grade indicate tendency for distant metastasis, mainly to the lungs.5,6 Approximately 25% of STS patients develop distant metastases, whereas lymphogenic spread is very rare, affecting only 2–3% of all STS patients.7–9 Besides STS subtype and grade, studies have identified anatomic site, tumor stage, size, and depth as predictors for survival.10 These parameters are included in several nomograms to estimate prognosis regarding local recurrence and survival.11 Hence, obtaining accurate information on these factors is crucial for formulating a multidisciplinary treatment plan.
The diverse presentation and localization of STS contribute to the challenges for optimal patient care. Timely diagnosis, for instance, may be difficult given the benign to malignant ratio of soft tissue tumors that has been estimated as 100:1. Furthermore, because of their rarity, physicians seldom encounter STS patients in their practice. Even if recognized as such, the complexities in the diagnostic workup and treatment of STS require adequate expertise and organization.7
Studies have emphasized the importance of guideline adherence in STS care as it results in better patient outcomes.12 – 14 In the Netherlands, national sarcoma guidelines were established in 1993, with updates in 2003 and 2011.15 Key recommendations (which became affirmed after our study period through the international guidelines issued by the European Society for Medical Oncology, ESMO) concerned the adequate pathology reporting of the main prognostic sarcoma characteristics, complete diagnostic workup of large (>5 cm), deeply located STS (imaging supplemented with pathologic examination) and the use of adjuvant radiotherapy after R1 resection (microscopic residual disease) or tumor spill.16 – 18
The purpose of this nationwide survey was to acquire insight of the performance of hospitals on STS care (excluding gastrointestinal stromal tumor, GIST), thereby identifying reference points for furthering quality of sarcoma care. In addition the potential improvement due to centralization of STS care was explored by comparing performance between high- and low-volume hospitals, academic centers versus general hospitals, and sarcoma research centers versus other hospitals.
Patients and Methods
Database
The Netherlands Cancer Registry (NCR), founded in 1989, includes all newly diagnosed malignancies, currently covering 17 million inhabitants. The main source of notification is the nationwide network and registry of histo- and cytopathology (www.palga.nl) and case ascertainment was achieved by linkage with the central hospital discharge registry. Upon notification, registrars gather data on patient and tumor characteristics and primary treatment modalities by extracting information directly from the hospital files. The NCR reports on national cancer incidence, prevalence, survival and mortality (www.cijfersoverkanker.nl). Consent for the design, data abstraction process, as well as storage protocols for this study was obtained from the supervisory committee of the NCR.
NCR data include patients’ age at diagnosis, histological subtype of sarcoma on the basis of the World Health Organization (WHO) classification 2002, and tumor grade according to the grading system of the French Federation of Cancer Centers (FNCLCC) Sarcoma Group.4,19 Primary sites are translated to ICD-O topography codes and tumor stage (depth and size) is recorded according to the TNM system of the International Union Against Cancer (UICC) supplemented with the Extent of Disease code of the American Surveillance, Epidemiology and End Results (SEER) program if available.20 – 23 Therapy is coded in sequence of administration, with codes differentiating between treatment modalities (surgery, radiotherapy, systemic therapy). In case of surgical treatment, date of resection and residual disease status are recorded.
For this study, data were retrieved on adult STS patients (≥18 years) diagnosed during the period 2006–2011, excluding Kaposi sarcoma, GIST, and sarcoma of the skin (Supplementary Table 1). Hospitals performing sarcoma surgery were classified according to their mean annual number of resections over the total study period as either high-volume (≥10 resections) or low-volume (<10 resections) and in addition hospital type (general vs. academic hospitals) and sarcoma research centers versus other hospitals. Research centers were characterized by participation in the European Organization for Research and Treatment of Cancer (EORTC) sarcoma group, which amongst others implies expertise of dedicated multidisciplinary sarcoma teams and centralized pathology review. These centers are Netherlands Cancer Institute Amsterdam, University Medical Center Rotterdam, University Medical Center Groningen, University Medical Center Nijmegen, and University Medical Center Leiden.
Clinical Indicators
Guideline adherence was evaluated with indicators reflecting quality of STS care and for which data were available in the NCR. The quality of pathology reports was determined by the availability of information on sarcoma subtype, grade, and assessment of residual disease status after surgery.24 In assessing the reports on grading, we excluded sarcoma subtypes that are not graded by definition or for which grading was not recommended: MPNST, angiosarcoma, extraskeletal myxoid chondrosarcoma, alveolar soft part sarcoma, clear cell sarcoma, and epithelioid sarcoma.19 Histomorphological codes M8800–M8806 were considered sarcoma lacking specific subtyping, and the availability of grade was assessed separately in the subgroup of liposarcoma (excluding well-differentiated tumors), fibrosarcoma, and leiomyosarcoma. In rating adequate reporting of residual disease, M1 disease and retroperitoneal tumors were excluded from the analyses.
The quality of the diagnostic workup of STS was evaluated by estimating the proportion of possible “whoops” resections. Although the NCR data did not distinguish between planned and unplanned procedures, potentially unplanned procedures were defined as resections of either large (>5 cm) or deep tumors (located beneath the superficial fascia, or with invasion of or through the fascia) without prior histopathologic information. In these cases, the date of first histopathologic confirmation coincided with the date of surgery.
In evaluating the use of adjuvant therapy, analysis was focused on the proportion of patients receiving radiation therapy. In particular, the prevalent guidelines recommend provision of adjuvant radiotherapy to patients with R1 resections, irrespective of their tumors’ grade. For the evaluation of adjuvant treatment, cases with distant disease and retroperitoneal tumors were excluded. In addition, overall 5-year survival rates for high-grade, nonmetastasized tumors in surgically resected patients were estimated.
Statistical Analyses
Resection rates and rates of R1 resections were tabulated by subgroups of patient and STS characteristics, and differences for significance were tested by χ 2 tests. Potential prognostic factors for R1 resections were selected for evaluation in multivariable logistic regression on the basis of p < 0.1 in the univariable analyses. Odds ratios were calculated together with 95% likelihood ratio confidence intervals.
Performances on most clinical indicators were analyzed as binary variables and logistic regressions were again applied to estimate the impact of hospital types on management of STS patients. Overall survival rates and hazard ratios were estimated in proportional hazards models. To account for missing data, multiple imputations were performed by chained equations under the assumption of missingness being random, thereby creating 50 data sets for each estimation. In addition to crude pooled estimates, we also provided odds and hazard ratios based on the imputed data adjusted for relevant case mix factors. All tests were two-sided and p < 0.05 was considered statistically significant. Statistical analyses were performed using Stata (version 14.0; StataCorp, College Station, TX).
Results
Patient and Tumor Characteristics
In total 3317 patients, 1830 men (55%) and 1487 women (45%), were diagnosed with a primary STS (Table 1). The median age was 63 (interquartile range 50–75) years; 47% was >65 years. Tumors were mostly located in the extremities (47%), e.g., lower extremity (34%) and upper extremity (13%), followed by the trunk (36%), head and neck region (11%), and retroperitoneum (7%). The majority of tumors were high-grade (54%). In more than a quarter of cases (28%), no information on grade could be retrieved from the pathology report. Almost one-third of STS (32%) was considered small (<5 cm), 61% larger than 5 cm, and in 7% the tumor size could not be retrieved from the clinical or pathology report. Superficially and deeply located tumors were approximately evenly distributed (46 and 43%, respectively), whereas depth was not reported in 12%. Metastatic disease was encountered in 14%. Liposarcoma (20%) and leiomyosarcoma (21%) comprised the most prevalent histology (Table 2).
Table 1.
General characteristics and resection rates for adult patients (≥18 years) diagnosed with a soft tissue sarcoma (STS) in the Netherlands during the time period 2006–2011
| Total (n = 3317) | Resection (n = 2698; 81.3%) | R1 resection (n = 393; 20.4%)* | ||||||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | p | n | % | p | |
| Sex | 0.10 | 0.20 | ||||||
| Male | 1830 | 55.1% | 1507 | 55.9% | 205 | 52.2% | ||
| Female | 1487 | 44.9% | 1191 | 44.1% | 188 | 47.8% | ||
| Age at diagnosis (year) | <0.00 | <0.00 | ||||||
| 18–49 | 794 | 23.9% | 691 | 25.6% | 85 | 21.6% | ||
| 50–64 | 965 | 29.1% | 807 | 29.9% | 99 | 25.2% | ||
| 65–79 | 1036 | 31.2% | 823 | 30.5% | 130 | 33.1% | ||
| ≥80 | 522 | 15.7% | 377 | 14.0% | 79 | 20.1% | ||
| Median (interquartile range) | 63 (50–75) | 62 (49–74) | 66 (52–77) | |||||
| Primary tumor site | <0.00 | <0.00 | ||||||
| Head and neck | 354 | 10.7% | 293 | 10.9% | 44 | 11.2% | ||
| Trunk | 1192 | 35.9% | 856 | 31.7% | 98 | 24.9% | ||
| (Retro)peritoneum | 227 | 6.8% | 169 | 6.3% | 39 | 9.9% | ||
| Extremity | 1544 | 46.6% | 1380 | 51.2% | 212 | 53.9% | ||
| Upper | 431 | 13.0% | 392 | 14.5% | 75 | 19.1% | ||
| Lower | 1113 | 33.6% | 988 | 36.6% | 137 | 34.9% | ||
| Tumor grade | <0.00 | 0.95 | ||||||
| Low grade | 566 | 17.1% | 528 | 19.6% | 76 | 19.3% | ||
| High grade | 1838 | 55.4% | 1526 | 56.6% | 246 | 62.6% | ||
| Unknown | 913 | 27.5% | 644 | 23.9% | 71 | 18.1% | ||
| Tumor size** | <0.00 | <0.00 | ||||||
| ≤5 cm | 933 | 32.4% | 892 | 38.2% | 120 | 30.5% | ||
| >5 cm | 1752 | 60.9% | 1334 | 57.2% | 262 | 66.7% | ||
| Unknown | 193 | 6.7% | 107 | 4.6% | 11 | 2.8% | ||
| Depth of tumor** | <0.00 | 0.01 | ||||||
| Superficial | 1437 | 45.7% | 1355 | 52.5% | 179 | 45.6% | ||
| Deep | 1348 | 42.9% | 996 | 38.6% | 178 | 45.3% | ||
| Unknown | 360 | 11.5% | 232 | 9.0% | 36 | 9.2% | ||
| Stage | <0.00 | – | ||||||
| Localized disease | 2838 | 85.6% | 2525 | 93.6% | 393 | 100.0% | ||
| Distant metastases | 479 | 14.4% | 173 | 6.4% | – | – | ||
* Excluding R2 resections, cases for which residual disease could not be determined (RX) and metastatic disease
** Excluding cases for which extent of disease (EOD) stage could be determine
Table 2.
STS subtypes diagnosed in adult patients (≥18 years) in the Netherlands during the time period 2006–2011
| Sarcoma subtype (WHO 2002) | Total | Median age (interquartile range) | Male/female | |
|---|---|---|---|---|
| n | % | Year | %/% | |
| Liposarcoma | 668 | 20.1 | 60 (49–71) | 59.6/40.4 |
| Well-differentiated liposarcoma | 256 | 7.7 | 61 (53–71) | 58.6/41.4 |
| Myxoid liposarcoma | 158 | 4.8 | 46 (37–60) | 57.0/43.0 |
| Round cell liposarcoma | 10 | 0.3 | 47 (44–52) | 90.0/10.0 |
| Pleomorphic liposarcoma | 48 | 1.4 | 67 (60–76) | 58.3/41.7 |
| Dedifferentiated liposarcoma | 150 | 4.5 | 65 (56–73) | 59.3/40.7 |
| Mixed-type liposarcoma | 8 | 0.2 | 65 (48–74) | 75.0/25.0 |
| Liposarcoma nos | 38 | 1.1 | 70 (57–79) | 68.4/31.6 |
| Fibrosarcoma | 381 | 11.5 | 65 (55–76) | 55.6/44.4 |
| Well-differentiated fibrosarcoma | 83 | 2.5 | 60 (48–74) | 45.8/54.2 |
| Conventional fibrosarcoma | 66 | 2.0 | 65 (57–77) | 56.1/43.9 |
| Poorly differentiated fibrosarcoma | 135 | 4.1 | 68 (61–80) | 56.3/43.7 |
| Fibrosarcoma nos | 97 | 2.9 | 64 (48–75) | 62.9/37.1 |
| Leiomyosarcoma | 701 | 21.1 | 64 (53–75) | 56.5/43.5 |
| Well-differentiated leiomyosarcoma | 148 | 4.5 | 61 (50–73) | 58.8/41.2 |
| Conventional leiomyosarcoma | 180 | 5.4 | 64 (53–75) | 55.0/45.0 |
| Poorly differentiated/plesiomorphic/epithelioid leiomyosarcoma | 137 | 4.1 | 64 (57–75) | 51.8/48.2 |
| Leiomyosarcoma nos | 236 | 7.1 | 67 (54–76) | 58.9/41.1 |
| Rhabdomyosarcoma | 91 | 2.7 | 56 (42–65) | 67.0/33.0 |
| (Embryonal) rhabdomyosarcoma | 40 | 1.2 | 53 (35–64) | 75.0/25.0 |
| Alveolar rhabdomyosarcoma | 13 | 0.4 | 26 (21–42) | 53.8/46.2 |
| Pleomorphic rhabdomyosarcoma | 38 | 1.1 | 63 (54–71) | 63.2/36.8 |
| Epithelioid hemangioendothelioma | 15 | 0.5 | 48 (36–59) | 40.0/60.0 |
| Angiosarcoma | 199 | 6.0 | 68 (61–79) | 33.2/66.8 |
| Synovial sarcoma | 111 | 3.3 | 45 (32–59) | 55.0/45.0 |
| Malignant peripheral nerve sheath tumor (MPNST) | 165 | 5.0 | 50 (36–67) | 55.8/44.2 |
| Malignant fibrous histiocytoma (MFH)/pleomorphic undifferentiated sarcoma (PUS) | 338 | 10.2 | 71 (60–81) | 54.7/45.3 |
| Other sarcoma | 648 | 19.5 | 64 (49–76) | 54.5/45.5 |
| Total | 3317 | 100.0 | 63 (50–75) | 55.2/44.8 |
Most patients underwent a resection (81%). Patients who did not underwent surgery were relatively older (median age 69 vs. 62 years), more often a tumor in the trunk (54 vs. 32%), and less often in the extremities (26 vs. 51%). Overall, resected tumors exhibited more favorable characteristics: lower grade, relatively smaller, more often superficially located and mostly localized disease. Surgery was performed in 14% of initial stage IV disease. Forty percent of the 1081 operated patients received radiation therapy, whereas 191 patients received chemotherapy (7%) (data not shown).
One-fifth of the resections were R1 resections (20%), thereby excluding cases with macroscopic and unknown residual disease and those primarily diagnosed with distant disease. R1 resections occurred more often in elderly patients and with tumors located in the (retro)peritoneum or upper extremity and less often in the trunk and lower extremity. In addition, surgery for larger STS and deeply located tumors showed an elevated risk of positive resection margins. Among patients who had an R1 resection, the proportion receiving adjuvant radiotherapy was 47%.
Hospital Characteristics
During the study period, diagnostic work and resection of STS was performed in 96 hospitals. Among these were eight academic hospitals and one cancer center (classified in the analyses as academic hospital); five of these were considered sarcoma research centers. A strongly skewed distribution of case volumes by hospital was observed (Fig. 1). Overall, 12% of hospitals accounted for half of all STS resections; this proportion decreased from 16% in 2006 to 10% in 2011. Furthermore, 75% of resections were performed in one-third of hospitals, whereas 90% were performed in almost two-thirds. Sarcoma research centers represented the largest surgical volume.
Fig. 1.

Distribution of patients over hospitals performing STS surgery during the time period 2006–2011
Over time, no trend was detected for whether or not patients underwent surgery in high-volume hospitals. However, we did observe a significant trend within the high-volume group. While the proportion of patients treated in hospitals performing a yearly total of 10–19 resections decreased from 27% in 2006 to 2% in 2011, there was an increase for those treated in hospitals performing 20 or more resections annually (p = 0.01; Fig. 2a). Academic hospitals accounted for 46% of STS resections, and this proportion did not show a trend over time (Fig. 2b). In contrast, the proportion of patients treated in sarcoma research centers increased from 28 to 41% (p < 0.01; Fig. 2c).
Fig. 2.
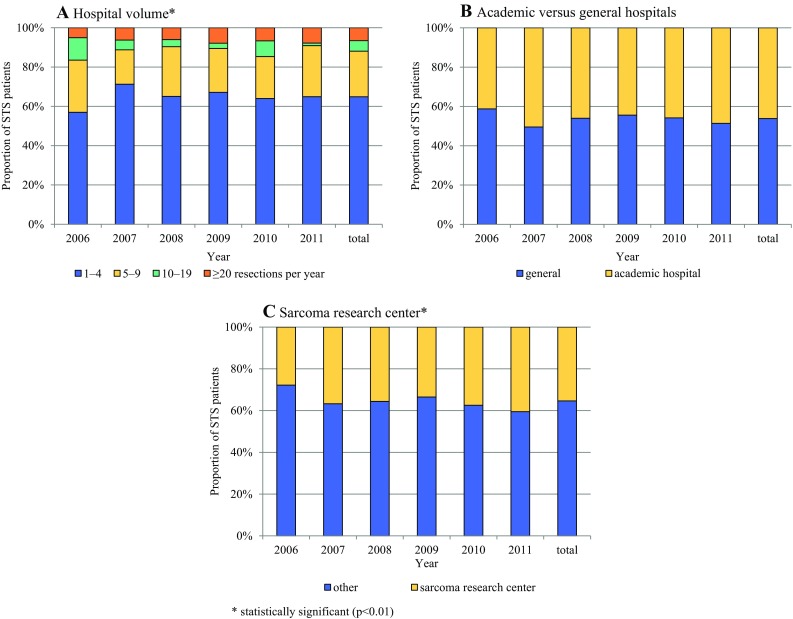
Trends over time according to hospital volume and hospital type with respect to STS surgery
Clinical Indicators
With respect to pathology reports, sarcoma subtype remained unknown in 20% of patients, tumor grade in 28% (when restricted to the subset of liposarcoma, fibrosarcoma, and leiomyosarcoma cases: 24%) and resection status in 24% (Table 3). High-volume pathology laboratories (compared with low-volume) and those located in academic hospitals (compared to those in general hospitals) and sarcoma research centers (compared with those in other hospitals) performed better in reporting tumor grade, resection status, whereas no difference was observed for reporting sarcoma subtype.
Table 3.
Variation in clinical indicators by hospital volume and hospital type, with estimations based on imputed data and adjusted for case mix factors (patients’ age, primary tumor site, sarcoma grade, size, and depth, and resection status if relevant)
| Overall | Hospital volume (≥10 cases vs. <10 cases) | Hospital type (academic vs. general) | Sarcoma research center (yes vs. no) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Crude | Adjusted | Crude | Adjusted | Crude | Adjusted | |||||
| % | OR | OR | 95% CI | OR | OR | 95% CI | OR | OR | 95% CI | |
| Pathology report: subtype** | ||||||||||
| Unknown subtype | 20.4% | 1.05 | 1.08 | (0.89–1.32) | 0.93 | 0.99 | (0.81–1.21) | 1.06 | 1.16 | (0.93–1.45) |
| Pathology report: grade** | ||||||||||
| Unknown grade | 27.5% | 0.85 | 0.96 | (0.80–1.13) | 0.53* | 0.65* | (0.53–0.78) | 0.54* | 0.68* | (0.54–0.84) |
| Liposarcoma, fibrosarcoma, leiomyosarcoma, excluding well-differentiated liposarcoma | 24.0% | 0.83 | 0.93 | (0.71–1.21) | 0.56* | 0.65* | (0.48–0.88) | 0.56* | 0.65* | (0.46–0.93) |
| Residual disease following resection | ||||||||||
| Unknown resection status** | 24.0% | 0.87 | 0.99 | (0.81–1.21) | 0.56* | 0.70* | (0.56–0.87) | 0.58* | 0.73* | (0.57–0.94) |
| Microscopic residual disease (R1) excluding M1 and (retro)peritoneal tumors | 20.0% | 1.23 | 1.22 | (0.95–1.57) | 1.41* | 1.41* | (1.09–1.82) | 1.53* | 1.56* | (1.22–2.00) |
| Macroscopic residual disease (R2) excluding M1 and (retro)peritoneal tumors | 3.0% | 1.00 | 0.67 | (0.37–1.21) | 1.24 | 0.76 | (0.42–1.38) | 1.23 | 0.94 | (0.52–1.69) |
| Possible “whoops” resection | ||||||||||
| Resection of deep or large tumors (>5 cm) without prior histopathologic confirmation | 32.0% | 0.19* | 0.19* | (0.15–0.24) | 0.18* | 0.18* | (0.14–0.23) | 0.22* | 0.20* | (0.16–0.26) |
| Radiotherapy excluding M1 and (retro) peritoneal tumors | ||||||||||
| No radiotherapy neoadjuvant or adjuvant | 58.1% | 0.39* | 0.56* | (0.47–0.68) | 0.36* | 0.53* | (0.43–0.65) | 0.38* | 0.53* | (0.43–0.64) |
| No adjuvant radiotherapy following R1 resection | 52.6% | 0.43* | 0.50* | (0.31–0.80) | 0.46* | 0.56* | (0.35–0.90) | 0.42* | 0.47* | (0.29–0.74) |
| Crude | Adjusted*** | Crude | Adjusted*** | Crude | Adjusted*** | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| % | HR | HR | 95%CI | HR | HR | 95% CI | HR | HR | 95%CI | |
| Overall survival surgically treated patients, excluding low-grade and M1 disease | ||||||||||
| 5-Year postdiagnosis | 61.3% | 1.12 | 1.15 | (0.99–1.34) | 1.15* | 1.14 | (0.97–1.33) | 1.10 | 1.08 | (0.93–1.27) |
CI confidence interval; OR odds ratio
* Statistically significant (p < 0.05)
** Comparison is calculated on the level of pathology laboratory
*** Additionally adjusted for adjuvant treatment
Following adjustment for case mix factors, resection rates of deep or large tumors without prior histopathologic confirmation seemed considerably higher in low-volume hospitals, general hospitals, and nonsarcoma research centers. In academic hospitals and sarcoma research centers, a larger proportion of operations comprised R1 resections.
The odds for sarcoma patients to receive radiotherapy appeared higher when surgery was performed in high-volume hospitals, academic hospitals, and sarcoma research centers. The same was true regarding adjuvant radiotherapy following R1 resection, although this effect was no longer significant between academic and general hospitals after adjustment for case mix factors. No differences in 5-year overall survival rates were detected between hospital categories.
Discussion
This survey indicates a need for improving STS care in the Netherlands, both in acquisition and reporting accurate diagnostic information and providing optimal STS treatment. Important STS tumor characteristics remained unmentioned in one-fifth of the pathology reports. The diagnostic workup of large or deeply located STS was incomplete in almost one-third. More than half of patients did not receive the required adjuvant radiation. This does not preclude, however, that patients and physicians may have opted for active surveillance to monitor the tumor’s biological behavior, provided that salvage surgery could be performed in the event of a recurrence.
For particular indicators (grade, resection status, “whoops” resection, and delivery of radiotherapy), outcomes were better when diagnostic and treatment occurred in high-volume hospitals, academic hospitals, and sarcoma research centers compared with other hospitals. Surgery in high-volume hospitals less often resulted in R2 resections. The introduction of evidence-based national STS guidelines produced little effect in contrast to bone sarcoma guidelines.25 Compliance to these guidelines was mostly moderate, particularly in smaller hospitals.14 Earlier surveys performed in France and the UK also showed disappointing compliance rates regarding STS guidelines, with overall estimates approximating 50% at best, and rates having remained fairly constant over time. 26 – 30 Such situations do appear to be successfully ameliorated by regional initiatives, for instance directed at improving the quality of pathology reports.13
To be sure, strict conformity to guidelines far from qualifies as optimal STS care. For instance, to preserve functionality of limbs affected by STS, positive margins may be justifiable.31 This would explain our counterintuitive finding that R1 resections more often occurred in academic hospitals and in sarcoma research centers: an R1 resection oftentimes represents the only treatment option for locally advanced STS.32 In case of residual disease, irrespective of it being microscopic or macroscopic cancer, an attempt to obtain adequate margins through reexcision should be evaluated, and the same holds for administration of adjuvant multimodality therapy. As for the observed omittance of adjuvant radiotherapy following an R1 resection, we could not rule out that wait-and-see policies were in fact pursued in such cases. In addition, treatment may have been withheld for both disease and patient-related factors (performance status, comorbidity).33
These considerations point to the limitations of this survey, the lack of information on patients’ performance status and comorbidities, the intent of treatment provided as well as the reasons for treatment omittance. As a consequence, case mix corrections may not have adequately accounted for the full variance of baseline characteristics across different hospital categories. Also, we were not able to conclusively infer from the cancer registry data whether resections without prior histopathologic confirmation indeed concerned unplanned excisions (“whoops” procedures). In addition, because no central pathology review was performed, misdiagnosed cases may have been included in the analyses. Nevertheless, the results seem largely valid for assessing STS care in the Netherlands; the analyses were performed on an unselected sample of Dutch STS patients, and main outcomes are consistent with other reports.
In describing the prospects for improving STS care, several challenges remain important to emphasize. Most importantly, with an incidence rate of approximately 3 cases per 100,000 inhabitants (European Standardized Rate), STS represent a group of uncommon tumors that show diverse presentations.1 A general practitioner in the Netherlands encounters on average one STS patient every twenty years and a general or orthopedic surgeon one in every four years. At the same time, as mentioned, benign soft-tissue tumors are more than 100 times as common as STS.34, 35 Misdiagnosis and inadequate treatment are likely to occur.
It is clear that optimal STS care requires extensive, multidisciplinary expertise, and well-organized care processes, and studies have confirmed benefit for management in sarcoma centers, or in hospitals working within specialized, dedicated STS networks.36 – 39
All in all, improvements in STS care may be achieved by having management primarily carried out in reference centers for sarcomas. Within reference networks, centers may share their multidisciplinary expertise and treat large numbers of patients. In this setting, centralized referral should be pursued as early as possible, preferably at the time of the clinical diagnosis of a suspected sarcoma. In practice, referral of all patients with a lesion likely to be a sarcoma would be recommended. This would mean referring all patients with an unexplained deep mass of soft tissues, or with a superficial lesion of soft tissues having a diameter of >5 cm.17 Although more centralized management may come with the cost of large numbers of patients being redirected to centers for benign abnormalities, potentially causing delayed treatment of those with proven STS, some have reported encouraging outcomes for more stringent referral patterns.40,41 In the Netherlands, national referral guidelines are already well-established for bone sarcoma.
While specific criteria for reference centers may vary from country to country, centralizing STS care should be based, among others, on the availability of state-of-the-art facilities for STS diagnostics and treatment, multidisciplinary expertise (employed in weekly tumor boards discussing new patients, for instance), and larger volumes of patients. Quality of STS care delivered should be monitored and outcomes reported on a regular basis. Also, centers are involved in ongoing clinical trials, for which patients’ enrollment is common.
In the Netherlands, centralizing STS care in five dedicated sarcoma centers, in analogy with the four national bone sarcoma centers, may enhance the development and implementation of new diagnostics (e.g., imaging technologies and improved STS subtyping according to tumors’ molecular makeup) and therapeutic strategies (introduction of new combined, pathway driven treatment modalities). Moreover, restricting the number of centers should facilitate a nationwide pathology review and reporting standard, and foster focused research initiatives on the mentioned themes. Hence, centralization may prove most beneficial for establishing disease-orientated sarcoma care, including participation in trials, which eventually should lead not only to more favorable clinical outcomes, but also to more efficient and cost-effective STS.42 It is time for integrated sarcoma care, in the Netherlands and in Europe.43
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgment
The authors thank the registrars of the Netherlands Comprehensive Cancer Organization (IKNL) for collecting the data in the Netherlands Cancer Registry (NCR) database, and the Dutch Cancer Society for supporting this survey. The survey formed part of the joint national program “The quality of cancer care initiative” of the Dutch Cancer Society and the Netherlands Comprehensive Cancer Organization (IKNL) under the chairmanship of Prof. Dr. C.J.H. van de Velde.
Disclosure
No conflict of interest declared.
Footnotes
Part of these results were presented at the European Cancer Congress, Amsterdam 2013.
Electronic supplementary material
The online version of this article (doi:10.1245/s10434-017-6003-3) contains supplementary material, which is available to authorized users.
References
- 1.Netherlands Cancer Registry Utrecht: IKNL; 2016 [cited 2016 March 20]. www.cijfersoverkanker.nl.
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F. Tumours of soft tissue and bone: pathology and genetics. World Health Organization Classification of Tumours. Lyon: IARC Press; 2013.
- 4.Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med. 2006;130(10):1448–1453. doi: 10.5858/2006-130-1448-GOSTSR. [DOI] [PubMed] [Google Scholar]
- 5.Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchere D, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91(10):1914–1926. doi: 10.1002/1097-0142(20010515)91:10<1914::AID-CNCR1214>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, de Mascarel A, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33(1):37–42. doi: 10.1002/ijc.2910330108. [DOI] [PubMed] [Google Scholar]
- 7.Weitz J, Antonescu CR, Brennan MF. Localized extremity soft tissue sarcoma: improved knowledge with unchanged survival over time. J Clin Oncol. 2003;21(14):2719–2725. doi: 10.1200/JCO.2003.02.026. [DOI] [PubMed] [Google Scholar]
- 8.Andreou D, Boldt H, Werner M, Hamann C, Pink D, Tunn PU. Sentinel node biopsy in soft tissue sarcoma subtypes with a high propensity for regional lymphatic spread–results of a large prospective trial. Ann Oncol. 2013;24(5):1400–1405. doi: 10.1093/annonc/mds650. [DOI] [PubMed] [Google Scholar]
- 9.Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217(1):72–77. doi: 10.1097/00000658-199301000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blay JY, Sleijfer S, Schoffski P, Kawai A, Brodowicz T, Demetri GD, et al. International expert opinion on patient-tailored management of soft tissue sarcomas. Eur J Cancer. 2014;50(4):679–689. doi: 10.1016/j.ejca.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 11.Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416–421. doi: 10.1097/SLA.0000000000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossi CR, Vecchiato A, Mastrangelo G, Montesco MC, Russano F, Mocellin S, et al. Adherence to treatment guidelines for primary sarcomas affects patient survival: a side study of the European CONnective TIssue CAncer NETwork (CONTICANET) Ann Oncol. 2013;24(6):1685–1691. doi: 10.1093/annonc/mdt031. [DOI] [PubMed] [Google Scholar]
- 13.Jansen-Landheer ML, Krijnen P, Oostindier MJ, Kloosterman-Boele WM, Noordijk EM, Nooij MA, et al. Improved diagnosis and treatment of soft tissue sarcoma patients after implementation of national guidelines: a population-based study. Eur J Surg Oncol. 2009;35(12):1326–1332. doi: 10.1016/j.ejso.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Nijhuis PH, Schaapveld M, Otter R, Hoekstra HJ. Soft tissue sarcoma–compliance with guidelines. Cancer. 2001;91(11):2186–2195. doi: 10.1002/1097-0142(20010601)91:11<2186::AID-CNCR1248>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 15.Practice guideline ‘Soft tissue sarcoma’ 2.0 Utrecht: IKNL; 2011 [updated 2011 November 1; cited 2016 March 24]. http://www.oncoline.nl/wekedelentumoren.
- 16.ESMO/European Sarcoma Network Working Group. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(7)vii92–9. [DOI] [PubMed]
- 17.Casali PG, Blay JY, Bertuzzi A, Bielack S, Bjerkehagen B, Bonvalot S, Boukovinas I, Bruzzi P, Dei Tos AP, Dileo P, Eriksson M. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(3):12. doi: 10.1093/annonc/mdu254. [DOI] [PubMed] [Google Scholar]
- 18.Koshy M, Rich SE, Mohiuddin MM. Improved survival with radiation therapy in high-grade soft tissue sarcomas of the extremities: a SEER analysis. Int J Radiat Oncol Biol Phys. 2010;77(1):203–209. doi: 10.1016/j.ijrobp.2009.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fletcher CDM, Unni KK, Mertens F. Tumours of Soft Tissue and Bone: Pathology and Genetics. World Health Organization Classification of Tumours. Lyon: IARC Press; 2002.
- 20.Fritz A, Percy C, Jack A, Shanmugaratnam K, Sobin L, Parkin DM, et al. ICD-O International Classification of Diseases for Oncology. Geneva: World Health Organization; 2000. [Google Scholar]
- 21.Sobin LH, Gospodarowicz MK, Wittekind C. TNM classification of malignant tumours. International Union Against Cancer (UICC) Chichester: Wiley; 2010. [Google Scholar]
- 22.Sobin LH, Wittekind C. TNM classification of malignant tumours International Union Against Cancer (UICC) New York: Wiley; 2002. [Google Scholar]
- 23.Young JLJ, Roffers SD, Ries LAG, Fritz AG, Hurlbut AA, editors. SEER summary staging manual—2000: codes and coding instructions. Bethesda: National Cancer Institute; 2001. [Google Scholar]
- 24.Association of Directors of Anatomic and Surgical Pathology Recommendations for the reporting of soft tissue sarcoma. Virchows Arch. 1999;434(3):187–191. doi: 10.1007/s004280050326. [DOI] [PubMed] [Google Scholar]
- 25.Ligier K, Maynou C, Leroy X, Robin YM, Martin P, Clisant S, et al. Improvement of the initial management of sarcomas after the dissemination of evidence-based guidelines depends on the primary sarcoma location: a population-based study. BMC Cancer. 2015;15:218. doi: 10.1186/s12885-015-1225-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathoulin-Pelissier S, Chevreau C, Bellera C, Bauvin E, Saves M, Grosclaude P, et al. Adherence to consensus-based diagnosis and treatment guidelines in adult soft-tissue sarcoma patients: a French prospective population-based study. Ann Oncol. 2014;25(1):225–231. doi: 10.1093/annonc/mdt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrier L, Buja A, Mastrangelo G, Vecchiato A, Sandona P, Ducimetiere F, et al. Clinicians’ adherence versus non adherence to practice guidelines in the management of patients with sarcoma: a cost-effectiveness assessment in two European regions. BMC Health Serv Res. 2012;12:82. doi: 10.1186/1472-6963-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glencross J, Balasubramanian SP, Bacon J, Robinson MH, Reed MW. An audit of the management of soft tissue sarcoma within a health region in the UK. Eur J Surg Oncol. 2003;29(8):670–675. doi: 10.1016/S0748-7983(03)00134-3. [DOI] [PubMed] [Google Scholar]
- 29.Clasby R, Tilling K, Smith MA, Fletcher CD. Variable management of soft tissue sarcoma: regional audit with implications for specialist care. Br J Surg. 1997;84(12):1692–1696. doi: 10.1002/bjs.1800841213. [DOI] [PubMed] [Google Scholar]
- 30.Ray-Coquard I, Thiesse P, Ranchere-Vince D, Chauvin F, Bobin JY, Sunyach MP, et al. Conformity to clinical practice guidelines, multidisciplinary management and outcome of treatment for soft tissue sarcomas. Ann Oncol. 2004;15(2):307–315. doi: 10.1093/annonc/mdh058. [DOI] [PubMed] [Google Scholar]
- 31.Kandel R, Coakley N, Werier J, Engel J, Ghert M, Verma S. Surgical margins and handling of soft-tissue sarcoma in extremities: a clinical practice guideline. Curr Oncol. 2013;20(3):54. doi: 10.3747/co.20.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis JJ, Leung D, Espat J, Woodruff JM, Brennan MF. Effect of reresection in extremity soft tissue sarcoma. Ann Surg. 2000;231(5):655–663. doi: 10.1097/00000658-200005000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farshadpour F, Schaapveld M, Suurmeijer AJ, Wymenga AN, Otter R, Hoekstra HJ. Soft tissue sarcoma: why not treated? Crit Rev Oncol Hematol. 2005;54(1):77–83. doi: 10.1016/j.critrevonc.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Rydholm A, Berg NO, Persson BM, Akerman M. Treatment of soft-tissue sarcoma should be centralised. Acta Ortho Scand. 1983;54(3):333–339. doi: 10.3109/17453678308996581. [DOI] [PubMed] [Google Scholar]
- 35.Myhre-Jensen O. A consecutive 7-year series of 1331 benign soft tissue tumours. Clinicopathologic data. Comparison with sarcomas. Acta Ortho Scand. 1981;52(3):287–293. doi: 10.3109/17453678109050105. [DOI] [PubMed] [Google Scholar]
- 36.Heudel PE, Cousin P, Lurkin A, Cropet C, Ducimetiere F, Collard O, et al. Territorial inequalities in management and conformity to clinical guidelines for sarcoma patients: an exhaustive population-based cohort analysis in the Rhone-Alpes region. Int J Clin Oncol. 2014;19(4):744–752. doi: 10.1007/s10147-013-0601-2. [DOI] [PubMed] [Google Scholar]
- 37.Cutts S, Andrea F, Piana R, Haywood R. The management of soft tissue sarcomas. Surgeon. 2012;10(1):25–32. doi: 10.1016/j.surge.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Abellan JF, Lamo de Espinosa JM, Duart J, Patino-Garcia A, Martin-Algarra S, Martinez-Monge R, et al. Nonreferral of possible soft tissue sarcomas in adults: a dangerous omission in policy. Sarcoma. 2009;2009:827912. doi: 10.1155/2009/827912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez JC, Perez EA, Moffat FL, Livingstone AS, Franceschi D, Koniaris LG. Should soft tissue sarcomas be treated at high-volume centers? An analysis of 4205 patients. Ann Surg. 2007;245(6):952–958. doi: 10.1097/01.sla.0000250438.04393.a8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pencavel TD, Strauss DC, Thomas GP, Thomas JM, Hayes AJ. Does the two-week rule pathway improve the diagnosis of soft tissue sarcoma? A retrospective review of referral patterns and outcomes over five years in a regional sarcoma centre. Ann R Coll Surg Engl. 2010;92(5):417–421. doi: 10.1308/003588410X12664192075972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Styring E, Billing V, Hartman L, Nilbert M, Seinen JM, Veurink N, et al. Simple guidelines for efficient referral of soft-tissue sarcomas: a population-based evaluation of adherence to guidelines and referral patterns. J Bone Joint Surg Am. 2012;94(14):1291–1296. doi: 10.2106/JBJS.K.01271. [DOI] [PubMed] [Google Scholar]
- 42.Seinen J, Ikkersheim D, Heineman EF, Hoekstra HJ. Sarcoomzorg UMCG overstijgt de afdeling. Medisch Contact. 2012;67(9):547–549. [Google Scholar]
- 43.Blay J-Y, Le Cesne A, Penel N, Bompas E, Duffaud F, Chevreau C, et al. Improved sarcoma management in a national network of reference centers: analysis of the NetSarc network on 13,454 patients treated between 2010 and 2014. ASCO Meet Abstr. 2016;34(15 suppl):11013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.