

The nervous system shows a tremendous amount of plasticity. More recently there has been an appreciation for compensatory actions that stabilize output in the face of perturbations to normal activity. In this study we demonstrate that neurons of the crustacean stomatogastric ganglion generate apparent compensatory responses to loss of peptide neuromodulation, adding to the repertoire of mechanisms by which the stomatogastric nervous system can regulate and stabilize its own output.
Keywords: stomatogastric nervous system, homeostatic plasticity, neuropeptide, neuromodulation
Abstract
We studied the changes in sensitivity to a peptide modulator, crustacean cardioactive peptide (CCAP), as a response to loss of endogenous modulation in the stomatogastric ganglion (STG) of the crab Cancer borealis. Our data demonstrate that removal of endogenous modulation for 24 h increases the response of the lateral pyloric (LP) neuron of the STG to exogenously applied CCAP. Increased responsiveness is accompanied by increases in CCAP receptor (CCAPr) mRNA levels in LP neurons, requires de novo protein synthesis, and can be prevented by coincubation for the 24-h period with exogenous CCAP. These results suggest that there is a direct feedback from loss of CCAP signaling to the production of CCAPr that increases subsequent response to the ligand. However, we also demonstrate that the modulator-evoked membrane current (IMI) activated by CCAP is greater in magnitude after combined loss of endogenous modulation and activity compared with removal of just hormonal modulation. These results suggest that both receptor expression and an increase in the target conductance of the CCAP G protein-coupled receptor are involved in the increased response to exogenous hormone exposure following experimental loss of modulation in the STG.
NEW & NOTEWORTHY The nervous system shows a tremendous amount of plasticity. More recently there has been an appreciation for compensatory actions that stabilize output in the face of perturbations to normal activity. In this study we demonstrate that neurons of the crustacean stomatogastric ganglion generate apparent compensatory responses to loss of peptide neuromodulation, adding to the repertoire of mechanisms by which the stomatogastric nervous system can regulate and stabilize its own output.
compensatory plasticity in the nervous system has been the subject of intense study for many years, with homeostatic plasticity of synaptic signaling garnering the lion’s share of the attention (Lee et al. 2014; Turrigiano 2012). For example, bidirectional synaptic scaling is a prevalent strategy for maintaining target levels of activity across nervous systems (Ibata et al. 2008; Lee et al. 2014), and mechanisms vary widely at both the pre- and postsynaptic levels (Maffei and Turrigiano 2008; Turrigiano 2008). There are also abundant examples of homeostatic plasticity of intrinsic excitability, primarily via feedback tuning of ionic conductance relationships to maintain a target level of excitability (Gasselin et al. 2015; Schulz 2006). While substantial progress has been made in our understanding of fundamental cellular properties underlying homeostatic regulation, less is understood about whether and how compensation for loss of neurohormonal inputs occurs in single identified neurons.
Neuromodulation represents an important level of flexibility in circuit function and adaptability, and all circuits likely are modulated by several compounds at all times (Bucher and Marder 2013; Harris-Warrick 2011; Nadim and Bucher 2014). Neuromodulation predominantly acts through second messenger systems activated via G protein-coupled receptors (GPCRs; Civelli 2012) and can be in the form of local but diffuse release from modulatory projection neurons or can act fairly globally in the form of neurohormones. Because modulation can act on multiple aspects of intrinsic excitability and synaptic function across neurons (Bucher and Marder 2013; Nadim and Bucher 2014), compensatory mechanisms for loss or change in modulatory state occur via multiple subcellular targets and often involve plasticity via GPCRs (Cai et al. 2002; Husch et al. 2012; Staunton et al. 1982).
The crustacean stomatogastric nervous system (STNS) has been an important model for the fundamental understanding of neuromodulation for many decades (Daur et al. 2016; Marder and Bucher 2007; Stein 2009). It consists of multiple central pattern generator networks contained within the single stomatogastric ganglion (STG), the most extensively studied being the pyloric network responsible for controlling the filtering apparatus of the stomach (Maynard and Dando 1974). The pyloric rhythm is a triphasic, continuously active motor pattern generated by three main neuron types: the single anterior burster (AB) and paired pyloric dilator (PD) pacemaker neurons and the follower cells, the single lateral pyloric (LP) and multiple pyloric (PY) neurons. Pyloric activity is dependent on neuromodulators reaching the STG (Marder and Eisen 1984; Miller and Selverston 1982), either through descending axons via the stomatogastric nerve (stn) or as neurohormones through the hemolymph (Billimoria et al. 2005; Chen et al. 2009). The majority of neuromodulators affecting STG neurons are neuropeptides (Christie et al. 2010) that have distinct subsets of cell targets but largely converge via GPCR signaling on one modulator-activated inward current, IMI (DeLong et al. 2009; Golowasch and Marder 1992; Swensen and Marder 2000). However, only recently has the first neuropeptide receptor, for the neurohormone crustacean cardioactive peptide (CCAP), been characterized from a molecular and expression perspective in cells of the STG (Garcia et al. 2015)—although new sequencing projects promise to expand the molecular characterization of neuropeptide signaling in this model system (Christie et al. 2015; Northcutt et al. 2016).
The loss of descending modulation to the STG, often referred to as “decentralization,” triggers changes in STG properties that are viewed as homeostatic or compensatory. Pyloric output in some species such as the lobster Jasus lalandii falls completely silent after decentralization and recovers in the absence of modulation days later (Thoby-Brisson and Simmers 1998, 2000). In the crab Cancer borealis, loss of descending modulation causes variable effects on the pyloric output (Hamood et al. 2015) and results in changes in gene expression (Temporal et al. 2012, 2014), intrinsic excitability (Khorkova and Golowasch 2007; Schulz and Lane 2017; Thoby-Brisson and Simmers 2002), and responses to subsequent exogenous application of neuromodulators such as the neuropeptide proctolin (Nahar et al. 2012). However, while compensatory changes in excitability via dopaminergic modulation have been studied extensively in the STG (Krenz et al. 2015; Kvarta et al. 2012; Rodgers et al. 2011a, 2013), less is known about how loss of peptide modulation alters response to subsequent modulation in specific neurons of the STG, and by what mechanism these changes may be acting. In this study, we investigate changes in responses of the LP neuron of the STG to the peptide hormone CCAP as a result of loss of endogenous modulation.
MATERIALS AND METHODS
Animals.
Adult Jonah crabs (C. borealis) were purchased from The Fresh Lobster Company (Gloucester, MA). Animals were kept at 12°C in artificial seawater until ready for use. All dissections were performed in physiological saline (in mM): 440 NaCl, 26 MgCl2 13 CaCl2, 11 KCl, and 10 HEPES, pH 7.4 (all chemicals obtained from Fisher Scientific). The STNS (Fig. 1) of each animal was dissected out and pinned on a Sylgard-coated dish (Dow Corning) containing chilled physiological saline. To impale cells for electrophysiological recordings, the STG was desheathed with a fine tungsten needle or iridectomy scissors (Fine Science Tools).
Fig. 1.
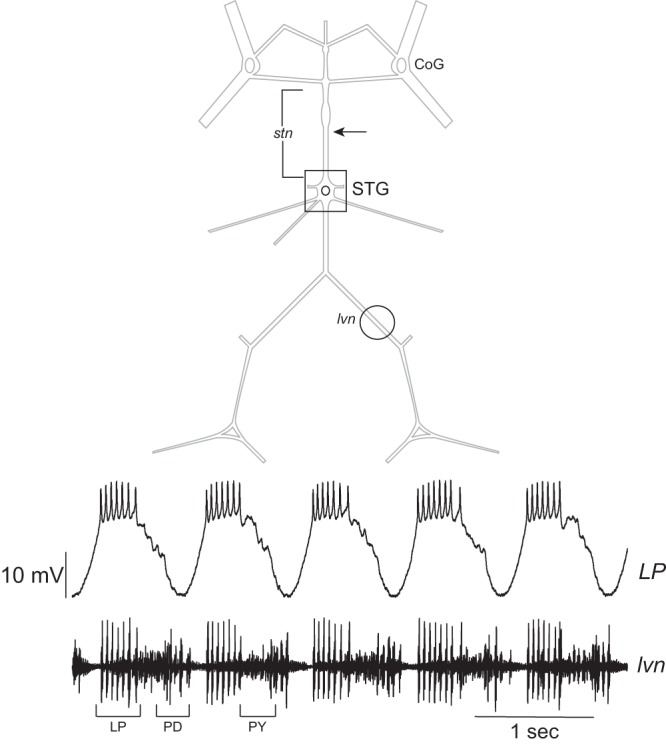
Schematic and representative LP recordings of Cancer borealis stomatogastric ganglion (STG). Simultaneous intracellular recording of a lateral pyloric (LP) neuron and extracellular recording from the lateral ventricular nerve (lvn) showing the triphasic pyloric rhythm. Arrow denotes the approximate location of where the stomatogastric nerve (stn) was cut in preparations that had descending modulation removed. CoG, commissural ganglion; PD, pyloric dilator neuron; PY, pyloric neuron.
Focus on LP as a target neuron and CCAP as a neurohormone.
Because the STG receives neuromodulatory inputs through one primary bundle of axons contained within the stn, it is difficult to limit experimental manipulation to a specific peptide modulator that is delivered via neurons projecting through the stn. However, we can specifically eliminate hormonal modulation by simply culturing intact STNS preparations in physiological saline that is devoid of hormonal modulators. A well-characterized peptide hormone, CCAP, is known to only be delivered to the STG through the hemolymph (Billimoria et al. 2005; Chen et al. 2009; Christie et al. 1995; Li et al. 2003). We can therefore perform experimentally controlled manipulation of hormonal CCAP exposure by simply incubating STNS preparations in physiological saline containing CCAP. Furthermore, CCAP is known to target a very specific subset of cells in the pyloric network, namely LP, the AB neuron, and the inferior cardiac (IC) neuron, whereas other peptide modulators have more diffuse and numerous cellular targets (Swensen and Marder 2001). Finally, CCAP is the only peptide modulator for which a receptor has been identified and characterized at a molecular level in the STG (Garcia et al. 2015). Therefore the use of CCAP in these experiments represents a relatively focal opportunity to study a well-controlled modulatory compound about which we have a good deal of a priori knowledge.
We also decided to focus our studies on a specific cellular target of CCAP in the STG, the LP neuron. LP is one of the few single-numbered cells in the STG, allowing us to look at the entirety of the effects on this specific cell type in each ganglion. It is one of the larger cells in the ganglion and easily identified by a distinctive waveform in intracellular recordings. The output of the LP neurons is easily measured in recordings of the lateral ventricular nerve (lvn), as the LP action potentials are the highest-amplitude spikes in extracellular recordings of pyloric output (see Fig. 1). The activity of LP and its response to CCAP are well described (Garcia et al. 2015; Swensen and Marder 2001; Weimann et al. 1997) and easily quantified as the bursting and spike frequency characteristics of this cell both in the ongoing pyloric rhythm and in response to current injection. Finally, because of the ease of identifying LP neurons and their relatively large somata (Ransdell et al. 2010), harvesting individual LP neurons for molecular analyses is relatively straightforward as well.
Electrophysiological recordings.
Somatic intracellular recording was used to identify neurons. Cell identification was done by matching action potentials and burst phasing from intracellular recordings with extracellular recordings of pyloric motor output (see Fig. 1). Extracellular recordings were obtained with stainless steel pin electrodes in Vaseline wells placed on the lvn (Fig. 1). Extracellular signals were amplified and filtered with a differential AC amplifier (A-M Systems). Intracellular recordings were made with glass electrodes containing 3 M KCl (8–17 MΩ) and Axoclamp 2A amplifiers (Molecular Devices). Data were acquired with Digidata 1440A (Molecular Devices) and Micro1401 mk2 (Cambridge Electronic Design) data acquisition boards and subsequently analyzed in Clampfit (version 10.2; Axon Instruments) or Spike2 (version 7; Cambridge Electronic Design). Current clamp was done with single-electrode impalements in bridge mode and was driven by, and voltages were recorded with, pCLAMP software (version 9.2.1.9; Axon Instruments). Current injections were performed in 1-nA increments from 1 to 5 nA for 2 s. Electrodes were bridged before neurons were impaled.
Experimental groups and LP activity measurements.
Responses of LP to different concentrations of CCAP were measured in five different experimental groups. Acute controls were STNS preparations that were removed from the animal and used within 4 h of the initial dissection. “24-h stn-intact” preparations were dissected out of the animal and maintained in organotypic culture for 24 h with the stn intact. These preparations exhibited normal rhythmic activity for the 24 h before the experiment but experience loss of hormonal modulation for 24 h. “24-h stn-cut” preparations were dissected from the animal, and a Vaseline well was placed on the stn. Tetrodotoxin (TTX; Alomone Laboratories) was used at 10−6 M to block action potentials on the stn before the nerve was cut with scissors approximately midway between the STG and the nerves that branch toward the commissural ganglia (see Fig. 1). These “decentralized” preparations were then maintained in organotypic culture for 24 h before the experiment. The loss of descending neuromodulation often leads to a reduction or loss of rhythmic bursting activity (Hamood et al. 2015). “24-h CCAP-incubated” preparations were treated identically to the 24-h stn-intact preparations but in addition were coincubated in organotypic culture with 10−7 M CCAP for 24 h before the experiment. “24-h cycloheximide (CHX)” preparations were treated identically to the 24-h stn-intact preparations but in addition were coincubated in organotypic culture with 10 µg/ml (Han et al. 2006) CHX (ACROS Organics) for 24 h before the experiment. “24-h CCAP+TTX-incubated” preparations were used only for CCAPr mRNA analyses, as their activity is compromised by addition of TTX, which does not wash out completely. These preparations were coincubated for 24 h in 10−7 M CCAP and 10−7 M TTX to decouple effects of CCAP application and activity on CCAPr mRNA levels.
Regardless of the experimental group a preparation belonged to, all CCAP response measurements were conducted under identical conditions subsequent to the experimental manipulations described above. All experimental groups were returned to physiological saline, and the stn (if still intact) was blocked with TTX (10−6 M) and cut. This decentralized state ensured that the only modulator responses measured were those to CCAP added under experimental control. If a preparation was incubated in either CCAP or CHX subsequent to the experiment, these preparations were washed in physiological saline for a minimum of 1 h before the experiment being performed. We also waited a minimum of 1 h after stn transection before carrying out any experiment.
To perform the experiments, we perfused increasing concentrations of CCAP from 10−9 M to 10−6 M at a rate of ~1 ml/min at 12°C, beginning with a control (no CCAP) saline perfusion. Each perfusion lasted 20 min, during which network and LP activity were continuously recorded. CCAP also targets the pacemaker AB neuron of the STG, and so most of the experimental preparations responded to CCAP by generating rhythmic motor output. From this rhythmic output, the LP burst characteristics reported in Figs. 2 and 3 were collected via both intracellular LP recordings as well as extracellular recordings of the lvn (see Fig. 1). To minimize time spent in experimental CCAP perfusion and maximize the health of the preparations, CCAP application was performed without washes in between. In the preparations used for frequency-current (f-I) analysis reported in Fig. 4, the CCAP perfusion at each concentration level was allowed to proceed for 20 min and LP activity was continuously recorded. At the end of 20 min, we applied three replicates of current pulses from 1 nA to 5 nA in 1-nA increments for 2 s each. We took the average of the three responses at each current level for our analysis. While these current injections were sometimes performed on top of ongoing spontaneous network activity as a result of CCAP activation of the pyloric rhythm, they provided us with an opportunity to compare current-evoked spiking across preparations in an unbiased fashion. Because these current injections were performed in bridge mode with a single electrode, we made no measurements of membrane potential during current injections but rather only measured spike frequencies during the current steps.
Fig. 2.

Long-term (24 h) decentralization enhances responses to CCAP application in the LP neuron of the pyloric network. Acute control (n = 7) and 24-h stn-cut (n = 6) preparations were compared with respect to their responses to 4 different concentrations of CCAP. All preparations had pyloric rhythms restored as a result of CCAP application. Left: data represented as means ± SD. In these CCAP-evoked rhythms, there were significant increases in LP burst duration and number of spikes per burst in 24-h stn-cut preparations relative to control. Results of 2-way repeated-measures ANOVAs are reported. **Significant differences (P < 0.01) between the 2 groups at a given concentration. Right: representative traces. Dashed lines indicate how LP cells with multiple bursts within a pyloric cycle were analyzed only for the primary burst following termination of PD inhibition.
Fig. 3.

Changes in LP burst responses to CCAP application are due at least in part directly to loss of hormonal CCAP modulation. Data represented as means ± SD. There were significant differences in LP burst duration and number of spikes per burst across 5 different experimental manipulations. Results of 2-way repeated-measures ANOVAs are reported. Different superscripted letters indicate significant (P < 0.05) differences among groups as revealed by post hoc comparisons (i.e., groups labeled with different letters show significant differences from one another). Because no significant effect of group was detected for burst frequency and spike frequency, post hoc analyses were not performed. No interaction effect was found between group and concentration for burst duration, so we report results of post hoc analyses for the group factor only. Because a significant interaction effect was detected for spikes per burst, we report differences among groups at each concentration where the effect was found (i.e., 10−7 M and 10−6 M CCAP exposure). Sample sizes as indicated in key.
Fig. 4.

Changes in LP spiking responses to CCAP application as measured by f-I analysis. Data represented as means ± SE. Spike frequencies were measured in all 5 experimental groups (n = 5 per group) during 5 different current injection amplitudes (from 1 nA to 5 nA, duration = 2 s) in the absence of CCAP (NO CCAP) as well as in 4 different concentrations of CCAP. Data are represented as both raw spike frequency (top) and Δ spike frequency from the “NO CCAP” state (bottom). Results of 2-way ANOVA are shown. Post hoc comparisons among experimental groups are denoted as in Fig. 3 (groups labeled with different superscripted letters show significant differences from one another). Because no interaction effects were detected, we report the post hoc results for the group factor only. Representative recordings of an LP cell from each experimental group are shown at bottom left. These traces were generated from a 3-nA current injection for 2 s in 10−7 M CCAP.
LP burst characteristics were extracted either from intracellular recordings or from the lvn recordings. LP spikes on the extracellular traces are almost always the highest-amplitude spikes and therefore are easily extracted by threshold voltage levels (see Figs. 1 and 2). In many of our experimental preparations during CCAP-evoked activity, LP displayed multiple burst events in a single pyloric cycle. When this was the case, we only extracted the initial LP burst activity for our analysis (see Fig. 2). The end of the initial LP burst was defined analytically as an increase in the time between spikes of the burst >200% and confirmed by examination of the recording for clear PY burst activity (see Fig. 1) that resulted in inhibition of LP spiking. This was the most conservative way to address the potential influence of our experimental manipulations changing synaptic properties in the ganglion and leading to multiple bursts of a given unit in a single pyloric burst cycle.
Measurements of neuromodulator-evoked mixed inward current.
Two-electrode voltage-clamp recordings were used to measure the modulator-activated inward current (IMI) in different concentrations of CCAP (Garcia et al. 2015). IMI measurements were done in the presence of 100 nM TTX to block voltage-gated sodium channels, 200 μM CdCl2 to block L-type calcium currents, 20 mM tetraethylammonium to block high-threshold K+ currents, and 1 μM picrotoxin to block inhibitory glutamatergic synapses. All blockers were from Sigma-Aldrich. Concentration dependence of IMI was measured as established previously (Garcia et al. 2015). In short, we held the membrane potential at −20 mV and applied CCAP (Bachem) in increasing concentrations from 0.1 nM to 1 μM. Each concentration was washed in for 6 min and washed out for 8–15 min. Current amplitudes were calculated as the maximal difference relative to the holding current before CCAP application. In some experiments, the neuropeptide proctolin (Bachem) was applied in combination with CCAP to test for occlusion effects. The differences between the baseline holding current and deflections caused by each concentration were plotted and fit to a three-parameter sigmoid.
C. borealis CCAP receptor mRNA quantification.
Collection of LP neurons and subsequent quantitative PCR were performed as described previously (Temporal et al. 2014). Briefly, to harvest LP neurons a Vaseline well was constructed around the STG and the ganglion was digested with 1 mg/ml collagenase-dispase (Roche) in physiological saline for ~5 min, until adherent glial cells detached and the neurons became loosened from one another within the ganglion. The STG was then washed multiple times with physiological saline, which was gradually replaced with a solution of ice-cold 70% ethylene glycol and 30% C. borealis saline. The area of the dish outside the petroleum jelly well was filled with distilled water, and the entire dish was frozen at −20°C for 1 h. The individual LP somata were harvested with handheld forceps, and each individual neuron was placed in RNA Lysis Buffer (Zymo Research) and stored at −80°C until RNA extraction. Total RNA was extracted from individual neurons with the Zymo RNA Microprep Kit (Zymo Research) and reverse transcribed with the SuperScript III first-strand synthesis kit (Invitrogen). The cDNA was precipitated overnight, washed, and resuspended in water for use as template in SYBR Green (SABiosciences, Frederick, MD)-based qPCR performed on a Rotorgene RG-3000 (Corbett Research) real-time PCR machine. C. borealis CCAP receptor (CbCCAPr) primers were previously validated and used for quantification as described in Garcia et al. (2015). Primer sequences were as follows: forward 5′-GGTGGCTCTGACTGTCTTCCTCTT-3′; reverse 5′-AAGGGGAAGGTGATGACGGTCACT-3′. Primer sequences for ion channel qPCR were as previously reported (Temporal et al. 2014).
Statistics.
All statistical tests were performed with SigmaPlot v11.0 (Systat). All data were confirmed to be of normal distribution as required by the statistical analyses employed. Effects of CCAP on LP output in ongoing network activity (Figs. 2 and 3) were analyzed via two-way (group × concentration) repeated-measures ANOVA. When no interaction effects were detected between the group and concentration factors, post hoc t-tests (with Dunn-Šidák corrections for multiple comparisons) are reported for differences among experimental groups and not each level of group by concentration. This takes into account that differences in the concentration factor reflect the expected result that responses to CCAP increase with increasing concentration and keeps false discovery rates as low as possible by minimizing multiple comparisons. The same logic is applied for analyses in Fig. 4 of effects of group and current level for spiking activity of LP neurons in different concentrations of CCAP. Differences in mean mRNA levels across cells (Fig. 5) were analyzed with one-way ANOVAs followed by post hoc t-tests with Dunn-Šidák corrections for multiple comparisons. Quantitative relationships between CCAPr mRNAs and LP output were analyzed with Pearson’s correlation test. IMI as a function of the log of the CCAP concentration was fit with three-parameter sigmoid functions of the following form: , where a is the maximal current (Imax), x0 the half-maximal effective concentration (EC50, IMI), and b the slope factor.
Fig. 5.

Effects of loss of modulation on CCAPr mRNA levels. A: changes in LP CCAPr mRNA levels in all 5 experimental groups are shown as means ± SD. There were significant differences in LP CCAPr mRNA levels across the 5 different experimental manipulations. Results of 1-way ANOVAs are reported. **Significant differences (P < 0.01) between groups; groups designated with ** show significant differences from those not marked with asterisks but not from one another. Sample sizes as indicated in each bar. B: changes in LP spikes per burst across experimental groups. Data from Fig. 3 are replotted to compare the patterns of CCAPr mRNA expression with output features of LP neurons during ongoing rhythmic activity. Statistics and keys as described in A. C: correlation between mean CCAPr mRNA levels and LP burst output (mean spikes per burst) in LP cells across experimental groups. Data are from A and B, plotted as a correlation of means. Results of a Pearson correlation test show a highly significant correlation between these 2 measured variables.
RESULTS
Effects of loss of modulation on LP responses to CCAP.
We first analyzed the effects of application of four concentrations of CCAP (from 10−9 M to 10−6 M) on the output of LP neurons 24 h after removal of descending neuromodulatory inputs via stn transection. The stn was transected on all preparations before CCAP perfusion. This ensures that modulation from the anterior commissural and esophageal ganglia is removed and the only source of modulation is the CCAP provided exogenously. For this experiment, we only measured the activity of LP in preparations where rhythmic activity was restored by CCAP (Weimann et al. 1997). Repeated-measures ANOVA determined that there was a significant effect of concentration but not decentralization state on both burst frequency and spike frequency within the burst of LP neurons (Fig. 2). These results indicate that CCAP is effectively targeting LP cells in these preparations but that decentralization does not influence these output features. However, there was a significant effect of both concentration and decentralization on LP burst duration and the number of spikes within LP bursts (Fig. 2); decentralized preparations showed an increase in both LP burst duration and number of spikes per burst. CCAP application in decentralized preparations often produced LP activity with multiple bursts per cycle (Fig. 2; see representative recordings), consistent with an increase in LP excitability, a decrease in the efficacy of the PY-to-LP synapse, or both. We analyzed only the primary burst of LP in these cases, to ensure that the effects we were seeing were due to the initial LP excitability (Fig. 2) and not ineffective PY synapse activity. Together these data suggest that loss of modulator input to LP increases the sensitivity of the cell to subsequent modulation.
The 24-h stn-cut manipulation represents multiple physiological manipulations relative to normal STG function. First, because these experiments are done as organotypic cultures, all hormonal modulation is removed in these experiments. CCAP is solely released as a neurohormone, and so organotypic culture removes endogenous CCAP modulation as a result. Second, stn transection removes all other descending neuromodulation from the anterior ganglia, which represents a substantial diversity of neuromodulatory impacts. Finally, stn transection can also substantially alter the activity of the preparations, ranging from modest changes in output to complete cessation of network activity (Hamood et al. 2015; Thoby-Brisson and Simmers 2000). Because in this study we are focusing on the impacts of CCAP modulation, we designed experiments that allowed us to disentangle the impacts of the direct loss of hormonal CCAP from neuromodulation and activity-dependent effects in this experimental system. Specifically, we can maintain both neuromodulation and activity states of STG preparations while depriving them of hormonal CCAP by incubating in organotypic culture for 24 h with the stn intact.
For all experimental groups, there was a significant effect of concentration on output features, demonstrating that CCAP is effectively modulating these experimental preparations. These 24-h stn-intact preparations (those with no endogenous hormonal modulation for 24 h) when challenged with exogenous CCAP show no significant difference in either burst frequency or LP spike frequency within the burst compared with acute controls and 24-h stn-transected preparations (Fig. 3). Conversely, 24-h stn-intact preparations do exhibit a significant increase in response to CCAP that is similar to that of the 24-h decentralized preparations: both of these groups have significant increases in LP burst duration and the number of LP spikes per burst compared with acute control preparations (Fig. 3). To determine whether these differences could be attributed solely to the loss of hormonal CCAP, we repeated the experiment with another group of preparations that were maintained in organotypic culture for 24 h with their stns intact but coincubated these preparations in 10−7 M CCAP for 24 h. In these 24-h CCAP-incubated preparations, coculture with CCAP eliminated the changes associated with decentralization and loss of hormonal modulation (Fig. 3), suggesting that loss of CCAP is directly responsible for subsequent changes in response to CCAP modulation. Finally, to begin to investigate the level at which changes in response to CCAP occur with loss of modulation, we blocked de novo protein synthesis in a group of preparations that were maintained in organotypic culture for 24 h with their stns intact by coculturing these preparations with CHX for 24 h. These CHX-treated preparations showed no changes in response to CCAP after 24 h (Fig. 3), suggesting that the effects seen in preparations lacking modulation require de novo protein synthesis.
To further probe the impacts of these experimental manipulations on LP neurons, we followed up the analyses of burst characteristics within the context of network activity in the same five experimental groups with f-I response analyses. Once again, the stn was transected on all preparations before CCAP perfusion and current injection. In each of the experimental groups, we performed five current-clamp steps from 1 nA to 5 nA during perfusion of four different CCAP concentrations (10−9 M to 10−6 M). The groups used were as described above. The results of these experiments are shown in Fig. 4 and are represented for each concentration of CCAP as both raw LP spike frequency at each current step level and the change in spike frequency relative to control (physiological saline with no CCAP added).
For all analyses in these current injection experiments, there was a significant effect of both experimental group and current level at every concentration of CCAP used, including the control physiological saline (Fig. 4). Control current injection experiments, analyzed at the level of raw spike frequency, reveal an overall impact of decentralization and CHX treatment on excitability of LP cells: both 24-h stn-cut and 24-h CHX-treated groups show less spiking activity in control saline relative to the other three experimental groups. In 10−9 M CCAP, all of the experimental groups except for the CHX-treated preparations showed equivalent responses; CHX-treated preparations show lower responses in 10−9 M CCAP than the remaining groups, a trend that persists at all concentrations of CCAP (Fig. 4, top). Across the increasing CCAP concentrations from 10−8 M to 10−6 M a pattern begins to emerge, with the 24-h stn-cut and 24-h stn-intact preparations showing similar strong responses to increasing CCAP concentrations, acute controls and CCAP-incubated preparations showing similar but more moderate responses, and the CHX-treated group showing the weakest responses to CCAP (Fig. 4, top).
When the f-I data are analyzed as a change in spike frequency relative to control, a similar picture emerges, but with evidence to suggest that the most profound changes in response to CCAP are found in the 24-h stn-cut group. Across all concentrations, and particularly at 10−9 M and 10−8 M CCAP levels, the 24-h stn-cut group shows a stronger change in spike frequency than any of the other groups, which show largely equivalent effects (Fig. 4, bottom). At 10−7 M the 24-h stn-intact group begins to show a higher level of response in this analysis than the remaining three groups, and at 10−6 M both the 24-h stn-cut and 24-h stn-intact groups show similar elevated responses relative to the other three groups.
To briefly summarize the results of the LP output studies, our data suggest that loss of modulation, either hormonal (24-h stn-intact group) or neuromodulation (24-h stn-cut group) causes an increase in the subsequent response to CCAP modulation in LP neurons 24 h later. This effect can be directly attributed in part to the loss of CCAP, as coincubation of preparations with CCAP (24-h CCAP-incubated group) prevents the changes in response seen in the 24-h stn-intact group. However, there may be other factors that influence these responses, including activity-dependent changes in sensitivity to CCAP, as the 24-h stn-cut group showed the most profound response to CCAP when looking at the change in spike frequency in the f-I analyses. Finally, the changes seen in responses to CCAP require de novo protein synthesis, as treatment with CHX abolishes the changes seen.
Changes in CCAP receptor mRNA accompany changes in response to CCAP.
We hypothesize that LP neurons compensate for lost CCAP modulation by modifying receptor expression. The CbCCAPr is a GPCR with high sequence similarity to other arthropod CCAP receptors and mammalian neuropeptide S receptors (Garcia et al. 2015). To test for changes in expression, we quantified CbCCAPr mRNA abundance in individual LP neurons after each experiment (Fig. 5). Loss of modulation in the 24-h stn-cut and 24-h stn-intact groups resulted in a significant increase in CCAPr mRNA in LP cells relative to acute controls (Fig. 5A). Incubation with 10−7 M CCAP for 24 h (stn intact) prevented this increase in CCAPr mRNA, resulting in LP cells with CCAPr mRNA levels similar to acute controls. We also investigated whether spiking activity in LP cells incubated in CCAP may play a role in mediating this response by coincubating stn-intact preparations in 10−7 M CCAP and TTX for 24 h. While these preparations cannot have their output activity measured, loss of activity in the presence of CCAP does not result in increased CCAPr mRNA levels (Fig. 5A), suggesting that there is a direct feedback of CCAPr activation on steady-state mRNA levels in LP cells.
Because the patterns of CCAPr mRNA levels across experimental groups (Fig. 5A) were strongly reminiscent of the patterns of activity changes seen in these cells (Fig. 5B), we then determined whether a relationship exists between CCAPr mRNA levels and LP output characteristics. At a population level, there is a very strong correlation across experimental groups between CCAPr mRNA and LP output, specifically LP spikes per burst (Fig. 5C). However, if CCAPr mRNA levels are ultimately causal with respect to physiological measures of LP output, this relationship should hold across individual cells regardless of their experimental group. We therefore analyzed the correlation between CCAPr mRNA levels and LP spikes per burst in individual neurons across experimental groups (Fig. 6). At three different concentrations of CCAP (10−8 M to 10−6 M) there was a significant correlation between CCAPr mRNA levels and LP output at both the individual neuron level (Fig. 6, left) and the population level across experimental groups (Fig. 6, right). These results strongly suggest a direct relationship between CCAPr mRNA abundance and the response to CCAP in a given neuron and that responses to loss of CCAP modulation include effects on steady-state mRNA levels.
Fig. 6.

Correlation between CCAPr mRNA levels and number of spikes per burst in individual LP neurons. Over the course of our experiments, we were able to successfully get high-quality recordings of both activity and CCAPr mRNA from the same LP neuron in 15 preparations across 4 experimental groups. Left: correlated CCAPr mRNA and spikes per burst from pooled neurons across experimental groups at 3 different concentrations of CCAP). Right: correlations among mean levels of CCAPr mRNA and LP spikes per burst from the 4 experimental groups. Mean levels contain data in addition to the neurons those represented in plots on left, as cells were included in the appropriate mean level if they produced CCAPr mRNA data, LP output data, or both. Results of Pearson tests are shown. No significant correlation was found between spike number and CCAPr mRNA at 10−9 M CCAP. Data for pooled means at 10−6 M CCAP (bottom right) are replotted from Fig. 5C.
Effects of loss of modulation on modulator-evoked current in LP cells.
Together, the results from Figs. 5 and 6 suggest that there is an activity-independent influence of loss of modulation on CCAPr mRNA levels and that these mRNA levels are directly correlated with the cell’s response to CCAP. CCAP activates a neuromodulator-evoked mixed inward cation current (IMI) that strongly influences the excitability of motor neurons in the STG (Garcia et al. 2015; Golowasch and Marder 1992; Swensen and Marder 2000). We hypothesize that because loss of modulation changes levels of CCAPr mRNA there ultimately is an effect on IMI current amplitude that results in the changes we see in LP activity and dose-dependent response to CCAP. To test this, we measured and compared CCAP-evoked IMI (IMI-CCAP) in LP neurons from acute control, 24-h stn-intact, 24-h stn-cut, and 24-h CCAP-incubated preparations (Fig. 7A). We measured IMI-CCAP by voltage-clamping cells at −20 mV and measuring the current amplitude evoked by different CCAP concentrations (see Garcia et al. 2015).
Fig. 7.
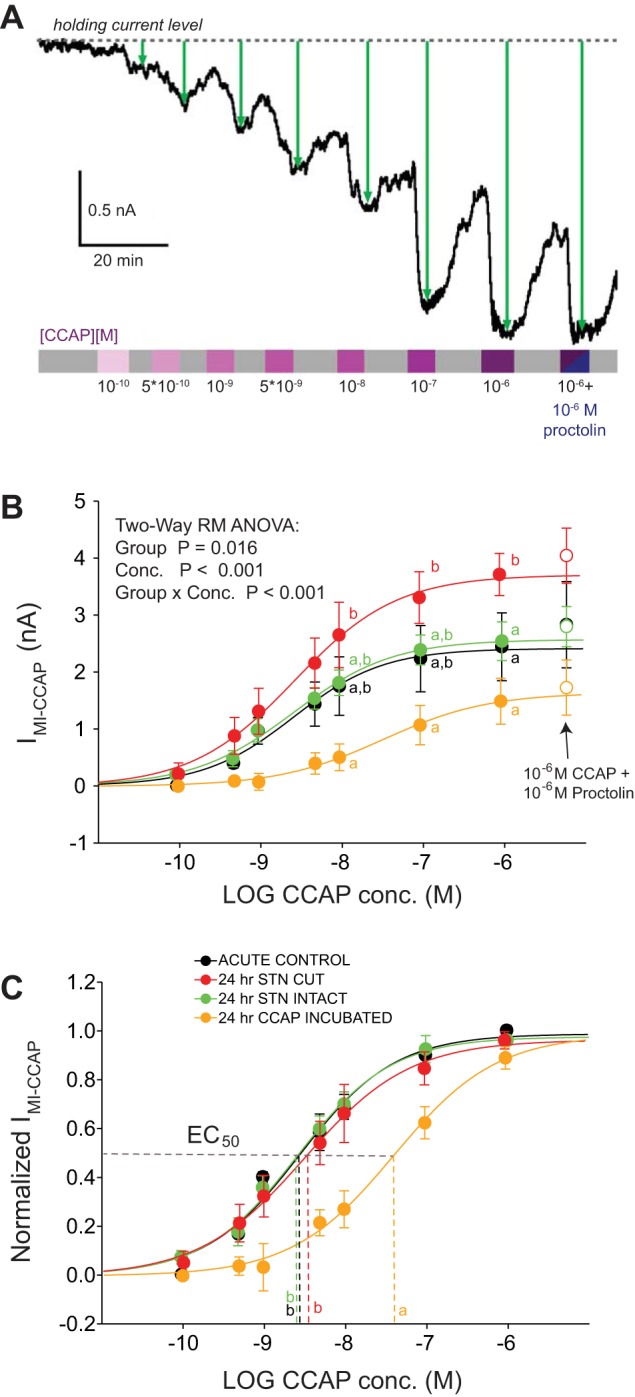
IMI-CCAP in LP cells differs in amplitude and concentration dependence across experimental groups. A: representative recording of CCAP-evoked IMI measurements. LP membrane potential was held via 2-electrode voltage clamp at −20 mV, and CCAP was applied in increasing concentrations from 0.1 nM to 1 μM for 6 min with a saline wash between each concentration. Current amplitudes were calculated as the maximal difference from the holding current before CCAP application (green line). In some experiments, proctolin was applied in combination with CCAP to test for occlusion effects. B: mean current values at different CCAP concentrations from measurements at −20 mV in 4 experimental groups (n = 5 per group). Sigmoid fits were generated from mean current levels averaged across individual experiments. Maximum current levels varied significantly across experimental groups as revealed by 2-way repeated-measures ANOVA. Post hoc analyses reveal that the maximal current elicited at 10−6 M was significantly larger in 24-h stn-cut preparations than in all 3 of the other groups. Groups designated with different superscripted letters show significant differences from one another for a given concentration of CCAP. Perfusion of proctolin (10−6 M) and CCAP (10−6 M) after the 10−6 M CCAP perfusion showed that proctolin caused no further significant increase in the current elicited for any of the 4 experimental groups (open circles), suggesting that CCAP occludes the effect of proctolin. C: same data as in A, but normalized to the maximal current in each experiment. Values for EC50 were significantly different across groups (P < 0.005, 1-way ANOVA—data were log transformed before analysis to match requirement for normality), with the EC50 smaller in all 3 groups relative to the CCAP-incubated group. Groups designated with different superscripted letters show significant differences from one another for EC50 of CCAP.
Figure 7B shows mean IMI-CCAP values in four experimental groups of LP cells as a function of CCAP concentration. Two-way repeated-measures ANOVA showed significant effects of both concentration (P < 0.001) and group (P = 0.016) as well as a significant interaction effect that prompted post hoc pairwise analyses. Post hoc analyses revealed that maximal current values at 10−6 M CCAP were significantly higher in the 24-h stn-cut group than the three other groups (Fig. 7B), consistent with higher CCAPr mRNA levels as well as the strongest response in the f-I analysis. Acute control and 24-h stn-intact groups show similar, intermediate levels of IMI-CCAP (Fig. 7B). CCAP-incubated preparations show lower levels of IMI-CCAP than any of the other groups (Fig. 7B), consistent with the low CCAPr mRNA levels seen in Fig. 5. Figure 7C shows the same data normalized to maximal current in each experimental group. The CCAP-incubated group also differed in concentration dependence, as both the EC50 and the slope factor in CCAP-incubated cells were significantly different from the other three groups (Fig. 7C). We therefore conclude that the lower expression level of CCAPr mRNA in CCAP-incubated preparations is accompanied by weaker activation of IMI-CCAP, as well as lower sensitivity to CCAP at virtually all concentrations.
Multiple neuromodulators can activate IMI in LP cells (Swensen and Marder 2000, 2001). In addition to CCAP, IMI is activated by the neuropeptide proctolin (Swensen and Marder 2001). To look for possible occlusion effects of multiple modulators that converge on IMI, after measuring the current response in 10−6 M CCAP we added 1 μM proctolin to the bath solution and measured evoked IMI again. We previously showed that 10−6 M CCAP elicits maximal IMI responses in LP that completely occlude the effect of proctolin (Garcia et al. 2015). Here we found that there also was no additional IMI evoked in any of the experimental groups with the subsequent application of proctolin, demonstrating that 10−6 M CCAP is able to maximally activate the current in each of the groups (Fig. 7B). We interpret this to mean that all available target ion channels that are responsible for IMI are activated by CCAP at 10−6 M, as addition of a converging neuromodulator cannot increase current responses. These results suggest that our 24 h stn-cut experimental group may show an increased number of channels responsible for IMI, in addition to increased CCAPr mRNA levels. However, we cannot rule out the possibility of changes in gating properties, unitary conductance, or both. These results also suggest that loss of modulators besides CCAP, activity changes, or both may increase the amount of IMI channels in these preparations relative to those in the acute control and 24-h stn-intact preparations.
Changes in ion channel mRNA accompany changes in response to CCAP.
In addition to changes in receptor expression and modulator-evoked currents, loss of modulation (and in turn activity) may influence excitability via voltage-dependent conductances as well. We explored this possibility by measuring mRNA abundances for five voltage-dependent ion channels in four experimental groups (see Fig. 8). We chose three calcium channels (Cb-CaV1, Cb-CaV2, and Cb-CaV3) that encode putative L-, P/Q-, and T-type channels, respectively, as well as two voltage-dependent K+ channels (shab and shal) that encode delayed rectified and A-type channels, respectively. We found that Cb-Cav3 was not expressed in appreciable amounts in LP cells (Fig. 8). Loss of modulation in the 24-h stn-cut and 24-h stn-intact groups resulted in a significant decrease in Cb-CaV1 mRNA and a significant increase in shal mRNA in LP cells relative to acute controls but no change in shab mRNA levels (Fig. 8). Incubation with 10−7 M CCAP for 24 h (stn intact) prevented these changes, resulting in LP cells with channel mRNA levels similar to acute controls (Fig. 8). These results are consistent with the overall decrease in excitability in the absence of modulator in LP cells of the 24-h stn-cut group (see Fig. 4, “NO CCAP”). However, the changes in expression in the 24-h stn-intact group are inconsistent with the lack of a change in their baseline control excitability.
Fig. 8.

Effects of loss of modulation on ion channel mRNA levels in LP neurons. Changes in LP voltage-dependent calcium and potassium channel mRNA levels in all 5 experimental groups are shown as means ± SD. There were significant differences in Cb-CaV1 (L-type calcium) and Shal (A-type potassium) mRNA levels across 4 different experimental manipulations. Results of 1-way ANOVAs are reported. Different letters above each bar indicate significant differences (P < 0.05) between groups. Sample sizes as indicated in each bar.
DISCUSSION
For many decades it has been known that denervated structures such as skeletal and smooth muscle, glands, and targets throughout the nervous system respond to loss of input by exhibiting increased sensitivity (Brown 1937; Cannon and Rosenblueth 1949; Nicholls 1956) and that this “denervation supersensitivity” (Cannon and Rosenblueth 1949) is due in part to an upregulation of postsynaptic receptor densities (Cai et al. 2002; Husch et al. 2012; Ko et al. 1977). However, this phenomenon is less well understood for modulation from both neural and hormonal sources, particularly with respect to plasticity of GPCRs (Hazell et al. 2012). In the STG, which is one of the best-studied neural networks in the context of neuromodulation, it is clear that loss of descending modulatory inputs results in alterations of STG network properties (Hamood et al. 2015; Khorkova and Golowasch 2007; Nahar et al. 2012; Temporal et al. 2012, 2014; Thoby-Brisson and Simmers 1998) and changes in responses to both subsequently restored descending projections and exogenously applied modulators (Nahar et al. 2012). However, the underlying physiological mechanisms of these changes in a given neuron type have not previously been investigated. Our data demonstrate that loss of modulation increases the response of LP neurons to the hormonal modulator CCAP. These changes are accompanied by changes in CCAPr mRNA levels in LP neurons, require de novo protein synthesis, and can be prevented by coincubation with CCAP. These results suggest that there is a direct feedback from loss of CCAP signaling to the production of CCAPr that increases subsequent response to the ligand. In addition, we demonstrated that the modulator-evoked current (IMI) activated by CCAP is greater in magnitude after loss of endogenous modulation and activity, suggesting multifaceted effects that ultimately influence the increase in response to modulator-accompanying decentralization of the STG.
The STG is modulated by a dizzying array of compounds (Stein 2009), including a substantial number of neuropeptides (Christie et al. 2010; Swensen and Marder 2000, 2001). CCAP is somewhat distinct among the myriad of modulators affecting STG output in that its source is purely hormonal, while the majority of compounds known to affect the STG are released into the STG neuropil from descending neuronal projections via the stn, often in addition to hormonal release. This makes our experimental capability to investigate the loss of CCAP relatively specific, as preparations that are maintained in organotypic culture, with or without intact descending stn projections, are subject to complete loss of CCAP modulation. Therefore we can decouple or specifically investigate loss of CCAP directly. Because we see increase in responsiveness to CCAP in LP cells in both stn-cut and stn-intact preparations maintained in culture for 24 h, and these effects are prevented by culturing with CCAP, we can reasonably conclude that there is a direct effect of loss of CCAP on LP responses to this ligand. However, there is evidence within multiple aspects of our data that suggests that loss of comodulation via stn transection, changes in activity associated with this decentralization, or both has a further impact on the response of LP to CCAP application. When we analyze our data as a change in spiking activity from baseline, we see a more profound change in response of the stn-cut preparations than those maintained in culture with the stn intact. This is due not to an increase in the maximum firing rate in decentralized preparations but rather a lower baseline level of excitability in these cells (see Fig. 4, “NO CCAP”). This results in a larger apparent effect of CCAP application in these preparations, even at concentrations as low as 10−9 M. However, with this difference measure, the 24-h stn-intact preparations ultimately do “catch up” in their response at 10−6 M. These results suggest that the maximal amount of depolarizing drive that can be generated by CCAP (presumably through IMI) is similar in 24-h stn-cut and 24-h stn-intact preparations but that a shift in sensitivity to lower concentrations in the stn-cut preparations may reflect changes in receptor density, IMI channel density, and/or changes in the signaling pathways between receptor and channel.
A change in CCAP receptor density is supported by our molecular data. We quantified CCAPr mRNA (Garcia et al. 2015) in individual LP neurons across our experimental groups and found that both 24-h stn-cut and 24-h stn-intact groups had significantly higher levels of CCAPr mRNA than control cells. Furthermore, coincubation with CCAP, even in the presence of TTX, prevented this increase in receptor mRNA, suggesting a direct feedback from CCAP receptor signaling to steady-state mRNA levels for the receptor. While measuring mRNA levels is not equivalent to measuring functional receptor protein, we saw a striking correlation between CCAPr mRNA levels and response to CCAP in LP cells across experimental groups, suggesting that there is a direct link between receptor mRNA abundance and physiology. The lack of a change in response to CCAP in 24-h stn-intact preparations cultured with CHX demonstrates that de novo protein synthesis is necessary to realize the effects of loss of CCAP, further supporting a link between CCAPr mRNA, receptor synthesis, and change in response to CCAP. Again, this is indirect evidence, as CHX inhibits all protein synthesis, and therefore we cannot attribute this effect to CCAPr specifically.
Our data suggest that decentralization, a manipulation that removes all modulation as well as altering LP activity, has a more profound sensitization of subsequent response to CCAP than loss of only hormonal modulation. This could be the result of a more profound change in CCAPr density, IMI density, or both. Our mRNA data demonstrate that there is not a greater increase in CCAPr mRNA between decentralized and stn-intact preparations, suggesting that IMI may also be affected by decentralization. During our experimental IMI measurements, exposure to a second neuropeptide known to activate this current (proctolin) did not result in significantly greater evoked IMI than CCAP alone in any of our experimental groups, yet decentralized preparations had significantly greater IMI amounts than any other experimental group. These data suggest that IMI itself also may change as a result of loss of neuromodulation, changes in activity, or both. Our experiments are unable to definitively determine which, if either, of these effects is at play in this system but are suggestive of a more complex multifaceted impact of decentralization beyond influencing receptor density.
Taken together, our data demonstrate that loss of the hormonal modulator CCAP causes an increase in subsequent responsiveness to this hormone and suggest that this is mediated through a direct effect of CCAP on receptor signaling activity to affect receptor density in part via increased steady-state receptor mRNA levels. However, the underlying mechanism we propose, while consistent with our data, is inferential. Furthermore, the experiments and our subsequent interpretation focus solely on the two end points of signaling: the receptor and the target. It is plausible that signaling pathways between these end points are also modified by loss of modulation and/or activity changes (Krenz et al. 2015). Nevertheless, our data demonstrate that feedback processes at the level of individual neurons and peptide hormone modulation can affect subsequent sensitization of response to these signals. Furthermore, the single cell-level data that combine electrophysiology and receptor mRNA abundance indicate that cellular sensitivity to a modulatory substance can be directly correlated with receptor expression itself but do not rule out other aspects of the signaling pathway or target channels.
In this study, we have demonstrated a seemingly “compensatory” change in neuropeptide signaling. However, applying a compensatory perspective to neuromodulation is less straightforward, as many modulatory events are the result of transient and episodic activation of sensory feedback (Blitz et al. 2004; Hamood et al. 2015; Hedrich et al. 2009), which can be quiescent for relatively long periods of time (even days to weeks). Therefore, our results raise the question of how a modulatory feedback mechanism works if there is not a reliable pattern of activation for these kinds of signaling compounds. One possibility is that CCAP is not released episodically but is present at a baseline level of modulatory tone all the time (Chen et al. 2009). One example of such a tonic modulatory influence is the well-described influence of dopamine on LP cells in the STG of the lobster Panulirus interruptus. In the micromolar range, low-affinity D1R receptors acutely respond to dopamine to decrease IA, which phase-advances LP in the pyloric rhythm (Krenz et al. 2013). However, tonic dopamine exposure also acts through high-affinity receptors at lower concentrations (in the nanomolar range) to regulate IH in an activity-dependent manner, compensating for decreased IA via a concomitant calcium/calcineurin-dependent decrease in IH that restores LP phase (Krenz et al. 2013, 2015; Rodgers et al. 2011b). These findings represent a mechanism by which modulation can maintain ratios of ionic conductances and that tonic low levels of modulation can potentiate activity-dependent regulation of conductances (Rodgers et al. 2011a). Some of our results parallel these effects of dopamine. For example, high-affinity dopamine receptors also act over longer timescales to increase ionic conductances via gene regulatory mechanisms at the level of transcription and translation (Krenz et al. 2014; Rodgers et al. 2013), just as we see changes at the mRNA level for channels and receptors in our results. Furthermore, our results show that exposure to tonic (24 h) CCAP subsequently enhances the firing response of LP cells to low concentrations of CCAP (see Fig. 4, “10−9 M CCAP”), even though this set of experimental conditions does not lead to enhanced levels of IMI (see Fig. 7). One possible interpretation of these data is that tonic CCAP exposure also alters ionic conductance ratios in a way that helps to stabilize LP output over time. Other peptide modulators such as proctolin and CabTRPIa that target STG neurons and activate IMI (Swensen and Marder 2000) are thought to be constitutively released through tonic activity of stn fibers (Billimoria et al. 2005). Therefore, there may be a reliable level of feedback for a subset of modulatory compounds that allow for compensatory tuning of receptor and IMI density, as well as ion channel expression and membrane conductance. Further studies will be required to examine the influence of CCAP on ionic conductances in a more comprehensive fashion.
GRANTS
This work was supported by National Institutes of Health Grants MH-46742 (D. J. Schulz and E. Marder) and NS-083319 (D. Bucher) and grants from the University of Missouri Research Board and Research Council (D. J. Schulz).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.M.L., V.J.G., D.B., and D.J.S. conceived and designed research; K.M.L., V.J.G., and S.T. performed experiments; K.M.L., V.J.G., S.T., D.B., and D.J.S. analyzed data; K.M.L., V.J.G., D.B., and D.J.S. interpreted results of experiments; K.M.L., V.J.G., D.B., and D.J.S. prepared figures; K.M.L. and D.J.S. drafted manuscript; K.M.L., V.J.G., D.B., and D.J.S. edited and revised manuscript; K.M.L., V.J.G., S.T., D.B., and D.J.S. approved final version of manuscript.
REFERENCES
- Billimoria CP, Li L, Marder E. Profiling of neuropeptides released at the stomatogastric ganglion of the crab, Cancer borealis with mass spectrometry. J Neurochem 95: 191–199, 2005. doi: 10.1111/j.1471-4159.2005.03355.x. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Beenhakker MP, Nusbaum MP. Different sensory systems share projection neurons but elicit distinct motor patterns. J Neurosci 24: 11381–11390, 2004. doi: 10.1523/JNEUROSCI.3219-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. The actions of acetylcholine on denervated mammalian frog’s muscle. J Physiol 89: 438–461, 1937. doi: 10.1113/jphysiol.1937.sp003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D, Marder E. SnapShot: Neuromodulation. Cell 155: 482–482.e1, 2013. doi: 10.1016/j.cell.2013.09.047. [DOI] [PubMed] [Google Scholar]
- Cai G, Wang HY, Friedman E. Increased dopamine receptor signaling and dopamine receptor-G protein coupling in denervated striatum. J Pharmacol Exp Ther 302: 1105–1112, 2002. doi: 10.1124/jpet.102.036673. [DOI] [PubMed] [Google Scholar]
- Cannon W, Rosenblueth A. The Supersensitivity of Denervated Structures. New York: Macmillan, 1949. [Google Scholar]
- Chen R, Ma M, Hui L, Zhang J, Li L. Measurement of neuropeptides in crustacean hemolymph via MALDI mass spectrometry. J Am Soc Mass Spectrom 20: 708–718, 2009. doi: 10.1016/j.jasms.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie AE, Chi M, Lameyer TJ, Pascual MG, Shea DN, Stanhope ME, Schulz DJ, Dickinson PS. Neuropeptidergic signaling in the American lobster Homarus americanus: new insights from high-throughput nucleotide sequencing. PLoS One 10: e0145964, 2015. doi: 10.1371/journal.pone.0145964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie AE, Skiebe P, Marder E. Matrix of neuromodulators in neurosecretory structures of the crab Cancer borealis. J Exp Biol 198: 2431–2439, 1995. [DOI] [PubMed] [Google Scholar]
- Christie AE, Stemmler EA, Dickinson PS. Crustacean neuropeptides. Cell Mol Life Sci 67: 4135–4169, 2010. doi: 10.1007/s00018-010-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O. Orphan GPCRs and neuromodulation. Neuron 76: 12–21, 2012. doi: 10.1016/j.neuron.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daur N, Nadim F, Bucher D. The complexity of small circuits: the stomatogastric nervous system. Curr Opin Neurobiol 41: 1–7, 2016. doi: 10.1016/j.conb.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong ND, Kirby MS, Blitz DM, Nusbaum MP. Parallel regulation of a modulator-activated current via distinct dynamics underlies comodulation of motor circuit output. J Neurosci 29: 12355–12367, 2009. doi: 10.1523/JNEUROSCI.3079-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia VJ, Daur N, Temporal S, Schulz DJ, Bucher D. Neuropeptide receptor transcript expression levels and magnitude of ionic current responses show cell type-specific differences in a small motor circuit. J Neurosci 35: 6786–6800, 2015. doi: 10.1523/JNEUROSCI.0171-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasselin C, Inglebert Y, Debanne D. Homeostatic regulation of h-conductance controls intrinsic excitability and stabilizes the threshold for synaptic modification in CA1 neurons. J Physiol 593: 4855–4869, 2015. doi: 10.1113/JP271369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golowasch J, Marder E. Proctolin activates an inward current whose voltage dependence is modified by extracellular Ca2+. J Neurosci 12: 810–817, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamood AW, Haddad SA, Otopalik AG, Rosenbaum P, Marder E. Quantitative reevaluation of the effects of short- and long-term removal of descending modulatory inputs on the pyloric rhythm of the crab, Cancer borealis. eNeuro 2: 1–25, 2015. doi: 10.1523/ENEURO.0058-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DW, Patel N, Watson RD. Regulation of protein synthesis in Y-organs of the blue crab (Callinectes sapidus): involvement of cyclic AMP. J Exp Zool A Comp Exp Biol 305: 328–334, 2006. doi: 10.1002/jez.a.263. [DOI] [PubMed] [Google Scholar]
- Harris-Warrick RM. Neuromodulation and flexibility in central pattern generator networks. Curr Opin Neurobiol 21: 685–692, 2011. doi: 10.1016/j.conb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell GG, Hindmarch CC, Pope GR, Roper JA, Lightman SL, Murphy D, O’Carroll AM, Lolait SJ. G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei—serpentine gateways to neuroendocrine homeostasis. Front Neuroendocrinol 33: 45–66, 2012. doi: 10.1016/j.yfrne.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich UB, Smarandache CR, Stein W. Differential activation of projection neurons by two sensory pathways contributes to motor pattern selection. J Neurophysiol 102: 2866–2879, 2009. doi: 10.1152/jn.00618.2009. [DOI] [PubMed] [Google Scholar]
- Husch A, Van Patten GN, Hong DN, Scaperotti MM, Cramer N, Harris-Warrick RM. Spinal cord injury induces serotonin supersensitivity without increasing intrinsic excitability of mouse V2a interneurons. J Neurosci 32: 13145–13154, 2012. doi: 10.1523/JNEUROSCI.2995-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57: 819–826, 2008. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Khorkova O, Golowasch J. Neuromodulators, not activity, control coordinated expression of ionic currents. J Neurosci 27: 8709–8718, 2007. doi: 10.1523/JNEUROSCI.1274-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko PK, Anderson MJ, Cohen MW. Denervated skeletal muscle fibers develop discrete patches of high acetylcholine receptor density. Science 196: 540–542, 1977. [DOI] [PubMed] [Google Scholar]
- Krenz WD, Hooper RM, Parker AR, Prinz AA, Baro DJ. Activation of high and low affinity dopamine receptors generates a closed loop that maintains a conductance ratio and its activity correlate. Front Neural Circuits 7: 169, 2013. doi: 10.3389/fncir.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz WD, Parker AR, Rodgers EW, Baro DJ. Dopaminergic tone persistently regulates voltage-gated ion current densities through the D1R-PKA axis, RNA polymerase II transcription, RNAi, mTORC1, and translation. Front Cell Neurosci 8: 39, 2014. doi: 10.3389/fncel.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz WD, Rodgers EW, Baro DJ. Tonic 5 nM DA stabilizes neuronal output by enabling bidirectional activity-dependent regulation of the hyperpolarization activated current via PKA and calcineurin. PLoS One 10: e0117965, 2015. doi: 10.1371/journal.pone.0117965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvarta MD, Harris-Warrick RM, Johnson BR. Neuromodulator-evoked synaptic metaplasticity within a central pattern generator network. J Neurophysiol 108: 2846–2856, 2012. doi: 10.1152/jn.00586.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Soares C, Béïque JC. Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology 78: 31–37, 2014. doi: 10.1016/j.neuropharm.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Li L, Kelley WP, Billimoria CP, Christie AE, Pulver SR, Sweedler JV, Marder E. Mass spectrometric investigation of the neuropeptide complement and release in the pericardial organs of the crab, Cancer borealis. J Neurochem 87: 642–656, 2003. doi: 10.1046/j.1471-4159.2003.02031.x. [DOI] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci 28: 4377–4384, 2008. doi: 10.1523/JNEUROSCI.5298-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol 69: 291–316, 2007. doi: 10.1146/annurev.physiol.69.031905.161516. [DOI] [PubMed] [Google Scholar]
- Marder E, Eisen JS. Electrically coupled pacemaker neurons respond differently to same physiological inputs and neurotransmitters. J Neurophysiol 51: 1362–1374, 1984. [DOI] [PubMed] [Google Scholar]
- Maynard DM, Dando MR. The structure of the stomatogastric neuromuscular system in Callinectes sapidus, Homarus americanus and Panulirus argus (Decapoda Crustacea). Philos Trans R Soc Lond B Biol Sci 268: 161–220, 1974. doi: 10.1098/rstb.1974.0024. [DOI] [PubMed] [Google Scholar]
- Miller JP, Selverston AI. Mechanisms underlying pattern generation in lobster stomatogastric ganglion as determined by selective inactivation of identified neurons. II. Oscillatory properties of pyloric neurons. J Neurophysiol 48: 1378–1391, 1982. [DOI] [PubMed] [Google Scholar]
- Nadim F, Bucher D. Neuromodulation of neurons and synapses. Curr Opin Neurobiol 29: 48–56, 2014. doi: 10.1016/j.conb.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahar J, Lett KM, Schulz DJ. Restoration of descending inputs fails to rescue activity following deafferentation of a motor network. J Neurophysiol 108: 871–881, 2012. doi: 10.1152/jn.00183.2012. [DOI] [PubMed] [Google Scholar]
- Nicholls JG. The electrical properties of denervated skeletal muscle. J Physiol 131: 1–12, 1956. doi: 10.1113/jphysiol.1956.sp005440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcutt AJ, Lett KM, Garcia VB, Diester CM, Lane BJ, Marder E, Schulz DJ. Deep sequencing of transcriptomes from the nervous systems of two decapod crustaceans to characterize genes important for neural circuit function and modulation. BMC Genomics 17: 868, 2016. doi: 10.1186/s12864-016-3215-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransdell JL, Faust TB, Schulz DJ. Correlated levels of mRNA and soma size in single identified neurons: evidence for compartment-specific regulation of gene expression. Front Mol Neurosci 3: 116, 2010. doi: 10.3389/fnmol.2010.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EW, Fu JJ, Krenz WD, Baro DJ. Tonic nanomolar dopamine enables an activity-dependent phase recovery mechanism that persistently alters the maximal conductance of the hyperpolarization-activated current in a rhythmically active neuron. J Neurosci 31: 16387–16397, 2011a. doi: 10.1523/JNEUROSCI.3770-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EW, Krenz WD, Jiang X, Li L, Baro DJ. Dopaminergic tone regulates transient potassium current maximal conductance through a translational mechanism requiring D1Rs, cAMP/PKA, Erk and mTOR. BMC Neurosci 14: 143, 2013. doi: 10.1186/1471-2202-14-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EW, Krenz WD, Baro DJ. Tonic dopamine induces persistent changes in the transient potassium current through translational regulation. J Neurosci 31: 13046–13056, 2011b. doi: 10.1523/JNEUROSCI.2194-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz DJ. Plasticity and stability in neuronal output via changes in intrinsic excitability: it’s what’s inside that counts. J Exp Biol 209: 4821–4827, 2006. doi: 10.1242/jeb.02567. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Lane BJ. Homeostatic plasticity of excitability in crustacean central pattern generator networks. Curr Opin Neurobiol 43: 7–14, 2017. doi: 10.1016/j.conb.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton DA, Magistretti PJ, Koob GF, Shoemaker WJ, Bloom FE. Dopaminergic supersensitivity induced by denervation and chronic receptor blockade is additive. Nature 299: 72–74, 1982. doi: 10.1038/299072a0. [DOI] [PubMed] [Google Scholar]
- Stein W. Modulation of stomatogastric rhythms. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 195: 989–1009, 2009. doi: 10.1007/s00359-009-0483-y. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Marder E. Multiple peptides converge to activate the same voltage-dependent current in a central pattern-generating circuit. J Neurosci 20: 6752–6759, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Marder E. Modulators with convergent cellular actions elicit distinct circuit outputs. J Neurosci 21: 4050–4058, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temporal S, Desai M, Khorkova O, Varghese G, Dai A, Schulz DJ, Golowasch J. Neuromodulation independently determines correlated channel expression and conductance levels in motor neurons of the stomatogastric ganglion. J Neurophysiol 107: 718–727, 2012. doi: 10.1152/jn.00622.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temporal S, Lett KM, Schulz DJ. Activity-dependent feedback regulates correlated ion channel mRNA levels in single identified motor neurons. Curr Biol 24: 1899–1904, 2014. doi: 10.1016/j.cub.2014.06.067. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Neuromodulatory inputs maintain expression of a lobster motor pattern-generating network in a modulation-dependent state: evidence from long-term decentralization in vitro. J Neurosci 18: 2212–2225, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Transition to endogenous bursting after long-term decentralization requires de novo transcription in a critical time window. J Neurophysiol 84: 596–599, 2000. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Long-term neuromodulatory regulation of a motor pattern-generating network: maintenance of synaptic efficacy and oscillatory properties. J Neurophysiol 88: 2942–2953, 2002. doi: 10.1152/jn.00482.2001. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4: a005736, 2012. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435, 2008. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimann JM, Skiebe P, Heinzel HG, Soto C, Kopell N, Jorge-Rivera JC, Marder E. Modulation of oscillator interactions in the crab stomatogastric ganglion by crustacean cardioactive peptide. J Neurosci 17: 1748–1760, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]