

Abstract
Background
Inflammation has been identified as a hallmark of high-risk Group A (GpA) ependymoma (EPN). Chronic interleukin (IL)-6 secretion from GpA tumors drives an immune suppressive phenotype by polarizing infiltrating monocytes. This study determines the mechanism by which IL-6 is dysregulated in GpA EPN.
Methods
Twenty pediatric GpA and 21 pediatric Group B (GpB) EPN had gene set enrichment analysis for MSigDB Hallmark gene sets performed. Protein and RNA from patients and cell lines were used to validate transcriptomic findings. GpA cell lines 811 and 928 were used for in vitro experiments performed in this study.
Results
The nuclear factor-kappaB (NF-κB) pathway is a master regulator of IL-6 and a signaling pathway enriched in GpA compared with GpB EPN. Knockdown of NF-κB led to significant downregulation of IL-6 in 811 and 928. NF-κB activation was independent of tumor necrosis factor alpha (TNF-α) stimulation in both cell lines, suggesting that NF-κB hyperactivation is mediated through an alternative mechanism. Leucine zipper downregulated in cancer 1 (LDOC1) is a known transcriptional repressor of NF-κB. In many cancers, LDOC1 promoter is methylated, which inhibits gene transcription. We found decreased LDOC1 gene expression in GpA compared with GpB EPN, and in other pediatric brain tumors. EPN cells treated with 5AZA-DC, demethylated LDOC1 regulatory regions, upregulated LDOC1 expression, and concomitantly decreased IL-6 secretion. Stable knockdown of LDOC1 in EPN cell lines resulted in a significant increase in gene transcription of v-rel avian reticuloendotheliosis viral oncogene homolog A, which correlated to an increase in NF-κB target genes.
Conclusion
These results suggest that epigenetic silencing of LDOC1 in GpA EPN regulates tumor biology and drives inflammatory immune phenotype.
Keywords: ependymoma, inflammation, LDOC1, methylation, NF-κB
Importance of the study
Ependymoma is an aggressive pediatric brain tumor with a high risk of recurrence and death. Two-thirds are located in the posterior fossa, and recent studies have identified 2 distinct molecular subgroups: Group A (GpA) and Group B (GpB). Inflammation has been identified as a hallmark of high-risk GpA through chronic secretion of IL-6 that polarizes the infiltrating immune cells toward a pro-tumor phenotype. Although fusion genes have been identified as drivers of supratentorial EPN, no fusions or mutations have been found in GpA. The present study is the first to identify the molecular mechanism of the inflammatory phenotype of GpA. Epigenetic silencing of LDOC1 in GpA leads to constitutively active NF-κB signaling independent of TNF-α stimulation. This results in chronic IL-6 secretion and subsequent downstream pro-tumor immune polarization. Targeting this pathway would not only potentially affect the tumor but also reverse the inflammatory immune phenotype.
Ependymoma (EPN) is the third most common central nervous system tumor in children and is associated with poor survival and long-term morbidity.1 Recent studies have identified 2 distinct molecular subgroups of posterior fossa (PF) EPN: Group A (GpA) and Group B (GpB), which exhibit different biological phenotypes and clinical outcomes.2–4 One significant difference between these 2 groups is their immunophenotype, with GpA EPN being defined by an inflammatory myeloid cell infiltration and exhausted T-cell phenotype.3 In contrast, GpB patients have a more immune activated, antitumor immunophenotype, likely contributing to tumor elimination and an overall more favorable outcome.3 We recently described chronic IL-6 secretion from tumors as the driver of the GpA EPN immunophenotype. This inflammatory cytokine activates signal transducer and activator of transcription 3 (STAT3) signaling in infiltrating myeloid cells, inducing the secretion of interleukin (IL)-8, another pro-inflammatory cytokine. IL-8 secretion further perpetuates the inflammatory phenotype in newly recruited myeloid cells through upregulation of additional cytokines, like IL-10 and IL-1β, and downregulation of immune activating cell surface markers human leukocyte antigen D-related and cluster of differentiation (CD)64.5 The underlying mechanism of IL-6 dysregulation in GpA EPN remains unknown.
The nuclear factor-kappaB (NF-κB) pathway is a complex signaling cascade involved in a variety of cancer cell functions, including proliferation, anti-apoptosis, metastasis, and inflammation.6–8 The NF-κB complex comprises 5 subunits: v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA) (p65), RelB, and c-Rel, which form a heterodimer with NF-κB1 (p50) or NF-κB2 (p52).9 In its inactive state, the dimer is restricted to the cytoplasm. Upon activation, the dimer translocates to the nucleus to initiate transcription of inflammatory response genes, including IL-6. The relationship between NF-κB and IL-6 has been described in glioblastoma (GBM), another aggressive CNS tumor.10 Recently, NF-κB signaling was shown to be a Hallmark signature in supratentorial ependymoma (ST EPN) through a RELA fusion protein that leads to a constitutively active NF-κB pathway.11,12 However, no fusion proteins have been identified in the 2 PF EPN subgroups.
Leucine zipper downregulated in cancer 1 (LDOC1) was originally identified as being suppressed in pancreatic and gastric cancer cells but has since been shown to regulate NF-κB transcription in additional cancer types.13,14 In a variety of cancers, methylation of the LDOC1 promoter leads to decreased LDOC1 protein expression.15–17 In patients with chronic lymphocytic leukemia, increased LDOC1 transcription has been correlated with improved survival.18 GpA EPN has been distinguished from GpB EPN by globally increased DNA methylation.19 We sought to determine if chronic IL-6 secretion in GpA EPN is regulated by the inverse relationship between NF-κB and LDOC1. The present study identifies potential epigenetic dysregulation underlying IL-6 overexpression in GpA EPN.
Materials and Methods
Study Approval
Primary patient tumor and normal brain samples were obtained from Children’s Hospital Colorado and collected in accordance with local and federal human research protection guidelines and institutional review board regulations (COMIRB 95-500 and 09-0906). Informed consent was obtained for all specimens collected.
Transcriptomic Analysis
The primary study cohort included: 19 primary GpA EPNs, 20 primary GpB EPNs, 9 ST EPNs, 5 myxopapillary EPNs, 41 high-grade gliomas, 32 medulloblastomas, 23 pilocytic astrocytomas, 26 craniopharyngiomas, and 20 atypical rhabdoid/teratoid tumors. Thirty-three normal brain samples from infra- and supratentorial anatomic sites obtained from autopsy or epilepsy surgery were included as controls. Tumor samples were analyzed using the Human Genome U133plus2 Array (Affymetrix) platform, and EPN subgrouping was determined using nonnegative matrix factorization clustering analysis as previously described.3 Gene set enrichment analysis (GSEA) was performed as previously described (GSE 66354).5 Our findings were validated using 2 independent patient cohorts from St Jude Children’s Research Hospital (GSE 21687) and the Toronto Hospital for Sick Children (GSE 27279).
Differential expression between diagnostic tumor groups was calculated using the Bioconductor Limma function.20 The Limma function performs pairwise comparisons between a target group and each of the other user-defined groups in the dataset. It employs an Empirical Bayes approach to calculate a moderated t-statistic, as well as calculating a false discovery rate (FDR) that accounts for multiple testing both within and across groups. Statistical significance was defined as FDR <0.05 and mean fold difference ≥1.5.
Cell Lines
EPN cell line 811 was established from the fourth recurrence in a patient with metastatic GpA EPN. EPN cell line 928 was established from the second recurrence of a patient also with metastatic GpA EPN. These unique cell lines are positive for EPN high-risk factor chromosome 1q gain (1q+). These cell lines were maintained under normal culture conditions in Optimem media supplemented with 15% fetal bovine serum (FBS) (O15; Invitrogen). Medulloblastoma cell line DAOY (American Type Culture Collection [ATCC]) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 1% sodium pyruvate, 1% nonessential amino acids, and 1% penicillin/streptomycin. The diffuse intrinsic pontine glioma cell line (DIPG iv), generously provided by Michelle Monje at the University of California San Francisco, was cultured as previously described.21 Human embryonic kidney (HEK) 293FT cells (ATCC) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cell line authentication was performed using short tandem repeat profiling and comparison with known cell line DNA profiles.
Short Hairpin RNA Transfection
Genetic knockdown of RELA (The RNAi Consortium [TRC] numbers TRCN0000014683 and TRCN0000014684) and LDOC1 (TRC numbers TRCN0000118177 and TRCN0000118179) was performed using a Sigma Aldrich pLKO system. Short hairpin (sh)RNA plasmids were packaged into virus by transfection of HEK 293FT cells with TransIT-LT1 Reagent (Mirus) and pVSV-G, pRRE, and pRSV packaging plasmids. Viral supernatant was harvested and centrifuged to remove debris. Polybrene was added to virus and to EPN cell lines prior to viral transduction. Virus was removed after 24 hours, and transduced cells were positively selected with puromycin. Knockdown was confirmed using quantitative real-time (qRT)-PCR and/or western blot.
Immunofluorescence
Experiments for confocal imaging were plated on poly-d-lysine coated chamber slides (Corning, 354632). The following day, cells were treated with 10 μM 5-aza-2ʹ-deoxycytidine (5AZA-DC) and replaced every 3 days for a total of 7 days. Tumor necrosis factor alpha (TNF-α) was added 1 hour prior to fixing in 4% paraformaldehyde. Primary antibody used was LDOC1 (Abcam Anti-LDOC1 antibody ab86126) or RELA (Cell Signaling, 6956S, clone D14E12). Secondary antibodies were conjugated with Alexa Fluor 488 (green) and 561 (red). Prior to staining, cells were permeabilized in 0.1% Triton-X and then blocked in 5% bovine serum albumin in 0.05% Triton-X. Nuclei were stained using 4′,6′-diamidino-2-phenylindole (DAPI) (ProLong Gold antifade reagent with DAPI, Life Technologies). Images were captured at 40x oil objective using a 3I Marianas inverted spinning disk confocal microscope and Evolve 16 bit electron-multiplying charge-coupled camera. Quantification was performed using ImageJ.
Cytokine Secretion
Media were harvested from cell lines and centrifuged to remove cellular debris. IL-6 was measured using Quantikine ELISA human IL-6 (R&D Systems) according to the manufacturer’s instructions. Viably frozen disaggregated tumor samples were thawed and flow sorted to separate immune cells from tumor cells as previously described.3 A high sensitivity Milliplex Map kit (Millipore) was used to measure the concentration of TNF-α as per manufacturer’s instructions.
Western Immunoblots
Nuclear and cytoplasm lysate extraction was performed using the Nuclear Extraction Kit (Abcam, ab113474) according to manufacturer’s instructions. For cell lines, 1 million cells and for patient samples 20 mg snap frozen tissue were used for extraction. Western blotting for proteins was performed using 15% Tris-HCL Criterion Prescast Gels (BioRad, 3450020) and transferred to polyvinylidene difluoride Immobilon-P Transfer Membranes (Millipore, IPVH0010). Primary antibodies used were RELA (Cell Signaling, 8242, clone D14E12), LDOC1 (Abcam, ab86126), and Lamin A/C (Cell Signaling, 4777, clone 4C11); and β-actin (Cell Signaling, 12262, clone 8H10D10) was used as protein loading control. Membranes were blocked in Tris-buffered saline Tween 5% milk or bovine serum albumin depending on the manufacturer’s recommendation. Bands were visualized with Immobilon Western Chemiluminescent horseradish peroxidase substrate (Millipore) on a Syngene G Box imager. Densitometry measurements were made using ImageJ software. All experiments were performed 3 times and representative blots are shown.
Neurosphere Growth Assay
Cells were treated with either shRELA or empty vector (shNT) virus and puromycin selected for 5 days. After selection, cells were harvested and plated at 1000 cells per well in O15 on a Corning round bottom ultra-low attachment plate and the plate was centrifuged to collect cells at the base of the well. Neurosphere growth was monitored on the IncuCyte Zoom, which takes real-time images of wells every 12 hours for 10 days. Area was calculated by measuring the diameter in both directions of the neurosphere.
Cell Proliferation
Cell proliferation (cell index) was checked by the xCelligence Real-Time Cell Analyzer (Roche). Cells were seeded in triplicate at 5000 cells/well in the E-Plate 96, a specialized 96-well plate used with the Real-Time Cell Analyzer instrument. Each of the 96 wells on the E-Plate 96 contains an integral sensor electrode array so that cells inside each well can be monitored and assayed. Cells were treated with lentivirus containing shRELA constructs or shNT for 24 hours. Virus media was replaced with fresh O15 and cell index was monitored for 72 hours. In order to validate knockdown, cells were plated in 6-well format and treated with viral supernatant in the same cell number to virus volume ratio as the 96-well E-Plate. Cells were harvested at the end of the xCelligence experiment and the knockdown validated by western blot.
Organotype Slice Culture
Slice cultures from primary tumor samples were maintained on Millicell Culture Inserts (Millipore) according to manufacturer’s protocol. Briefly, 3 approximate 0.33 cm slices of primary tumor sample were placed onto a cell culture insert and maintained in specialized slice culture media (Neurobasal A media containing B27, glutamax, L-glutamine, HEPES [4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid], and fibroblast growth factor). Collected media were stored at −80°C. Following the final media collection, we performed IL-6 enzyme-linked immunosorbent assay (ELISA) as described above. In this format, cultures remain viable for up to 10 days as measured by Ki67 staining on embedded tissue.
NF-κB Activity Assay
RNA was collected from LDOC1 knockdown and shNT cells following 5 days of puromycin selection. One microgram of RNA was converted into cDNA using Applied Biosystems reverse transcription kit according to manufacturer’s protocol. The cDNA was loaded onto Human NFkB Signaling Targets RT2 Profiler PCR Array (Qiagen) per manufacturer’s protocol. Experiments were run in triplicate and files were analyzed using software provided by Qiagen (www.SABiosciences.cin/pcrarraydataanalysis.php). Cycle threshold values were normalized using the 5 housekeeping genes provided on the plate according to the analysis software recommendations. The genes that were upregulated compared with shNT are reported.
Statistical Analyses
Statistical analyses were performed using R bioinformatics, Prism GraphPad v6, and Microsoft Excel software. For all tests, statistical significance was defined as P < 0.05 using Student’s t-test.
Results
NF-κB Activity Is a Hallmark of GpA EPN
We sought to identify the mechanism for chronic IL-6 secretion in GpA EPN by performing a nonbiased GSEA to identify candidate transcription factors overexpressed by GpA compared with GpB. EPN samples in our cohort were designated into 3 molecular subgroups by nonnegative matrix factorization clustering of transcriptomic profiles: ST, GpA, and GpB EPN (Supplementary Fig. S1). Transcriptomic profiles of primary GpA and GpB EPN were analyzed for enrichment using MSigDB Hallmark gene sets. In GpA EPN, NF-κB signaling genes (HALLMARK_TNF_SIGNALING_VIA_NFKB) were the most enriched of the 50 Hallmark gene sets (normalized enrichment score [NES] = 2.88; Supplementary Table S1; Fig. 1A). We validated these findings using 2 independent pediatric brain tumor datasets. In the dataset from the Toronto Hospital for Sick Children, NF-κB was the second most enriched Hallmark gene set (NES = 2.46; Supplementary Table S2; Supplementary Fig. S2A). Additionally, in the St Jude Children’s Research Hospital cohort, NF-κB was in the top 10 most enriched Hallmark gene sets in GpA compared with GpB (NES = 2.48; Supplementary Table S3; Supplementary Fig. S2B). NF-κB signaling regulates a variety of cellular functions by initiating transcription of response genes, many of which are cytokines. Importantly, aberrant NF-κB activity was recently identified as a hallmark of ST EPN, driven by a RELA fusion gene.11,12 Fusion genes have not been identified in PF EPN; however, RELA gene expression was significantly higher in GpA EPN compared with GpB (Fig. 1B).
Fig. 1.

NF-κB is hyperactivated in GpA EPN. (A) MSigDB HALLMARK_TNF_SIGNALING_NFKB gene set enriched in GpA (n = 19) to GpB (n = 20) EPN. (B) RELA gene expression significantly higher in GpA versus GpB. * P < 0.05. (C) Nuclear and cytoplasmic lysates collected from snap frozen EPN patient samples. Ratio of nuclear RELA to cytoplasmic RELA densitometry (n = 9 each GpA and GpB). A representative blot is shown. (D) Immunofluorescence staining of RELA baseline (left) and after 1 h TNF-α stimulation (right) in 2 GpA EPN cell lines, 928 and 811. Images are representative of 3 independent experiments. RELA is stained red and the nucleus is stained with DAPI in blue.
The activity of the NF-κB protein complex is dictated in part by subcellular localization. When inactive, the RELA/NF-κB1 dimer is restricted to the cytoplasm by inhibitor of kappa B (IkB). Upon activation of the canonical pathway by TNF-α, the IKK kinase complex phosphorylates IkB, releasing IkB from the dimer. The dimer is then phosphorylated and translocates to the nucleus to induce transcription. GpA patients had a greater ratio of nuclear versus cytoplasmic RELA protein than GpB (Fig. 1C). However, there was no significant difference in tumor secreted TNF-α between subgroups (Supplementary Fig. S2C). This suggests that GpA patients have NF-κB activity that is independent of TNF-α stimulation. To validate these findings we measured nuclear NF-κB in GpA cell lines compared with 2 non-EPN cell lines: DAOY and DIPG iv. In an unstimulated state, both EPN cell lines have significantly higher baseline nuclear NF-κB compared with DAOY and DIPG iv (Fig. 1D and Supplementary Fig. S3). We observed that when stimulated with TNF-α, NF-κB translocated into the nucleus in the remaining cells. These data suggest that there is underlying NF-κB activity that is independent of TNF-α stimulation in GpA EPN.
NF-κB Regulates IL-6 and Is Required for EPN Cell Growth
We have previously shown that NF-κB transcription factor signaling is the most upregulated transcription factor target gene set in GpA EPN compared with GpB.5 Within this gene set, IL-6 was the second most upregulated gene in GpA compared with GpB (fold change = 13.41, FDR q = 0.00038).5 In our working model, IL-6 drives the inflammatory immune microenvironment found in GpA EPN.5 We validated these findings in the datasets from the Toronto Hospital for Sick Children (NES = 1.48) and St Jude Children’s Hospital (NES = 1.73; Supplementary Fig. S4A and B). To determine if NF-κB regulates IL-6 in vitro, we used lentiviral delivery of 2 shRNA constructs targeting the RELA subunit, a critical component of the functioning NF-κB complex (Fig. 2A). IL-6 secretion was significantly reduced after RELA knockdown in 811 using both shRNA constructs (Fig. 2B). In 928, only partial knockdown of RELA was achieved using one construct. This knockdown was sufficient to hinder biological function but was not sufficient to reduce IL-6 secretion (Fig. 2A and B). This suggests that in order to block the immunological effect of NF-κB signaling, complete inhibition of the pathway is required. Cell survival and proliferation are also functions regulated by NF-κB. Consistent with this, loss of RELA led to decreased neurosphere size compared with shNT in both cell lines (Fig. 2C). In addition, cell proliferation was significantly hindered after RELA knockdown in EPN cell lines (Fig. 2D). These data are consistent with our hypothesis that NF-κB regulates both the tumor-specific functions and immune phenotype of GpA EPN.
Fig. 2.

NF-κB regulates IL-6 secretion and tumor cell survival in GpA EPN cell lines. (A) Western blot validation of RELA knockdown using lentiviral delivery of shRNA targeting RELA in 811 and 928. (B) IL-6 secretion after RELA knockdown. IL-6 secretion was normalized to nontarget shRNA (NT). (C) Neurosphere area after 10 days of growth following RELA knockdown. Experiments were repeated in triplicate. * denotes P < 0.05. (D) Cell proliferation after RELA knockdown measured using the xCelligence system.
LDOC1 Gene and Protein Expression Is Downregulated in GpA EPN
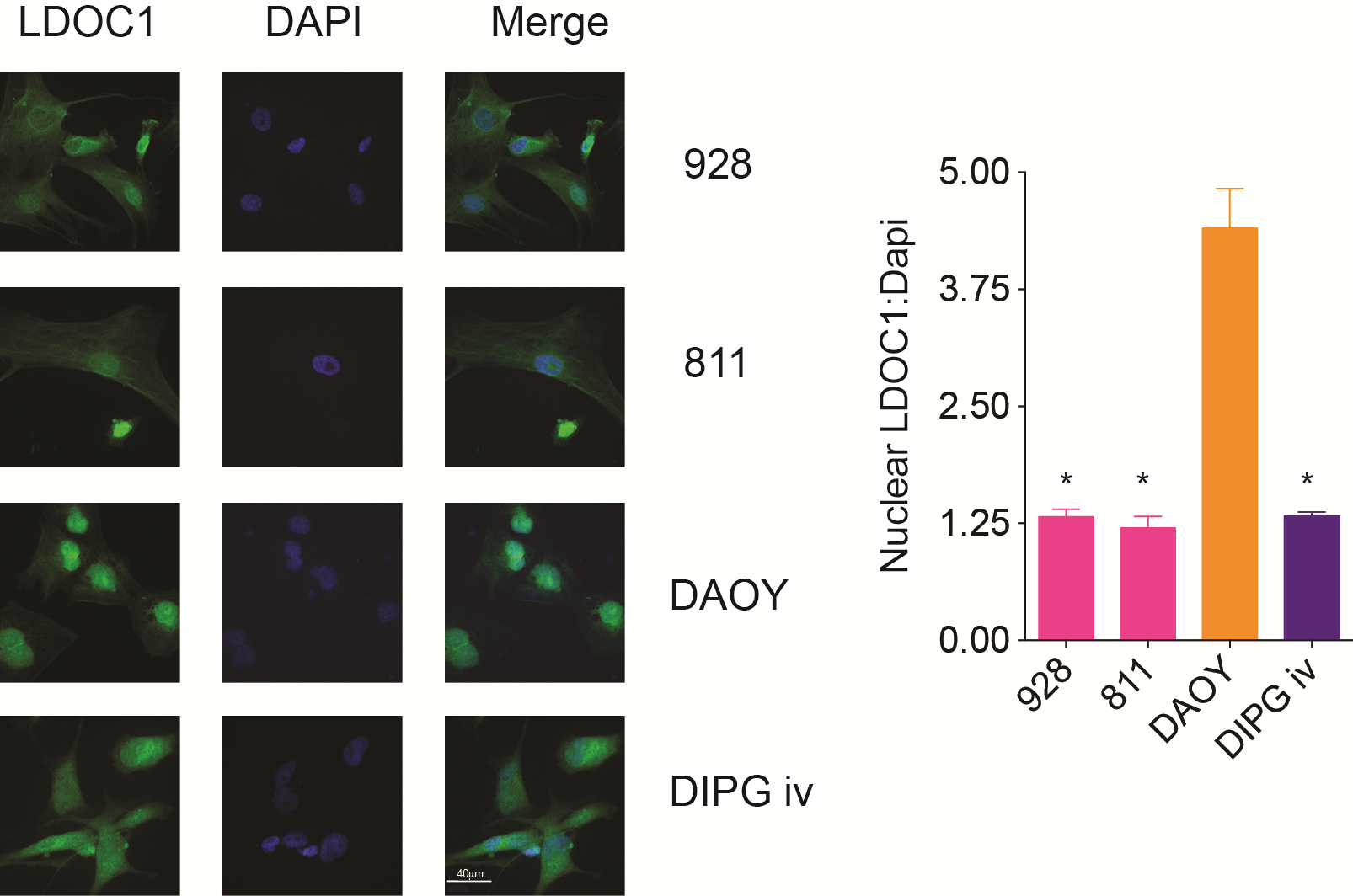
Using our transcriptomic database (GSE 66354) of pediatric brain tumors and normal brain (n = 228), we ran a Limma analysis comparing GpA EPN tumors pairwise to all other pediatric brain tumors, including GpB and ST EPN. Limma analysis revealed that of the over 20000 genes, LDOC1 was the only gene significantly underexpressed in GpA EPN compared with all other pediatric brain tumors, including GpB and ST EPN, and normal brain (Fig. 3A). In the Toronto dataset, LDOC1 was significantly downregulated in GpA compared with GpB EPN (P = 0.0013) and downregulation trended toward significance in the St Jude dataset (P = 0.0914) (Supplementary Fig. S5A and B). LDOC1 is downregulated in a variety of immune suppressive tumor types, including cervical, ovarian, gastric, pancreatic, and papillary thyroid cancers.14–17 LDOC1 loss is also associated with poor outcome in chronic lymphocytic leukemia.18 We compared LDOC1 gene expression with survival in PF EPN, independently of the molecular subgroup. For this analysis, we ranked our PF EPN patients based on LDOC1 expression and divided the patients into the top one-third expressing, middle one-third, and bottom one-third of patients. We found that patients with high and mid mRNA levels of LDOC1 both have significantly (P = 0.04) longer progression-free survival (PFS) compared with patients with low mRNA expression (Fig. 3B). There was a median PFS of 51 months for high LDOC1, 54.6 months for mid, and 23.7 months for low LDOC1 patients. Hazard ratio comparing low expressors to high was 2.7 with 95% CI of 1.046 to 7.306. For low expression compared with mid, the hazard ratio was 2.8 with 95% CI of 1.033 to 7.624. To rule out confounding factors in this study we also calculated the effect of molecular subgroup, extent of resection, patient age, and other clinical factors on survival (Supplementary Table S4). None of these factors are significantly associated with PFS in our study cohort, providing further evidence of the importance of LDOC1 in EPN biology. Similar to the gene expression, LDOC1 protein levels are significantly lower in GpA compared with GpB tumor samples (Fig. 3C). We also found that LDOC1 transcription levels were significantly lower in EPN cell lines compared with commonly used pediatric brain tumor cell lines (Fig. 3D). We validated LDOC1 mRNA expression level with LDOC1 protein levels and found a decrease in LDOC1 protein expression in both GpA EPN cell lines compared with DIPG iv and DAOY cell lines (Fig. 3E). In addition to decreased protein levels, we observed less nuclear LDOC1 in the EPN cell lines compared with DAOY (Supplementary Fig. S6), indicating the possibility that LDOC1 is deactivated.14 We did not observe enhanced nuclear LDOC1 in DIPG iv despite having higher levels of LDOC1 protein.
Fig. 3.

LDOC1 is identified as the only gene underexpressed in GpA EPN. (A) Limma analysis of 228 patient samples comparing GpA with all other pediatric brain tumors and normal brain identified LDOC1 underexpressed in GpA. (B) PFS for LDOC1 mRNA expression in PF EPN. Median survival for high expression patient was 51 months, for mid-expression 54.6 months, and for low expression patients 23.7 months. P-value determined using Mantel‒Cox test. (C) LDOC1 protein expression in GpA (n = 8) and GpB (n = 7) EPN. Densitometry ratio of LDOC1 to actin. *P < 0.05. (D) LDOC1 gene transcript comparing GpA cell lines with common pediatric brain tumor cell lines. (E) LDOC1 protein expression in GpA EPN cell lines compared with DAOY and DIPG iv. Densitometry ratio of LDOC1 to actin.
LDOC1 Is Epigenetically Silenced in GpA EPN
Several studies have shown that LDOC1 expression is regulated through DNA methylation.15–17 GpA EPN harbors a cytosine-phosphate-guanine (CpG) island methylation phenotype that is not seen in GpB.19 We therefore hypothesized that DNA methylation served as the primary mechanism of LDOC1 silencing in GpA EPN. To test this hypothesis, we first treated EPN cell lines with DNA methyl transferase inhibitor, 5AZA-DC. After 7 days of treatment with 10 μM 5AZA-DC, we measured LDOC1 mRNA. In both cell lines, LDOC1 mRNA was significantly increased after 5AZA-DC treatment compared with untreated controls (Fig. 4A). Additionally, we saw a significant increase in nuclear LDOC1 protein levels by immunofluorescence (Fig. 4B and C). This suggests that LDOC1 transcription is inhibited through DNA promoter methylation in GpA EPN cell lines.
Fig. 4.

LDOC1 gene expression is silenced through DNA methylation. (A) LDOC1 gene expression measured by qRT-PCR after 7 days of 5AZA-DC treatment. Cells were treated with 10 μM 5AZA-DC refreshed every 3 days. Experiments were completed in triplicate and * denotes P < 0.05. (B) Immunofluorescence staining of LDOC1 (green) after 7 days of 10 μM 5AZA-DC refreshed every 3 days. Experiments were completed in triplicate and images are representative. (C) Quantification of LDOC1 expression after 5AZA-DC treatment ratio of nuclear LDOC1 to DAPI signal.
To confirm LDOC1 methylation in GpA EPN, genomic DNA from GpA and GpB EPN were bisulfite converted, cloned, and sequenced. We identified 12 predicted CpG islands in the LDOC1 promoter region, which were hypermethylated in GpA compared with GpB tumors (Supplementary Fig. S7A). As we have previously published, EPNs are heterogeneous tumors comprising up to 25% immune cells that can contribute to the overall genomic landscape.22 Therefore, we used our EPN cell lines as a model for tumor-specific methylation, as the culture technique for our patient-derived cell lines do not contain immune cells. We confirmed methylation of the predicted LDOC1 promoter CpG islands in the cell lines. Cell line 928 had higher methylation than 811 cells, which was consistent with 811 having higher LDOC1 gene expression (Supplementary Fig. S7B). Furthermore, we performed an Illumina 850K methylation array following 5AZA-DC treatment in 811 and 928. It was found that 5AZA-DC had a greater demethylating effect on LDOC1, driving the increase in gene and protein expression (Supplementary Fig. S8A). These data give further evidence that DNA methylation is the likely mechanism in silencing of LDOC1 in GpA EPN.
LDOC1 Suppresses NF-κB Transcription and Activation Leading to Downregulation of IL-6
LDOC1 has been identified as a negative transcriptional regulator of NF-κB.13,14 To determine whether LDOC1 is negatively regulating NF-kB in EPN we used 5AZA-DC treatment to demethylate and consequently increase LDOC1 expression. We found that after 5AZA-DC treatment of both EPN cell lines, RELA transcription (Fig. 5A) was significantly reduced. We also measured a reduction in IL-6 secretion in 5AZA-DC treated cells (Fig. 5B). Treatment by 5AZA-DC also inhibited nuclear translocation of NF-κB, even after TNF-α stimulation (Fig. 5C and D). These data indicate that LDOC1 expression is negatively correlated wih IL-6 secretion and NF-κB activity. However, 5AZA-DC is a global inhibitor of methylation (Supplementary Fig. S8B) and is not specific to LDOC1. To address this in part we examined changes in methylation of RELA and IL-6 in response to 5AZA-DC treatment. We found that the changes in methylation in RELA and IL-6 following 5AZA-DC were less than LDOC1, suggesting that the changes in RELA expression and IL-6 secretion are more likely due to the derepression of LDOC1 and not a direct effect of 5AZA-DC treatment on these genes (Supplementary Fig. S8C).
Fig. 5.

5AZA-DC treatment led to decreased RELA transcription and suppression of NF-κB activity. (A) Treatment with 10 μM 5AZA-DC of EPN cell lines for 7 days with refreshing every 3 days. RELA gene transcription by qRT-PCR normalized to untreated (UT) control. (B) IL-6 secretion measured by ELISA (right) in pg/mL. Experiments were repeated in triplicate and * denoted P < 0.05. (C) Quantification of RELA expression after 5AZA-DC treatment ratio of nuclear RELA to DAPI signal. (D) Immunofluorescence staining of RELA after 10 μM 5AZA-DC treatment; 100 ng/mL TNF-α stimulation for 1 h. RELA staining in green and nucleus stained with DAPI (blue).
To further explore the correlation between LDOC1 and IL-6, we utilized patient-derived ex vivo organotype cultures to measure IL-6 secretion. These cultures are the closest in vitro model for GpB patients, as no GpB cell lines or short-term cultures are available. In patient samples with low LDOC1 mRNA expression, IL-6 secretion was significantly higher than in patients with high LDOC1 mRNA (Fig. 6A). Interestingly, in this cohort of patients IL-6 secretion was correlated with LDOC1 expression and not the molecular subgrouping. Conversely when LDOC1 was knocked down in 811 and 928 using 2 shRNA constructs, IL-6 was significantly increased (Fig. 6B–C). We also observed a significant increase in RELA transcription following LDOC1 knockdown (Fig. 6D). To further validate that NF-κB activation is negatively regulated by LDOC1, we measured RNA expression of 84 NF-κB signaling targets following LDOC1 knockdown. In both cell lines, we saw upregulation of most NF-κB targets with one or both constructs (Fig. 6E) compared with shNT. More specifically, 24 targets associated with cell cycle regulation, cell survival, and immune regulation were upregulated in both cell lines with both shRNA constructs (Supplementary Table S5), including IL-6, RELA, and STAT3, a gene we had previously identified as a key component on GpA biology.5 Collectively, these data show that suppression of LDOC1, through DNA methylation, leads to derepression of NF-κB and increased IL-6 production, which underlies both the tumor biology and immunobiology of poor outcome PF EPN.
Fig. 6.

LDOC1 regulates RELA transcription and activation of NF-κB signaling in GpA EPN. (A) IL-6 secretion measured from ex vivo patient-derived organotype slice cultures. Red indicates patients with low LDOC1 gene expression and blue denotes patients with high LDOC1. Replicate points denote cultures when material allowed for more than 1 well to be plated. (B) LDOC1 knockdown by lentiviral delivery of shRNA targeting LDOC1. Knockdown was validated by qRT-PCR. (C) IL-6 secretion by ELISA from cells after LDOC1 knockdown. (D) RELA gene expression after LDOC1 knockdown by qRT-PCR, which was normalized to shNT. Experiments were completed in triplicate and * denotes P < 0.05. (E) NF-κB target genes upregulated following LDOC1 knockdown. Experiments were performed in triplicate. Venn diagram displays number of genes upregulated with each construct compared with shNT.
Discussion
EPN is a devastating pediatric brain tumor with limited available treatment modalities and a high rate of relapse.23,24 In recent years, 2 molecularly and clinically distinct subgroups of PF EPN have been described.2–4,25 Progress in improving treatment options for PF EPN has been hindered due to the lack of relevant preclinical in vitro and in vivo models. This has made it difficult to advance our understanding of the biology of these tumors. Surgery and radiation at relapse does not favor GpA patients.26 We have demonstrated phenotypic differences of immune genes between PF EPN subgroups in which GpA exhibit a pro-tumor, inflammatory microenvironment contrast to the immune-activated GpB.3 We also previously reported that the mechanism driving the inflammatory phenotype in GpA EPN is chronic tumor secreted IL-6 that polarizes infiltrating monocytes and myeloid cells to an M2-like state.5 We here demonstrated that IL-6 secretion in GpA EPN is regulated by constitutive NF-κB activation.
Here we show that NF-κB is derepressed in GpA EPN through epigenetic silencing of LDOC1, which is a negative transcriptional regulator of NF-κB. In ST EPN harboring the C11orf95-RELA fusion, the NF-κB signaling pathway is activated independently of TNF-α stimulation.11,12 In GpA EPN, no gene fusions or mutations that might underlie NF-kB derepression have been reported to date. Our present findings provide a potential mechanism for this GpA associated phenotype, showing that NF-κB is derepressed in GpA EPN through epigenetic silencing of LDOC1, which is a negative transcriptional regulator of NF-κB.
The prognostic ability of histopathological grading criteria and other immunohistochemistry markers to risk-stratify patients are still both inconclusive and contentious in ependymoma. Chromosome 1q+, a known negative prognostic factor,3,4 is found more frequently in GpA EPN. LDOC1 gene expression was a predictor of PFS independently of EPN subgrouping. These data suggest that LDOC1 expression could be used as a predictive marker for determining outcome of a patient as well as whether a patient will respond to new chemotherapy trials.
The mechanism identified in these studies provides a potential target for development of therapies to improve future trials in this high-risk EPN subgroup. We show that targeting NF-κB leads to decreased neurosphere growth and proliferative potential. NF-κB signaling is also regulating chronic IL-6 secretion from the GpA tumor cells, which is responsible for the inflammatory phenotype observed in this tumor. However, data in the 928 cell line showed that partial inhibition of the NF-κB signaling is sufficient to hinder biological phenotype but was unable to suppress the immunological effect of the pathway. NF-κB activity is also associated with chemotherapy resistance in a variety of cancers. Chemotherapy is thought to activate the pathway by inhibiting IkB kinase, a key regulator in NF-κB activity.27 In ependymoma specifically, STAT3 signaling is associated with chemotherapy resistance,28 and we showed that STAT3 was one of the NF-κB target genes upregulated after LDOC1 knockdown. We therefore hypothesize that NF-κB activation in GpA EPN may also contribute to the chemotherapy resistance at relapse. Further investigation is needed to establish whether NF-κB signaling in GpA underlies chemotherapy resistance. Targeting the NF-κB/IL-6 pathway may deliver a multifaceted antitumor effect by decreasing tumor cell survival, reversing the inflammatory immune microenvironment, and potentially sensitizing tumor cells to chemotherapy.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was funded by Hyundai Hope on Wheels, the Tanner Seabaum Foundation, and the Morgan Adams Foundation. J.M.L. is supported by an elope, Inc. St. Baldrick’s Foundation Scholar Award and NIH/NCI (K08CA193982). The Genomics and Microarray Core receives direct funding support from the National Cancer Institute (P30CA046934). The Advance Light Microscopy Core is supported in part by NIH/NCATS Colorado CTSI (UL1 TR001082).
Conflict of interest statement. None.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank the University of Colorado Denver Genomics and Microarray Core and University of Colorado Denver Advanced Light Microscopy Core Facility.
References
- 1. Ridley L, Rahman R, Brundler MA, et al. ; Children’s Cancer and Leukaemia Group Biological Studies Committee. Multifactorial analysis of predictors of outcome in pediatric intracranial ependymoma. Neuro Oncol. 2008;10(5):675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wani K, Armstrong TS, Vera-Bolanos E, et al. ; Collaborative Ependymoma Research Network. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012;123(5):727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffman LM, Donson AM, Nakachi I, et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014;127(5):731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Griesinger AM, Josephson RJ, Donson AM, et al. Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in Group A ependymoma. Cancer Immunol Res. 2015;3(10):1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010;86(2):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dutta J, Fan Y, Gupta N, Fan G, Gélinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25(51):6800–6816. [DOI] [PubMed] [Google Scholar]
- 8. Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. [DOI] [PubMed] [Google Scholar]
- 9. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. [DOI] [PubMed] [Google Scholar]
- 10. McFarland BC, Hong SW, Rajbhandari R, et al. NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS One. 2013;8(11):e78728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature. 2014;506(7489):451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Figarella-Branger D, Lechapt-Zalcman E, Tabouret E, et al. Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor-kappaB signaling. Neuro Oncol. 2016;18(7):919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagasaki K, Schem C, von Kaisenberg C, et al. Leucine-zipper protein, LDOC1, inhibits NF-kappaB activation and sensitizes pancreatic cancer cells to apoptosis. Int J Cancer. 2003;105(4):454–458. [DOI] [PubMed] [Google Scholar]
- 14. Zhao S, Wang Q, Li Z, et al. LDOC1 inhibits proliferation and promotes apoptosis by repressing NF-kappaB activation in papillary thyroid carcinoma. J Exp Clin Cancer Res. 2015;34:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buchholtz ML, Brüning A, Mylonas I, Jückstock J. Epigenetic silencing of the LDOC1 tumor suppressor gene in ovarian cancer cells. Arch Gynecol Obstet. 2014;290(1):149–154. [DOI] [PubMed] [Google Scholar]
- 16. Buchholtz ML, Jückstock J, Weber E, Mylonas I, Dian D, Brüning A. Loss of LDOC1 expression by promoter methylation in cervical cancer cells. Cancer Invest. 2013;31(9):571–577. [DOI] [PubMed] [Google Scholar]
- 17. Lee CH, Wong TS, Chan JY, et al. Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. J Pathol. 2013;230(3):298–309. [DOI] [PubMed] [Google Scholar]
- 18. Duzkale H, Schweighofer CD, Coombes KR, et al. LDOC1 mRNA is differentially expressed in chronic lymphocytic leukemia and predicts overall survival in untreated patients. Blood. 2011;117(15):4076–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- 21. Monje M, Mitra SS, Freret ME, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Griesinger AM, Birks DK, Donson AM, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191(9):4880–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bouffet E, Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv Syst. 1999;15(10):563–570. [DOI] [PubMed] [Google Scholar]
- 24. Wright KD, Gajjar A. Current treatment options for pediatric and adult patients with ependymoma. Curr Treat Options Oncol. 2012;13(4):465–477. [DOI] [PubMed] [Google Scholar]
- 25. Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramaswamy V, Hielscher T, Mack SC, et al. Therapeutic impact of cytoreductive surgery and irradiation of posterior fossa ependymoma in the molecular era: a retrospective multicohort analysis. J Clin Oncol. 2016;34(21):2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 2010;1805(2):167–180. [DOI] [PubMed] [Google Scholar]
- 28. Phi JH, Choi SA, Kim SK, Wang KC, Lee JY, Kim DG. Overcoming chemoresistance of pediatric ependymoma by inhibition of STAT3 signaling. Transl Oncol. 2015;8(5):376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.