

Abstract
Irpex lacteus, a cosmopolitan white-rot fungus, degrades lignin and lignin-derived aromatic compounds. In this study, we report the high-quality draft genome sequence of I. lacteus F17, isolated from a decaying hardwood tree in the vicinity of Hefei, China. The genome is 44,362,654 bp, with a GC content of 49.64% and a total of 10,391 predicted protein-coding genes. In addition, a total of 18 snRNA, 842 tRNA, 15 rRNA operons and 11,710 repetitive sequences were also identified. The genomic data provides insights into the mechanisms of the efficient lignin decomposition of this strain.
Electronic supplementary material
The online version of this article (10.1186/s40793-017-0267-x) contains supplementary material, which is available to authorized users.
Keywords: Short genome report, Genome sequence, Irpex lacteus F17, White-rot fungus, Hardwood tree, Lignin decomposition
Introduction
Irpex lacteus, a white-rot fungus with biotechnological potential, is currently considered the most important lignocellulose-degrading organism because of its potential to degrade lignin and bioremediate other lignin-related pollutants (such as industrial dyes and aromatic pollutants) [1–3]. Lignocellulose, which is the most abundant renewable biomass in terrestrial environments, is composed of three major components: cellulose, hemicellulose, and lignin [4]. Among them, lignin is a highly irregular and heterogeneous biopolymer, which makes it recalcitrant to degradation. Compared with other wood-decay fungi, I. lacteus plays an important role in the efficient enzymatic conversion of renewable biomass, and it shows remarkable resistance to pollutant toxicity in water and soil environments [5]. I. lacteus is known to remove various aromatic compounds, including endocrine disruptors, synthetic dyes, and polycyclic aromatic hydrocarbons [1, 6], and it can also be used to obtain ethanol via the biological pre-treatment of lignocellulose [7].
I. lacteus is a cosmopolitan species that is widespread in Europe, North America, and Asia [8–10]. The fungus produces hydrolases, such as exo- and endo-cellulases, and extracellular oxidative enzymes, such as LiP, MnP, as well as Lac [11, 12], thereby showing a pattern of ligninolytic enzymes that is typical of white-rot fungi. Starting in the 1960’s, several studies by Japanese researchers mainly focused on the activities of the exo- and endo-cellulases, as well as an exo-cellulase gene, from I. lacteus [13]. Subsequently, the LiP and MnP of I. lacteus were isolated and characterized, and the biotechnological applicability of this fungus has drawn considerably interests in recent years [5]. Recently, we have degraded and detoxicated the synthetic dyes by using manganese peroxidase isolated from I. lacteus F17 [14, 15]. However, the genome sequence of I. lacteus has not been reported. Thus, the genomic traits of I. lacteus are required to reveal and elucidate the ligninolytic potential of the type strain of white-rot fungi. Here, the genome sequence of I. lacteus F17 is presented. To the best of our knowledge, this is the first high-quality draft genome sequence of I. lacteus available so far.
Organism information
Classification and features
The sequenced strain of I. lacteus F17 was isolated from a decaying hardwood tree in May 2009 in the vicinity of Hefei, China (Table 1). Figure 1a shows the growth status of I. lacteus F17 which was cultured on PDA medium (200 g/L of potato extract, 20 g/L of glucose, and 20 g/L of agar) after 5 days at 28 °C. The strain grew faster and formed a white colony with a diameter of 6.8 cm. The micrograph of I. lacteus F17 mycelia grown on PDA after 3 days was obtained by OLYMPUS BX51 (Fig. 1b). The mycelia were picked up from an agar plate using a tiny tweezer, mounted on glass slides, and then stained with an appropriate amount of fungal staining solution mixed with lactic acid, carbolic acid and cotton blue (lactic acid 10 mL, carbolic acid 10 g, glycerol 20 mL, cotton blue 0.02 g, distilled water 10 mL) for light microscopic examination (400×).
Table 1.
Classification and general features of Irpex lacteus F17 [19]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Fungi | TAS [5] | |
| Phylum Basidiomycota | TAS [5] | ||
| Class Basidiomycetes | TAS [5] | ||
| Order Polyporales | TAS [5] | ||
| Family Polyporaceae | TAS [5] | ||
| Genus Irpex | TAS [14] | ||
| Species Irpex lacteus | TAS [14] | ||
| Strain: F17 | TAS [14] | ||
| Gram stain | n/a | n/a | |
| Cell shape | Filaments | TAS [5] | |
| Motility | Non-motile | TAS [5] | |
| Sporulation | Basidiospore | NAS | |
| Temperature range | Not reported | n/a | |
| Optimum temperature | 28 °C | NAS | |
| pH range; Optimum | Not reported | n/a | |
| Carbon source | Potato, Glucose | TAS [14, 15] | |
| MIGS-6 | Habitat | Dead wood, hardwood tree | TAS [5, 14] |
| MIGS-6.3 | Salinity | Not reported | n/a |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [14, 15] |
| MIGS-15 | Biotic relationship | Free-living | TAS [5] |
| MIGS-14 | Pathogenicity | Not reported | n/a |
| MIGS-4 | Geographic location | Mountain Dashu, Hefei, China | TAS [14, 15] |
| MIGS-5 | Sample collection | May 2009 | TAS [14] |
| MIGS-4.1 | Latitude | 31.85 | NAS |
| MIGS-4.2 | Longitude | 117.27 | NAS |
| MIGS-4.4 | Altitude | 284 m | NAS |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [33]
Fig. 1.
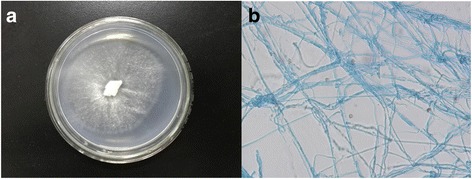
a: Colony of I. lacteus F17 grown on PDA medium for 5 days at 28 °C; b: Micrograph of I. lacteus F17 mycelia using optical microscope with 400× magnification. Mycelia were stained with lactophenol cotton blue stain solution
I. lacteus F17 resides in the Eukaryota, in the Fungal Kingdom, and it belongs to the family Polyporaceae, order Polyporales, class Basidiomycetes, Phylum Basidiomycota. Several other white-rot fungi with important biological function are members of the Polyporales, including Phanerochaete chrysosporium, Dichomitus squalens, Trametes versicolor, Polyporus brumalis, and Ceriporiopsis subvermispora. I. lacteus F17 has been identified and classified based on its Internal Transcribed Spacer region in our previous study [14]. The 18S rRNA gene data of I. lacteus F17 and several other Polyporales species were aligned using ClustalW [16]. Phylogenetic analysis based on the nearest neighbor joining method was performed using the MEGA6 package [17]. The confidence levels for the individual branches were determined by bootstrap analysis with 1000 replicates. The final phylogenetic tree was visualized with TreeView [18]. I. lacteus F17 is phylogenetically closely related to C. subvermispora (Fig. 2).
Fig. 2.

Phylogenetic tree based on 18S rRNA gene showing phylogenetic position of I .lacteus F17. Sequences were subjected to phylogenetic analysis using CLUSTALW [16] and MEGA 6.0 [17] to construct a nearest neighbor joining tree. The GenBank accession numbers for each strain are listed in parenthesis. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) was shown next to the branches. The GenBank accession numbers for each strain were listed in parenthesis. The tree was drawn by TreeView [18], and the scale bar represents 0.1 nucleotide substitution per nucleotide position
Genome sequencing information
Genome project history
I. lacteus F17 was selected for sequencing due to its bioremediation of organic pollutants and application to enzymatic biotechnologies. The genome of this strain was sequenced by SMRT technology, and genome assembly and annotation were performed at the Beijing Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). The whole genome shotgun project was started in May 2016, finished in August 2016 and has been submitted to NCBI under the accession number of MQVO00000000. Table 2 summarized the project data. The project information was in compliance with MIGS version 2.0 [19].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | High-quality draft |
| MIGS-28 | Libraries used | Illumina:350 bp small fragment library PacBio: 20 kb SMRT Bell library |
| MIGS 29 | Sequencing platforms | Illumina HiSeq PE150 PacBio RSII |
| MIGS 31.2 | Fold coverage | Illumina: 20× PacBio: 70× |
| MIGS 30 | Assemblers | SOAP denovo SMRT 2.3.0 |
| MIGS 32 | Gene calling method | PASA/Cufflinks/Augustus 2.7 |
| Locus Tag | BSQ47 | |
| Genbank ID | MQVO00000000 | |
| Genbank Date of Release | February 06, 2017 | |
| GOLD ID | NA | |
| BIOPROJECT | PRJNA354901 | |
| MIGS 13 | Source Material Identifier | F17 |
| Project relevance | Biotechnology, mycology |
Growth conditions and genomic DNA preparation
I. lacteus F17 was deposited at the CCTCC under the accession number of 10.1601/strainfinder?urlappend=%3Fid%3DCCTCC+AF+2014020. The strain was grown on PDA slants for 5 days at 28 °C, at which time the mycelia were scraped from the medium and lysed by liquid nitrogen grinding. The genomic DNA was extracted using the sodium dodecyl sulfate method. The harvested DNA was analyzed by agarose gel electrophoresis and purified using AMpure PB magnetic beads and then quantified by a Qubit® 2.0 fluorometer (Thermo Scientific, USA). In the end, the total amount of 28 μg DNA with a final concentration higher than 50 ng/μL and a A260/A280 ratio of 1.9 was placed in dry ice and sent to the sequencing.
Genome sequencing and assembly
A fungal survey by Illumina massively parallel sequencing technology was first used to make an evaluation for the fine mapping and assembly optimization of the fungal genome preassembling. Then the genome of I. lacteus F17 was sequenced by using PacBio’s SMRT technology. For the Illumina sequencing, the genome was sequenced using a single 350 bp insert genomic DNA library that was generated on a HiSeq 4000 PE150 system (Illumina, San Diego, CA, USA). For the PacBio sequencing, the genomic DNA was sheared into 20 kb fragments using a g-TUBE (Covaris, Woburn, MA, USA), and it was sequenced on an RSII system (PacBio, Menlo Park, CA, USA) after constructing the SMRT Bell library. The average sequencing depth of the 350 bp library was 20×, whereas the depth of the PacBio library was 70×.
Two assembly strategies were used respectively after filtering low-quality reads. A fungal survey produced 1564 Mb of clean data from 1700 Mb of raw data using SOAP denovo technology [20]. The PacBio subreads which were assembled into a primary assembly were completed with the Hierarchical Genome Assembly Process (Pacific Biosciences). A total of 3494 Mb of clean data were detected from the genome of I. lacteus F17 using samtools to fix the errors from the PacBio. The low quality reads were filtered by the SMRT 2.3.0 technology [21], and the filtered reads were assembled to generate one contig without gaps. A total of 317 contigs with an N50 of 1.15 Mb were generated from I. lacteus F17 genome. Finally, a 44.36 Mb draft genome of I. lacteus F17 was obtained. In addition, we used BUSCO [22] to assess the completeness of I. lacteus F17 genome and the genome has an estimated completeness of 86.9%, which indicated that we obtained a high-quality genome assembly in this study.
Genome annotation
By combining three types of genotype calling, including de novo PASA prediction of Transdecoder/Glimmer/Snap based on transcriptome data, Cufflinks prediction based on transcriptome data and de novo Augustus (version 2.7) [23], a total number of 10,391 protein coding genes were predicted. The interspersed repetitive sequences were predicted using the RepeatMasker [24]. The tandem repeats were analyzed by the Tandem Repeats Finder [25] and the tRNA genes were predicted by the tRNAscan-SE [26]. The rRNA genes were analyzed by the rRNAmmer [27] and the snRNA were predicted by BLAST against the Rfam [28, 29] database. In the end, 18 snRNA, 842 tRNA, 15 rRNA operons and a total of 11,710 repetitive sequences were identified in the genome. Seven databases, including Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, COG, Non-Redundant Protein Database, Transporter Classification Database, Swiss-Prot, and Pfam database were employed to predict gene functions. A whole genome BLAST search (E-value less than 1e-5, minimal 2 alignment length percentage larger than 40%) was performed against above seven databases. All putative proteins were compared to the entries in the CAZy database using a BLAST search. Secreted proteases were predicted with SignalP 4.1 [30] and TMHMM 2.0 [31], respectively. Other proteins that are important in wood-decay (oxidoreductases) and connected to fungal secondary metabolism were also predicted, according to a previously published method [4, 32].
Genome properties
The draft genome sequence was based on an assembly of 317 contigs amounting to 44,362,654 bp, with a GC content of 49.64% (Table 3). From the genome, 875 RNAs (including 18 snRNA, 842 tRNA, and 15 rRNA operons), as well as 11,710 repetitive sequences, were detected. In addition, a total of 10,661 genes were predicted, of which 10,391 are protein coding genes. Table 4 presented the distribution of genes into COGs functional categories. Of the last, 2065 genes (19.37%) were assigned to COG functional categories, the most abundant of them lies in the COG category named “Posttranslational modification, protein turnover, chaperones” (245 proteins) followed by “Translation, ribosomal structure and biogenesis” (215 proteins), “General function prediction only” (211 proteins), “Energy production and conversion” (168 proteins), “Nucleotide transport and metabolism” (144 proteins), “RNA processing and modification” (121 proteins), and “Intracellular trafficking and secretion” (116 proteins).
Table 3.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 44,362,654 | 100.00 |
| DNA coding (bp) | 15,030,327 | 33.88 |
| DNA G + C (bp) | 22,021,621 | 49.64 |
| DNA scaffolds | – | |
| Total genes | 10,661 | 100.00 |
| Protein coding genes | 10,391 | 97.47 |
| RNA genes | 875 | 8.21 |
| Pseudo genes | unknown | |
| Genes in internal clusters | unknown | |
| Genes with function prediction | 7532 | 70.65 |
| Genes assigned to COGs | 2065 | 19.87 |
| Genes with Pfam domains | 6287 | 58.97 |
| Genes with signal peptides | 761 | 7.1 |
| Genes with transmembrane helices | 2752 | 25.81 |
| CRISPR repeats | 0 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 215 | 2.07 | Translation, ribosomal structure and biogenesis |
| A | 121 | 1.16 | RNA processing and modification |
| K | 83 | 0.80 | Transcription |
| L | 56 | 0.54 | Replication, recombination and repair |
| B | 42 | 0.40 | Chromatin structure and dynamics |
| D | 63 | 0.61 | Cell cycle control, Cell division, chromosome partitioning |
| V | 4 | 0.04 | Defense mechanisms |
| T | 114 | 1.10 | Signal transduction mechanisms |
| M | 22 | 0.21 | Cell wall/membrane/envelope biogenesis |
| N | 0 | 0.00 | Cell motility |
| U | 116 | 1.12 | Intracellular trafficking and secretion |
| O | 245 | 2.36 | Posttranslational modification, protein turnover, chaperones |
| C | 168 | 1.62 | Energy production and conversion |
| G | 78 | 0.75 | Carbohydrate transport and metabolism |
| E | 144 | 1.39 | Amino acid transport and metabolism |
| F | 44 | 0.42 | Nucleotide transport and metabolism |
| H | 45 | 0.43 | Coenzyme transport and metabolism |
| I | 86 | 0.83 | Lipid transport and metabolism |
| P | 64 | 0.62 | Inorganic ion transport and metabolism |
| Q | 28 | 0.27 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 211 | 2.03 | General function prediction only |
| S | 66 | 0.64 | Function unknown |
| – | 8374 | 80.59 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
A total of 320 CAZyme-encoding genes were identified, including 53 CBMs, 161 GHs, 30 glycosyl transferases, four polysaccharide lyases, 64 AAs, and eight carbohydrate esterases (Additional file 1: Table S1). In conclusion, I. lacteus F17 possesses more CAZy families than other fungi (Additional file 2: Table S2), especially in the families AA3 (17 copies), AA9 (21 copies), CBM1 (34 copies), and GH5 (24 copies), which are all involved in plant cell wall degradation.
Insights from the genome sequence
Until now, this is the first draft genome sequence of the genus Irpex. The phylogenetic analysis based on the 18S rRNA gene data confirms its closest relationship of I. lacteus F17 to C. subvermispora. Annotation of the I. lacteus F17 genome indicates that this strain possesses 320 carbohydrate-active enzymes, 191 lignin-related oxidoreductases, 568 secreted proteases, and six secondary metabolism gene clusters (Additional file 3: Table S3), all of which confirm its high lignin decomposition ability. Fifteen enzymes were classified as probable ligninolytic enzymes, including a Lac, an LiP, and 13 MnPs, one of which was identified previously [14]. Interestingly, both I. lacteus F17 and C. subvermispora have the largest number of MnPs, even greater than that of P. chrysosporium (five MnPs), as determined by comparing 34 basidiomycetes, including 26 fungal species belonging to the Polyporales, as well as eight species in Agaricales, Russulales, Hymenochaetales, and Corticiales, respectively (Additional file 4: Table S4). A high number of MnP isozymes suggest that I. lacteus F17 has a good ability to degrade lignin and other organic pollutants.
Conclusions
In this study, we characterized the genome of I. lacteus F17 that was isolated from a decaying hardwood tree in the vicinity of Hefei, China. Notably, this is a first discovered sequenced strain, and we found it has lots of lignocellulose decomposition related genes. The genome sequencing information not only revealed its ligninolytic enzyme diversity, but also contributed to a better understanding of the efficient lignin decomposition of this strain. In summary, I. lacteus F17 has become one of model ligninolytic basidiomycetes whose availability of genomic sequences will facilitate future genetic engineering to degrade lignin and other organic pollutants.
Additional files
Table S1. Total CAZy families in I. lacteus F17. (XLSX 17 kb)
Selection of the CAZy families involved in plant cell wall degradation. (XLS 40 kb)
Gene contents in oxidoreductases, secreted proteases and secondary metabolism in the genomes of I. lacteus F17. (DOCX 15 kb)
Comparison of the number of MnPs from 34 fungal species belonging to the Polyporales and eight other fungi. (XLS 38 kb)
Acknowledgments
Funding
This research was supported by the National Natural Science Foundation of China (31570102, 31070109).
Abbreviations
- AA
Auxiliary activities
- BLAST
Basic local alignment search tool
- CAZy
Carbohydrate-active enzymes
- CBM
Carbohydrate-binding modules
- CCTCC
China Center for Type Culture Collection
- COG
Clusters of orthologous groups
- GH
Glycoside hydrolases
- Lac
Laccase
- LiP
Lignin peroxidase
- MnP
Manganese peroxidase
- PacBio
Pacific Bioscience
- PDA
Potato dextrose agar
- SMRT
Single Molecule Real-Time
Authors’ contributions
MWY participated in the sequence alignment and drafted the manuscript. WML carried out the laboratory experiments. ZHD participated in the sequence alignment. YLZ participated in the design of the study and performed the statistical analysis. RJ conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40793-017-0267-x) contains supplementary material, which is available to authorized users.
Contributor Information
Mengwei Yao, Email: 1033605128@qq.com.
Wenman Li, Email: 18206383201@163.com.
Zihong Duan, Email: 1294643776@qq.com.
Yinliang Zhang, Email: ylzhang@ahu.edu.cn.
Rong Jia, Email: ahdxjiarong@126.com.
References
- 1.Kasinath A, Novotný C, Svobodová K, Patel K, Šašek V. Decolorization of synthetic dyes by Irpex lacteus in liquid cultures and packed-bed bioreactor. Enzyme Microb Tech. 2003;32:167–173. doi: 10.1016/S0141-0229(02)00279-X. [DOI] [Google Scholar]
- 2.Šašek V, Novotný Č, Vampola P. Screening for efficient organopollutant fungal degraders by decolorization. Czech Mycol. 1998;50:303–311. [Google Scholar]
- 3.Song HG. Biodegradation of aromatic hydrocarbons by several white-rot fungi. J Microbiol. 1997;35:66–71. [Google Scholar]
- 4.Morin E, Kohler A, Baker A, Foulongne M, Lomard V, Nagy L, Ohm R, Patyshakuliyeva A, Burn A, Aerts A, Bailey A, Billette L, Coutinho P, Deakin G, Doddapaneni H, Floudas D, Grimwood J, Hilden K, Kues U, Labutti K, Lapidus A, Lindquist E, Lucas S, Murat C, Riley R, Salamov A, Schmutz J, Subramanian V, Wosten H, Xu J, Eastwood D, Foster G, Sonnenberg D, Cullient D, Vries R, Lundell T, Hibbert D, Henrissat B, Burton K, Kerrigan R, Challen M, Grigoriev L, Martin F. Genome sequence of the button mushroom Agaricus bisporus revals mechanisms governing adapatation to a humic-rich ecological niche. Proc Natl Acad Sci U S A. 2012;109:17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novotny C, Cajthaml T, Svobodova K, Susla M, Šašek V. Irpex lacteus, a white-rot fungus with biotechnological potential—review. Folia Microbiol. 2009;54(5):375–390. doi: 10.1007/s12223-009-0053-2. [DOI] [PubMed] [Google Scholar]
- 6.Baborová P, Möder M, Baldrian P, Cajthamlová K, Cajthaml T. Purification of a new manganese peroxidase of the white-rot fungus Irpex lacteus, and degradation of polycyclic aromatic hydrocarbons by the enzyme. Res Microbiol. 2006;157(3):248–253. doi: 10.1016/j.resmic.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Garcia M, Lopez-Abelairas M, Lu-Chau TA, Lema J. Fungal pretreatment of agricultural residues for bioethanol production. Ind Crop Prod. 2016;89:486–492. doi: 10.1016/j.indcrop.2016.05.036. [DOI] [Google Scholar]
- 8.Kellner H, Luis P, Pecyna M, Barbi F, Kapturska D, Kruger D, Rzak D, Marmeisse R, Marmeisse R, Vandenbol M, Hofrichter M. Widespread occurrence of expressed fungal secretory peroxidases in forest soils. PLoS One. 2014;9(4):e95557. doi: 10.1371/journal.pone.0095557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novotný Č, Erbanová P, Cajthaml T, Dosoretz RC, Sasek V. Irpex lacteus, a white rot fungus applicable to water and soil bioremediation. Appl Microbiol Biot. 2000;54(6):850–853. doi: 10.1007/s002530000432. [DOI] [PubMed] [Google Scholar]
- 10.Qin X, Zhang J, Zhang X, Yang Y. Induction, purification and characterization of a novel manganese peroxidase from Irpex lacteus CD2 and its application in the decolorization of different types of dye. PLoS One. 2014;9(11):e113282. doi: 10.1371/journal.pone.0113282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanda T, Wakabayashik N. Purification and properties of an endocellulase of avicelase type from Irpex lacteus (Polyporus tulipiferae) J Biochem. 1976;79(5):977–988. doi: 10.1093/oxfordjournals.jbchem.a131165. [DOI] [PubMed] [Google Scholar]
- 12.Cajthaml T, Erbanova P, Kollmann A, Novotny Č, Šasek V, Mougin C. Degradation of PAHs by ligninolytic enzymes of Irpex lacteus. Folia Microbiol. 2008;3(53):289–294. doi: 10.1007/s12223-008-0045-7. [DOI] [PubMed] [Google Scholar]
- 13.Nisizawa K, Hashimoto Y. Cellulose-splitting enzymes. VI. Difference in the specificities of cellulase and β-glucosidase from Irpex lacteus. Arch Biochem Biophys. 1959;81(1):211–222. doi: 10.1016/0003-9861(59)90191-2. [DOI] [PubMed] [Google Scholar]
- 14.Chen WT, Zheng LL, Jia R, Wang N. Cloning and expression of a new manganese peroxidase from Irpex lacteus F17 and its application in decolorization of reactive black 5. Process Biochem. 2015;50(11):1748–1759. doi: 10.1016/j.procbio.2015.07.009. [DOI] [Google Scholar]
- 15.Yang XT, Zheng JZ, Lu YM, Jia R. Degradation and detoxification of the triphenylmethane dye malachite green catalyzed by crude manganese peroxidase from Irpex lacteus F17. Environ Sci Pollut Res. 2016;23(10):9585–9597. doi: 10.1007/s11356-016-6164-9. [DOI] [PubMed] [Google Scholar]
- 16.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ. Higgins1 DG. ClustalW and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 17.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Page RDM. TreeView: an application to display phylogenetic trees on personal computers. Computer Applic Biosci. 1996;12(4):357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 19.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33(6):623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 22.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 23.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 24.Saha S, Bridges S, Magbanua ZV, Peterson DG. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008;36(7):2284–2294. doi: 10.1093/nar/gkn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37(Database issue):136–140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: Inference of RNA alignments. Bioinformatics. 2009;25(10):1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen TN, Brunak S, Von HG, Nielsen H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat Meth. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 31.Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 32.Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martinez AT, Otillar R, Spatafora JW, Yadav JS, Aerts A, Benoit I, Boyd A, Carlson A, Copeland A, Coutinho PM, Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Gorecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kuees U, TKA K, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, DJ ML, Morgenstern I, Morin E, Murat C, Nagy LG, Nolan M, Ohm RA, Patyshakuliyeva A, Rokas A, Ruiz-Duenas FJ, Sabat G, Salamov A, Samejima M, Schmutz J, Slot JC, John FS, Stenlid J, Sun H, Sun S, Syed K, Tsang A, Wiebenga A, Young D, Pisabarro A, Eastwood DC, Martin F, Cullen D, Grigoriev IV, Hibbett DS. The paleozoicorigin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Total CAZy families in I. lacteus F17. (XLSX 17 kb)
Selection of the CAZy families involved in plant cell wall degradation. (XLS 40 kb)
Gene contents in oxidoreductases, secreted proteases and secondary metabolism in the genomes of I. lacteus F17. (DOCX 15 kb)
Comparison of the number of MnPs from 34 fungal species belonging to the Polyporales and eight other fungi. (XLS 38 kb)