

Abstract
Background
Alcohol consumption is an established risk factor for breast cancer and the association generally appears stronger among estrogen receptor (ER)-positive tumors. However, the biological mechanisms underlying this association are not completely understood.
Methods
We analyzed messenger RNA (mRNA) microarray data from both invasive breast tumors (N = 602) and tumor-adjacent normal tissues (N = 508) from participants diagnosed with breast cancer in the Nurses’ Health Study (NHS) and NHSII. Multivariable linear regression, controlling for other known breast cancer risk factors, was used to identify differentially expressed genes by pre-diagnostic alcohol intake. For pathway analysis, we performed gene set enrichment analysis (GSEA). Differentially expressed genes or enriched pathway-defined gene sets with false discovery rate (FDR) <0.1 identified in tumors were validated in RNA sequencing data of invasive breast tumors (N = 166) from The Cancer Genome Atlas.
Results
No individual genes were significantly differentially expressed by alcohol consumption in the NHS/NHSII. However, GSEA identified 33 and 68 pathway-defined gene sets at FDR <0.1 among 471 ER+ and 127 ER- tumors, respectively, all of which were validated. Among ER+ tumors, consuming 10+ grams of alcohol per day (vs. 0) was associated with upregulation in RNA metabolism and transport, cell cycle regulation, and DNA repair, and downregulation in lipid metabolism. Among ER- tumors, in addition to upregulation in RNA processing and cell cycle, alcohol intake was linked to overexpression of genes involved in cytokine signaling, including interferon and transforming growth factor (TGF)-β signaling pathways, and translation and post-translational modifications. Lower lipid metabolism was observed in both ER+ tumors and ER+ tumor-adjacent normal samples. Most of the significantly enriched gene sets identified in ER- tumors showed a similar enrichment pattern among ER- tumor-adjacent normal tissues.
Conclusions
Our data suggest that moderate alcohol consumption (i.e. 10+ grams/day, equivalent to one or more drinks/day) is associated with several specific and reproducible biological processes and pathways, which adds potential new insight into alcohol-related breast carcinogenesis.
Electronic supplementary material
The online version of this article (doi:10.1186/s13058-017-0901-y) contains supplementary material, which is available to authorized users.
Keywords: Alcohol, Prospective, Epidemiology, Breast tumor, Gene expression
Background
Alcohol consumption is an established breast cancer risk factor [1]. Large prospective cohort studies have reported a modest but significant increase in risk (8–9%) per 10 g of alcohol consumed per day [1, 2]. Specifically, in the Nurses’ Health Study (NHS) with long-term average alcohol consumption, the risk increased by 15% (95% confidence interval (CI) 1.06–1.24) for 5.0–9.9 g/day of alcohol and by 51% (95% CI 1.35–1.70) for at least 30 g/day of alcohol, compared to women who did not drink [3]. The positive association was observed in both estrogen receptor (ER)-positive (ER+) and ER-negative (ER-) tumors but appeared stronger with ER+ than with ER- tumors [3, 4].
The mechanism underlying the alcohol and breast cancer association is not completely understood. One major hypothesis is that this association is mediated, at least in part, through estrogen metabolism [5, 6]. Other hypothesized mechanisms include the generation of acetaldehyde and reactive oxygen species (ROS) during alcohol metabolism [7]. Acetaldehyde has been classified as a carcinogen by the International Agency for Research on Cancer (IARC) [8] and, after alcohol administration, accumulation of acetaldehyde was observed in rat mammary tissue in experimental studies [9, 10]. Ethanol oxidation can lead to generation of ROS in rat mammary tissue [9, 10] and ROS promotes many aspects of tumor development and progression [11]. In addition, disruption of folate metabolism and DNA and/or histone hypomethylation have been hypothesized to be involved in alcohol-mediated carcinogenesis [8]. However, despite these hypotheses, no definitive mechanisms have yet been identified.
Assessment of molecular and/or genetic markers in breast tumor tissues may provide insights into the underlying mechanism(s) for established breast cancer risk factors. Recent studies evaluating breast tumor genome-wide gene expression profiling have identified molecular signatures associated with several established risk factors, such as body mass index (BMI) [12] and parity [13]. However, to date, no studies have assessed alcohol-related molecular signatures in breast tumors. To help unravel the underlying mechanisms of alcohol consumption and breast cancer risk, we evaluated the association between pre-diagnostic alcohol consumption and genome-wide gene expression in breast tumor and tumor-adjacent normal tissue in the prospective NHS and NHSII, and further validated our results in an independent validation dataset obtained from The Cancer Genome Atlas (TCGA) [14]. We hypothesized that the biological pathways underlying the association between alcohol and breast cancer could vary by tumor ER status and thus conducted the analysis by tumor ER expression.
Methods
Study population
The NHS was established in 1976 when 121,700 US female registered nurses, aged 30–55 years, completed an initial mailed questionnaire, and the NHSII was established in 1989, when 116,429 US female registered nurses, aged 25–42 years, completed and returned an initial questionnaire. Both cohorts have been followed biennially by mailed questionnaire to update information on exposure status and ascertain newly diagnosed diseases, including cancers. All women reporting incident diagnoses of breast cancer were asked for permission to review their medical records; cases for which pathology reports were obtained were confirmed by medical record review (>99%).
For this analysis, we included invasive breast cancer cases with both sufficient RNA from formalin-fixed paraffin-embedded (FFPE) tumor blocks for expression profiling and with available blood samples (the latter criterion to maximize the utility of the subset of cases that could be arrayed). Upon meeting the two criteria, in the NHS, we identified 532 invasive postmenopausal cases diagnosed in 1990–2004 which were a subset of the Cancer Genetic Markers of Susceptibility (CGEMS) initiative [15]; in the NHSII, invasive cases, regardless of menopausal status, diagnosed in 1995–2009 in the NHSII (N = 280) were included. Archived FFPE breast tumor blocks were obtained from the cohort tumor tissue repository; details of breast tumor tissue block collection have been described previously [16, 17]. Although only a subset of all the eligible cases were included in the TMA (primarily because either the tumor blocks had been destroyed by the hospital or there was insufficient tumor in the block), in each cohort, the characteristics of participants included in the TMA were very similar to those of all the eligible cases, including alcohol consumption and other breast cancer risk factors (e.g. first-degree family history, BMI and parity). The study was approved by the Committee on the Use of Human Subjects in Research at the Brigham and Women’s Hospital.
Assessment of alcohol exposure and other covariates
The assessment of alcohol consumption has been reported in detail elsewhere [3]. Briefly, information was first collected in 1980 in the NHS and in 1991 in the NHSII when participants reported their average frequency of intake for each alcoholic beverage (i.e. beer, wine, and liquor) during the previous 12 months through a semi-quantitative food frequency questionnaire, which was updated every 2–4 years thereafter in each cohort. Alcohol intake (grams per day) was calculated as the sum of the daily number of drinks multiplied by the average alcohol content of each beverage type (12.8 g per beer, 11.0 g per glass of wine, and 14.0 g per serving of liquor). We then calculated cumulative average intake by averaging alcohol consumption over time using all available information beginning in 1980 (NHS) or 1991 (NHSII). We also evaluated recent alcohol intake using information from the questionnaire cycle before diagnosis (i.e. 2–4 years before diagnosis). Cumulative average and recent alcohol consumption were highly correlated (Spearman r = 0.87) and results were very similar when using either cumulative average or recent alcohol; thus we presented results from recent alcohol intake. Covariate data, including parity, family history of breast cancer, BMI (weight(kg)/height(m)2), menopausal status and menopausal hormone therapy (MHT) use were obtained from the NHS or NHSII questionnaire at baseline and subsequent biennial questionnaires; for BMI, menopausal status and MHT use, the information taken from the most recent questionnaire was used.
Gene expression microarray and quality control analysis
RNA was extracted from multiple cores of 1 or 1.5 mm taken from tumor (N = 3 cores) or adjacent normal (N = 5 cores) tissues from FFPE blocks using the Qiagen AllPrep RNA isolation kit. Tumor-adjacent normal tissue was generally > 1 cm from the tumor edge. Since FFPE samples are known to have variable yields, tissues from all the cores from the same patient were placed into one microtube to maximize RNA yield. Total RNA was used to synthesize double-stranded complementary DNA which was then fragmented and hybridized to Affymetrix Glue Grant Human Transcriptome Array [18] (HTA 3.0v1 pre-release version from Affymetrix, Santa Clara, CA, USA). We included four independent breast tumor samples as technical replicates (identified from Beth Israel Deaconess Medical Center, Boston, MA, USA) in each assay plate; the correlation of these replicates across all arrays was ≥ 0.93.
Gene expression data were normalized and summarized using robust multiarray average (RMA; Affymetrix Power Tools (APT) v1.18.0). Out of the total 1324 tumor and tumor-adjacent normal specimens (934 and 390 in the NHS and NHSII, respectively), we excluded 131 (14%) and 43 (11%) from the NHS and NHSII, respectively, with an area under the curve (AUC) < 0.55 (evaluated using APT probeset summarization-based metrics) and further excluded 40 that failed the non-outlier analysis by arrayQualityMetrics v3.24.0 [19], leaving 1110 samples (602 tumors and 508 tumor-adjacent normal samples) for analysis. Although tumor specimens from the NHS were generally older than those from the NHSII, the proportions filtered out according to RNA quality (i.e. 14% vs. 11%) were similar in the two cohorts. Non-specific filtering by median expression levels was used to remove the bottom 25% of expressed probes, leaving 25,979 gene-level annotated transcript clusters included in the analysis. Gene expression data were deposited into the Gene Expression Omnibus [GEO: GSE93601].
We also assessed biological concordance (i.e. probe expression concordance with protein markers measured by immunohistochemical (IHC) staining) for select probes. We confirmed the correlation between probes for ESR1, PGR, and ERBB2 with IHC markers, ER, progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), in tumors to confirm biological reproducibility of the data (Additional file 1: Figure S1).
Statistical analysis
We performed analyses at the level of both probes and pathway-defined gene sets (Fig. 1). All analyses were conducted separately in ER+ tumors, ER+ tumor-adjacent normal tissues, and ER- tumors and ER- tumor-adjacent normal tissues. We conducted multivariable linear regression using the R Bioconductor package linear models for microarray data (LIMMA) [20] for 25,979 probes. To maximize power, samples from the NHS and NHSII were pooled in all analyses (although NHS and NHSII samples were run on different plates, all samples were normalized together) and we adjusted for microarray plate, thus controlling for both cohort and plate, in the regression models. Alcohol consumption was defined as a three-category variable: 0, > 0 to < 10 and 10+ g/day. Factors correlated with alcohol consumption and/or those known to affect tumor gene expression were evaluated as potential covariates in the regression models. Age at diagnosis, year of diagnosis, microarray plate, first-degree family history of breast cancer and recent BMI were included in the final models presented here. Although smoking status is often correlated with alcohol intake, smoking was not adjusted for in the analysis because so few women (~8%) were current smokers 2–4 years before diagnosis. In the single-probe analysis (N = 25,979 probes), individual probes were considered significantly differentially expressed by alcohol intake using a false discovery rate (FDR) threshold: FDR <0.1 for tumors and FDR <0.05 for tumor-adjacent normal tissues (due to the lack of a validation dataset of tumor-adjacent normal samples, a more stringent FDR threshold was applied).
Fig. 1.
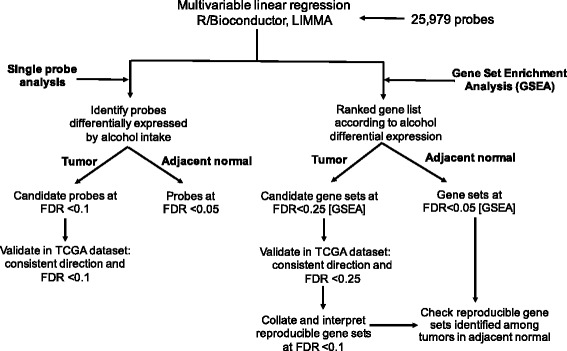
Analysis strategy for identifying differentially expressed probes or enriched pathway-defined gene sets in the Nurses’ Health Study (NHS) and the NHSII. FDR false discovery rate, LIMMA linear models for microarray data
To incorporate biological knowledge into the analysis, we further performed gene set enrichment analysis (GSEA) [21] to identify pathway-defined gene sets that varied by alcohol intake. Gene sets were collected from the Molecular Signatures Database (MSigDB) (http://www.broadinstitute.org/gsea/msigdb/), including 217 from BioCarta, 186 from Kyoto Encyclopedia of Genes and Genomes (KEGG), 674 from Reactome, and 825 from Gene Ontology (GO) biological process; those with < 15 genes or > 500 genes were filtered out, leaving 1293 pathway-defined gene sets in the analysis. The four pathway databases were included because each has a distinct but also complementary approach to capture known biological pathways [22]. We used the GSEA “Pre-ranked” function and imported ranked gene lists according to the alcohol-associated t statistic from the regression models. In the pathway analysis, only probes that are annotated as a gene (N = 15,407 probes) were included in the GSEA. Briefly, all the genes were first ranked according to the alcohol-associated t statistic; an enrichment score was then calculated for each gene set. The enrichment score corresponds to a weighted Kolmogorov-Smirnov-like statistic and reflects the extent to which the gene set is overrepresented at the extreme (i.e. top or bottom) of the entire ranked list [21]. If the enrichment score is positive (e.g. the gene set is overrepresented by top ranked genes), then the gene set is considered upregulated while it is considered downregulated if the score is negative. In the discovery stage, among tumors, gene sets at FDR <0.25 were considered significantly enriched. Again, a more stringent FDR threshold (i.e. FDR <0.05) was applied to tumor-adjacent normal samples. We further performed leading-edge subset analysis to identify the core set (i.e. key genes) of the gene set that accounted for the enrichment signal [21].
Validation analysis
The validation dataset consisted of RNA sequencing (RNA-Seq) data from 166 invasive breast tumors, a subset of breast tumor samples from TCGA that had pre-diagnostic alcohol consumption (generally defined as recent intake) and comparable covariate data. For the validation dataset, we originally contacted six TCGA sites with the largest number of potential cases and four of them agreed to collect or provide already available breast cancer risk factor data, including the University of Pittsburgh, Roswell Park Cancer Institute, the Mayo Clinic and Memorial Sloan Kettering Cancer Center. A total of 220 invasive cases had RNA-Seq data and at least some of the key covariates (e.g. BMI or alcohol or parity), of which 166 had complete information on alcohol consumption and covariates that were required for adjustment in the regression models. TCGA RNA-Seq data were previously processed using the MapSplice algorithm [23] to perform the alignment and RNA-Seq by expectation maximization (RSEM) [24] to estimate gene abundance. The expression dataset included 20,531 genes; in the differential expression analysis, genes with low expression (i.e. < 25th percentile) according to median counts per million were removed, leaving 15,398 unique genes. The common genes in the NHS/NHSII and the TCGA dataset accounted for approximately 84% of all the genes in each dataset. The RNA-Seq data were normalized using the trimmed mean of M-values [25] and log-transformed with associated precision weights using Voom. Multivariable linear regression implemented through R/Bioconductor LIMMA was then used to identify genes that were differentially expressed by recent alcohol intake and we further performed GSEA using similar methods as in the NHS/NHSII.
To validate the significantly enriched pathway-defined gene sets identified in the NHS/NHSII, we required that these gene sets showed a consistent direction (i.e. same upregulation or downregulation) of enrichment and an FDR <0.25 in the TCGA dataset (Fig. 1). Among those replicated gene sets, we only reported gene sets at FDR <0.1 in the NHS/NHSII. No validation dataset of breast normal or tumor-adjacent normal samples with alcohol consumption information was available and thus it was not feasible for us to replicate our results in further datasets.
Results
In the NHS and NHSII, the average alcohol intake was relatively low (mean 6.4 g/day, SD 11.4). Approximately 34% of the women had no recent alcohol consumption and 45% women consumed < 10 g of alcohol per day and only 21% women consumed 10+ g/day of alcohol. Age at diagnosis and parity were roughly evenly distributed across categories of recent alcohol intake. Women with higher alcohol intake were less likely to have a first-degree family history of breast cancer, had lower BMI and were diagnosed in more recent years (Table 1). Among women with natural menopause or bilateral oophorectomy, those with alcohol intake at least 10 g/day were less likely to use MHT compared to women with no or lower alcohol intake. Out of the 602 tumor specimens, 445 (74%) had matched adjacent normal tissues. The characteristics of women with tumor-adjacent normal samples were similar to those with only tumor specimens available (data not shown). Alcohol consumption and other risk factors such as age at diagnosis and BMI were similar between women diagnosed with ER+ tumors and those with ER- tumors (Table 2). Compared to ER+ tumors, ER- tumors tended to be larger, moderately or poorly differentiated, and diagnosed at a later stage.
Table 1.
Characteristics of patients with invasive breast cancer according to recent alcohol consumption in the NHS and the NHSII
| Recent alcohol consumption, g/day | ||||||
|---|---|---|---|---|---|---|
| 0 | > 0 to < 10 | 10+ | ||||
| N = 206 | N = 267 | N = 126 | ||||
| Mean | SD | Mean | SD | Mean | SD | |
| Age at diagnosis, years | 61.6 | 9.6 | 59.9 | 9.0 | 62.3 | 9.8 |
| BMI at diagnosis, kg/m2 | 27.3 | 5.7 | 25.7 | 4.6 | 24.5 | 4.3 |
| Parity | 2.6 | 1.7 | 2.7 | 1.7 | 2.5 | 1.9 |
| Cumulative average alcohola, g/day | 0.8 | 2.1 | 4.2 | 4.0 | 19.4 | 12.0 |
| Recent alcohol, g/day | 0 | 0 | 3.7 | 2.6 | 22.9 | 16.0 |
| N | % | N | % | N | % | |
| First-degree family history | 38 | 18.4 | 45 | 16.9 | 13 | 10.3 |
| Menopausal at diagnosis | ||||||
| Premenopausal | 32 | 15.5 | 56 | 21.0 | 15 | 11.9 |
| Postmenopausal | 169 | 82.0 | 203 | 76.0 | 108 | 85.7 |
| Unknown | 5 | 2.4 | 8 | 3.0 | 3 | 2.4 |
| Current MHT useb | 86 | 52.4 | 117 | 60.0 | 46 | 43.8 |
| Year of diagnosis | ||||||
| 1990‒1999 | 122 | 59.2 | 157 | 58.8 | 66 | 52.4 |
| 2000‒2004 | 66 | 32.0 | 90 | 33.7 | 52 | 41.3 |
| 2005‒2009 | 18 | 8.7 | 20 | 7.5 | 8 | 6.3 |
NHS Nurses’ Health Study, BMI body mass index, MHT menopausal hormone therapy
aCalculated as average alcohol consumption over time prior to breast cancer diagnosis using all available exposure information
bCurrent MHT use among postmenopausal women only
Table 2.
Study population and tumor characteristics by ER status in the NHS and the NHSII
| ER+ tumors N = 471 |
ER- tumors N = 127 |
|||
|---|---|---|---|---|
| Mean | SD | Mean | SD | |
| Age at diagnosis, years | 61.4 | 9.6 | 59.1 | 8.9 |
| BMI at diagnosis, kg/m2 | 26.0 | 5.0 | 26.1 | 5.1 |
| Parity | 2.6 | 1.8 | 2.9 | 1.8 |
| Recent alcohol, g/day | 6.5 | 11.7 | 5.8 | 10.0 |
| Cumulative average alcohol, g/day | 6.4 | 9.5 | 5.5 | 8.2 |
| N | % | N | % | |
| First-degree family history | 79 | 16.8 | 16 | 12.6 |
| Year of diagnosis | ||||
| 1990‒1999 | 251 | 53.3 | 91 | 71.7 |
| 2000 − 2004 | 182 | 38.6 | 26 | 20.5 |
| 2005‒2009 | 38 | 8.1 | 10 | 7.9 |
| Tumor size | ||||
| 0.1‒2.0 cm | 365 | 77.5 | 68 | 53.5 |
| 2.1‒4.0 cm | 75 | 15.9 | 46 | 36.2 |
| > 4.0 cm | 23 | 4.9 | 7 | 5.5 |
| Unknown | 8 | 1.7 | 6 | 4.7 |
| Lymph node involvement | ||||
| None | 349 | 74.1 | 95 | 74.8 |
| 1‒3 positive nodes | 85 | 18.0 | 22 | 17.3 |
| > 3 positive nodes | 33 | 7.0 | 8 | 6.3 |
| Metastatic at diagnosis | 4 | 0.8 | 1 | 0.8 |
| Unknown | 0 | 0 | 1 | 0.8 |
| Grade | ||||
| Well-differentiated | 124 | 26.3 | 6 | 4.7 |
| Moderately differentiated | 243 | 51.6 | 35 | 27.6 |
| Poorly differentiated | 89 | 18.9 | 67 | 52.8 |
| Unknown | 15 | 3.2 | 19 | 15.0 |
| Stagea | ||||
| I | 305 | 64.8 | 57 | 44.9 |
| II | 122 | 25.9 | 59 | 46.5 |
| III | 40 | 8.5 | 9 | 7.1 |
| IV | 4 | 0.8 | 1 | 0.8 |
| Unknown | 0 | 0 | 1 | 0.8 |
ER estrogen receptor, NHS Nurses’ Health Study, BMI body mass index
aStaging was based on tumor size and lymph node involvement
Patients with invasive breast cancer in the validation dataset (i.e. TCGA) were younger at diagnosis, had a higher BMI, were less likely to drink alcohol and were diagnosed more recently (i.e. 2005–2009), compared to those in the NHS/NHSII (Additional file 2: Table S1). In addition, the TCGA dataset included a greater percentage of premenopausal women than the NHS/NHSII dataset (38% vs. 17%); among postmenopausal women, those in the TCGA dataset were less likely to use MHT than women in the NHS/NHSII dataset. While the majority of the TCGA tumors were stage II or III, about 60% of the tumors in the NHS/NHSII were stage I. Similar to the NHS/NHSII, in TCGA, women with higher alcohol intake (i.e. 1+ drink per day) had a lower BMI, were less likely to have a first-degree family history of breast cancer, and tended to be premenopausal/perimenopausal and were diagnosed in more recent years (i.e. 2008–2011), compared to women with lower alcohol intake (Additional file 2: Table S2).
In the single-probe analysis, after adjusting for multiple comparisons, no probes were significantly differentially expressed by recent alcohol consumption (i.e. 10+ vs. 0 g/day) in tumor or tumor-adjacent normal samples (Additional file 2: Table S3 and Additional file 3: Figure S2). When comparing alcohol intake < 10 vs. 0 g/day, two probes showed significantly decreased expression in ER- tumors only (FDR = 0.05, Additional file 2: Table S3); however, no such significantly decreased expression was observed when comparing alcohol intake of 10+ vs. 0 g/day.
In contrast to the single-probe analysis, we observed significant enrichment for 239 pathway-defined gene sets (FDR <0.25) among ER+ tumors when comparing recent alcohol intake of 10+ g/day with 0 g/day, including 220 upregulated and 19 downregulated gene sets (Fig. 2). Out of the 220 upregulated gene sets, 63 (28.6%) were replicated in TCGA, of which 28 were at FDR <0.1 (Table 3 and Additional file 2: Table S4); out of the 19 downregulated gene sets, 11(57.9%) were replicated, of which 5 were at FDR <0.1 (Table 3). Among the replicated and significantly enriched (FDR <0.1) gene sets in ER+ tumors, alcohol intake (i.e. 10+ vs. 0 g/day) was associated with overexpression of genes involved in RNA metabolism and transport (e.g. REACTOME_METABOLISM_OF_RNA), cell cycle (e.g. GO_MEIOTIC_CELL_CYCLE), DNA repair (e.g. REACTOME_DOUBLE_STRAND_BREAK_REPAIR), downregulation of lipid metabolism (i.e. REACTOME_LIPID_DIGESTION_MOBILIZATION_AND_TRANSPORT) and PPAR signaling pathway (i.e. KEGG_PPAR_SIGNALING_PATHWAY). As there were multiple pathway-defined gene sets linking to similar biological processes, we noted that these gene sets contained both common and distinct genes. For instance, the leading-edge subset analysis revealed that among the three DNA repair related gene sets, there were six common genes (i.e. ATM, LIG1, NBN, RAD50, RAD52 and RPA1) which accounted for 12%, 43% and 10% of the key genes (i.e. leading-edge subsets) in the gene set GO_DNA_REPAIR, REACTOME_DOUBLE_STRAND_BREAK_REPAIR and GO_RESPONSE_TO_DNA_DAMAGE_STIMULUS, respectively. In contrast to ER+ tumors, among ER+ tumor-adjacent normal specimens, there was no enrichment for cell cycle related gene sets, and several gene sets of RNA processing were significantly enriched but downregulated (Fig. 3a). However, in both ER+ tumors and tumor-adjacent normal, alcohol consumption was associated with significant downregulation in lipid metabolism and in the PPAR signaling pathway. The PPAR signaling pathway consists of three subfamilies (i.e. alpha, gamma and delta) and the leading-edge subset analysis among ER+ tumors revealed that PPARG specifically was among the core genes that accounted for the enrichment signal.
Fig. 2.
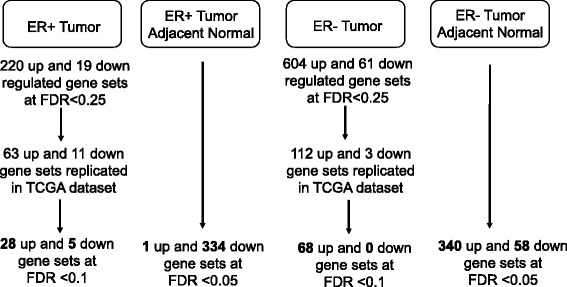
Number of enriched pathway-defined gene sets by alcohol (10+ vs. 0 g/day) in the Nurses’ Health Study (NHS) and the NHSII. ER estrogen receptor, FDR false discovery rate, TGCA The Cancer Genome Atlas
Table 3.
Enriched gene setsa by recent alcohol consumptionb in ER+ tumors in the NHS and the NHSII
| Pathway-defined gene set | Number of enriched genes | NES | FDR |
|---|---|---|---|
| Upregulated | |||
| REACTOME_NONSENSE_MEDIATED_DECAY_ENHANCED_BY_THE_EXON_JUNCTION_COMPLEX | 106 | 2.56 | <0.0001 |
| REACTOME_INFLUENZA_LIFE_CYCLE | 134 | 2.49 | <0.0001 |
| REACTOME_METABOLISM_OF_RNA | 248 | 2.14 | 0.001 |
| REACTOME_NEP_NS2_INTERACTS_WITH_THE_CELLULAR_EXPORT_MACHINERY | 26 | 1.99 | 0.007 |
| REACTOME_TRANSPORT_OF_MATURE_MRNA_DERIVED_FROM_AN_INTRONLESS_TRANSCRIPT | 31 | 1.98 | 0.008 |
| REACTOME_TRANSPORT_OF_RIBONUCLEOPROTEINS_INTO_THE_HOST_NUCLEUS | 26 | 1.97 | 0.008 |
| REACTOME_ANTIVIRAL_MECHANISM_BY_IFN_STIMULATED_GENES | 61 | 1.93 | 0.014 |
| POSITIVE_REGULATION_OF_TRANSCRIPTION_FROM_RNA_POLYMERASE_II_PROMOTER | 61 | 1.93 | 0.015 |
| REACTOME_INTERACTIONS_OF_VPR_WITH_HOST_CELLULAR_PROTEINS | 31 | 1.90 | 0.018 |
| MRNA_PROCESSING_GO_0006397 | 67 | 1.89 | 0.019 |
| REACTOME_SYNTHESIS_OF_GLYCOSYLPHOSPHATIDYLINOSITOL_GPI | 17 | 1.89 | 0.019 |
| MEIOTIC_CELL_CYCLE | 24 | 1.89 | 0.020 |
| REACTOME_TRANSPORT_OF_MATURE_TRANSCRIPT_TO_CYTOPLASM | 51 | 1.86 | 0.025 |
| RNA_PROCESSING | 159 | 1.85 | 0.027 |
| REACTOME_PROCESSING_OF_CAPPED_INTRON_CONTAINING_PRE_MRNA | 132 | 1.83 | 0.028 |
| REACTOME_METABOLISM_OF_NON_CODING_RNA | 45 | 1.83 | 0.029 |
| REACTOME_DOUBLE_STRAND_BREAK_REPAIR | 18 | 1.84 | 0.030 |
| PROTEIN_RNA_COMPLEX_ASSEMBLY | 60 | 1.84 | 0.030 |
| REACTOME_REGULATION_OF_GLUCOKINASE_BY_GLUCOKINASE_REGULATORY_PROTEIN | 26 | 1.78 | 0.048 |
| Downregulated | |||
| REACTOME_LIPID_DIGESTION_MOBILIZATION_AND_TRANSPORT | 38 | −2.12 | 0.017 |
| REACTOME_SMOOTH_MUSCLE_CONTRACTION | 21 | −2.13 | 0.024 |
| REACTOME_MUSCLE_CONTRACTION | 40 | −2.06 | 0.025 |
| KEGG_PPAR_SIGNALING_PATHWAY | 58 | −2.03 | 0.025 |
ER estrogen receptor, NHS Nurses’ Health Study, NES normalized enrichment score, FDR false discovery rate
aOnly gene sets replicated in The Cancer Genome Atlas (TCGA) dataset and FDR <0.05 are shown
bEnriched gene sets for comparison of recent alcohol consumption 10+ vs. 0 g/day
Fig. 3.
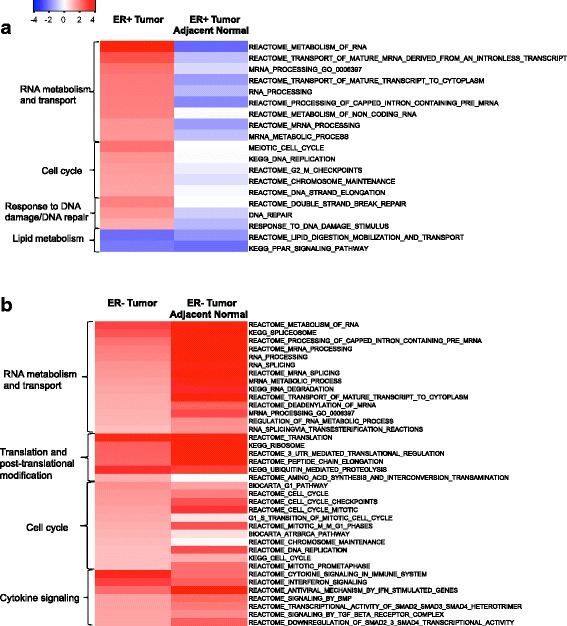
a, b Replicated enriched pathway-defined gene sets (false discovery rate (FDR) <0.1) by alcohol consumption in the Nurses’ Health Study (NHS) and the NHSII. Replicated significantly enriched gene sets according to recent alcohol intake (i.e. 10+ vs. 0 g/day) in estrogen receptor (ER)+ tumors and the same gene sets in tumor-adjacent normal tissues (a), and in ER- tumors and the same gene sets in tumor-adjacent normal tissues (b), NHSI/II. The primary biological processes observed are shown: N = 19 and 38 gene sets for ER+ and ER- tumors, respectively; -log10(FDR)*direction: red indicates upregulation and blue downregulation
For ER- tumors, we observed significant enrichment for 665 pathway-defined gene sets (FDR <0.25) when comparing recent alcohol intake of 10+ g/day with 0 g/day, including 604 upregulated and 61 downregulated gene sets (Fig. 2). Out of the 604 upregulated gene sets, 112 (18.5%) were replicated in TCGA, of which 68 were at FDR <0.1 (Table 4 and Additional file 2: Table S5); out of the 61 downregulated gene sets, 3 (4.9%) were replicated but none were at FDR <0.1. The 68 reproducible and significantly (FDR <0.1) upregulated gene sets identified among ER- tumors demonstrated that, in addition to the upregulation in RNA processing and cell cycle regulation, alcohol intake was also linked to strong enrichment in cytokine signaling (e.g. REACTOME_INTERFERON_SIGNALING and REACTOME_SIGNALING_BY_TGF_BETA_RECEPTOR_COMPLEX) and translation and post-translational modification (e.g. KEGG_UBIQUITIN_MEDIATED_PROTEOLYSIS). Among the cytokine signaling pathways, four gene sets were related to TGF-β/SMAD/BMP signaling; four common genes (i.e., SMAD4, SMURF2, UBE2D3, and UBE2D1) were observed among the leading-edge subsets of these four gene sets, and the overlapping genes accounted for about 15%, 50%, 21% and 29% of the leading-edge subset of REACTOME_SIGNALING_BY_TGF_BETA_RECEPTOR_COMPLEX, REACTOME_DOWNREGULATION_OF_SMAD2_3_SMAD4_TRANSCRIPTIONAL_ACTIVITY, and REACTOME_TRANSCRIPTIONAL_ACTIVITY_OF_SMAD2_SMAD3_SMAD4_HETEROTRIMER, and REACTOME_SIGNALING_BY_BMP, respectively. Similar significant enrichment was also observed among ER- tumor-adjacent normal tissues (Fig. 3b).
Table 4.
Enriched gene setsa by recent alcohol consumptionb in ER- tumors in the NHS and the NHSII
| Pathway-defined gene set | Number of enriched genes | NES | FDR |
|---|---|---|---|
| Upregulated | |||
| REACTOME_TRANSLATION | 144 | 2.31 | 0.0002 |
| REACTOME_CYTOKINE_SIGNALING_IN_IMMUNE_SYSTEM | 218 | 2.32 | 0.0003 |
| KEGG_UBIQUITIN_MEDIATED_PROTEOLYSIS | 124 | 2.38 | 0.001 |
| REACTOME_METABOLISM_OF_MRNA | 206 | 2.25 | 0.001 |
| PROTEIN_RNA_COMPLEX_ASSEMBLY | 60 | 2.25 | 0.001 |
| REACTOME_INTERFERON_SIGNALING | 129 | 2.21 | 0.001 |
| REACTOME_METABOLISM_OF_RNA | 248 | 2.18 | 0.001 |
| REACTOME_SRP_DEPENDENT_COTRANSLATIONAL_PROTEIN_TARGETING_TO_MEMBRANE | 108 | 2.17 | 0.001 |
| REACTOME_INFLUENZA_LIFE_CYCLE | 134 | 2.13 | 0.002 |
| KEGG_SPLICEOSOME | 122 | 2.10 | 0.002 |
| KEGG_RIBOSOME | 84 | 2.08 | 0.003 |
| BIOCARTA_CDC42RAC_PATHWAY | 15 | 2.08 | 0.003 |
| REACTOME_3_UTR_MEDIATED_TRANSLATIONAL_REGULATION | 104 | 2.07 | 0.003 |
| REACTOME_NONSENSE_MEDIATED_DECAY_ENHANCED_BY_THE_EXON_JUNCTION_COMPLEX | 106 | 2.05 | 0.003 |
| REACTOME_PEPTIDE_CHAIN_ELONGATION | 85 | 2.03 | 0.004 |
| REACTOME_ANTIVIRAL_MECHANISM_BY_IFN_STIMULATED_GENES | 61 | 2.01 | 0.005 |
| RIBONUCLEOPROTEIN_COMPLEX_BIOGENESIS_AND_ASSEMBLY | 79 | 1.97 | 0.006 |
| REACTOME_PROCESSING_OF_CAPPED_INTRON_CONTAINING_PRE_MRNA | 132 | 1.97 | 0.006 |
| REACTOME_INFLUENZA_VIRAL_RNA_TRANSCRIPTION_AND_REPLICATION | 101 | 1.94 | 0.008 |
| REACTOME_MRNA_PROCESSING | 151 | 1.92 | 0.009 |
| RNA_PROCESSING | 159 | 1.88 | 0.013 |
| BIOCARTA_G1_PATHWAY | 25 | 1.84 | 0.017 |
| RNA_SPLICING | 87 | 1.82 | 0.020 |
| REACTOME_MRNA_SPLICING | 104 | 1.80 | 0.023 |
| REACTOME_CELL_CYCLE | 328 | 1.79 | 0.024 |
| REACTOME_CELL_CYCLE_CHECKPOINTS | 97 | 1.78 | 0.026 |
| CELLULAR_COMPONENT_ASSEMBLY | 272 | 1.75 | 0.030 |
| REGULATION_OF_TRANSCRIPTION_FROM_RNA_POLYMERASE_II_PROMOTER | 271 | 1.75 | 0.030 |
| MRNA_METABOLIC_PROCESS | 78 | 1.74 | 0.031 |
| REACTOME_HIV_INFECTION | 183 | 1.74 | 0.031 |
| RANSCRIPTION_FROM_RNA_POLYMERASE_II_PROMOTER | 427 | 1.73 | 0.032 |
| REACTOME_SIGNALING_BY_BMP | 21 | 1.72 | 0.032 |
| REACTOME_TRANSCRIPTIONAL_ACTIVITY_OF_SMAD2_SMAD3_SMAD4_HETEROTRIMER | 33 | 1.72 | 0.033 |
| REACTOME_ACTIVATION_OF_THE_MRNA_UPON_BINDING_OF_THE_CAP_BINDING_COMPLEX_AND_EIFS_AND_SUBSEQUENT_BINDING_TO_43S | 56 | 1.71 | 0.034 |
| REACTOME_SIGNALING_BY_TGF_BETA_RECEPTOR_COMPLEX | 56 | 1.70 | 0.037 |
| REACTOME_CELL_CYCLE_MITOTIC | 255 | 1.70 | 0.037 |
| NUCLEAR_EXPORT | 31 | 1.68 | 0.041 |
| REACTOME_DOWNREGULATION_OF_SMAD2_3_SMAD4_TRANSCRIPTIONAL_ACTIVITY | 18 | 1.68 | 0.041 |
| POSITIVE_REGULATION_OF_NUCLEOBASENUCLEOSIDENUCLEOTIDE_AND_NUCLEIC_ACID_METABOLIC_PROCESS | 143 | 1.68 | 0.042 |
| KEGG_RNA_DEGRADATION | 55 | 1.67 | 0.042 |
| KEGG_ADHERENS_JUNCTION | 70 | 1.67 | 0.043 |
| G1_S_TRANSITION_OF_MITOTIC_CELL_CYCLE | 25 | 1.67 | 0.045 |
| REACTOME_TRANSPORT_OF_MATURE_TRANSCRIPT_TO_CYTOPLASM | 51 | 1.66 | 0.045 |
| POSITIVE_REGULATION_OF_TRANSCRIPTION | 134 | 1.65 | 0.048 |
| Downregulated | |||
| None | |||
ER estrogen receptor, NHS Nurses’ Health Study, NES normalized enrichment score, FDR false discovery rate
aOnly gene sets replicated in The Cancer Genome Atlas (TCGA) dataset and FDR <0.05 are shown
bEnriched gene sets for comparison of recent alcohol consumption 10+ vs. 0 g/day
In both ER+ and ER- tumors, alcohol intake was associated with upregulation in gene sets involved in RNA metabolism and transport and cell cycle. For instance, “REACTOME_METABOLISM_OF_RNA” was the top ranked pathway under the category of RNA metabolism and transport in both ER+ and ER- tumors (Fig. 3a and b). However, despite some overlapping gene sets within each category, there were some specific gene sets in either ER+ or ER- tumors. For example, among cell cycle related gene sets, in ER+ tumors, genes involved in the G2/M phase checkpoint were overexpressed while in ER- tumors, genes involved in the G1 or G1/S transition were upregulated (Fig. 3a and b). Further, as we hypothesized that the biological mechanism of the alcohol and breast cancer association may vary between ER+ and ER- tumors, we also noted that there were several ER+ or ER- tumor-specific gene sets. Among ER+ tumors, alcohol intake was linked to gene sets involved in upregulation of DNA repair but this not observed among ER- tumors. In addition, genes related to lipid metabolism were down-expressed in ER+ tumors but not in ER- tumors. On the other hand, upregulation in cytokine signaling was only observed among ER- tumors.
Among ER+ tumor-adjacent normal tissues, we observed significant enrichment for 335 pathway-defined gene sets (FDR <0.05), of which 1 was upregulated and 334 were downregulated by recent alcohol intake (i.e. 10+ vs. 0 g/day); among ER- tumor-adjacent normal tissues, 340 and 58 pathway-defined gene sets were significantly (FDR <0.05) upregulated and downregulated, respectively. Table 5 presents the top 10 ranked upregulated or downregulated pathway-defined gene sets identified in the NHS/NHSII. Among ER+ tumor-adjacent normal tissues, only one gene set (i.e. olfactory transduction) was significantly upregulated at FDR <0.05; the top ranked downregulated gene sets included mitochondrial respiratory electron transport and TCA cycle, WNT signaling pathway, integrin pathway and focal adhesion, and fatty acids/triacylglycerol/ketone body metabolism. Among ER- tumor-adjacent normal tissues, the top ranked upregulated gene sets, such as RNA metabolism and translation, were also seen among those top ranked in ER- tumors; the strongest enrichment for downregulated gene sets included neuroactive ligand-receptor interaction, GPCR ligand binding, and cytochrome P450 arranged by substrate type.
Table 5.
Top 10 enriched gene setsa by alcohol consumptionb in tumor-adjacent normal samples in the NHS and the NHSII
| Pathway-defined gene set | Number of enriched genes | NES | FDR |
|---|---|---|---|
| ER+ tumor-adjacent normal tissues | |||
| Upregulated | |||
| KEGG_OLFACTORY_TRANSDUCTION | 153 | 1.97 | 0.04 |
| Downregulated | |||
| REACTOME_TCA_CYCLE_AND_RESPIRATORY_ELECTRON_TRANSPORT | 123 | −2.33 | <0.001 |
| REACTOME_SIGNALING_BY_WNT | 60 | −2.33 | <0.001 |
| REACTOME_PYRUVATE_METABOLISM_AND_CITRIC_ACID_TCA_CYCLE | 39 | −2.30 | <0.001 |
| REACTOME_CTNNB1_PHOSPHORYLATION_CASCADE | 15 | −2.28 | <0.001 |
| KEGG_FOCAL_ADHESION | 186 | −2.27 | <0.001 |
| BIOCARTA_RHO_PATHWAY | 30 | −2.27 | <0.001 |
| BIOCARTA_INTEGRIN_PATHWAY | 36 | −2.26 | <0.001 |
| BIOCARTA_PYK2_PATHWAY | 27 | −2.26 | <0.001 |
| KEGG_PATHOGENIC_ESCHERICHIA_COLI_INFECTION | 51 | −2.25 | <0.001 |
| REACTOME_FATTY_ACID_TRIACYLGLYCEROL_AND_KETONE_BODY_METABOLISM | 151 | −2.24 | 1.55E-04 |
| ER- tumor-adjacent normal | |||
| Up-regulated | |||
| REACTOME_METABOLISM_OF_MRNA | 206 | 2.87 | <0.001 |
| REACTOME_METABOLISM_OF_RNA | 248 | 2.79 | <0.001 |
| REACTOME_NONSENSE_MEDIATED_DECAY_ENHANCED_BY_THE_EXON_JUNCTION_COMPLEX | 106 | 2.66 | <0.001 |
| REACTOME_INFLUENZA_LIFE_CYCLE | 134 | 2.65 | <0.001 |
| REACTOME_3_UTR_MEDIATED_TRANSLATIONAL_REGULATION | 104 | 2.64 | <0.001 |
| REACTOME_TRANSLATION | 144 | 2.63 | <0.001 |
| REACTOME_SRP_DEPENDENT_COTRANSLATIONAL_PROTEIN_TARGETING_TO_MEMBRANE | 108 | 2.62 | <0.001 |
| REACTOME_TRANSPORT_OF_MATURE_TRANSCRIPT_TO_CYTOPLASM | 51 | 2.61 | <0.001 |
| REACTOME_PROCESSING_OF_CAPPED_INTRON_CONTAINING_PRE_MRNA | 132 | 2.60 | <0.001 |
| REACTOME_MRNA_PROCESSING | 151 | 2.59 | <0.001 |
| Down-regulated | |||
| KEGG_NEUROACTIVE_LIGAND_RECEPTOR_INTERACTION | 192 | −2.52 | <0.001 |
| REACTOME_GPCR_LIGAND_BINDING | 297 | −2.49 | <0.001 |
| REACTOME_TRANSPORT_OF_GLUCOSE_AND_OTHER_SUGARS_BILE_SALTS_AND_ORGANIC_ACIDS_METAL_IONS_AND_AMINE_COMPOUNDS | 76 | −2.36 | <0.001 |
| REACTOME_CLASS_A1_RHODOPSIN_LIKE_RECEPTORS | 212 | −2.29 | 4.14E-04 |
| REACTOME_POTASSIUM_CHANNELS | 86 | −2.30 | 5.18E-04 |
| REACTOME_CYTOCHROME_P450_ARRANGED_BY_SUBSTRATE_TYPE | 37 | −2.20 | 2.04E-03 |
| GENERATION_OF_A_SIGNAL_INVOLVED_IN_CELL_CELL_SIGNALING | 27 | −2.14 | 3.22E-03 |
| REACTOME_VOLTAGE_GATED_POTASSIUM_CHANNELS | 36 | −2.14 | 3.29E-03 |
| G_PROTEIN_SIGNALING_COUPLED_TO_CYCLIC_NUCLEOTIDE_SECOND_MESSENGER | 83 | −2.12 | 3.50E-03 |
| REACTOME_G_ALPHA_I_SIGNALLING_EVENTS | 147 | −2.12 | 3.50E-03 |
ER estrogen receptor, NHS Nurses’ Health Study, NES normalized enrichment score, FDR false discovery rate
aTop 10 ranked gene sets at FDR <0.05 are shown for upregulation and downregulation, respectively
bEnriched gene sets for comparison of recent alcohol consumption 10+ vs. 0 g/day
As several enzymes, such as alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH), are known to play an important role in alcohol metabolism, we specifically examined the expression of genes involved in alcohol metabolism in tumors and adjacent normal tissues. Most of these genes, including ADH1B, ALDH1A1, ADH1C and ALDH2, were significantly down-expressed in ER+ or ER- tumors compared to tumor-adjacent normal tissues (Table 6), although none showed significant differential expression by alcohol intake in either tissue type. For instance, among the seven alcohol metabolism genes included in our data, ADH1B showed the most reduced expression in ER+ or ER- tumors (fold change 0.40).
Table 6.
Differential expression of alcohol metabolism genes in tumor and tumor-adjacent normal tissues in the NHS and the NHSII
| ER+ tumors vs. tumor-adjacent normal tissues | ER- tumors vs. tumor-adjacent normal tissues | |||||||
|---|---|---|---|---|---|---|---|---|
| Probeset ID | Entrez ID | Symbol | Log2(FC) | t valuea | FDRb | Log2(FC) | t valuea | FDRb |
| TC0401141 | 125 | ADH1B | −1.33 | −21.1 | 2.92E-61 | −1.32 | −11.0 | 1.59E-14 |
| TC0901044 | 216 | ALDH1A1 | −0.61 | −16.7 | 2.77E-44 | −0.58 | −8.6 | 6.93E-11 |
| TC0401142 | 126 | ADH1C | −0.25 | −14.6 | 3.80E-36 | −0.29 | −7.6 | 3.08E-09 |
| TC1100272 | 847 | CAT | −0.34 | −10.2 | 1.99E-20 | −0.32 | −4.2 | 6.20E-04 |
| TC1200702 | 217 | ALDH2 | −0.18 | −7.0 | 6.53E-11 | −0.20 | −4.0 | 1.01E-03 |
| TC0401137 | 128 | ADH5 | −0.11 | −4.8 | 7.69E-06 | −0.04 | −0.7 | 6.00E-01 |
| TC1000709 | 1571 | CYP2E1 | −0.02 | −1.9 | 8.21E-02 | −0.02 | −0.6 | 7.02E-01 |
ER estrogen receptor, NHS Nurses’ Health Study, FDR false discovery rate, FC fold change
aThe t values were obtained from paired t tests of tumor and adjacent normal tissues: 357 pairs of ER+ tumor and ER+ tumor-adjacent normal tissues, and 86 pairs for ER- tumor and ER- tumor-adjacent normal, respectively
bFDR was calculated across all the 25,979 probes
As more than half (i.e. 60%) of the tumors in the NHS/NHSII were stage I tumors while the majority of the tumors in TCGA were stage II or III, we further conducted stratified analyses according to tumor stage in secondary analyses. Specifically, we performed GSEA among stage II/III ER+ tumors in the NHS/NHSII and further validated in stage II/III ER+ tumors in TCGA; we were not able to conduct similar analysis among stage II/III ER- tumors because of the limited case numbers in TCGA. We found that the replicated enrichment signals in stage II/III ER+ tumors were very similar to those in all ER+ tumors in the NHS/NHSII (Additional file 2: Table S6).
Discussion
To our knowledge, this is the first epidemiologic study to assess the association between pre-diagnostic alcohol consumption and breast tumor genome-wide gene expression. In the differential gene expression analysis by recent alcohol consumption, we did not find individual genes significantly upregulated or downregulated by alcohol after accounting for multiple comparisons. However, gene set analysis identified reproducible enriched pathway-defined gene sets in breast tumors. Specifically, recent alcohol intake of at least 10 g/day was linked to increased proliferation and lower lipid metabolism in ER+ tumors; among ER- tumors, in addition to an increase in proliferation, some further signals, including upregulation in cytokine signaling, such as interferon (IFN) and TGF-β signaling pathways, were noted.
Cohort studies generally support a stronger positive association among ER+ tumors than among ER- tumors [26, 27]. A strong enrichment signal observed from GSEA was increased proliferation in ER+ tumors. Several of the significantly upregulated gene sets, including cell cycle regulation (e.g. mitosis and G2/M checkpoint) and DNA repair are closely related to proliferation [28]. In addition, RNA processing (e.g. RNA splicing or transport) has been shown to affect cell cycle and proliferation [29], although increased RNA processing also may be a consequence of proliferation. Our finding is consistent with experimental studies in which ethanol promoted proliferation in ER+ breast tumor cell lines [30–32]. In ER+ tumors, we also observed downregulation of lipid metabolism, including the PPAR-gamma signaling pathway. PPAR-gamma signaling plays an essential role in adipocyte differentiation and expression of adipocyte specific genes, and also regulates lipid metabolism, cell proliferation and differentiation, glucose homeostasis and inflammation [33]. Further, in experimental studies, PPAR-gamma inhibited proliferation in ER+ breast cancer cell lines [34] and ethanol inhibited PPAR-gamma dependent transcriptional activation [35]. Taken together, downregulation of the PPAR-gamma signaling pathway is consistent with the observed increase in proliferation in our data. In addition, lower lipid metabolism was observed among ER+ tumor-adjacent normal tissues in the current dataset. If replicated, this finding suggests that alcohol consumption disrupts lipid metabolism, providing another possible link to alcohol-related breast pathogenesis.
Among the hypothesized mechanisms through which alcohol consumption increases breast cancer risk, particularly ER+ disease, the most studied pathway is estrogen metabolism with supporting evidence from intervention studies that alcohol drinking is associated with increases in circulating estrogens [5, 6]. In experimental studies, ER-mediated estrogen signaling can increase cell proliferation that in turn can induce genetic mutations [36, 37], while estrogen metabolites, independent of ER signaling, also can cause DNA damage [37]. Although our data are consistent with estrogen/ER signaling mediated increased proliferation and DNA damage, we did not find alcohol intake to be associated with increased expression of specific estrogen-related genes or gene sets in ER+ tumors or tumor-adjacent normal tissue. The reason is unclear. To what extent alcohol-associated estrogen metabolism occurs in breast tissue in cancer-free women and whether it would be preserved in breast tissue during tumor progression is not known. In a recent study that explored parity-associated gene expression signatures, the signature identified in normal breast tissue was preserved in ER+ but not in ER- breast tumors [13]. Further, as shown in alcohol intervention studies with a crossover study design [5, 6], the alcohol-associated increases in circulating estrogen or estrogen metabolite levels are a relatively acute alcohol effect. Whether alcohol-induced estrogen metabolism occurring in breast tissue is similarly short term or more long lasting is not known, and could have influenced our ability to detect an association.
Among ER- tumors, recent alcohol consumption was also linked to increases in proliferation. In addition to cell cycle upregulation, significant increases in translational and post-translational modification were observed, which may be associated with alterations in cell cycle and regulation of cell growth [38]. However, a prior experimental study reported that ethanol only induced proliferation in ER+ but not ER- breast cancer cell lines [31]. The observation of alcohol-related proliferation in both ER+ and ER- tumors in our data suggests that alcohol-induced proliferation may not exclusively act through estrogen metabolism, as no pathway-defined gene sets related to estrogen metabolism were significantly enriched by alcohol intake.
Compared to the enrichment signals observed among ER+ tumors, a distinct enrichment was found in ER- tumors: alcohol intake was associated with upregulation in cytokine signaling including IFN signaling and TGF-β signaling pathways. Alcohol is known to modulate the immune system in a complex way. In animal models, chronic ethanol exposure was shown to alter cytokine levels (e.g. TNF-α, TGF-β, IL-6) in a variety of tissues, including lung, liver and brain [39], although breast tissue was not assessed. In a population study of over 1300 women, circulating IL-6 levels significantly increased among women consuming at least one alcoholic drink per day, while no increase was reported among women with light alcohol intake (i.e. less than one drink/day) [40]. In addition, cytokines play an important role in breast tumor growth and progression [41, 42]. Expression levels of multiple cytokines were higher in ER- compared to ER+ breast tumors, including IFN-γ, TNF-α, and IL-6 and IL-8 [43]. Further, breast tumor ER expression may be an important mediator of the transition of TGF-β from tumor suppression to tumor promotion: loss of ER expression (i.e. in ER- tumor cells) and loss of hormonally controlled growth may lead to an increased tumor promoting effect of TGF-β [44]. Interestingly, the significantly enriched pathway-defined gene sets identified in ER- tumors had a consistent enrichment pattern and even stronger enrichment signals among ER- tumor-adjacent normal tissues, while no similarities were observed in enrichment signals between ER+ tumors and ER+ tumor-adjacent normal tissues. One possible reason is that ER- tumors in this dataset were on average more advanced than ER+ tumors (i.e. larger in size, higher in grade and at a more advanced stage); thus, although the tumor-adjacent normal tissues were defined as generally > 1 cm from the tumor edge, ER- tumors may have more strongly influenced adjacent normal tissues. However, the fact that adjacent normal tissues may contain information on the environment surrounding the tumors may not directly relate to alcohol consumption; thus, the exact reason for the consistent enrichment pattern in ER- tumors and adjacent normal tissues is not clear. As this is the first ever assessment of alcohol intake and gene expression in both tumor and adjacent normal tissues, replication of this finding in other studies will be important.
Although none of the known alcohol metabolism genes were differentially expressed by alcohol intake in breast tumors in this study, several of these genes, including ADH1B, ADH1C, ALDH2 and ALDH1A1 were substantially downregulated in tumors compared to tumor-adjacent normal tissues, regardless of tumor ER expression. Our results are consistent with a previous study that observed class I ADH (including ADH1A, ADH1B and ADH1C) to be more highly expressed, at both mRNA and protein levels, in normal breast tissues from cancer-free women than in invasive breast tumor tissues [45]. Despite no significant enrichment of pathway-defined gene sets involved in alcohol metabolism according to alcohol intake, it was interesting to see that recent alcohol consumption (i.e. 10+ g/day) was marginally associated with significant downregulation in retinol metabolism (i.e. KEGG_RETINOL_METABOLISM) among ER+ tumors (FDR = 0.11; also replicated in TCGA) because ADH enzymes are also involved in retinol metabolism [46]. Abnormal retinoid metabolism has been observed in several cancers, including breast cancer [47].
Our study, using tumor genome-wide gene expression profiling provided novel insights of alcohol-related molecular pathways in breast tumors. The evaluation was conducted within large prospective cohort studies with detailed data on alcohol consumption, covariates, and cancer diagnosis and tumor characteristics. In addition, this study has a sizable number of breast tumor and tumor-adjacent normal specimens. Further, our results, using microarrays, were validated in another platform using RNA-Seq. Finally, in addition to single-probe analysis, we conducted pathway analysis (i.e. GSEA), which has several advantages over single-gene analysis. For example, it makes interpretation easier by focusing on pathways and biological processes rather than single high-scoring genes which may be poorly annotated. GSEA also makes it possible to detect modest expression changes in individual genes as it can increase the signal-to-noise ratio. However, one drawback in this GSEA is that the P value estimation using gene permutation under GSEA “Preranked”’ function does not take into account correlation among genes [48].
Our study also has limitations. One limitation is the use of FFPE tissues for gene expression profiling, because FFPE can make retrieval of RNA challenging due to chemical modification of RNA and related RNA degradation. However, archived FFPE samples have been shown comparable to fresh-frozen samples in assessing differential expression in lung, colon and kidney tissues [49]. In addition, our results were validated in a dataset derived from fresh-frozen breast tumor samples that were part of TCGA. However, there were differences in patient characteristics (e.g. age) and tumor characteristics (i.e. tumor stage) between the NHS and TCGA datasets. Another limitation is the lack of a validation dataset for tumor-adjacent normal tissues. Further, women participating in this study had relatively low levels of alcohol consumption and thus we were not able to evaluate the effect of moderate to heavy alcohol intake on tumor gene expression. Indeed, in epidemiologic studies, compared to women without recent alcohol intake, those with recent consumption 10–20 g/day had only ~10% increased breast cancer risk while those consuming at least 30 g/day had > 30% increased risk [3]. Finally, the tumor gene expression profiling here may be a mixed profiling of malignant epithelial and stromal cells. Laser capture microdissection can be used to isolate specific cell types; however, it was not feasible considering the large sample size in this study.
Conclusions
Our data suggest that alcohol consumption is associated with increased proliferation and lower lipid metabolism among ER+ breast tumors while among ER- breast tumors, alcohol consumption is not only linked to increased proliferation but also upregulation in cytokine signaling, particularly IFN and TGF-β signaling. Future studies of gene expression profiling in normal breast tissues from cancer-free women are of particular interest. Alcohol consumption is not considered a prognostic factor for breast cancer recurrence or death [50]. Assessment of the effect of alcohol consumption on normal breast tissues, together with profiling data from breast tumors and/or tumor-adjacent normal tissues will be critical in further elucidating the alcohol-related breast carcinogenesis. Furthermore, as alcohol is known to impact one-carbon metabolism and induce aberrant DNA methylation [51], integrating DNA methylation and gene expression data may provide deeper insights into the underlying biology of the association between alcohol and breast cancer.
Additional files
ESR1, PGR, ERBB2 mRNA expression by IHC ER, PR, and HER2, respectively, in the NHS and the NHSII. (PDF 273 kb)
Comparison of study population characteristics in the NHS/NHSII and TCGA. Table S2. Characteristics of invasive breast cancer cases by recent alcohol consumption in the TCGA. Table S3. Top 10 ranked differentially expressed probes by recent alcohol consumption in the NHS and the NHSII. Table S4. Enriched gene sets (FDR 0.05–0.1) by recent alcohol consumption in ER+ tumors in the NHS and the NHSII. Table S5. Enriched gene sets (FDR 0.05–0.1) by recent alcohol consumption in ER- tumors in the NHS and the NHSII. Table S6. Enriched gene sets by alcohol consumption in stage II/III ER+ tumors in the NHS and the NHSII. (DOCX 56 kb)
Differentially expressed probes (N = 25,979) by alcohol consumption in the NHS and the NHSII. The top four figures show recent alcohol intake > 0 - < 10 vs. 0 g/day and the bottom four figures show recent alcohol intake 10+ vs. 0 g/day. (PDF 1418 kb)
Acknowledgements
We would like to thank the participants and staff of the Nurses’ Health Study and the Nurses’ Health Study II for their valuable contributions and the cancer registries in the following states for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA and WY. The authors assume full responsibility for analyses and interpretation of these data.
Funding
This study was supported in part by a Komen Foundation Grant SAC110014 and the NIH NCI U19/GAME-ON DRIVE (CA148065) initiative, UM1 CA186107, P01 CA87969, and UM1 CA176726.
Availability of data and materials
Gene expression data are publicly available through the Gene Expression Omnibus [GEO:GSE93601].
Abbreviations
- ADH
Alcohol dehydrogenase
- ALDH
Aldehyde dehydrogenase
- AUC
Area under the curve
- BMI
Body mass index
- CGEMS
Cancer Genetic Markers of Susceptibility
- CI
Confidence interval
- ER
Estrogen receptor
- FDR
False discovery rate
- FFPE
Formalin-fixed paraffin-embedded
- GO
Gene Ontology
- GSEA
Gene set enrichment analysis
- HER2
Human epidermal growth factor receptor 2
- IFN
Interferon
- IHC
Immunohistochemistry
- IL
Interleukin
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LIMMA
Linear models for microarray data
- MHT
Menopausal hormone therapy
- MSigDB
Molecular Signatures Database
- NES
Normalized enrichment score
- NHS
Nurses’ Health Study
- PR
Progesterone receptor
- RMA
Robust multiarray average
- RNA-Seq
RNA sequencing
- RSEM
RNA-Seq by expectation maximization
- TCGA
The Cancer Genome Atlas
- TGF
Transforming growth factor
- TNF
Tumor necrosis factor
Authors’ contributions
Conception and design: SEH; development of methodology: JW, YJH, AH, VJC, AHB and SEH; acquisition of data: DJH, SEH, CBA, TAK, FM and CMV; analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): JW, YJH, SEH and AHB; writing, review and/or revision of the manuscript: JW, YJH, AHE, RMT, AH, VJC, CBA, VPA, AB, FJC, TAK, FM, CMV, DJH, AHB and SEH; administrative, technical, or material support: DJH and SEH; study supervision: SEH and AHB. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Committee on the Use of Human Subjects in Research at the Brigham and Women’s Hospital. Informed consent was obtained from participating women.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13058-017-0901-y) contains supplementary material, which is available to authorized users.
Contributor Information
Jun Wang, Phone: 323-442-8112, Email: junw@usc.edu.
Yujing J. Heng, Email: yheng@bidmc.harvard.edu
A. Heather Eliassen, Email: nhahe@channing.harvard.edu.
Rulla M. Tamimi, Email: nhrmt@channing.harvard.edu
Aditi Hazra, Email: AHAZRA@bwh.harvard.edu.
Vincent J. Carey, Email: stvjc@channing.harvard.edu
Christine B. Ambrosone, Email: Christine.Ambrosone@RoswellPark.org
Victor P. de Andrade, Email: victor.andrade@accamargo.org.br
Adam Brufsky, Email: brufskyam@upmc.edu.
Fergus J. Couch, Email: Couch.Fergus@mayo.edu
Tari A. King, Email: TKING7@PARTNERS.ORG
Francesmary Modugno, Email: modugnof@upmc.edu.
Celine M. Vachon, Email: Vachon.Celine@mayo.edu
David J. Hunter, Email: dhunter@hsph.harvard.edu
Andrew H. Beck, Email: andy.beck@pathai.com
Susan E. Hankinson, Email: shankinson@schoolph.umass.edu
References
- 1.Jung S, Wang M, Anderson K, Baglietto L, Bergkvist L, Bernstein L, van den Brandt PA, Brinton L, Buring JE, Eliassen AH, et al. Alcohol consumption and breast cancer risk by estrogen receptor status: in a pooled analysis of 20 studies. Int J Epidemiol. 2016;45(3):916–928. doi: 10.1093/ije/dyv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith-Warner SA, Spiegelman D, Yaun SS, van den Brandt PA, Folsom AR, Goldbohm RA, Graham S, Holmberg L, Howe GR, Marshall JR, et al. Alcohol and breast cancer in women: a pooled analysis of cohort studies. JAMA. 1998;279(7):535–540. doi: 10.1001/jama.279.7.535. [DOI] [PubMed] [Google Scholar]
- 3.Chen WY, Rosner B, Hankinson SE, Colditz GA, Willett WC. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA. 2011;306(17):1884–1890. doi: 10.1001/jama.2011.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li CI, Chlebowski RT, Freiberg M, Johnson KC, Kuller L, Lane D, Lessin L, O'Sullivan MJ, Wactawski-Wende J, Yasmeen S, et al. Alcohol consumption and risk of postmenopausal breast cancer by subtype: the women's health initiative observational study. J Natl Cancer Inst. 2010;102(18):1422–1431. doi: 10.1093/jnci/djq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorgan JF, Baer DJ, Albert PS, Judd JT, Brown ED, Corle DK, Campbell WS, Hartman TJ, Tejpar AA, Clevidence BA, et al. Serum hormones and the alcohol-breast cancer association in postmenopausal women. J Natl Cancer Inst. 2001;93(9):710–715. doi: 10.1093/jnci/93.9.710. [DOI] [PubMed] [Google Scholar]
- 6.Reichman ME, Judd JT, Longcope C, Schatzkin A, Clevidence BA, Nair PP, Campbell WS, Taylor PR. Effects of alcohol consumption on plasma and urinary hormone concentrations in premenopausal women. J Natl Cancer Inst. 1993;85(9):722–727. doi: 10.1093/jnci/85.9.722. [DOI] [PubMed] [Google Scholar]
- 7.Seitz HK, Pelucchi C, Bagnardi V, La Vecchia C. Epidemiology and pathophysiology of alcohol and breast cancer: update 2012. Alcohol Alcohol. 2012;47(3):204–212. doi: 10.1093/alcalc/ags011. [DOI] [PubMed] [Google Scholar]
- 8.Secretan B, Straif K, Baan R, Grosse Y, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, et al. A review of human carcinogens − part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009;10(11):1033–1034. doi: 10.1016/S1470-2045(09)70326-2. [DOI] [PubMed] [Google Scholar]
- 9.Castro GD, Delgado de Layno AM, Fanelli SL, Maciel ME, Diaz Gomez MI, Castro JA. Acetaldehyde accumulation in rat mammary tissue after an acute treatment with alcohol. J Appl Toxicol. 2008;28(3):315–321. doi: 10.1002/jat.1281. [DOI] [PubMed] [Google Scholar]
- 10.Fanelli SL, Maciel ME, Diaz Gomez MI, Delgado de Layno AM, Bietto FM, Castro JA, Castro GD. Further studies on the potential contribution of acetaldehyde accumulation and oxidative stress in rat mammary tissue in the alcohol drinking promotion of breast cancer. J Appl Toxicol. 2011;31(1):11–19. doi: 10.1002/jat.1555. [DOI] [PubMed] [Google Scholar]
- 11.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuentes-Mattei E, Velazquez-Torres G, Phan L, Zhang F, Chou PC, Shin JH, Choi HH, Chen JS, Zhao R, Chen J et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J Natl Cancer Inst. 2014;106(7). [DOI] [PMC free article] [PubMed]
- 13.Rotunno M, Sun X, Figueroa J, Sherman ME, Garcia-Closas M, Meltzer P, Williams T, Schneider SS, Jerry DJ, Yang XR, et al. Parity-related molecular signatures and breast cancer subtypes by estrogen receptor status. Breast Cancer Res. 2014;16(4):R74. doi: 10.1186/bcr3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed]
- 15.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41(5):579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamimi RM, Baer HJ, Marotti J, Galan M, Galaburda L, Fu Y, Deitz AC, Connolly JL, Schnitt SJ, Colditz GA, et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Res. 2008;10(4):R67. doi: 10.1186/bcr2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Zhang X, Beck AH, Collins LC, Chen WY, Tamimi RM, Hazra A, Brown M, Rosner B, Hankinson SE. Alcohol consumption and risk of breast cancer by tumor receptor expression. Horm Cancer. 2015;6(5–6):237–246. doi: 10.1007/s12672-015-0235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu W, Seok J, Mindrinos MN, Schweitzer AC, Jiang H, Wilhelmy J, Clark TA, Kapur K, Xing Y, Faham M, et al. Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci USA. 2011;108(9):3707–3712. doi: 10.1073/pnas.1019753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics − a bioconductor package for quality assessment of microarray data. Bioinformatics (Oxford, England). 2009;25(3):415–6. [DOI] [PMC free article] [PubMed]
- 20.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed]
- 21.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11(12):843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 23.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(18):e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romieu I, Scoccianti C, Chajes V, de Batlle J, Biessy C, Dossus L, Baglietto L, Clavel-Chapelon F, Overvad K, Olsen A, et al. Alcohol intake and breast cancer in the European prospective investigation into cancer and nutrition. Int J Cancer. 2015;137(8):1921–1930. doi: 10.1002/ijc.29469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SY, Kolonel LN, Lim U, White KK, Henderson BE, Wilkens LR. Alcohol consumption and breast cancer risk among women from five ethnic groups with light to moderate intakes: the Multiethnic Cohort Study. Int J Cancer. 2014;134(6):1504–1510. doi: 10.1002/ijc.28476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Blackinton JG, Keene JD. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol. 2014;34:44–54. doi: 10.1016/j.semcdb.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan S, Meng Q, Gao B, Grossman J, Yadegari M, Goldberg ID, Rosen EM. Alcohol stimulates estrogen receptor signaling in human breast cancer cell lines. Cancer Res. 2000;60(20):5635–5639. [PubMed] [Google Scholar]
- 31.Singletary KW, Frey RS, Yan W. Effect of ethanol on proliferation and estrogen receptor-alpha expression in human breast cancer cells. Cancer Lett. 2001;165(2):131–137. doi: 10.1016/S0304-3835(01)00419-0. [DOI] [PubMed] [Google Scholar]
- 32.Candelaria NR, Weldon R, Muthusamy S, Nguyen-Vu T, Addanki S, Yoffou PH, Karaboga H, Blessing AM, Bollu LR, Miranda RC, et al. Alcohol regulates genes that are associated with response to endocrine therapy and attenuates the actions of tamoxifen in breast cancer cells. PloS One. 2015;10(12):e0145061. doi: 10.1371/journal.pone.0145061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123(6):993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 34.Dong JT. Anticancer activities of PPARgamma in breast cancer are context-dependent. Am J Pathol. 2013;182(6):1972–1975. doi: 10.1016/j.ajpath.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Petersen RK, Larsen SB, Jensen DM, Christensen J, Olsen A, Loft S, Nellemann C, Overvad K, Kristiansen K, Tjonneland A, et al. PPARgamma-PGC-1alpha activity is determinant of alcohol related breast cancer. Cancer letters. 2012;315(1):59–68. doi: 10.1016/j.canlet.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 37.Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99(Pt A):61–6. [DOI] [PubMed]
- 38.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3(3):179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 39.Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, Qin L, Szabo G, Wheeler M, Zou J. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30(4):720–730. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- 40.Volpato S, Pahor M, Ferrucci L, Simonsick EM, Guralnik JM, Kritchevsky SB, Fellin R, Harris TB. Relationship of alcohol intake with inflammatory markers and plasminogen activator inhibitor-1 in well-functioning older adults: the Health, Aging, and Body Composition study. Circulation. 2004;109(5):607–612. doi: 10.1161/01.CIR.0000109503.13955.00. [DOI] [PubMed] [Google Scholar]
- 41.Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interf Cytokine Res. 2015;35(1):1–16. doi: 10.1089/jir.2014.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolini A, Carpi A, Rossi G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006;17(5):325–337. doi: 10.1016/j.cytogfr.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissiere F, Laune D, Roques S, Lazennec G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9(1):R15. doi: 10.1186/bcr1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buck MB, Knabbe C. TGF-beta signaling in breast cancer. Ann NY Acad Sci. 2006;1089:119–126. doi: 10.1196/annals.1386.024. [DOI] [PubMed] [Google Scholar]
- 45.Triano EA, Slusher LB, Atkins TA, Beneski JT, Gestl SA, Zolfaghari R, Polavarapu R, Frauenhoffer E, Weisz J. Class I alcohol dehydrogenase is highly expressed in normal human mammary epithelium but not in invasive breast cancer: implications for breast carcinogenesis. Cancer Res. 2003;63(12):3092–3100. [PubMed] [Google Scholar]
- 46.Kedishvili NY, Stone CL, Popov KM, Chernoff EA. Role of alcohol dehydrogenases in steroid and retinoid metabolism. Adv Exp Med Biol. 1997;414:321–329. doi: 10.1007/978-1-4615-5871-2_37. [DOI] [PubMed] [Google Scholar]
- 47.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 48.Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics (Oxford, England). 2007;23(8):980–7. [DOI] [PubMed]
- 49.Abdueva D, Wing M, Schaub B, Triche T, Davicioni E. Quantitative expression profiling in formalin-fixed paraffin-embedded samples by affymetrix microarrays. J Mol Diagn. 2010;12(4):409–417. doi: 10.2353/jmoldx.2010.090155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ali AM, Schmidt MK, Bolla MK, Wang Q, Gago-Dominguez M, Castelao JE, Carracedo A, Garzon VM, Bojesen SE, Nordestgaard BG, et al. Alcohol consumption and survival after a breast cancer diagnosis: a literature-based meta-analysis and collaborative analysis of data for 29,239 cases. Cancer Epidemiol Biomarkers Prev. 2014;23(6):934–945. doi: 10.1158/1055-9965.EPI-13-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7(8):599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ESR1, PGR, ERBB2 mRNA expression by IHC ER, PR, and HER2, respectively, in the NHS and the NHSII. (PDF 273 kb)
Comparison of study population characteristics in the NHS/NHSII and TCGA. Table S2. Characteristics of invasive breast cancer cases by recent alcohol consumption in the TCGA. Table S3. Top 10 ranked differentially expressed probes by recent alcohol consumption in the NHS and the NHSII. Table S4. Enriched gene sets (FDR 0.05–0.1) by recent alcohol consumption in ER+ tumors in the NHS and the NHSII. Table S5. Enriched gene sets (FDR 0.05–0.1) by recent alcohol consumption in ER- tumors in the NHS and the NHSII. Table S6. Enriched gene sets by alcohol consumption in stage II/III ER+ tumors in the NHS and the NHSII. (DOCX 56 kb)
Differentially expressed probes (N = 25,979) by alcohol consumption in the NHS and the NHSII. The top four figures show recent alcohol intake > 0 - < 10 vs. 0 g/day and the bottom four figures show recent alcohol intake 10+ vs. 0 g/day. (PDF 1418 kb)
Data Availability Statement
Gene expression data are publicly available through the Gene Expression Omnibus [GEO:GSE93601].