

Abstract
The pyrrole-2-aminoimidazole (P-2-AI) alkaloids are a growing family of marine alkaloids, now numbering well over 150 members, with high topographical and biological information content. Their intriguing structural complexity, rich and compact stereochemical content, high N to C ratio (~1:2), and increasingly studied biological activities are attracting a growing number of researchers from numerous disciplines world-wide. This review surveys advances in this area with a focus on the structural diversity, biosynthetic hypotheses with increasing but still rare verifying experimental studies, asymmetric syntheses, and biological studies including cellular target receptor isolation studies of this stimulating and exciting alkaloid family.
1 Introduction
The ability of the exquisite biosynthetic machinery found in living organisms to first construct a simple building block with multi-faceted reactivity such as acetylCoA (1)/malonyl CoA(2), dimethylallylpurophosphate (3), or shikimic acid (4) and utilize it to construct a plethora of fascinating chemical structures including terpenes, polyketides, and alkaloids respectively, is awe-inspiring (e.g. 7-10, Figure 1). The energy expenditure in generating such complex structures, including biosynthesis of the complex biosynthetic enzymes required for their synthesis, suggests that these molecules must serve a purpose and indeed these genetically-encoded small molecules1 are often associated with biological activity. The generation of the pyrrole-2-amino imidazole (P-2-AI) family of alkaloids, hypothesized to originate from clathrodin/oroidin (5/6) (Figure 1) (a.k.a. clathrodin- or oroidin-derived alkaloids), is another amazing example of such a feat of complex biosynthetic machinery found in marine sponges or their symbionts to construct an arsenal of fascinating and potenitally therapeutically useful bioactive molecules (e.g. palau’amine (10)). While detailed molecular-level ecology studies of these natural products may ultimately reveal their utility for the producing organism, if any, the present value of such natural products can be found in the resultant inspiration given to isolation chemists to tackle their isolation and structure elucidation, to synthetic chemists in assaulting their synthesis by development of novel strategies and methods, biochemists and both cellular and molecular biologists to understand their bioactivity at the molecular level, and ultimately pharmaceutical companies to translate these natural products into potential drugs for therapies to combat various human diseases.
Fig. 1.

Common biosynthetic building blocks including dimethylallylpyrophosphate (3), acetyl and malonyl CoA (1,2), shikimic acid (4) leading to the steroid lanosterol (8), the polyketide brefeldin A (7), and the alkaloid podophylotoxin (9), respectively and the subject of this review, new building blocks, clathrodin and oroidin (5,6), leading to complex P-2-AIs including palau’amine (10).
The P-2-AI alkaloids constitute a biogenetically unique natural product family which to date have been exclusively isolated from marine sponges. Now numbering more than 150 members, the last decade has witnessed significantly increased interest in this alkaloid family along several lines of investigation due to the continued isolation of more complex members with novel and unique molecular architecture and biological activity. These marine alkaloids correlate well with the phylogenetic relationships within families of sponges belonging to Agelasida and Axinellida (Halichondrida) orders. In addition to further understanding of their ecological roles, the biosynthetic origin of the P-2-AIs remains a very intriguing question. The technically restricted use and low incorporation levels of isotopically labeled precursors employed in early sponge feeding experiments2 left room for speculation on the biogenetic origin of early biosynthetic intermediates e.g. clathrodin (5) or oroidin (6). While biosynthetic details are completely lacking, biomechanistic analysis of isolated P-2-AIs and successful synthetic strategies premised on speculative biosynthetic pathways have provided some indirect evidence regarding biosynthetic feasibility. From the standpoint that inherent reactivity of biosynthetic intermediates may offer clues about actual biosynthetic pathways leading to various complex P-2-AIs, isolation of an increasing number of putative biosynthetic intermediates has also been extremely important in developing biosynthetic hypotheses. The evolution of their biogenetic pathways appears to be based in large part on the very interesting ambivalent reactivity embedded in the 2-aminoimidazole nucleus (Figure 2). Thus, great effort has been devoted to unveiling the mechanistic and stereochemical details of reactions involving these pivotal intermediates.
Fig. 2.
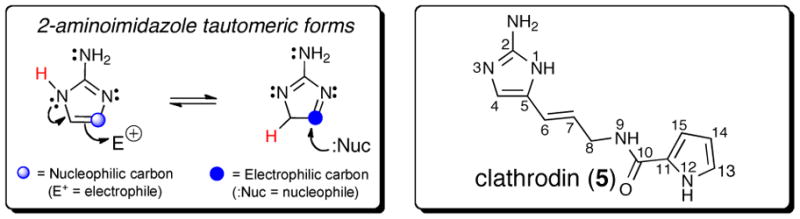
Dual reactivity of the common 2-aminoimidazole nucleus found in the P-2-AIs metabolites and numbering used in this review of the postulated precursor, clathrodin (5), containing the 2-aminoimidazole nucleus.
Several reviews describing biosynthetic hypotheses of the so-called bromopyrrole or pyrrole-imidazole alkaloids isolated from Agelas genus (Agelasida order) were reviewed by Braekman in 1992.3 In the present review, this family is named pyrrole-2-aminoimidazole (henceforth P-2-AIs), to highlight both the participatory heterocycles in presumed biosynthetic events and the contribution of the amino group to the reactivity pattern of the imidazole ring.
In this review, we have introduced a numbering system that is meant to amplify the importance of the 2-aminoimidazole nucleus, which is key to the molecular diversity of the P-2-AIs (Figure 2). The carbon chain begins with the aminoimidazole and numbering continues to the branched pyrrole-2-carboxamide. This numbering system is also applied to dimeric structures and partially truncated natural products and atoms keep their original numbering for biomechanistic reasons. The use of trivial names is also adopted in some cases for practical reasons.
Following early insights by Faulkner and Clardy (oroidin),4 Rinehart (sceptrin),5 Horne (mauritiamine, axinohydantoin, slagenins),6 and Kinnel and Scheuer (palau’amine)7 on possible biosynthetic pathways toward specific natural products, a comprehensive account on the chemical reactivity and hypothetical chemical pathways leading to all the complex P-2-AI metabolites known to date was reported by Al-Mourabit in 2001.8 Extension of these hypotheses to include stereochemical relationships, especially in light of the structural revision of palau’amine (10) and extension to the recently isolated tetrameric P-2-AI members, was reported by Köck and Baran in a 2007 mini-review.9 Several review articles have compiled synthetic advances within the P-2-AI metabolites and partial descriptions of biosynthetic aspects of selected members of this family.10
The present review focuses on the structural diversity of the P-2-AIs in the context of discussing possible biosynthetic origins of these alkaloids and when available, substantiating synthetic studies that demonstrate the proposed reactivity. A review of only asymmetric synthesis of P-2-AIs is included giving the relevance to subsequent pharmacological and biological studies including scarce but growing structure-activity and biological receptor isolation studies, which are also described.
2 Structural Diversity, Proposed Biosynthetic Hypotheses, and Biosynthetic Studies of the Pyrrole-2-Aminoimidazole (P-2-AIs) Alkaloids
To conceive and eventually discover the operative overall biosynthetic pathways leading to the complex molecular architectures of these alkaloids, several lines of investigation have been undertaken. These studies are aimed at understanding the origin and the reactivity of simple precursors e.g. clathrodin (5), oroidin (6), debromodispacamide B (11) (Figure 3) and possible pathways for their conversion to higher order P-2-AIs. These include:
Fig. 3.

Putative biosynthetic progression from simple amino acids to monomeric, dimeric, and tetrameric P-2-AIs, degradation products and possible shunt metabolites.
Biochemical investigations involving incorporation studies linking early precursors, presumably amino acids, to monomers like clathrodin or debromodispacamide and their brominated derivatives (e.g. oroidin) and incorporation studies of labeled putative intermediates into higher order P-2-AIs.
Chemical reactivity studies of presumed precursors including clathrodin, oroidin, or debromodispacamide B to higher order P-2-AIs including the exquisite control exerted by the unique multifaceted chemical behavior of the 2-aminoimidazole nucleus (vide supra).
Synthetic studies premised on presumed biosynthetic pathways (‘bioinspired’ syntheses) to determine the feasibility of such transformations while also providing unique and often highly concise routes to these complex molecules.
Based on the combination of newly isolated P-2-AI members and ‘bioinspired’ synthetic studies, highly analogous biosynthetic hypotheses have been posited by several groups that appear to be converging. The global biosynthetic pathways include formation of presumed building blocks (e.g. clathrodin (5) and oroidin (6)) from amino acid precursors and further transformation and elaboration of these building blocks into more complex P-2-AIs (Figure 3). Furthermore, degradation of more or less elaborated P-2-AI members could explain the isolation of several simpler P-2-AI derivatives. Hence, aminoimidazolyl propeneamine (AAPE) (12) is possibly a case in point for such degradation, as it is a “misleading” compound with respect to biosynthetic origins. While it was considered to be an early precursor for oroidin (6) through acylation with 4,5-dibromopyrrole-2-carboxylic acid, the pseudodipeptide route is favored over amide hydrolysis of oroidin.11,12 In addition, the bromopyrrole unit is found in other seemingly related but distinctly different natural products (e.g. clathramides, hanishin,13 and manzacidins) raising the possibility that these are shunt metabolites resulting from mixing of clathrodin-utilizing and alternative biosynthetic pathways (Figure 3).
2.1. Biosynthesis from simple early precursors: pre-clathrodin events
The majority of reported biosynthetic hypotheses for formation of the P-2-AI building blocks, clathrodin (5) and its brominated derivatives, originate from proteinogenic amino acids. The postulated key steps rely on several presumed precursors isolated from phylogenetically related sponges or co-isolated together with other higher order P-2-AIs. While it is reasonable to speculate that the pyrrole-2-carboxylic acid unit (13) (Figure 3) is derived from proline oxidation (Scheme 1a),14 several alternative possibilities have been suggested for the origin of the 2-amino-5-(3-amino) propylimidazole unit including diketopiperazine intermediates (Scheme 1b).
Scheme 1.

Possible routes to debromodispacamide B (11) and clathrodin (5) via (a) Pathway A: pyrrole oxidation and nucleophilic addition to an activated acyl pyrrole (b) Pathway B: dipeptide-based synthesis through diketopiperazines and intramolecular rearrangements
Kitagawa,11 and later Braeckman3 proposed that proline, ornithine, and guanidine are probable precursors of both the bromopyrrole and 2-aminoimidazolinone moieties found in P-2-AIs. Following this proposal, Kerr reported the incorporation of the 14C-labeled proline, ornithine and histidine into the cyclized oroidin derivative stevensine (16) (Scheme 2).2 How the incorporation of histidine into oroidin (6) could proceed remains an interesting question. Unfortunately, only low levels of radioactivity and specific incorporation levels were observed with amino acids in the Kerr experiment ([U-14C]-His, 1460 dpm (0.026%) and [U-14C]-ornithine, 1300 dpm (0.024%), and these results along with the lack of in vivo biosynthetic studies, still leaves the question unanswered. Based on some synthetic studies and the isolation of a C11N5 diketopiperazine, verpacamide C (18), Al-Mourabit and coworkers proposed proline or arginine as early precursors for both the pyrrole-carboxylic acid and the 2-aminoimidazole moieties.12 As there is no direct biochemical evidence for the structure of the early precursors involved in the formation of the 2-aminoimidazole and 2-aminoimidazolone motifs, hypotheses on the conversion of proline, ornithine, histidine, arginine, or lysine into the P-2-AI metabolites remain purely speculative at the present time. The ideas put forward with a particular focus on the origin of the 2-aminoimidazole moiety are summarized in Scheme 2.
Scheme 2.

Proposed biosyntheses of the P-2-AIs building blocks
In the Kitagawa/Braekman proposal suggesting ornithine as the precursor, it was envisioned that the α-amino acid function was transformed into 2-aminoimidazolinone precursors via a guanidine intermediate (Scheme 2, path a).3,11 This proposal was recently supported by the isolation of the latonduines (14)15 although the absence of a true 2-aminoimidazole nucleus leaves the question of their belonging to P-2-AI alkaloids open. In addition, the pyrrole-2-carboxylic acid is a very common acylating agent present in several marine organisms and microorganisms, and its presence alone is certainly not sufficient evidence for a relationship with P-2-AI biosynthesis.16 Based on the isolation of a pyrrole-lysine pseudo-dipeptide (15) (Scheme 2), Köck and Lindel proposed a lysine precursor in which the terminal amine was converted into a guanidine and then into a 2-aminoimidazole after oxidative hydroxylation.17 Lindel subsequently synthesized aminohomohistidine from α-hydroxy-(L)-lysine demonstrating possible chemistry enabling synthesis of the 2-aminoimidazole moiety from an oxidized lysine motif.18 In addition, Lindel and coworkers reported the biomimetic synthesis of cyclooroidin itself in good yield.19 Following the first experimental biosynthetic study toward stevensine (16) performed by Kerr,2 further progress towards substantiating biomechanistic speculations experimentally is lacking and the quest to provide support for or against various proposals could uncover unexpected and thus intriguing biosynthetic processes. One such study involving the synthesis of 15N-labelled oroidin (6) has recently appeared (vide infra).20
2.2. Post-clathrodin/oroidin biosynthetic events including controlled cyclizations
To date, no experimental biosynthetic studies have been reported testing the incorporation of linear derivatives like clathrodin (5) or oroidin (6) into polycyclized, complex P-2-AI members despite the almost assumed biosynthetic connnection. However, recently Molinski and Romo reported the first investigations in this direction aimed at monitoring the incorporation of synthetic 15N-labeled oroidin (6) into complex P-2-AIs by both 15N NMR and FT-MS.20 While this initial report only described the sensitivity of these methods, planned sponge-feeding experiments may provide the first experimental evidence for the common assumption that clathrodin/oroidin are precursors to more complex P-2-AIs.
The hypothetical chemical pathways described below are primarily based on isolated P-2-AIs, the established reactivity of clathroidin/oroidin, and in some cases successful biomimetic or more accurately ‘bioinspired’ reaction sequences given that the biosynthesis of these natural products has not yet been elucidated.
2.3. Polycyclic P-2-AI monomers
Since the landmark ‘bioinspired’ synthesis of dibromophakellin (20) from dihydrooroidin (21) by Büchi in 1982,21 oxidative strategies taking advantage of the reactivity of the 2-aminoimidazole nucleus have been widely employed for the synthesis of several P-2-AI monomeric members. In extensions of Büchi’s seminal work, Horne reported marked improvements for the oxidative cyclizations to the phakellins and also the rearrangement of dibromophakellin (20) to dibromoisophakellin (22) at 130 °C involving a net N→C migration presumably involving an iminium intermediate 24.22 This process is conceptually similar to that described by Buchi for the synthesis of parazoanthoxanthin23 and later revisited and elegantly expanded by Horne.6c Feldman utilized sulfur oxidation by way of a Pummerer reaction to access the core structures of dibromophakellstatin and dibromoagelaspongin and proposed a key spiro intermediate 24 that overlaps with biomechanistic speculations.24 Horne also reported the non-propensity of spiro derivatives to cyclize.22,25
Inspired by these early findings and by initial mechanistic speculations by Rinehart and others,5 the comprehensive analysis of the formation of monomeric and dimeric polycyclic P-2-AIs published in 2001 by Al Mourabit and Potier8 suggests a biosynthetic route starting from oroidin (6) and derivatives. This proposal involves only proton-mediated isomerizations into tautomeric forms I-IV followed by intramolecular cyclizations (Scheme 4)
Scheme 4.

Proton-mediated isomerization of clathrodin/oroidin and further polycyclizations: alternative biosynthetic hypothesis for the formation of dibromophakellin and dibromoagelaspongin
It was envisioned that cyclization of the pyrrole onto transient aminoimidazole tautomer 26 (tautomer type IV, Scheme 4) could generate bicyclic, spirocyclic, and macrocyclic pre-transannular cyclization intermediates (e.g. 28). Subsequent transannular cyclizations of the latter species could give rise to the tetracyclic phakellins and agelaspongins. Baran recently provided evidence for this hypothesis in a recent synthesis of palau’amine in which the “phakellin” substructure was forged through a transannular cyclization of a “macropalau’amine” precursor (vide infra).26 It is possible that macrocyclization/transannular cyclization induced by tautomer IV is arguably one of the major pathways followed in the formation of several polycyclic P-2-AI metabolites. Interestingly, the phakellin-expanded derivative 28 (Scheme 4) was also obtained by Sharma in 1978 as a degradation product of dibromophakellin (20).27 Formation of partially cyclized intermediates like the hypothetical pre-phakellin spiro intermediate 2921 and agelaspongin intermediate 3028 can also be accounted for by similar mechanisms. In fact, tautomers III and IV (Scheme 5) form the basis and can account for a general chemical pathway involved in P-2-AI polycyclic monomers. Dibromoisophakellin (22), hymenin (31) and cyclooroidin (32) could be formed through a similar transient intermediate derived from oroidin (6). Cyclooroidin, agesamides A-B,29 and oxocyclostylidol30 presumably arising from cyclization of the fleeting tautomer III are less frequent, likely due to the low intrinsic diversity derived from potential intramolecular cyclizations of this tautomer.
Scheme 5.

Intramolecular cyclizations of presumed precursors including tautomers and pre-transannular intermediates leading to bi-, tri-, and tetracyclic monomers
Cylindradine A (33), potentially generated via an ‘ipso’ rearrangement of dibromophakellin (20) was also recently isolated (Scheme 6).31
Scheme 6.

‘ Ipso’ rearrangement of dibromophakellin (20) to cylindradine A (33)
The biologically and structurally interesting agelastatins A-F32 are intriguing examples of P-2-AI alkaloids derived from C8-C4 and N12-C7 cyclizations (Scheme 7). A pre-agelastatin intermediate (38) generated by an initial C8-C4 cyclization proposed by Al-Mourabit8 is clearly supported by the recent isolation of the dimeric natural product nagelamide J (37) by Kobayashi.33 The agelastatin pathway further demonstrates the general applicability of the biogenetic proposal based on tautomeric forms of clathodrin/oroidin and the great potential for P-2-AI metabolites to undergo oxidative couplings.
Scheme 7.

Proposed biogenesis of agelastatins and the related dimeric nagelamides
2.4. Polycyclic P-2-AI dimers
One of the first biosynthetic proposals for the generation of higher order P-2-AIs was advanced by Kinnel and Scheuer7 who envisioned a Diels-Alder cycloaddition between a hypothetical ‘dehydrophakellin’ dienophile (42) and AAPE (12) (Scheme 8). A subsequent chloroperoxidase-initiated oxidation/ring-contraction event would deliver the hexacyclic core (43) of the originally assigned structure of palau’amine. Inspired by this hypothesis, successful Diels-Alder/oxidative ring contraction sequences on related systems were reported independently by Romo34 and subsequently by Lovely.35 Furthermore, the isolation of cis-12-chloro-11-hydroxybromoisophakellin (44)36 resulting from chlorohydration of a hypothetical ‘dehydroisophakellin’ (isomeric form of 42) provides further credence to the proposed biosynthetic Diels-Alder reaction. However, it should be noted that the cis-stereochemistry of the chlorohydrin found in this metabolite suggests chlorohydroxylation of a trans-alkene found in oroidin (6) followed by cyclization to the phakellin core and points to an alternative Diels-Alder process leading to the trans-ring fusion (vide infra). The relative stereochemistry of this phakellin chlorohydrin is identical to that of girolline (73, vide infra),37 presumably derived from chlorohydroxylation of AAPE (12).
Scheme 8.
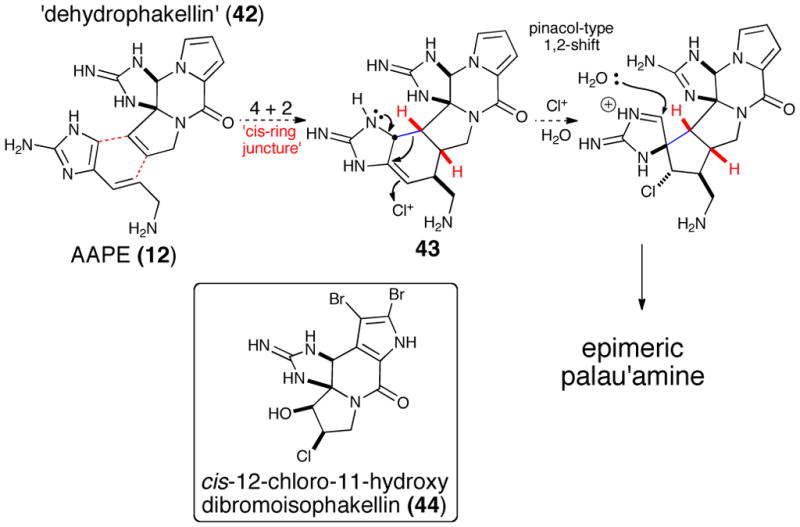
Original Kinnel/Scheuer biosynthetic hypothesis for palau’amine involving a [4+2] cycloaddition/chorination/ring contraction sequence. (Inset: An isolated phakellin derivative supporting the proposed dehydrophakellin.)
A universal chemical pathway for the formation of more complex, dimeric, polycyclic P-2-AI members known up to 2000, involving tautomeric forms I-IV of clathrodin (5) (and brominated derivatives), was proposed in 2001 by Al-Mourabit and Potier (Scheme 9).8 Analysis of the ambivalent reactivity of the central intermediate clathrodin (5) led a logical pathway that provides insights into the dimerization process of P-2-AIs members that extends from hypotheses already described above leading to bicyclic, tricyclic, and tetracyclic P-2-AIs.
Scheme 9.

Tautomerism in the building blocks of the P-2-AIs
The decisive first C-C bond enables the possibility of numerous cascades of cyclization reactions and various modes of C-C and N-C dimerization involving tautomers I-IV (Scheme 9) with nucleophilic and electrophilic reactive positions dependent on the tautomer engaged in the dimerization step. In general, C13 and C14 are nucleophilic sites albeit exclusively towards halogenation, N1, N3, N17, N9, N12, C15 and O16 typically act as nucleophiles for intra- or inter-molecular cyclizations, while C4, C5, C6 and C7 are ambivalent. The dual reactivity of these latter reactive sites is then modulated by the pre-organization and presumed substrate-biosynthetic enzyme interactions resulting in the predominance of one of the transient tautomers I-IV. Subsequent investigations, including molecular mechanics calculations, concluded that the tautomeric interconversion of I-IV is more easily explained in acidic conditions.38 This reactivity then plausibly leads to the various dimerizations and further polycyclizations observed (Scheme 10).
Scheme 10.

Alternative “first-bond” dimerization pathways from clathrodine precursors
The fact that all the dimeric P-2-AI members isolated during the last decade fit in the general chemical pathway suggested for their formation is a testament to the soundness of this proposal. In addition, the revision of palau’amine’s relative configuration gave even more consistency to the overall hypothesis,9 revealing complete uniformity of stereochemical relationships within the P-2-AI dimer subclass (Scheme 11). The tremendous structural variety exhibited in the many dimers isolated in recent years provides a wealth of valuable clues on the mechanisms involved in their formation. Among the possible initial C-C dimerizations, a C7-C7′ dimer provides the highest potential for generating the greatest structural diversity, and when analyzing various congeners having a C7-C7′ connection, it becomes apparent that all could be derived from the 1st multi-faceted intermediate 51 (Scheme 10) and more advanced 2nd multi-faceted intermediate 53 (Scheme 11) via different modes of intramolecular oxidative cyclizations. It should be noted that the C7-C7′ dimerization leading to the intermediate 51 was initially proposed by Rinehart, who suggested a unified mechanism for the formation of both the ageliferins and the sceptrins from clathrodine monomers.39 Alternative initial C-C bond formations can give rise to the primary dimers mauritiamine (46) and the nagelamides (45) and the newly isolated polycyclic benzosceptrins (47) and stylissazoles (69, vide infra, Scheme 14). In his synthesis of mauritiamine,6a Horne demonstrated the propensity of amino-imidazole derivatives to dimerize and although a plausible mechanism was not detailed, it could reasonably proceed according to these biomimetic suggestions. The first C7-C7′ “intermediate A” 51 was also invoked in the formation of sceptrin, and the ageliferins (48) through a non-concerted mechanism (Scheme 10).8 Furthermore, the C7-C7′ “intermediate A” (51) could undergo an enzymatic chloro-hydroxylation to give chlorinated adduct (54), followed by intramolecular cyclization generating the 2nd multifaceted C7-C7′ intermediate B (53) (Scheme 11) which features the spirocyclic subunit common to numerous complex P-2-AIs. The different modes of proposed oxidative cyclizations summarized below, would then deliver the variety of complex molecular architectures observed in these metabolites.
Scheme 11.

Generation of molecular complexity in ‘higher order’ dimeric P-2-AIs from common multifaceted C7-C7′ precursors (intermediate B, 53)
Scheme 14.

Proposed biogenetic pathway for stylissazoles A-C (69A-C)
It is important to note that the 2nd multifaceted intermediate 53, a putative common precursor to several higher order P-2-AIs, could also be derived via a modified “Diels-Alder”/oxidative ring contraction sequence as originally proposed by Kinnel and Scheuer (vide supra, Scheme 8). However, the revision of the azabicyclo[3.3.0]octane core of palau’amine to the trans-ring fusion40 requires that if a [4+2] cycloaddition does occur to give a cyclohexene adduct, delivering an ageliferin-type intermediate 57, it must involve a non-cyclic dienophile (e.g. clathrodin) possessing E olefin stereochemistry (Scheme 11, i.e. 57→56→53). This sequence would provide the observed trans relative stereochemistry in the [3.3.0] bicyclic core common to all higher order P-2-AIs including palau’amine (10), axinellamine (59), massadine (60), and styloguanidine (61).
The presence of similar structural motifs and functionalities in several P-2-AIs regardless of their monomeric or dimeric nature suggests common biosynthetic transformations leading to their formation. A particular illustration is found in the palau’amines and the regioisomeric styloguanidines that could arise from macrocyclic fleeting intermediates (“macrophakellin” and “macroisophakellin” respectively in Scheme 11), which then collapse in a transannular fashion generating the strained trans-azabicyclo[3.3.0]octane core of these alkaloids. Variations of Al-Mourabit’s postulated biosynthetic intermediate B 53 were exploited by Baran for the synthesis of axinellamine A and B,41 massadine chloride and massadine42 and very recently for palau’amine, elegantly utilizing a “macropalau’amine” precursor (vide infra).
A C4-C6′ connection and the following cascade were successfully performed recently by Harran, including the challenging chlorination event required for the cascade (Scheme 12).43 The oxidative chlorination process formed the imbedded spirocyclopentane motif observed in palau’amine (10) and congeners. Harran’s approach was based on the biosynthetic proposal invoking an initial C7-C7′ bond formation (e.g. 51) leading to dimeric dispacamide-type intermediates.44 Interestingly, dispacamide-type dimers 63 indeed undergo an oxidative chlorination/cyclization event installing the highly advanced chlorinated cyclopentane intermediate 64. The reaction sequence demonstrated the potential for chemoselectivity to initiate cyclization once the C7-C7′ bond was installed.
Scheme 12.
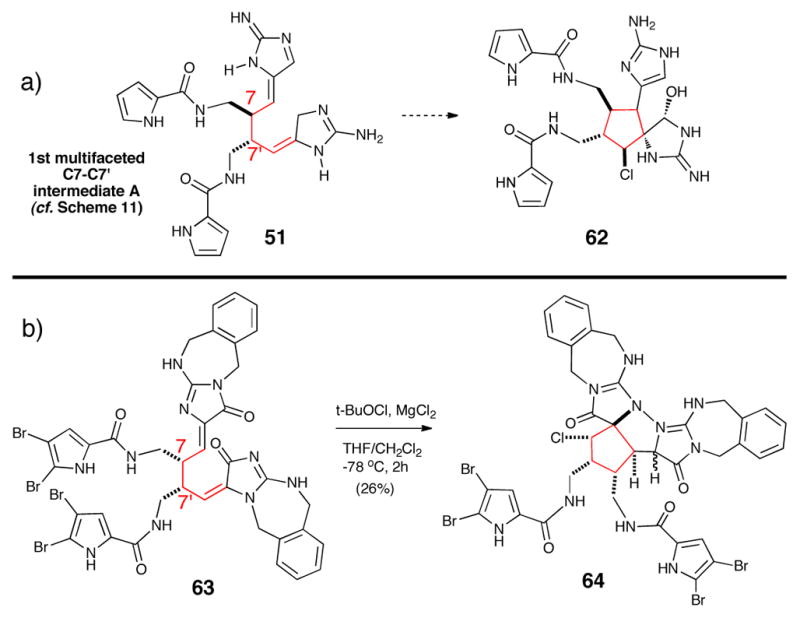
a) Proposed biosynthetic process for cyclopentane formation. b) Bioinspired approach by Harran mimicking this putative biosynthetic process.43
While massadine stands out in this sub-class as it is the only metabolite featuring a pyran core and a hydroxyl group in place of the otherwise ubiquitous chlorine, speculations on its biogenesis were rather scarce. In 2006, during studies toward palau’amine and congeners, the Romo group reported that an unexpected reaction of a cyclopentyl chloride 65a, and subsequently a cyclopentyl iodide 65b, with azide surprisingly proceeded with retention of configuration (Scheme 13).45 This group concluded that a mechanism involving neighboring group participation of the spiroaminoimidazole might be in operation involving an aziridinium species. It was further proposed that a similar process could account for the biosynthesis of massadine from an earlier chlorinated precursor.
Scheme 13.

a) Proposed aziridinium intermediate suggested by an observed retention of configuration displacement of chloride by azide. b) Romo’s proposed biosynthesis of massadine based on this observation (ref. 37)
In 2007 Baran and Köck provided evidence for this pathway by isolating the unprecedented massadine chloride and reported its rapid conversion into massadine in an aqueous medium at 60 °C. They also advanced a rationale for why this peculiar behavior is not observed in related chlorocylopentane-bearing P-2-AI metabolites; namely, this displacement was possible with chloromassadine due to a more favorable geometric disposition for displacement due to the dihedral angle between the aminoimidazole C-N bond and the C-Cl bond.46 This presumed ‘biomimetic transformation’ was exploited by Baran in his total synthesis of massadine.42 Furthermore, it could be envisioned that a similar mechanism is involved in the dimerization of massadine leading to the stylissadines, the only tetrameric metabolites that have been isolated to date.
In 2010, isolation of three novel P-2-AI dimeric members, stylissazoles A-C (69A-C) demonstrate the first examples of dimerization involving solely N-C bond formation (Scheme 14).47 These members provide another dimension to the molecular diversity of P-2-AI metabolites and further highlight the unique dual reactivity of the vinylogous 2-aminoimidazole precursors. The co-occurrence of stylissazoles A-C give biosynthetic clues to this new dimerization/oxidation sequence. Scheme 14 shows a proposed biogenesis suggesting that the building blocks of the molecule are clathrodin and the brominated hymenidin. The biogenetic scheme also suggests that stylissazole C might arise from stylissazole B or from a common “pre-stylissazole B/C”. The pathway would presumably start from clathrodin (tautomer I) and hymenidin (tautomer IV) for stylissazole A and tautomer III for stylissazole B-C in a 1,2 or 1,4 aza-Michael fashion to produce the decisive first N3′-C4 and N3′-C7 bounds. The formation of the N-C connected stylissazoles A-C dimers likely involves N-C bond construction in the first step.
The dimeric P-2-AI examples given here suggest that various modes of C-C and N-C dimerization involving tautomers I-IV are possible. Nucleophilic and electrophilic reacting centers can vary based on the tautomer engaged in the dimerization step. Unlike other classes of alkaloids, the group of P-2-AI alkaloids shows an unprecedented use of multifaceted intermediates that enable a high degree of complexity and molecular diversity (Scheme 15). The enzymatic stabilization leading to the selection of the reacting tautomers from relatively simple molecules is an interesting question to be answered. The dynamic hydrogen-bonding interactions of monomers like clathrodin with the host enzyme will likely unveil mechanistically interesting and highly ordered biochemical catalytic system.
Scheme 15.

Summary of biogenetic proposals for P-2-AIs dimers
3 Enantioselective Chemical Syntheses of the Pyrrole-Imidazole Alkaloids (P-2-AIs)
Given the immense structural diversity and architectural complexity of P-2-AIs, it is understandable that efforts toward the asymmetric, total synthesis of these marine alkaloids has been a very prolific domain of study for chemical synthesis. Numerous synthetic groups continue to be active in the area and a significant amount of progress has been achieved recently, including the first completed enantioselective synthesis of monomeric P-2-AIs and racemic syntheses of long elusive, complex dimeric alkaloids. A detailed and exhaustive account of these synthetic endeavors in this area goes beyond the scope of the present review, which has a different focus. Furthermore, a majority of the synthetic work toward P-2-AIs is the subject of several recent reviews.9,48 The enantioselective synthesis of natural products is paramount to subsequent studies of bioactivity including pharmacology, cellular receptor isolation, and structure-activity studies (SAR) for potential drug discovery and development. For this reason and due to space limitations, only enantioselective routes to P-2-AIs will be described with the exception of the recent synthesis of racemic palau’amine by Baran. However mention must be made of outstanding contributions to this area by Overman,49 Harran,43 Horne, Ohta,50 Carreira,51 Chen,52 and Lovely, 35,53 Gin,54 Gleason,55 and Williams.56
One of the earliest marine P-2-AIs isolated is girolline (73) (a.k.a. girodazole), which despite lacking a pyrrole sub-unit is always included in any discussion about P-2-AIs as it is proposed to be derived from the same simple precursor, AAPE. Shortly after its isolation, synthetic efforts by Ahond and Commerçon led to the elucidation of the absolute configuration of girolline.57 In his approach Commerçon58 employed an Evan’s aldol strategy to establish the chlorohydrin relative and absolute stereochemistry (i.e. chlorohyrdin 77). Subsequently, Ahond and Poupat59 disclosed a chiral pool approach that generated the targeted stereochemical relationship via elaboration of D-arabinose (80).
Many of the simpler “linear” P-2-AIs (vide supra) either lack stereogenic centers (e.g. oroidin and analogues) or are naturally occurring as racemates (e.g. midpacamide,60 dispacamides C and D,61 and tauroacidin B62). Yet other hydroxylated metabolites which showed optical rotation were revealed, upon further investigation, to exist as a non-racemic mixture of enantiomers like tauroacidin62 and mukanadin A, which was isolated two years previously and found to be identical to dispacamide D.63 Debromodispacamide D possessing a single carbinol stereogenic center was also isolated as a racemate, and its synthesis in optically active form began with hydroxyproline (Scheme 17)64 and subsequent stability studies were instrumental in confirming that racemization does not occur during the purification process. The key step was an intriguing presumably biomimetic oxidative reaction that generated the aminoimidazolone moiety via the fragmentation of a proposed spirocyclic dioxetanone (86) intermediate.
Scheme 17.

Concise, enantioselective synthesis of debromodispacamide D by Al Mourabit using a biomimetic oxidation.63 EDCI = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, DMAP = N,N-dimethylaminopyridine, m.s. = molecular sieves, DMF = N,N-dimethylformamide
A presumed intramolecular oxidative cyclization also introduces elements of stereochemistry in cyclooroidin and the slagenins. Following two racemic syntheses, the absolute configuration of natural cyclooroidin was confirmed by asymmetric synthesis in 2006 by Pelloux-Leon and Minassian,65 expanding on their previous synthesis of the longamide B (89) related from (L)-aspartic acid methyl ester (90, Scheme 18).66 After conversion of the carboxylate moiety of longamide B (89) into the corresponding mixed anhydride, sequential treatment with diazomethane and aqueous hydrogen bromide delivered the optically active bromoketone 93. Subsequent reaction with Boc-guanidine directly generated the aminoimidazole ring, and cleavage of the carbamate protecting group delivered cyclooroidin (32).
Scheme 18.

Enantioselective synthesis of cyclooroidin by Pelloux-Leon and Minassian.65 HOBT = N-hydroxybenzotriazole, DCC = dicyclohexylcarbodiimide, DMF = N-N-dimethylformamide, THF = tetrahydrofuran, NBS = N-bromosuccinimide. IBX = Iodoxybenzoic acid, TFA = trifluoroacetic acid
The absolute configuration of the slagenins was established by Jiang, who completed the first total synthesis of the antipodes of natural slagenins B and C (Scheme 19a).67 After Noyori reduction of ethyl-4-chloroacetoacetate, the newly generated alcohol was protected as the TBS ether. Azide displacement of the terminal chloride was then followed by conversion of the ethyl ester to glyoxal 97. One pot intermolecular urea cyclization/intamolecular THF annulation then generated a 50% yield of a 9:5 (α:β) mixture of inseparable diastereomeric bicyclic cores of the slagenins. Finally, azide reduction and acylation of the resulting primary amines with 4-bromopyrrole carboxylic acid delivered the slagenin optical antipodes, which could be separated by column chromatography.
Scheme 19.

Enantioselective syntheses of slagenins. a) Jiang’s synthesis of the antipodes of slagenins B and C.61 b) Gurjar’s synthesis of the natural enantiomers. 63 BINAP = binaphthyl; TBS = t-butyldimethylsilyl; Im = imidazole, DMF = N,N-dimethylformamide, DMDO = dimethyldioxirane; THF = tetrahydrofuran; AIBN = azoisobutyronitrile. TBAF = tetrabutylammonium fluoride; Ts = p-toluenesulfonyl.
The following year Jiang68 and Gurjar69 independently but nearly simultaneously arrived at the synthesis of the natural enantiomers starting from L-xylose (not shown) and L-arabinose (Scheme 19b) respectively. In Gurjar’s approach starting from known L-arabinose derivative 99, Barton-McCombie deoxygenation of the free hydroxyl, was followed by azide displacement on the activated primary alcohol and selective protection and oxidation to generate furanone 100. Intermolecular urea cyclization in the presence of hydrogen fluoride and methanol, then generated an inseparable mixture of the bicyclic cores of natural slagenin B and C (94B, C). The natural products were then completed by installation of the pyrrole side chain as described above.
While no significant biological activity was initially reported for phakellin (103), one of the simplest tetracyclic P-2-AI congeners, several groups actively pursued its synthesis for at least two reasons. One driving force was the possibility that the isolation of dibromophakellin by Sharma, during his searching for an elusive potent antibiotic activity found in extracts of Phakellia flabellata70 was in fact due to dibromophakellin as suggested later by Kinnel and Scheuer based on their finding palau’amine possessed greater bioactivity compared to dibromopalau’amine.7 A second driving force was the possibility of using developed phakellin annulation strategies in synthetic approaches toward palau’amine (see section V). The synthesis of (+)-phakellin by Romo71 began with L-proline and trichloroacetylpyrrole to construct the tricyclic ketopiperazine. Installation of the pendant guanidine then set the stage for the key microwave-mediated oxidative cyclization of 104 leading to the formation of (+)-phakellin (103). The strategy employed by Nagasawa72 for the synthesis of (+)-phakellin started with the construction of hydroxyketopiperazine (107) from 4-hydroxyproline (106) and pyrrole carboxylic acid (13). Overman rearrangement of the trichloroacetylimidate transient intermediate (109) obtained from treatment with trichloroacetonitrile then installed the tertiary amide precursor (102) to the required guanidine (110). Intramolecular cyclization of pendant guanidine 110 finally delivered (+)-phakellin (103).
The tetracyclic phakellstatins, which appear to be hydrolyzed versions of phakellins, have attracted significant interest because of their anticancer activity (vide infra) and since their isolation a number of racemic syntheses have been reported.21,73 The first asymmetric synthesis of (+)-phakellstatin was completed by Romo in 2003 and led to the enantiomeric natural product (Scheme 21a).74 Romo’s synthesis employed a desymmetrization strategy of a diketopiperazine (114) derived from dimerization of proline and a Hoffman rearrangment simultaneously introduced the C10-N9 bond and formed the final cyclic urea. The synthesis confirmed the absolute configuration of the natural product. In 2007, Lindel disclosed the a synthesis of the natural (−)-enantiomer starting with the construction of optically active tricyclic enamide 119 from hydroxyproline (106) and dibromopyrrole carboxylic acid (Scheme 21b).75 The key step involved a diastereoselective, intermolecular 3-component imidazolone cyclization with enamide 119 proceeding through a presumed transient bis-carbamate intermediate 120 to deliver cyclic urea 121. Deprotections, bromination, and deoxygenation delivered (−)-phakellstatin ((−)-112).
Scheme 21.

Enantioselective syntheses of dibromophakellstatin. a) Romo’s synthesis of (+)- phakellstatin.74 b) Lindel’s synthesis of (−)-phakellstatin.75 KHMDS = potassium hexamethyldisilylazide, Bn = benzyl; DMDO = dimethyldioxirane; py=pyridine; Cbz = benzyloxycarbonyl; DMAP = dimethylaminopyridine, DIAD = diisopropylazodicarboxylate; TFA = trifluoroacetate; TBS = t-butyldimethylsilyl; DIBAL = diisobutylaluminum hydride, Ms = methanesulfonyl; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; THF = tetrahydrofuran; AIBN = azoisobutyronitrile; Ts = p-toluenesulfonyl.
Of all monomeric P-2-AIs, the agelastatins have garnered the most synthetic interest and this is undoubtedly due to its highly potent antitumor activity. Eight asymmetric syntheses have been reported to date along with a comparable number of racemic approaches (Scheme 22).76 While unique and creative, each synthesis features an appended acyl-pyrrole moiety, which enables a late-stage cyclization of the pyrrole and a pendant urea onto the C ring. These ring closures construct the B and D rings respectively, delivering the tetracyclic core of agelastatin. Thus, the primary differences in synthetic approaches are primarily the routes taken to the optically active, functionalized cyclopentane(C ring). Enantioselective strategies to agelastatin along with starting materials employed are summarized (Scheme 22) and briefy discussed below.
Scheme 22.

Feldman was the first to synthesize the natural enantiomers of agelastatin A and B.77 In his strategy, acetylene 123, derived from alkylative coupling of (R)-epichlorohydrin and TMS-acetylene, was further elaborated to oxazolidinone 124. This served as the susbstrate for a key intramolecular CH insertion of an alkylidene carbene generated from acetylenic stannane 124 to generate cyclopentene 125, an intermediate efficiently elaborated to the ABC ring system of the agelastatins. In 2003 Hale reported a formal synthesis of agelastatin A78 involving enantioselective construction of an advanced oxazolidinone 128 previously reported as a racemate by Weinreb.79 Several manipulations of the known aziridine 126 led to diene 122, which was readily converted to cyclopentene 7 via ring-closing metathesis (RCM). The following year, Hale and coworkers devised a new end game strategy from cyclopentene 128 involving a 9-step route to generate several hundred milligrams of enantiomerically pure agelastatin A.80 In Davis’s 2005 synthesis the key intermediate cyclopentenone 131 was forged by alkylation of sulfinimine 129 and ring closing metathesis of the advanced allyl-enone 130. 81 The asymmetric allylic alkylation (AAA) protocol reported by Trost in 2006, 82 and again in 2009 83 rapidly assembled the pyrrole-cyclopentene precursor 134 to the ABC ring system of agelastatin A. Remarkably, the target was then completed efficiently in only an additional 4 steps. In 2007 Ichikawa constructed the functionalized C ring 135 via a [3,3]-sigmatropic rearrangement of an allylic isocyanate (136), which proceeded very efficiently with complete transfer of chirality.84 The synthesis disclosed by Tanaka and Yoshimitsu in 2008 commenced with the hetero Diels-Alder cycloaddition of cyclopentadiene and acylnitroso dienophile 139, followed by reductive N-O cleavage and enzymatic resolution, which delivered the enantiomerically pure C ring precursor 138.85 The same approach was also utilized in their second generation synthesis completed the following year.86 Also in 2009, the strategy employed by Chida featured an elegant sequential Overman/Mislow-Evans rearrangement of bis-trichloroimidate 141 followed by ring-closing metathesis that established cyclopentene 140.87 In 2009, DuBois disclosed an 11-step synthesis of (−)-agelastatin A,88 hinging on a novel catalytic intramolecular aziridination method applied to sulfonamide 144 derived from heterobicyclic 145, followed by sequential regioselective ring openings to afford the completely functionalized C ring 143.
Sceptrin (146), which bears an intriguing cyclobutane, is one of the simplest “dimeric” P-2-AIs and it has been proposed as the natural precursor of the more complex and highly sought-after members of the family (e.g. palau’amine). After two racemic syntheses appeared virtually simultaneously from the groups of Baran and Birman.89 The latter group revised their original approach and prepared the key optically active cyclobutane intermediate through the enzymatic resolution of the previously reported Diels Alder adduct 156, followed by photo-induced oxaquadricyclane rearrangement (152→ 153→154; Scheme 23). Introduction of the bromopyrrole side-chains and aminoimidazole annulation, then completed the first and only enantioselective synthesis of sceptrin (146) to date.90 Expanding on early investigations in the Baran laboratory that led to the discovery that sceptrin could be induced to undergo ring expansion in an aqueous medium at high temperatures, this group disclosed the first enantioselective synthesis of (−)-ageliferin (157) and its optical antipode (not shown) from sceptrin (146, Scheme 24).90
Scheme 23.

Enantioselective synthesis of sceptrin (146) by Baran.90 PLE = pig liver esterase, DMT-MM = 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride, TsOH = p-toluenesulfonic acid, DIBAL = diisobutylaluminum hydride, MsCl = methanesulfonyl chloride, DMF = N, N-dimethylformamide, THF = tetrahydrofuran.
Scheme 24.
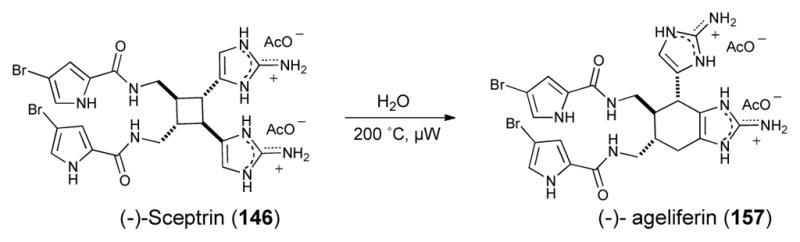
Conversion of sceptrin (146) to ageliferin (157) by Baran.
Baran’s recent synthesis of the long elusive palau’amine91 represents an important milestone in complex P-2-AI synthesis and sets the bar for future endeavors in the this field. This synthesis, while racemic, is included to demonstrate another ‘bioinspired’ strategy involving a transannular stitching to form the strained [3.3.0] core from a macrocyclic precursor, originally posited by Al-Mourabit.8 Starting from the previously reported chlorocyclopentane precursor 162, guanidine annulation and a key silver-mediated oxidation of the unprotected aminoimidazole generated the spirocyclic core 165 of the target. The latter step is notable for its high chemoselectivity and for enabling the use of a latent cyclic hydroxy guanidine in the form of an aminoimidazoline. Following construction of the pendant aminoimidazole, bromination of the aminoimdazole enabled coupling of the acylpyrrole precursor 167 to deliver bis-azide 168. Reduction of the azide functionalities, site-selective amide coupling with the pyrrole carboxylate moiety and the proximal primary amine delivered a “macropalau’amine” advanced intermediate 169. Finally, acid promoted transannular cyclization afforded the strained azabicyclo [3.3.0] core found in palau’amine (10). The inability to perform a biomimetic oxidative biscyclization for late-stage phakellin annulation is instructive. The latter difficulty was recently overcome by the groups of Feldman92 and Chen93 through a phakellin annulation process of a 6-membered versus 5-membered ring leading to a trans azabicyclo [3.4.0] core.
4 Biological Activity and Pharmacology Including Cellular Targets of the P-2-AI Alkaloids
Marine organisms and in particular sponges, and their associated symbiots, have become a fertile ground in the search for natural products that can combat cancer and viruses.94 Notable among such ‘leads’ from marine sources are the nucleosides spongothymidine and spongouridine which led to the development of Ara-C, used clinically for leukemia and lymphoma, and Ara-A, an antiviral agent. More recently, the anticancer agents ectineiasidin (ET-743) (PharmaMar) and bryostatin derivative (Eisai, Inc.) were approved as anticancer agents for clinical use.
In the course of isolating P-2-AIs, as with most natural products, bioassay guided fractionation is typically used to identify the most compelling and potent isolates possessing a particular bioactivity. Some of the most interesting P-2-AI’s from the standpoint of established bioactivity are described below. However, the initial bioactivity identified for a given natural product may not be the most imporant or the bioactivity with the greatest potential for drug development. It is for this reason that isolation and identification of exact cellular targets for natural products continues to be a very important goal in harvesting the full biological potential of isolated bioactive natural products. Unfortunately, in the case of P-2-AIs, very few direct cellular targets have been identified and therefore, this remains an important area for further research. However, some studies aimed at further elucidating details regarding the biological effects of several P-2-AIs including SAR studies have been reported and these are summarized below. In this section, the various P-2-AIs are categorized and described with respect to the bioactivity which has been most extensively studied.
4.1 Oroidin, Ageliferin, and Derivatives: Potential Antibacterial (Antibiofilm) and Antifouling Agents
The Melander group has extensively studied the potential of dibromoageliferin, oroidin, and related P-2-AIs as inhibitors of bacterial biofilm formation, an important approach for combating bacterial infections.95 A series of oroidin derivatives were prepared including ‘reverse amide’ derivatives 171 that maintained and in some cases exceeded the antibacterial activity of the native natural product (Scheme 100). N-alkylation of the pyrrole nitrogen leading to N-methyloroidin (e.g.170) was found to increase inhibition and dispersion activity of the bacterium Acinetobacter baumannii.96
Ageliferin (157a), bromoageliferin (157b), and dibromageliferin (157c) were initially reported by Rhinehart97 and subsequently fully characterized structurally by Kobayashi and determined to be activators of actomysin ATPase.98 Two simplified ageliferin derivatives, trans-ageliferin (±)-172 (TAGE) and cis-ageliferin derivative (±)-173 (CAGE) synthesized as racemates, inhibited the ability of Pseudomonas aeruginosa strains to form biofilms but interestingly displayed differential toxicity toward planktonic P. aeurginosa bacteria.99 To date, the bioactivity of the optically active series has not been reported nor has the cellular target for these simplified ageliferin derivatives been described. Further simplified ageliferin derivatives removing all stereochemistry and leading to benzimidazole cores (e.g. 174) were described to also have useful antibacterial activity.
Other P-2-AIs Exhibiting Antibacterial and Antifouling Activity
A large number of P-2-AIs exhibit antibacterial activity towards various bacteria including S. aureus, E. coli and B. subtilis. Nakamuric acid (176) and its corresponding methyl ester (177) from Agelas nakamurai only exhibited antibacterial activity against B. subtilis, with 9 mm zones of inhibition. These P-2-AIs exhibit lower antibiotic activity compared to sceptrin (52) and ageliferin (157a)130 and these preliminary SAR studies suggested that structural changes in the guanidine moiety of the dimer can alter bioactivity. The presence of the oxoguanidine in oxysceptrins and absence of the guanidine in nakamuric acid and its methyl ester leads to a substantial decrease in activity or complete loss of antibiotic activity. A non cyclized, keramadine (175) also showed activity against S. aureus and B. subtilis100 A new oroidin dimer, mauritiamine (46), isolated from the marine sponge Agelas mauritiana exhibited moderate antibacterial activity against Flavobacterium marinotypicum with growth inhibitory zones of 10 and 15 mm at 10 μg/disk, respectively.101 Axinellamines B-D (59B,59D), was active against Helicobacter pylori at 1000 μM.102 Kobayashi’s group has isolated several dimeric bromopyrrole alkaloids nagelamides, ranging from nagelamide A-R (46A-46R) exhibiting antibacterial activity. Nagelamides A-H (46A-46H), nagelamide Q-R (46Q-46R), ageliferins (157a-c), and sceptrin (52) showed antimicrobial activity against bacteria, Micrococcus luteus, B. subtilis and E. coli (Figure 5).103,104 Nagelamide O (46O) was weakly active against B. subtilis, Micrococcus luteus and S. aureus (MIC 33.3 μg/mL each).105 Nagelamide J (37) was active against only Staphylococcus aureus (MIC, 8.35 μg/mL) and nagelamides K-L (46L-46L) against Micrococcus luteus (MIC, both 16.7 μg/mL).106
Fig. 5.

Structures of P-2-AIs with antibacterial activity
Palau’amine (10) isolated from Stylotella aurantium displayed antibacterial activity against S. aureus and B. subtilis among other bioactivities (vide infra).7 The degree of bromination on the pyrrole was shown to affect the biological activity with the monobromo and dibromo derivatives found to be less active compared to the non-brominated congener. The structurally related styloguanidine (61) isolated from the sponge Stylotella aurantium from the Yap sea, showed inhibitory activity against chitinase, an enzyme responsible for hydrolysis of chitin, using the bacterium Schwanella sp. at 2.5 mg/disk. Chitin plays an important role in a wide variety of organisms ranging from nutrition to defense and control of ecdysis in arthropods, therefore chitinase inhibitors have potential as antifungal and insecticidal agents.107 For example, the styloguanidines were found to inhibit the moulting of cyprid larvae of barnacles at 10 ppm demonstrating their potential as antifouling agents.
4.2 Girolline (aka girodazole): A Cellular Probe for Eukaryotic Protein Synthesis and a Potential Anticancer Agent
Girolline (73), also known as girodazole, was isolated from Pseudaxinyssa cantharella and Axinella sp. from New Caledonia in 1988.108 In 1991, this simple P-2-AI, a result of apparent chlorohydroxylation of 2-APPE, was recognized as a potent antitumor agent and found to exhibit significant cytotoxicity in vitro against several tumor cell lines and in vivo against several murine grafted tumors: leukemia (P388, L1210), mammary adenocarcinoma (MA 16/C) and hitiocytosarcoma (M5076).109 However, girolline did not exhibit any antitumour activity in a phase-I clinical trial, but had severe cardiovascular toxicity.110 Girolline is one of the first P-2-AIs for which a detailed molecular level understanding of its mode of action has been elucidated. Comparative studies revealed that, despite its relatively simple but potentially reactive structure, which embodies latent potential for epoxide or aziridine formation and thus potential action as an alkylating agent for example to nucleic acids, it was determined that girolline’s anticancer activity was not a result of alkylation on DNA or RNA. Rather, it involved inhibition of the termination steps of protein synthesis in vivo.111 It is interesting to note that several marine sponge isolates including mycalamide A,112 pateamine113,114, and others are also potent inhibitors of protein synthesis, a primary cellular function for survival. Girolline induced G2/M cell cycle arrest in several tumor cell lines and immunochemical analysis revealed that polyubiquitinated p53 was accumulated in girolline-treated cells, while other polyubiquitinated cellular proteins were not accumulated, indicating that the effect of girolline is specific for p53. On the other hand, girolline did not inhibit proteasome activity in vitro, and accumulation of polyubiquitinated p53 was scarcely detected in the presence of leptomycin B, an inhibitor of nuclear export. Thus, it was proposed that girolline affects the step of recruitment of polyubiquitinated p53 to the proteasome, postulating that it might be a novel inhibitor against the ubiquitin-dependent proteolytic pathway.115 To gain further insight into the mechanism of action of girolline in vivo, effects on translation and on cell cycle progression were investigated. Girolline neither induced an increase in translational read through nor affected the polysome profile of treated cells. Cell-cycle analysis shows that girolline induced a dose- and time-dependent arrest of cells in G2 phase. Altogether, these results suggest that girolline does not act on translation in vivo, but interferes with the control of the G2 checkpoint.116 However, recently the crystal structure of girolline in the E binding site of the 50S ribosomal subunit of Haloarcula Marismortui (Hma) was reported,117 an unprecedented site for antibiotic binding and shows hydrogen bonding with the hydroxyl, amino, and imidazole ring nitrogen of this target protein (Figure 6b).
Fig. 6.

(a) Structure and pharmacology of girolline (73, a.k.a. giradazole) (b) key interactions between girolline and E site binding domain of 50 S ribosomal subunit (with permission from Elsevier-still needed)
4.3 Hymenialdisines: Novel Kinase Inhibitors of the Ras-MAPK Signaling Cascade as Potential Treatments for Osteoarthritis
10Z-Debromohymenialdisine (182), was first isolated in 1980 from the Caribbean sponge Phakellia flabellata (Figure 7a).118 Its corresponding (E) isomer was isolated in 1996 from the sponge Stylotella aurantium.119 Debromohymenialdisine has been shown to slow joint deterioration and cartilage degradation associated with osteoarthritis in animal models and is currently under development as a promising new drug candidate.120,121 A number of reports centering on 10Z-debromohymenialdisine and the related analogue hymenialdisine122 have appeared describing the modulation of protein kinase C and the proinflammatory transcription factor, nuclear factor κB (NFκB).123,124,125 NFκB is believed to be involved in the regulated gene expression of the inflammatory enzyme, cyclooxygenase (COX-II).126 Several structural congeners of the hymenialdisine sub-group of P-2-AIs displayed inhibitor activity in the Ras-MAPK signaling cascade.127 In addition, 10Z-debromohymeialdisine inhibited checkpoint kinases Chk1 and 2 with IC50 values of 3 and 3.5 μM, respectively. A detailed study of the inhibitory activity of hymenaldisine towards kinases including the synthesis of >60 analogs led to the identification of 11 new kinase targets for this class of P-2-AIs (Figure 7b).128 In addition, an X-ray structure of CDK2 complexed with hymenialdisine A has been reported and showed that this inhibitor occupied the ATP binding pocket. Importantly, this structure also revealed that the pyrrole bromine was pointing out of the binding pocket,129 therefore two affinity resins were prepared by direct Sonogashira couplings at the bromopyrrole leading to amines 184 and 185 (control) which were then coupled to NHS-agarose resins (Figure 7c). Affinity chromatography experiments led to the identification of six binding proteins including the known high affinity interacting kinases, GSK3α/β and Mek1, in addition to two new interacting kinases, p90RSK and an unknown kinase thought to be an isoform of BIKe (BMP2-inducible kinase). (Z)-debromohymenialdisine and (Z)-hymenialdisine also proved to be active against human monocytic leukemia cells (MONOMAC- 6) with IC50 values of 2.4 and 0.2 μg/mL, respectively.130 The synthesis and utility of hymenialdisines as potential medicinal agents has been recently reviewed.131
Fig. 7.

(a) Structure of 10Z-hymenialdisine (182) and summary of pharmacology (b) derivatization sites for kinase specificity studies (c) amine derivatives 184/185 used for affinity chromatography experiments
4.3.1 Other P-2-AIs with Anticancer Potential but Unknown Cellular Targets
Several other P-2-AIs have been reported to have anticancer activity but details regarding their specific cellular targets are lacking. One potent anticancer P-2-AI is agelastatin A (122) and more extensive SAR data are available for this natural product due to isolation of several congeners and synthesis of derivatives (Figure 8). Agelastatin A (122)132 was first isolated in 1993 by Pietra from Agelas dendromorpha and displays significant cytotoxicity against both the L1210 and KB tumor cell lines as well as notable activity against a doxorubicin-resistant L1210 leukemia strain.133 (−)-Agelastatin A is 1.5 to 16 times more potent than cis-platin at inhibiting cell growth, its effects being most pronounced against human bladder, skin, colon, and breast carcinomas.134 Additional congeners, agelastatins C (122C) and D (122D), were isolated by Molinski in 1998 from the West Australian axinellid sponge Cymbastela sp.131 and found to selectively inhibit GSK-3 (glycogen synthase kinase-3), a kinase that may be involved in the onset of Alzheimer’s disease, with an IC50 of 12 μM.129 Structure-activity relationship studies show that unsubstituted OH-C(8a), H-N(5), and H-N(6) moieties are essential in the B/D transoid configuration for high cytotoxic activity. Reductive debromination of the pyrrole bromine also leads to a significant loss of potency.132 (−)-Agelastatin A showed a wide range of anticancer, antiangiogenic,135 antimetastatic,132 and even insecticidal properties. Its potent cytotoxicity was also observed against a panel of human cancer cell lines (IC50 0.097–0.703 μM) providing important SAR data primarily by detailed and methodical studies by Hale and coworkers.136 The latter studies revealed for the first time that agelastatin A down-regulates β-catenin expression while simultaneously up-regulating Tcf-4, causing the repression of osteopontin (OPN).134 Despite these promising biological activities, the cellular receptor of the agelastatins has not been reported and its potential as a drug candidate has not been fully explored.
Fig. 8.
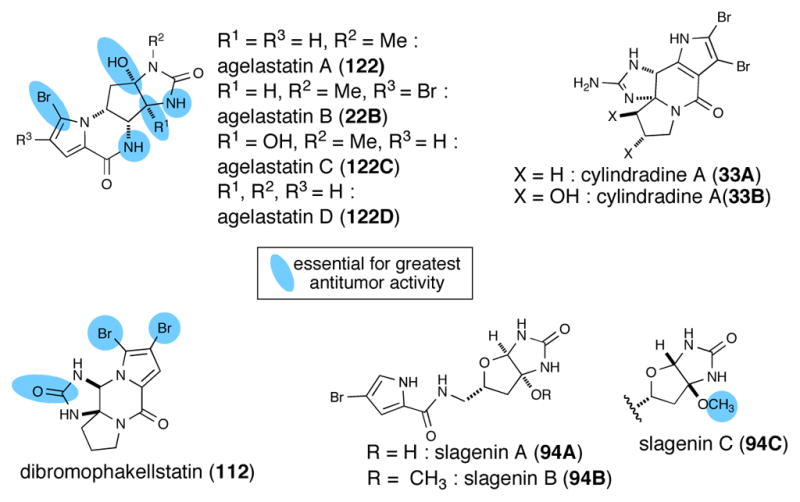
P-2-AIs with anticancer potential but unknown cellular targets and summary of known SAR data
Dibromophakellstatin (112) from the Republic of Seychelles sponge Phakellia mauritiana was identified as the most significant cancer cell growth inhibitory substance. In the original isolation, phakellstatin showed antineoplastic activity against melanoma Sk-MEL-5 and colon KM20L2 cell lines (ED50 0.28 μM for both cell lines)137 and antitumor activity was confirmed in another small panel of cancer lines by Lindel using racemic phakellstatin.138a Namely, antitumor activity was found against SF-268 (brain cancer, ED50 1.4 μM), NCI-H460 (large cell lung cancer, 2.1 μM), and KM20L2 (colon cancer, 0.4 μM) cell lines. In subsequent studies, Lindel reported potent cell growth inhibitory activity with dibromophakellstatin against a more extensive panel of 36 human cancer cell lines (ED50 0.28–3.9 μM).138b The greatest potency of racemic dibromophakellstatin was observed toward an ovarian (OVXF 899L, 0.60 μM, IC50), a glioblastoma (CNXF 498NL, 0.93 μM), a non-small lung (LXF 529L (0.96 μM), and uterine UXF 1138L (1.21 μM) cancer cell lines. Importantly, Lindel also claimed that the selectivity profile of rac-dibromophakellstatin may be indicative of a novel mechanism of action. Limited structure-activity studies of dibromophakellstatin to date have indicated that the bromides are essential for activity since their removal led to dramatic loss of antitumor activity. In addition, the lack of anticancer activity of the phakellins suggests that the cyclic urea is also of importance for activity. Lindel identified a dichlorocyclopropane derivative of phakellstatin that exhibited more potent (average IC50 7 μM against 36 cell lines tested), albeit less pronounced selectivity profile than dibromophakellstatin (average IC50 >30 μM). They also verified that only one enantiomer, (−)-phakellstatin, exhibited antitumor activity suggestive of a specific mechanism of action with a likely cellular macromolecular receptor.
Several other P-2-AIs have been reported as cytotoxic agents to various cancer cell lines. Slagenins B (94B) and C (94C), which share a bicyclic imidazolone with the agelastatins, exhibited cytotoxicity against leukemia L1210 cells in vitro (21 and 20 μM, respectively), however slagenin A (94A) exhibited greatly reduced activity (IC50 >29 μM, Figure 8).139 This limited SAR data for the slagenins suggests that the presence of the methoxyaminal rather than a free carbinolamine is more important than ring fusion stereochemistry. Recently, cylindradine A (33A) and B (33B) isolated from Axinella cylindratus showed moderate inhibitory activity against the murine leukemia cell lines P388 with IC50 values of 20 and 77 μM, respectively.31 In the original isolation of palau’amine (10), it was reported to be active against P-388 (IC50 0.1 μgmL−1), A549 (0.2μg/mL), HT-29 (2μg/mL) and KB (10 μg/mL) cell lines.7
4.4 Sceptrin and Related P-2-AI’s: Antibacterial Agents Targeting Actin
One of the more structurally interesting and potent antibacterial P-2-AIs is sceptrin (146), isolated from Agelas sceptrum and exhibiting activity against S. aureus (MIC 15 μg/mL), B. subtilis and Pseudomonas aeruginosa. The activity was considerably greater than that recorded for oroidin.140 In one study, sceptrin demonstrated a bacteriostatic rather than a bactericidal effect on exponentially growing E. coli cells.141 In 1991, Keifer and coworkers reported diacetate salts of seven P-2-AI alkaloids from the Caribbean sponge, Agelas conifera. Among these, debromosceptrin (180), sceptrin (146), ageliferin (46E), bromoageliferin (46F), dibromoageliferin (46G) inhibited growth of B. subtilis at 10 μg/disk with lower activites found for dibromosceptrin (181), debromooxysceptrin (179) and oxysceptrin (178). Sceptrin, ageliferin, and bromoageliferin were also found to have antibacterial activity against E. coli at 10 μg/disk.142,130 along with a previously observed antibiotic effect of sceptrin and ageliferin against B. subtilis, S. aureus, and E. coli. at a concentration of 0.2 μmol/disk.
Recently a binding protein of sceptrin from E. Coli was identified via a bidirectional affinity protocol.143 The process involved the use of resins with bound E. Coli derived proteins to first partially purify crude natural product extracts to separate protein-interactive natural products from an Agelas conifera extract, and following random alkylation of alcohol/amine functional groups of the partially purified natural product mixture with a coumarin-derived immunoaffinity fluorescent (IAF) tag 186 via ring cleavage of an acyl aziridine, forward affinity co-immunoprecipitation in a sandwich assay with an antibody that recognizes the IAF tag was utilized to identify binding protein(s) (Figure 9). In this manner, a 40 kDa protein was identified as the actin equivalent protein in E. Coli, MreB, and is in accord with the observed bacteriostatic effect of sceptrin and its ability to disrupt bacterial cell wall biosynthesis leading to spheroblast formation.141 The structure of the IAF-tagged sceptrin 187, which was not reported, would provide useful information regarding SAR of this P-2-AI subset by eliminating sectors of the natural product that do not bind to MreB. The full utility of this potentially powerful methodology for isolating cellular receptors of bioactive natural products, which does not require apriori structural information for a natural product, will be realized when an affinity resin for each protein in the human proteome becomes available. This will enable both a reverse affinity purification of crude natural product extracts for specific cellular protein target(s) of interest and a forward affinity purification using co-immunoprecipitation with the IAF tag.
Fig. 9.

Synthesis of a random (structure undefined) immunoaffinity fluorescence (IAF) tagged-sceptrin dervative 187 and its use in a sandwich assay using an IAF antibody (blue) to identify MreB (red) as an E. Coli binding protein of sceptrin.
4.5 Receptor Antagonists
Although the cyclic oroidin derivative dibromophakellin (20) was first isolated and characterized more than 30 years ago144, no investigation into its mode of action were reported. Indeed no significant biological activity was initially associated with this simple P-2-AI (Figure 10). Only recently Quinn,145 after re-isolation of dibromophakellin from the organic extract of the marine sponge Acanthella costata, discovered activity as an antagonist of the α2B adrenoceptor, a subtype of G-protein coupled receptor affecting analgesia, sedation, hypotension, regulation of hypothermia, gastric protection and gastric motility. This points the importance of re-screening known natural products using new assays.
Fig. 10.
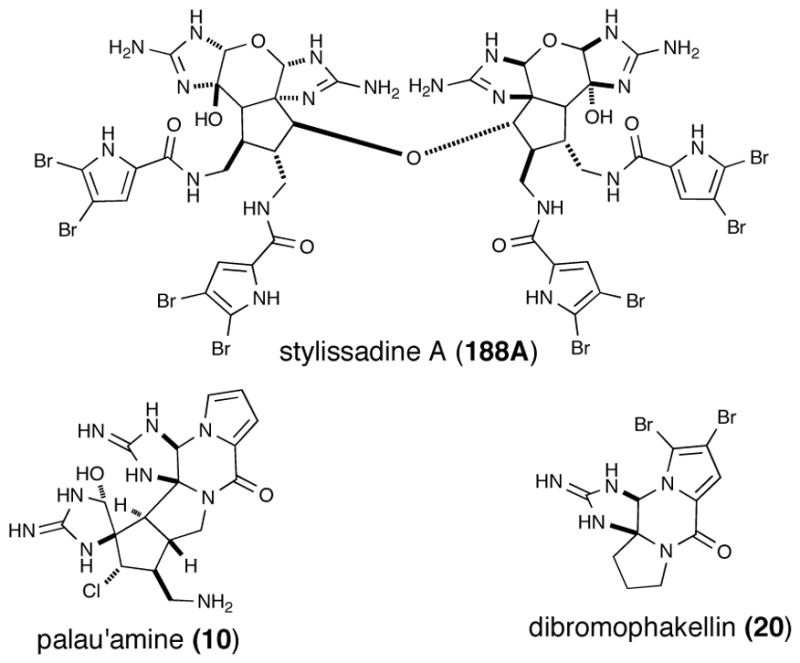
Structures of receptor antagonists, styillisadine A (188A) and dibromophakellin (20) and the immunosuppressive agent, palau’amine (10)
The recently isolated stylissadine A (188A) and B (188B),146 the first “tetrameric” PIAs, derived from dimerization of massadine, were shown to be specific antagonists of the P2X7 receptor with IC50 values of 0.7 and 1.8 μM, respectively Although the physiological function of P2X7 remains unknown it was advanced that it may have a role in inflammation and immunomodulation.
4.6 Immunosuppressive Agents
At the time of its initial isolation, palau’amine (10) exhibited one of the most potent and interesting biological activities, namely immunosuppressive activity in the mixed lymphocyte reaction with an IC50 value of < 42.8 nM) coupled with low acute toxicity (LD50 13 mg/kg (ip in mice) (Figure 10).7 This P-2-AI was at one time undergoing preclinical studies.147 A review article describing phage display also reported an attempted application of this technology to identify a cellular target for palau’amine, however the only isolated sequence corresponded to hNopp140, a presumed promiscuous non-specific binder, previously observed in phage display experiments and concluded to be an artifact.148 No further reports have reconfirmed biological activity nor described new bioactivities for palau’amine.
4.7 Phosphatase Inhibitors
The dimeric bromopyrrole alkaloids, nagelamides A-C, G, and H (46A-C, G, H), were shown to inhibit PP2A (IC50 12 to >50 μM) a protein phosphatase involved in the regulation of numerous cellular functions, incluing muscle contractions, neurotransmission cell proliferation, carcinongenesis, and apotosis (Figure 11a).103
Fig. 11.

Miscellaneous enzymatic inhibitory activities reported for several P-2-Ais including (a) phosphatase inhibitors, nagelamides A-C, G, and H (46A-C, G, H) (b) kinase inhibitors, konbu’acidin A (190) and spongiacidins A (189A) and B (189B), tauroacidins A (191A) and B (191B) and (c) a protein prenylation inhibitor, massadine (60).
4.8 Other P-2-AIs Exhibiting Kinase Inhibition
Konbu’acidin A (190) was found to inhibit cyclin-dependent kinase 4 (CDK4), a protein tyrosine kinase involved in cell cycling events, with an IC50 of 27 μM; however, this activity appears decoupled from cytotoxicity since at this concentration no toxicity was observed with L1210 or KB cells (IC50 > 27 μM; Figure 11b).149 Spongiacidins A and B ((189A, B) closely related to the hymenialdisines) isolated from Hymeniacidon sp., inhibited c-erbB-2 kinase (IC50 21 and 18 μM, respectively) and CDK4 (IC50 80 and 37 μM, respectively).150 However, spongiacidins C and D (189C, D) did not inhibit either kinase (IC50, >50 μg/mL). Taurocidins A and B (191A, B) isolated from the Okinawan sponge Hymeniacidon sp., also exhibited inhibitory activity against EGF receptor kinase and c-erB2 kinase with IC50 in the 28–45 μM range.151
4.9 Massadine: A Protein Prenylation Inhibitor
Massadine (60)152 isolated from the organic extract of the marine sponge Stylissa aff. massa, showed inhibitory activity of the geranylgeranyltransferase type I (GGTase I) enzyme, a Zn(II) metalloenzyme involved in protein prenylation of Candida albicans with an IC50 of 3.9 μM (Figure 11c).
Note Added in Revision
Several papers detailing further structural and synthetic studies have appeared since initial submission of this review. Collaborative work by Lindel, Kinnel and Köck153 provided strong evidence in support of the presumed absolute configuration of palau’amine. Several important contributions including a total synthesis of the unnatural enantiomer of cyclooroidin by Lovely154 as well as synthetic approaches to partial structures of palau’amine by Williams,155 Feldman,92 and Chen,93 and massadine by Carreira156 which are not described in further detail in this review. However, mention must be made of the elegant and efficient biomimetic strategy leading to all congeners of agelastatin that was recently described by Movassaghi (Scheme 26).157 Importantly, this synthesis follows one possible biosynthetic route premised on the isolation of the related P-2-AI, cyclooroidin (32).
Scheme 26.

Synthesis of all agelastatins isolated to date by Movassaghi;157 NBS = N-bromosuccinimide, THF = tetrahydrofuran, DTBMP = 2,6-di-tert-butyl-4-methylpyridine, CuTC = copper (I) thiophene-2- carboxylate. (* = overall yields calculated from reported step-wise yields)
Movassaghi’s synthesis commenced with pyrrole 195, prepared in one step from commercially available D-aspartic acid dimethyl ester 193. After bromination and acylation of the pyrrole, treatment with NaBH4 and p-toluenesulfonic acid delivered the key bicyclic intermediate 197 in 99% ee on >10 g scale. Subsequent coupling with either stannylurea 194B or 194A (the latter generated in situ from a triazone precursor) provided imidazolone 198, aptly dubbed “pre-agelastatin.” A final acidmediated cyclization delivered agelastatin A (122A) and B (122B) in modest yields. Further standard manipulations of these materials allowed access to the remaining members of this subset of P-2-AIs for the first time.
A recent report described the biological analysis of 14 P-2AIs and provided the first detailed study of the antiprotozoal potential of these alkaloids.158 Dibromopalau’amine (IC50 = 0.80 μM against T. brucei rhodesiense; IC50 = 1.9 μM against Leishmania donovani) and longamide B (IC50 = 12.6 μM) exhibited the most potent activity as trypanocidal and antileishmanial agents, while dispacamide B (3) and spongiacidin B (6) were found to be interesting antimalarial lead compounds. Cytotoxic activity was also reported toward mammalian (L6) cells for dibromopalau’amine and longamide B (cytotoxicity concentration (CC), CC50 = 7.7 μM and 28.4 μM, respectively).
5 Summary and Future Outlook
Bioactive natural products (i.e. genetically encoded small molecules1) continue to be a productive gold mine for discoveries in synthetic strategy development and for novel biosynthetic studies leading to novel mechanisms and enzymatic machinery. Furthermore, natural products provide inspiration for novel synthetic methods and insights into cellular function and perturbation or inhibition of aberrant cellular machinery leading to potential drugs for human ailments. This is not surprising given that these natural products have been designed to interact with various cellular receptors including the biosynthetic enzymes involved in their synthesis and tailoring. It is our hope that this review has conveyed this to be true for the continually growing family of pyrrole-2-aminoimidazole (P-2-AI) alkaloids. However, what should also be clear to the reader is that with respect to the P-2-AI family, we are only at the initial stages of discovery with respect to practical asymmetric syntheses important for further biological studies, biosynthetic studies, chemical genetics studies, and drug discovery and development. While over 150 members of this family are known, the recent discovery of the benzosceptrins, styllisazoles, and stylissadines, an apparent dimer of massadine, clearly suggests that further bioassay-guided searching for novel members of this family will continue to offer great dividends in terms of novel structure and function. Furthermore, the discovery of new bioactivities for ‘old’ P-2-AIs such as dibromophakellin and dibromopalau’amine demonstrates the importance of rescreening collections.
Future research in the P-2-AI alkaloid area is likely to reveal insights into the actual intermediates, biosynthetic machinery, and synthetic strategies used by P-2-AI generating-enzymes to produce the presumed universal precursors clathrodin/oroidin and the higher order P-2-AIs, e.g. ageliferin, axinellamine, palau’amine, stylissadine. The great utility of biosynthetic speculations guiding total synthesis endeavors to ultimately achieve high synthetic efficiency will continue to provide practical avenues for answering structure-activity questions by synthesis of even the most complex P-2-AIs. Furthermore, direct evidence for the numerous proposed biosynthetic pathways will undoubtedly uncover some interesting biosynthetic mechanisms and catalytic systems. While new methods for elucidating cellular receptors for natural products continue to be developed,159 it should be expected that the number of P-2-AI/cellular receptor complexes known will increase dramatically in the coming years beyond the few (only 3 to date) that are currently known. Undoubtedly, the future will reveal additional novel enantioselective synthetic strategies and biosynthetic, pharmacological, and cellular receptor isolation studies of this exciting family of natural products known as the pyrrole-2-aminoimidazole alkaloids.
Fig. 4.

(a) Structures of oroidin (6) and oroidin-inspired derivatives including ‘reverse amide’ derivatives displaying antibiofilm activity (b) Structure of the ageliferins and simplified derivatives demonstrating antibacterial activity
Scheme 3.

Büchi’s landmark ‘bioinspired’ synthesis of dibromophakellin (20) and more recent optimizations and isomerizations/rearrangements
Scheme 16.

Enantioselective syntheses of girolline (73, a.k.a. giradazole). a) Commerçon’s aldol approach.58 b) Ahond’s chiral pool strategy.59 Tr = triphenylmethyl; Boc = t-butoxycarbonyl, Im = imidazole, DMF = N,N-dimethylformamide, THF = tetrahydrofuran.
Scheme 20.

Enantioselective syntheses of phakellin. a) Romo’s synthesis of (+)-phakellin71 b) Nagasawa’s synthesis of (+)-dibromophakellin.72 (IBX = iodoxobenzoic acid; DMSO = Dimethyl sulfoxide. DPPA = diphenylphosphoryl azide; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; THF = tetrahydrofuran, Tces = trichloroethylsulfonyl, HMDS = hexamethyldisilazane; TBS = t-butyldimethylsilyl; EDCI = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; DMAP = dimethylaminopyridine; Ms = Methanesulfonyl; py = pyridine; DIBAL = diisobutylaluminum hydride; Boc = t-butoxycarbonyl, Tf = trifluoromethylsulfonyl)
Scheme 25.

Racemic synthesis of palau’amine by Baran.91 PMBCl = p-methoxybenzyl chloride, TBAI = tetrabutylammonium iodide, DMF = N, N-dimethylformamide, TMSOTf = trimethylsilyl trifluoromethanesulfonate, DIPEA = diisopropylethylamine, NBS = N-bromosuccinimide, THF = tetrahydrofuran, TFA = trifluoroacetic acid, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, IBX = 2-iodoxybenzoic acid, TFAA = trifluoroacetic anhydride, EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, HOBt = N-hydroxybenzotriazole.
References
- 1.Miller SJ, Clardy J. Nat Chem. 2009;1:261. doi: 10.1038/nchem.269. [DOI] [PubMed] [Google Scholar]
- 2.Andrade P, Willoughby R, Pomponi S, Kerr R. Tetrahedron Lett. 1999;40:4775. [Google Scholar]
- 3.Braekman JC, Daloze D, Stoller C, van Soest RWM. Biochem Syst Ecol. 1992;20:417. [Google Scholar]
- 4.Walker RP, Faulkner DJ, Van Engen D, Clardy J. J Am Chem Soc. 1981;103:6772. [Google Scholar]
- 5.Keifer P, Schwartz R, Moustapha LD, Koker ES, Hughes RG, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965. [Google Scholar]
- 6.(a) Olofson A, Yakushijin K, Horne DA. J Org Chem. 1997;62:7918. doi: 10.1021/jo9715682. [DOI] [PubMed] [Google Scholar]; (b) Sosa CB, Yakushijin K, Horne DA. Org Lett. 2000;2:3443–3444. doi: 10.1021/ol000233v. [DOI] [PubMed] [Google Scholar]; (c) Xu Y, Yakushijin K, Horne DA. J Org Chem. 1996;61:9569. [Google Scholar]
- 7.(a) Kinnel RB, Gehrken HP, Scheuer PJ. J Am Chem Soc. 1993;115:3376. [Google Scholar]; (b) Kinnel RB, Gehrken HP, Swali R, Skoropowski G, Scheuer PJ. J Org Chem. 1998;63:3281. [Google Scholar]
- 8.Al-Mourabit A, Potier P. Eur J Org Chem. 2001:237. [Google Scholar]
- 9.Köck M, Grube A, Seiple IB, Baran PS. Angew Chem Int Ed. 2007;46:6586. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]
- 10.a) Forte B, Malgesini B, Piutti P, Quartieri F, Scolaro A, Papeo G. Mar Drugs. 2009;7:705. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weinreb SM. Nat Prod Rep. 2007;24:931. doi: 10.1039/b700206h. [DOI] [PubMed] [Google Scholar]; c) Jin Z. Nat Prod Rep. 2006;23:464. doi: 10.1039/b502166a. [DOI] [PubMed] [Google Scholar]; d) Lindel T, Hoffmann H, Hochgurtel M, Pawlik JR. J Chem Ecol. 2000;26:1477. [Google Scholar]
- 11.Kitagawa I, Kobayashi M, Kitanaka K, Kido M, Kyogoku Y. Chem Pharm Bull. 1983;31:2321. [Google Scholar]
- 12.a) Travert N, Al-Mourabit A. J Am Chem Soc. 2004;126:10252. doi: 10.1021/ja047574e. [DOI] [PubMed] [Google Scholar]; b) Vergne C, Bouray-Esnault N, Perez T, Martin M-T, Adeline M-T, Huu Dau ET, Al-Mourabit A. Org Lett. 2006;8:2421. doi: 10.1021/ol0608092. [DOI] [PubMed] [Google Scholar]
- 13.Mancini I, Guella G, Amade P, Roussakis C, Pietra F. Tetrahedron Lett. 1997;38:6271. [Google Scholar]
- 14.Vaillancourt FH, Yeh A, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 15.Linington RG, Williams DE, Tahir A, Soest R, Andersen RJ. Org Lett. 2003;5:2735. doi: 10.1021/ol034950b. [DOI] [PubMed] [Google Scholar]
- 16.For a biosynthetic proposal for ageladine see: Fujita M, Nakao Y, Matsunaga S, Seiki M, Itoh Y, Yamashita J, Van Soest RWM, Fusetani N. J Am Chem Soc. 2003;125:15700. doi: 10.1021/ja038025w.
- 17.Lindel T, Hochgürtel M, Assmann A, Köck M. J Nat Prod. 2000;63:1566. doi: 10.1021/np000160o. [DOI] [PubMed] [Google Scholar]
- 18.Friedel M, Lindel T. Tetrahedron Lett. 2004;45:2779. [Google Scholar]
- 19.Po1verlein C, Breckle G, Lindel T. Org Lett. 2006;8:819. doi: 10.1021/ol0526219. [DOI] [PubMed] [Google Scholar]
- 20.Wang YG, Morinaka BI, Reyes JCP, Wolff JJ, Romo D, Molinski TF. J Nat Prod. 2010;73:428. doi: 10.1021/np900638e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foley LH, Büchi G. J Am Chem Soc. 1982;104:1776. [Google Scholar]
- 22.Barrios Sosa AC, Yakushijin K, Horne AD. J Org Chem. 2002;67:4498. doi: 10.1021/jo020063v. [DOI] [PubMed] [Google Scholar]
- 23.(a) Braun M, Büchi G. J Am Chem Soc. 1976;98:3049. doi: 10.1021/ja00426a080. [DOI] [PubMed] [Google Scholar]; (b) Braun M, Büchi G, Bushey DF. J Am Chem Soc. 1978;100:4208. [Google Scholar]
- 24.Feldman KS, Skoumbourdis AP, Fodor MD. J Org Chem. 2007;72:8076. doi: 10.1021/jo701487j. [DOI] [PubMed] [Google Scholar]
- 25.Xu Y, Yakushijin K, Horne AD. J Org Chem. 1996;61:9571. [Google Scholar]
- 26.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew Chem Int Ed. 2009;48:1. [Google Scholar]
- 27.Nakadai M, Harran PG. Tetrahedron Lett. 2006;47:3933. [Google Scholar]; Sharma G. Drugs Food Sea, Myth, Reality. Intl Symp Proc. 1978:203. [Google Scholar]
- 28.Feldman KS, Fodor MD. J Am Chem Soc. 2008;130:14964. doi: 10.1021/ja807020d. [DOI] [PubMed] [Google Scholar]; Picon S, Huu Dau E-T, Martin M-T, Retailleau P, Zaparucha A, Al-Mourabit A. Org Lett. 2009;11:2523. doi: 10.1021/ol900745c. [DOI] [PubMed] [Google Scholar]
- 29.Tsuda M, Yasuda T, Fukushi E, Kawabata J, Sekiguchi M, Fromont J, Kobayashi J. Org Lett. 2006;8:4235. doi: 10.1021/ol061464q. [DOI] [PubMed] [Google Scholar]
- 30.Grube A, Köck M. J Nat Prod. 2006;69:1212. doi: 10.1021/np050408f. [DOI] [PubMed] [Google Scholar]
- 31.Kuramoto M, Miyake N, Ishimaro Y, Ono N, Uno H. Org Lett. 2008;10:5465. doi: 10.1021/ol802263j. [DOI] [PubMed] [Google Scholar]
- 32.D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc, Chem Commun. 1993:1305. [Google Scholar]; Hong TW, Jimenez DR, Molinski TF. J Nat Prod. 1998;61:158. doi: 10.1021/np9703813. [DOI] [PubMed] [Google Scholar]; Tilvi S, Moriou C, Martin M-T, Gallard J-F, Sorres J, Patel K, Petek S, Debitus C, Ermolenko L, Al-Mourabit A. J Nat Prod. 2010;73:720. doi: 10.1021/np900539j. [DOI] [PubMed] [Google Scholar]
- 33.Araki A, Tsuda M, Kubota T, Mikami Y, Fromont J, Kobayashi J. Org Lett. 2007;9:2369. doi: 10.1021/ol0707867. [DOI] [PubMed] [Google Scholar]
- 34.Dilley AS, Romo D. Org Lett. 2001;3:1535. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]
- 35.Lovely CJ, Du H, He Y, Rasika Dias HV. Org Lett. 2004;6:735. doi: 10.1021/ol036403w. [DOI] [PubMed] [Google Scholar]
- 36.Tsukamoto S, Tane K, Ohta T, Matsunaga S, Fusetani N, Van Soest RWM. J Nat Prod. 2001;64:1576. doi: 10.1021/np010280b. [DOI] [PubMed] [Google Scholar]
- 37.Ahond A, Bedoya-Zurita M, Colin M, Fizames C, Laboute P, Lavelle F, Laurent D, Poupat C, Pusset M, Pusset J, Thoison O, Potier P. C R Acad Sci Paris, serie II. 1981;307:145. [Google Scholar]; Chiaroni A, Riche X, Ahond A, Poupat C, Pusset M, Potier P. C R Acad Sci Paris, serie II. 1981;312:49. [Google Scholar]
- 38.Wei Y, Zipse H. Eur J Org Chem. 2008;47:3811–3816. and references cited therein. [Google Scholar]
- 39.Kiefer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965. [Google Scholar]
- 40.Buchanan MS, Cattoll AR, Addepalli R, Avery VM, Hooper JNA, Quinn RJ. J Org Chem. 2007;72:2309. doi: 10.1021/jo062007q. [DOI] [PubMed] [Google Scholar]; Kobayashi H, Kitamura K, Nagai K, Nakao Y, Fusetani N, van Soest RWM, Matsunaga S. Tetrahedron Lett. 2007;48:2127. [Google Scholar]; Grube A, Köck M. Angew Chem Int Ed. 2007;46:2320. doi: 10.1002/anie.200604076. [DOI] [PubMed] [Google Scholar]; Buchanan MS, Carroll AR, Quinn RJ. Tetrahedron Lett. 2007;48:4573. [Google Scholar]
- 41.O’Malley DP, Yamaguchi J, Young IS, Seiple IB, Baran PS. Angew Chem Int Ed. 2008;47:3581. doi: 10.1002/anie.200801138. [DOI] [PubMed] [Google Scholar]
- 42.Su S, Seiple IB, Young IS, Baran PS. J Am Chem Soc. 2008;130:16490. doi: 10.1021/ja8074852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Q, Hurley P, Ding H, Roberts AG, Akella R, Harran PG. J Org Chem. 2009;74:5909. doi: 10.1021/jo900755r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrido-Hernandez H, Nakadai M, Vimolratana M, Li Q, Doundoulakis T, Harran PG. Angew Chem Int Ed. 2005;44:765. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]; Li Q, Hurley P, Ding H, Roberts AG, Akella R, Harran PG. J Org Chem. 2009;74:5909. doi: 10.1021/jo900755r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Dilley AS, Poullenec KG, Romo D. Tetrahedron. 2006;62:7155. [Google Scholar]
- 46.Grube A, Immel S, Baran PS, Köck M. Angew Chem Int Ed. 2007;46:6721. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]
- 47.Patel K, Laville R, Martin M-T, Tilvi S, Moriou C, Gallard J-F, Ermolenko L, Debitus C, Al-Mourabit A. Angew Chem Int Ed. 2010;48:4775. doi: 10.1002/anie.201000444. [DOI] [PubMed] [Google Scholar]
- 48.Reviews on total syn of P-2-AIs, see: Arndt HD, Riedrich M. Angew Chem Int Ed. 2008;47:4785. doi: 10.1002/anie.200801793.Weinreb SM. Nat Prod Rep. 2007;24:931. doi: 10.1039/b700206h.
- 49.Overman LE, Rogers BN, Tellew JE, Trenkle WC. J Am Chem Soc. 1997;119:7159. [Google Scholar]; Lanman BA, Overman LE. Heterocycles. 2006;70:557. doi: 10.3987/com-06-s(w)16. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lanman BA, Overman LE, Paulini R, White NS. J Am Chem Soc. 2007;129:12896. doi: 10.1021/ja074939x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawasaki I, Sakaguchi N, Fukushima N, Fujioka N, Nikaido F, Yamashita M, Ohta S. Tetrahedron Lett. 2002;43:4377. [Google Scholar]
- 51.Starr JT, Kock G, Carreira RM. J Am Chem Soc. 2000;122:8793. [Google Scholar]
- 52.Tan X, Chen C. Angew Chem Int Ed. 2006;45:4345. doi: 10.1002/anie.200601208. [DOI] [PubMed] [Google Scholar]
- 53.Sivappa R, Hernandez NM, He Y, Lovely CJ. Org Lett. 2007;9:3861. doi: 10.1021/ol0711568. [DOI] [PubMed] [Google Scholar]
- 54.Bultman MS, Ma J, Gin DY. Angew Chem Int Ed. 2008;47:6821. doi: 10.1002/anie.200801969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cernak TA, Gleason JL. J Org Chem. 2008;73:102. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]; Hudon J, Cernak TA, Ashenhurst JA, Gleason JL. Angew Chem. 2008;120:9017. doi: 10.1002/anie.200803344. [DOI] [PubMed] [Google Scholar]; Hudon J, Cernak TA, Ashenhurst JA, Gleason JL. Angew Chem Int Ed. 2008;47:8885. doi: 10.1002/anie.200803344. [DOI] [PubMed] [Google Scholar]
- 56.Namba K, Kaihara Y, Yamamoto H, Imagawa H, Tanino K, Williams RM, Nishizawa M. Chem J Eur. 2009;15:6560. doi: 10.1002/chem.200900622. [DOI] [PMC free article] [PubMed] [Google Scholar]; Williams RM, Burnett CM. ACS Symp Ser. 2009;1009:420. [PMC free article] [PubMed] [Google Scholar]
- 57.Bedoya-Zurita M, Ahond A, Poupat C, Potier P. Tetrahedron. 1989;45:6713. [Google Scholar]; Commerçon A, Gueremy CA. Tetrahedron Lett. 1991;32:1419. [Google Scholar]
- 58.Commerçon A, Paris JM. Tetrahedron Lett. 1991;32:4905. [Google Scholar]
- 59.Ahond A, Al Mourabit A, Bedoya-Zurita M, Heng R, Marques Braga R, Poupat C, Potier P. Tetrahedron. 1992;48:4327. [Google Scholar]
- 60.No optical rotation has ever been reported for natural midpacamide.
- 61.Cafieri F, Carnussio R, Fattorusso E, Taglialatela-Scafati O, Vallefuoco T. Bioorg Med Chem Lett. 1997;7:2283. [Google Scholar]
- 62.Kobayashi J, Inaba K, Tsuda M. Tetrahedron. 1997;53:16679. Tauroacidin A was determined to be a 6:4 mixture of 9S-9R isomers. [Google Scholar]
- 63.Uemoto H, Tsuda M, Kobayashi J. J Nat Prod. 1999;62:1581. doi: 10.1021/np9902542. Mukadin A was determined to be a 7:3 mixture of 9R-9S isomers. [DOI] [PubMed] [Google Scholar]
- 64.Vergne C, Appenzeller J, Ratinaud C, Martin M-T, Zaparucha A, Al-Mourabit A. Org Lett. 2008;10:493. doi: 10.1021/ol702866m. [DOI] [PubMed] [Google Scholar]
- 65.Patel J, Pelloux-Leon N, Minassian F, Vallée Y. Tetrahedron Lett. 2006;47:5561. [Google Scholar]
- 66.Patel J, Pelloux-Leon N, Minassian F, Vallée Y. J Org Chem. 2005;70:9081. doi: 10.1021/jo051555l. [DOI] [PubMed] [Google Scholar]
- 67.Jiang B, Liu J-F, Zhao S-Y. Org Lett. 2001;3:4011. doi: 10.1021/ol016689+. [DOI] [PubMed] [Google Scholar]
- 68.Jiang B, Liu J-F, Zhao S-Y. Org Lett. 2002;4:3951. doi: 10.1021/ol0268034. [DOI] [PubMed] [Google Scholar]
- 69.Gurjar MK, Bera S. Org Lett. 2002;4:3569. doi: 10.1021/ol020112q. [DOI] [PubMed] [Google Scholar]
- 70.Sharma GM, Burkholder PR. J Chem Soc Chem Comm. 1971:151. [Google Scholar]
- 71.Wang S, Romo D. Angew Chem Int Ed. 2008;47:1284. doi: 10.1002/anie.200703998. [DOI] [PubMed] [Google Scholar]
- 72.Imaoka T, Iwamoto O, Noguchi K, Nagasawa K. Angew Chem Int Ed. 2009;48:3799. doi: 10.1002/anie.200806233. [DOI] [PubMed] [Google Scholar]
- 73.Feldman KS, Skoumbourdis AP. Org Lett. 2005;7:929. doi: 10.1021/ol0500113. [DOI] [PubMed] [Google Scholar]; Jacquot DEN, Hoffmann H, Polborn K, Lindel T. Tetrahedron Lett. 2002;43:3699. [Google Scholar]; Chung R, Yu E, Incarvito CD, Austin DJ. Org Lett. 2004;6:3881. doi: 10.1021/ol0490532. [DOI] [PubMed] [Google Scholar]; Lu J, Tan X, Chen C. J Am Chem Soc. 2007;129:7768. doi: 10.1021/ja072844p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poullenec KG, Romo D. J Am Chem Soc. 2003;125:6344. doi: 10.1021/ja034575i. [DOI] [PubMed] [Google Scholar]
- 75.Zöllinger M, Mayer P, Lindel T. Synlett. 2007;17:2756. [Google Scholar]
- 76.For a review that includes racemic syntheses of agelastatin, see ref. 48.
- 77.Feldman KS, Saunders JC, Wrobleski MC. J Org Chem. 2002;67:7096. doi: 10.1021/jo026287v. [DOI] [PubMed] [Google Scholar]
- 78.Hale MM, Domostoj KJ, Tocher DA, Irving E, Scheinmann F. Org Lett. 2003;5:2927. doi: 10.1021/ol035036l. [DOI] [PubMed] [Google Scholar]
- 79.Stien D, Anderson GT, Chase CE, Koh Y-H, Weinreb SM. J Am Chem Soc. 1999;121:9574. [Google Scholar]
- 80.Domostoj MM, Irving E, Scheinmann F, Hale KJ. Org Lett. 2004;6:2615. doi: 10.1021/ol0490476. [DOI] [PubMed] [Google Scholar]
- 81.Davis FA, Deng J. Org Lett. 2005;7:621. doi: 10.1021/ol047634l. [DOI] [PubMed] [Google Scholar]
- 82.Trost BM, Dong G. J Am Chem Soc. 2006;128:6054. doi: 10.1021/ja061105q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trost BM, Dong G. Chem Eur J. 2009;15:6910. doi: 10.1002/chem.200900794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ichikawa Y, Yamaoka T, Nakano K, Kotsuki H. Org Lett. 2007;9:2989. doi: 10.1021/ol0709735. [DOI] [PubMed] [Google Scholar]
- 85.Yoshimitsu T, Ino T, Tanaka T. Org Lett. 2008;10:5457. doi: 10.1021/ol802225g. [DOI] [PubMed] [Google Scholar]
- 86.Yoshimitsu T, Ino T, Futamura N, Kamon T, Tanaka T. Org Lett. 2009;11:3402. doi: 10.1021/ol9012684. [DOI] [PubMed] [Google Scholar]
- 87.Hama N, Matsuda T, Sato T, Chida N. Org Lett. 2009;11:2687. doi: 10.1021/ol900799e. [DOI] [PubMed] [Google Scholar]
- 88.When PM, Du Bois J. Angew Chem Int Ed. 2009;48:3802. doi: 10.1002/anie.200806292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.(a) Baran PS, Zografos AL, O’Malley DP. J Am Chem Soc. 2004;126:3726. doi: 10.1021/ja049648s. [DOI] [PubMed] [Google Scholar]; (b) Birman VB, Jiang XT. Org Lett. 2004;6:2369. doi: 10.1021/ol049283g. [DOI] [PubMed] [Google Scholar]
- 90.O’Malley DP, Li K, Maue M, Zografos AL, Baran PS. J Am Chem Soc. 2007;129:4762. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 91.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew Chem Int Ed. 2010;49:1095. doi: 10.1002/anie.200907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feldman KS, Nuriye AY. OrgLett. 2010;12:4532. doi: 10.1021/ol1018322. [DOI] [PubMed] [Google Scholar]
- 93.Ma Z, Lu J, Wang X, Chen C. Chem Commun. 2011;47:427. doi: 10.1039/c0cc02214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.(a) Carté BK. BioScience. 1996;46:271. [Google Scholar]; (b) Sipkema D, Franssen MCR, Osinga R, Tramper J, Wijffels RH. Mar Biotechnol. 2005;7:142. doi: 10.1007/s10126-004-0405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Singh R, Sharma M, Joshi P, Rawat DS. Anti-Cancer Agents in Med Chem. 2008;8:603. [PubMed] [Google Scholar]
- 95.Huigens RW, Reyes S, Reed CS, Bunders C, Rogers SA, Steinhauer AT, Melander C. Bioorg Med Chem. 2010;18:663. doi: 10.1016/j.bmc.2009.12.003.and references cited. Rogers SA, Huigens RW, Melander C. J Am Chem Soc. 2009;131:9868. doi: 10.1021/ja9024676.Huigens RW, III, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C. J Am Chem Soc. 2007;129:6966. doi: 10.1021/ja069017t.
- 96.Richards JJ, Reed CS, Melander C. Bioorg Med Chem Lett. 2008;18:4325. doi: 10.1016/j.bmcl.2008.06.089. [DOI] [PubMed] [Google Scholar]
- 97.Rinehart KL. Pure Appl Chem. 1989;61:525. [Google Scholar]
- 98.Kobayashi J, Tsuda M. Tetrahedron. 1990;46:5579. [Google Scholar]
- 99.Huigens RW, III, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C. J Am Chem Soc. 2007;129:6966. doi: 10.1021/ja069017t. [DOI] [PubMed] [Google Scholar]
- 100.Cafieri F, Fattorusso E, Taglialatela-Scafati O. J Nat Prod. 1998;61:122. doi: 10.1021/np970323h. [DOI] [PubMed] [Google Scholar]
- 101.Sachiko T, Haruko K, Hiroshi H, Nobuhiro F. J Nat Prod. 1996;59:501. [Google Scholar]
- 102.Urban S, De Almeida Leone P, Carroll AR, Fechner GA, Smith J, Hooper JNA, Quinn RJ. J Org Chem. 1999;64:731. doi: 10.1021/jo981034g. [DOI] [PubMed] [Google Scholar]
- 103.Endo T, Tsuda M, Okada T, Mitsuhashi S, Shima H, Kikuchi K, Mikami Y, Fromont J, Kobayashi J. J Nat Prod. 2004;67:1262. doi: 10.1021/np034077n. [DOI] [PubMed] [Google Scholar]
- 104.Araki A, Kubota T, Aoyama K, Mikami Y, Fromont J, Kobayashi J. Org Lett. 2009;11:1785. doi: 10.1021/ol900328c. [DOI] [PubMed] [Google Scholar]
- 105.Yasuda T, Araki A, Kubota T, Ito J, Mikami Y, Fromont J, Kobayashi J. Org Lett. 2009;72:488. doi: 10.1021/np800645q. [DOI] [PubMed] [Google Scholar]
- 106.Araki A, Tsuda M, Kubota T, Mikami Y, Fromont J, Kobayashi J. Org Lett. 2008;10:2099. doi: 10.1021/ol8003904. [DOI] [PubMed] [Google Scholar]
- 107.Kato T, Shizuri Y, Izumida H, Yokoyama A, Endo M. Tetrahedron Lett. 1995;36:2133. [Google Scholar]
- 108.Ahond A, Zurita MB, Colin M, Fizames C, Laboute P, Lavelle F. Comptes Rendus de l’Academie des Sciences II. 1988;307:145. [Google Scholar]
- 109.Lavelle F, Zerial A, Fizames C, Rabault B, Curaudeau A. Invest New Drugs. 1991;9:233. doi: 10.1007/BF00176976. [DOI] [PubMed] [Google Scholar]
- 110.Catimel G, Coquard R, Guastalla J-P, Merrouche Y, Le Bail N, Alakl MK, Dumortier A, Foy M, Clavel M. Cancer Chemother Pharmacol. 1995;35:246. doi: 10.1007/BF00686555. [DOI] [PubMed] [Google Scholar]
- 111.Colson G, Rabault B, Lavelle F, Zerial A. Biochem Pharmacol. 1992;43:1717. doi: 10.1016/0006-2952(92)90701-j. [DOI] [PubMed] [Google Scholar]
- 112.Burres NS, Clement JJ. Cancer Res. 1989;49:2940. [PubMed] [Google Scholar]
- 113.Low W-K, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick W, Romo D, Liu JO. Mol Cell. 2005;20:709. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 114.Low W-K, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Rzasa RM, Shea HA, Li S, Park K, Ma G, Romo D, Liu JO. Methods in Enzymology. 2007;431:303. doi: 10.1016/S0076-6879(07)31014-8. [DOI] [PubMed] [Google Scholar]
- 115.Tsukamoto S, Yamashita K, Tane K, Kizu R, Ohta T, Matsunaga S, Fusetani N, Kawahara H, Yokosawa H. Biol Pharm Bull. 2004;27:699. doi: 10.1248/bpb.27.699. [DOI] [PubMed] [Google Scholar]
- 116.Diop D, Chauvin C, Salhi S, Poupat C, Ahond A, Jean-Jean O. CR Biologies. 2007;330:855. doi: 10.1016/j.crvi.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 117.Schroeder SJ, Blaha G, Tirado-Rives J, Steits TA, Moore PB. J Mol Biol. 2007;367:1471. doi: 10.1016/j.jmb.2007.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sharma GM, Buyer JS, Pomerantz MW. J Chem Soc, Chem Commun. 1980:435. [Google Scholar]
- 119.Williams DH, Faulkner D. J Nat Prod Lett. 1996;9:57. [Google Scholar]
- 120.Chipman S, Faulkner DJ. WO/1996/040147
- 121.OsteoArthritis Sciences, Inc. The Reagents of the University of California. Use of Debromohymenialdisine for Treating Osteoarthritis. 5,591,740. US Patent. 1997
- 122.Cimino G, De Rosa S, De Stefano S, Mazzarella L, Puliti R, Sodano G. Tetrahedron Lett. 1982;23:767. [Google Scholar]
- 123.Breton JJ, Chabot-Fletcher M. J Pharmacol Exp Ther. 1997;282:459. [PubMed] [Google Scholar]
- 124.SmithKline Beecham Corporation. Protein Kinase C Inhibitor. 5,616,577. US Patent. 1997
- 125.SmithKline Beecham Corporation. Medicament. 5,565,448. US Patent. 1996
- 126.Roshak A, Jackson JR, Chabot-Fletcher M, Marshall LA. JPharmacol Exp Ther. 1997;283:955. [PubMed] [Google Scholar]
- 127.Tasdemir D, Mallon R, Greenstein M, Feldberg LR, Kim SC, Collins K, Wojciechowics D, Mangalindan GC, Concepcion GP, Harper MK, Ireland CM. J Med Chem. 2002;45:529. doi: 10.1021/jm0102856. [DOI] [PubMed] [Google Scholar]
- 128.Wan Y, Hur W, Cho CY, Francisco YL, Adrian J, Lozach O, Bach S, Mayer T, Fabbro D, Meijer L, Gray NS. Chem Biol. 2004;11:247. doi: 10.1016/j.chembiol.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 129.Meijer L, Thunnissen A-MWH, White AW, Garnier M, Nikolic M, Tsai L–H, Walter J, Cleverley KE, Salinas PC, Wu Y-Z, Biernat J, Mandelkow E-M, Kim S-H, Pettit GR. Chem Biol. 2000;7:51. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 130.(a) Eder C, Proksch P, Wray V, Van Soest RWM, Ferdinandus E, Pattisina LA, Sudarsono J Nat Prod. 1999;62:1295. doi: 10.1021/np990071f. [DOI] [PubMed] [Google Scholar]; (b) Eder C, Proksch P, Wray V, Steube K, Bringmann G, Van Soest RWM, Ferdinandus SE, Pattisina LA, Wiryowidagdo S, Moka W. J Nat Prod. 1999;62:184. doi: 10.1021/np980315g. [DOI] [PubMed] [Google Scholar]
- 131.Nguyen TN, Tepe JJ. Curr Med Chem. 2009;16:3122. doi: 10.2174/092986709788803015. [DOI] [PubMed] [Google Scholar]
- 132.D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc Chem Comm. 1993;16:1305. [Google Scholar]
- 133.D’Ambrosio M, Guerriero A, Ripamonti M, Debitus C, Waikedre J, Pietra F. Helv Chim Acta. 1996;79:727. [Google Scholar]
- 134.Mason CK, McFarlane S, Johnston PG, Crowe P, Erwin PJ, Domostoj MM, Campbell FC, Manaviazar S, Hale KJ, El-Tanani M. Mol Cancer Ther. 2008;7:548. doi: 10.1158/1535-7163.MCT-07-2251. [DOI] [PubMed] [Google Scholar]
- 135.Pettit GR, Ducki S, Herald DL, Doubek DL, Schmidt JM, Chapuis J-C. Oncol Res. 2005;15:11. doi: 10.3727/096504005775082075. [DOI] [PubMed] [Google Scholar]
- 136.Hale K, Domostoj M. Strat Tact Org Syn. 2005;6:352. [Google Scholar]
- 137.Pettit GR, McNulty J, Herald DL, Doubek DL, Chapuis J-C, Schmidt JM, Tackett LP, Boyd MR. J Nat Prod. 1997;60:180. doi: 10.1021/np9606106. [DOI] [PubMed] [Google Scholar]
- 138.(a) Zollinger M, Kelter G, Fiebig H-H, Lindel T. Bioorg Med Chem Lett. 2007;17:346. doi: 10.1016/j.bmcl.2006.10.046. [DOI] [PubMed] [Google Scholar]; (b) Zöllinger M, Mayer P, Lindel T. J Org Chem. 2006;71:9431. doi: 10.1021/jo061813u. [DOI] [PubMed] [Google Scholar]
- 139.Tsuda M, Uemoto H, Kobayashi J. Tetrahedron Lett. 1999;40:5709. [Google Scholar]
- 140.Faulkner DJ. PATENT U.S. 4,370,484. CODEN: USXXAM US. 1983:3. Appl. No.: 242728.
- 141.Bernan VS, Roll DM, Ireland CM, Greenstein M, Maiese WM, Steinberg DA. J Antimicrob Chemother. 1993;32:539. doi: 10.1093/jac/32.4.539. [DOI] [PubMed] [Google Scholar]
- 142.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965. [Google Scholar]
- 143.Rodriguez AD, Lear MJ, La Clair JJ. J Am Chem Soc. 2008;130:7256. doi: 10.1021/ja7114019. [DOI] [PubMed] [Google Scholar]
- 144.Sharma G, Magdoff-Fairchild B. J Org Chem. 1977;42:4118. [Google Scholar]
- 145.Davis RA, Fechner, Sykes GA, Garavelas M, Pass A, Carroll DM, Addepalli AR, Avery R, Hooper VM, Quinn JN. Bioorg Med Chem. 2009;17:2497. doi: 10.1016/j.bmc.2009.01.065. [DOI] [PubMed] [Google Scholar]
- 146.Buchanan MS, Carroll AR, Addepalli R, Avery VM, Hooper JNA, Quinn RJ. J Org Chem. 2007;72:2309. doi: 10.1021/jo062007q. [DOI] [PubMed] [Google Scholar]
- 147.European Patent Application No. 94302770.6.
- 148.Piggott A, Karuso P. Mar Drugs. 2005;3:36. [Google Scholar]
- 149.Kobayashi J, Suzuki M, Tsuda M. Tetrahedron. 1997;53:15681. [Google Scholar]
- 150.Inaba K, Sato H, Tsuda M, Kobayashi J. J Nat Prod. 1998;61:693. doi: 10.1021/np970565h. [DOI] [PubMed] [Google Scholar]
- 151.Kobayashi J, Inaba K, Tsuda M. Tetrahedron. 1997;53:16679. [Google Scholar]
- 152.Nishimura S, Matsunaga S, Shibazaki M, Suzuki K, Furihata K, van Soest RWM, Fusetani N. Org Lett. 2003;5:2255. doi: 10.1021/ol034564u. [DOI] [PubMed] [Google Scholar]
- 153.Lindel T, Jacquot DEN, Zollinger M, Kinnel RB, McHugh S, Kock M. Tetrahedron Lett. 2010;51:6353. [Google Scholar]
- 154.Mukherjee S, Sivappa R, Yousufuddin M, Lovely CJ. Org Lett. 2010;12:4940–4943. doi: 10.1021/ol1020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Namba K, Inai M, Sundermeier U, Greshock TJ, Williams RM. Tetrahedron Lett. 2010;51:6557. doi: 10.1016/j.tetlet.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chinigo G, Breder A, Carreira EM. Org Lett. 2011;13:78. doi: 10.1021/ol102577q. [DOI] [PubMed] [Google Scholar]
- 157.Movassaghi M, Siegel DS, Han S. Chem Sci. 2010;1:561. doi: 10.1039/c0sc00351d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Scala F, Fattorusso E, Menna M, Taglialatela-Scafati O, Tierney M, Kaiser M, Tasdemir D. Mar Drugs. 2010;8:2162. doi: 10.3390/md8072162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.(a) Carlson EE. ACS Chem Biol. 2010;5:639. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Osada H, editor. Protein Targeting with Small Molecules: Chemical Biology Techniques and Applications. John Wiley & Sons; 2009. [Google Scholar]