

Abstract
Hi-C experiments study how genomes fold in 3D, generating contact maps containing features as small as 20bp and as large as 200-Mb. Here we introduce Juicebox, a tool for exploring Hi-C and other contact map data. Juicebox allows users to zoom in and out of Hi-C maps interactively, just as a user of Google Earth might zoom in and out of a geographic map. Maps can be compared to one another, or to 1D tracks or 2D feature sets.
Graphical abstract
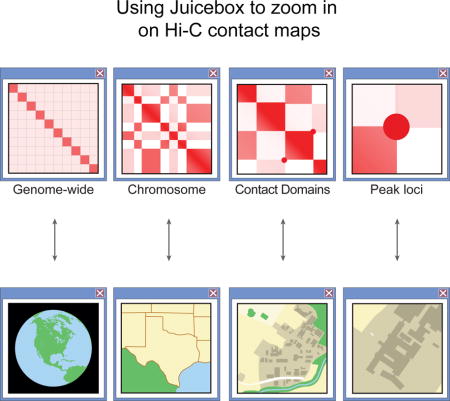
Hi-C is a widely employed method for studying how genomes are folded in three dimensions (Rao et al., 2014). In Hi-C, DNA loci that are spatially proximate in a system of interest (such as a cell nucleus or an in vitro DNA preparation) are ligated to one another and sequenced. This results in a Hi-C contact map: a list of pairs of genomic positions that were adjacent to each other in three-dimensional (3D) space. Typically, the pairwise interactions produced by Hi-C experiments are visualized as a heatmap: the linear genome is partitioned into loci of a fixed size, or resolution (e.g., 1-Mb or 1-Kb) and each entry in the two-dimensional (2D) heatmap corresponds to the number of contacts observed between a pair of loci during the experiment.
Developing adequate visualizations for Hi-C heatmaps is a challenge, because the size of the meaningful biological features they contain ranges over at least seven orders of magnitude: from loops anchored at 20-bp CTCF sites to territories that extend across 200-Mb chromosomes. At the highest meaningful resolutions, published Hi-C heatmaps contain trillions of entries, and only a tiny portion can be displayed at any given time. At coarser resolutions, more of the map can be shown, but the fine structure can no longer be resolved.
Several Hi-C visualization systems exist. Like a paper atlas, these browsers show the map at a single resolution (typically either 1-Mb or 100-Kb), either in its entirety (Servant et al., 2012; Paulsen et al., 2014; Lieberman-Aiden et al., 2009) or very close to the diagonal (Zhou et al., 2013). Users cannot zoom in and out in real time. Also, most of these systems are designed to show specific, previously published datasets, and they do not allow users to visualize their own experiments.
Here we introduce Juicebox, a tool for exploring Hi-C data and contact maps in general (Figure 1). Juicebox allows users to explore Hi-C heatmaps interactively, zooming in and out just as a user of Google Earth might zoom in and out of a geographic map; it integrates many technologies developed for the Integrative Genomics Viewer (Robinson et al., 2011) with a broad ensemble of methods specifically designed for handling 2D contact data. Individual maps can be normalized (corrected for experimental bias), compared to one-dimensional (1D) tracks (such as gene tracks or chromatin immunoprecipitation sequencing [ChIP-seq] data), and compared to 2D feature lists (such as loop and domain annotations). Multiple maps can be browsed side by side simultaneously and compared with one another in various ways, revealing both conservation and variation across cell types and species. Users can create their own heatmaps to explore their own experiments (Durand et al., 2016). Juicebox was an invaluable tool in making biological discoveries across many size scales in our group’s recent paper (Rao et al., 2014).
Figure 1. Juicebox enables exploration of contact maps at many resolutions.
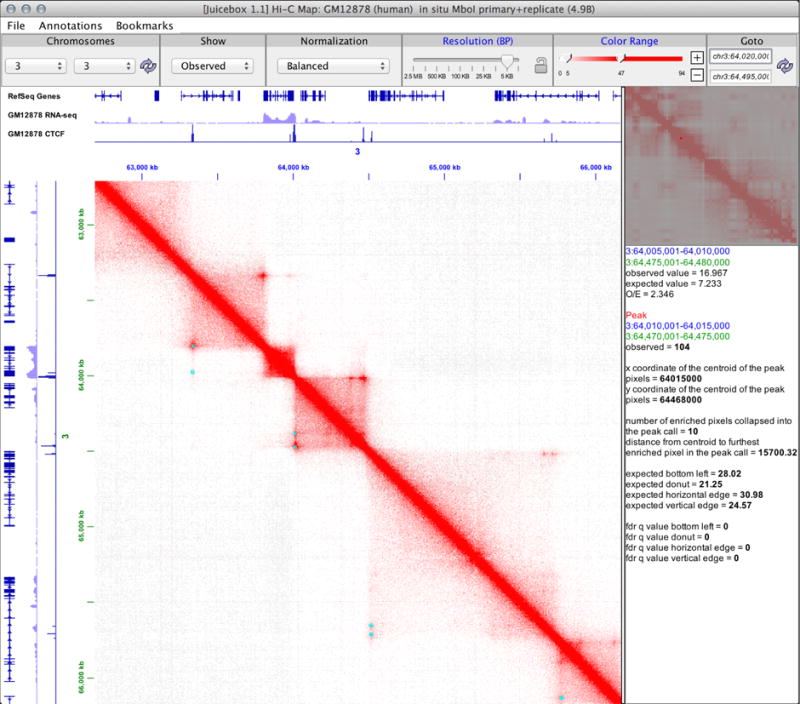
Here, we see a screenshot of Juicebox zoomed into 5-Kb resolution on chromosome 3. The toolbar at the top allows users to quickly navigate between different chromosomes, views, normalizations, and resolutions. Users can load one-dimensional tracks and compare them to features seen in the heatmap. Two-dimensional features can be superimposed on the main map. Several peaks are annotated (cyan), each peak indicating the presence of a chromatin loop. A peak in the CTCF track, indicating CTCF binding, is seen at most loop anchors. At the top right, a mini-map shows the whole chromosome at low resolution. Below, hover text shows additional data for one of the highlighted peaks.
In Juicebox—when using Hi-C maps of adequate depth and quality—increasingly small features can be resolved as the user zooms in. In a genome-wide view, chromosome territories are evident, as are chromosomal rearrangements such as translocations. Clicking on a particular chromosome zooms into its intrachromosomal map, optimized for the user’s monitor. The broad compartmentalization of the genome, which manifests as alternating long-range patterns, is typically visible at this resolution. On the X chromosome, two superdomains may also become apparent, partitioning the chromosome. These are accompanied by superloops, bright peaks many megabases away from the diagonal.
Zooming in further can be accomplished by double-clicking, by using the resolution slider, or by drawing a box around a region of interest. At 50-Kb resolution, subcompartments can be seen, reflecting finer differences in the long-range contact pattern. At 25-Kb resolution, contact domains appear: these are intervals containing loci, which preferentially form contacts with one another, and that form squares along the diagonal. Juicebox makes it easy to compare these structures to a large number of broad-source chromatin marks at once. We find that the epigenetic marks that decorate particular contact domains correlate strongly with differences in long-range contact pattern.
At 5-Kb resolution, chromatin loops are readily seen as bright peaks in which contact frequency is enhanced relative to the local neighborhood. These tend to lie at the corners of contact domains. Finally, at 1-Kb, the relationship between the loops and point-source epigenetic tracks can be interrogated. For instance, it becomes clear the chromatin loops are frequently anchored at convergent CTCF motifs, i.e., CTCF motifs that point toward one another (Rao et al., 2014). Juicebox can be used to zoom in much further, although maps with enough data to support such studies do not yet exist.
Juicebox is available as a Java application that can be downloaded and launched via aidenlab.org/juicebox. The code is open source and licensed under the MIT license, available at github.com/theaidenlab/Juicebox. Users can explore their own data or examine data from over 15 Hi-C, 5C, and CHIA-PET publications. We also provide the software and test datasets used to review this manuscript at http://dx.doi.org/10.17632/dj4nrsc552.1. We hope that allowing researchers to zoom inward will help studies of chromatin architecture to zoom onward.
Acknowledgments
This work was supported by an NIH New Innovator Award (1DP20D008540-01), the NHGRI Center for Excellence for Genomic Sciences (P50HG006193), an NVIDIA Research Center Award, an IBM University Challenge Award, a Google Research Award, a Cancer Prevention Research Institute of Texas Scholar Award (R1304), a McNair Medical Institute Scholar Award, the President’s Early Career Award in Science and Engineering, a grant from the NSF Physics Frontiers Center (Center for Theoretical Biological Physics), and a grant from the Welch Foundation to E.L.A.; an NIGMS grant (R01GM074024) to JPM; and an NHGRI grant (HG003067) to E.S.L. We thank Suhas Rao, Miriam Huntley, Elena Stamenova, and Olga Dudchenko for their help with testing Juicebox. The Center for Genome Architecture is grateful to Janice, Robert, and Cary McNair for support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: Author contributions are as follows: E.L.A. conceived of this project; J.T.R. and N.C.D. created the tool; J.T.R., N.C.D., M.S., and I.M. contributed to tool development; N.C.D., J.P.M., E.S.L, and E.L.A. prepared the manuscript.
References
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum N, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie B, Sabo P, Dorschner M, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J, Sandve GK, Gundersen S, Lien TG, Trengereid K, Hovig E. HiBrowse: multi-purpose statistical analysis of genome-wide chromatin 3D organization. Bioinformatics. 2014;30:1620–1622. doi: 10.1093/bioinformatics/btu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A Threedimensional Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttan M, Lander ES, Getz G, Mesirov JP. Integrative Genomics Viewer. Nature Biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen CJ, Heard E, Dekker J, Barillot E. HiTC: Exploration of high-throughput ‘C’ experiments. Bioinformatics. 2012;28:2843–2844. doi: 10.1093/bioinformatics/bts521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lowdon RF, Li D, Lawson HA, Madden PAF, Costello JF, Wang T. Exploring long-range genome interactions using the WashU EpiGenome Browser. Nature Methods. 2013;10:375–376. doi: 10.1038/nmeth.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]