

Abstract
The NMR structure of the peptidyl-tRNA hydrolase (PTH) domain from Pseudomonas syringae (P. syringae PTH domain) was solved by using recently devised protocol for high throughput protein structure determination. The P. syringae PTH domain belongs to a large Pfam family PF00472, which consists of at least 1549 proteins annotated as ‘hydrolysis domains of peptidyl-tRNA’. The structure of P. syringae PTH domain expands the ‘structural coverage’ of the PFam family.
Keywords: Protein translation, Peptidyl-tRNA hydrolase domain, Release factors, Structural genomics, GFT NMR
Introduction
The 100-residue peptidyl-tRNA hydrolase domain from the gram-negative bacterium Pseudomonas syringae infecting tomato, referred to in this paper as the ‘P. syringae PTH domain’, (gi|42602314, SwissProt/TrEMBL ID Q885L4_PSESM, access number Q885L4)1 constitutes the hydrolysis domain of peptide chain release factor (RF) encoded by gene PSPTO1818. The P. syringae PTH domain has no significant amino acid sequence similarity to any protein with known three-dimensional structure, and was selected by the Protein Structure Initiative-2 of the United States National Institutes of Health and assigned to the Northeast Structural Genomics consortium (NESG; http://www.nesg.org) (NESG target ID PsR211). The P. syringae PTH domain belongs to the large Pfam2 familiy PF00472 which currently contains 1549 members, all of which were identified as such PTH domains.3 Many members of this family appear to function in prokaryotic translation termination by hydrolyzing the peptidyl t-RNA ester after polypeptide synthesis is completed. A universally conserved GGQ sequence motif acts as the active site and mimics the aminoacyl stem of peptidyl t-RNA (notably, one of the Glu residues of the motif coordinates the water molecule required for hydrolysis).4,5
In general, releasing factors are classified based on their specific recognition of tRNA stop codons. Class 1 factors include RF-1 and RF-2 both of which recognize UAA. In contrast, UAG and UGA are specifically recognized only by, respectively, RF-1 and RF-2. Class 2 releasing factors are non codon specific GTP binding proteins and stimulate class 1 releasing factor activity. Five structures of class 1 release factors have thus far been solved: peptide chain RF-1 from E.coli (RF1_ECOLI; PDB ID: 2b3t; sequence identity of 20% with P. syringae PTH domain), peptide chain RF-1 from Thermotoga maritima (RF1_THEMA; PDB ID: 1rq0; sequence identity of 16% with P. syringae PTH domain), immature colon carcinoma transcript 1 protein from Mus Musculus (MOUSE) (ICT1_MOUSE; PDB ID: 1j26, sequence identity of 16% with P. syringae PTH domain), peptide chain RF-1 from Streptococcus mutans (RF1_STRMU; PDB ID: 1zbt, sequence identity of 15% with P. syringae PTH domain) and peptide chain RF-2 from E.coli (RF2_ECOLI; PDB ID: 1gqe; sequence identity of 13% with P. syringae PTH domain). Evidently, the low level of sequence identity across this very large protein domain family prevents the building of good quality homology model for the P. syringae PTH domain. Here we report the NMR solution structure solved by use of a protocol devised for high-throughput NMR protein structure determination.6
Materials and Methods
The P. syringae PTH domain was cloned, expressed and purified following standard protocols developed by the NESG for production of uniformly U-13C,15N-labeled protein samples.7 First, a domain comprising 130-residues was expressed, but the 2D [15N,1H] HSQC spectrum revealed that the C-terminal 30 residues are flexibly disordered in solution. Using these NMR data, the construct was optimized and the 100-residue domain was expressed for NMR structure determination. The corresponding segment of gene PSPTO1818 from P. syringae was cloned into a pET21 (Novagen) derivative, yielding the plasmid Psr211-1-100-21.2. The resulting construct contains eight nonnative residues at the C-terminus (LEHHHHHH) that facilitate protein purification. Escherichia coli BL21 (DE3) pMGK cells, a rare codon enhanced strain, were transformed with PsR211-1-100-21.2, and cultured in MJ9 minimal medium containing (15NH4)2SO4 and U-13C-glucose as sole nitrogen and carbon sources. U-13C, 15N P. syringae PTH domain was purified using an AKTAxpress (GE Healthcare) based two-step protocol consisting of IMAC (HisTrap HP) and gel filtration (HiLoad 26/60 Superdex 75) chromatography. The final yield of purified U-13C, 15N P. syringae PTH domain (> 98% homogenous by SDS-PAGE; 12.7 kDa by MALDI-TOF mass spectrometry) was about 15 mg/L. The final sample of U-13C, 15N labeled P. syringae PTH domain was prepared at a concentration of ~1.2 mM in 95% H2O/5% D2O solution containing 5mM CaCl2, 100 mM NaCl, 20mM NH4OAc, 10mM DTT, 0.02% NaN3, 5% D2O, pH 5.5. An isotropic overall rotational correlation time of ~6.5 ns was inferred from 15N spin relaxation times, indicating that the protein is monomeric in solution. This conclusion was further confirmed by an analytic gel-filtration (Agilent Technologies) followed by a combination of static light scattering and refractive index (Wyatt Technology).7
All NMR spectra were recorded at 25 °C on a Varian INOVA 750 spectrometer equipped with a cryogenic probe. Five through-bond correlated G-matrix Fourier transform8 (GFT) NMR experiments8–10 were collected for backbone and side chain resonance assignment (total measurement time: 51 hours), and a 3D 15N/13Caliphatic/13Caromatic-resolved [1H,1H]-NOESY spectrum10 (mixing time: 70 ms; measurement time: 24 hours) was acquired to derive 1H-1H distance constraints. Spectra were processed and analyzed with the programs NMRpipe11 and XEASY12, respectively. Sequence specific backbone (HN, Hα, N, Cα) and Hβ/Cβ resonance assignments were obtained with (4,3)D HNNCαβCα/CαβCα(CO)NHN4 and (4,3)D HαβCαβ(CO)NHN experiments using the program AUTOASSIGN.13 Side-chain assignments were accomplished by using aliphatic and aromatic (4,3)D HCCH. Overall, assignments were obtained for 99% of routinely assigned backbone (excluding the N-terminal NH3+, the Pro 15N and the 13C’ shifts) and 13Cβ, and for 98% of the side chain chemical shifts (excluding Lys NH3+, Arg NH2, OH, side chain 13C′ and aromatic quaternary 13C shifts; Table I). Stereo-specific assignments were obtained for 38% of the β-methylene groups exhibiting non-degenerate proton chemical shifts using glomsa module of CYANA.14 90% of the Val and Leu isopropyl moieties with non-degenerate chemical shifts (Table I) were obtained by using partially 13C labeled (5%).15 Chemical shifts were deposited in the BioMagResBank (accession code: 15471). Upper distance limit constraints for structure calculations were extracted from NOESY (Table I), and backbone dihedral angle constraints were derived from chemical shifts as described16 for residues located in regular secondary structure elements (Table I) by using the program TALOS. The programs CYANA14,17 and AUTOSTRUCTURE18 were used in parallel to automatically assign long-range NOEs. The final structure calculations were performed using version 2.1 of CYANA17 and CNS19 with the DYANA constraints (see: http://www.las.jp/prod/cyana/eg).
Table I.
Statistics of NMR Structure of P.syringae PTH domain
| Conformationally restricting distance constraints | |
| Intraresidue [I = j] | 448 |
| Sequential [(I – j) = 1] | 586 |
| Medium Range [1 < (I – j) ≤ 5] | 354 |
| Long Range [(I – j) > 5] | 541 |
| Total | 1929 |
| Dihedral angle constraints | |
| Φ | 51 |
| Ψ | 51 |
| Number of constraints per residue | 20.3 |
| Number of long-range constraints per residue | 5.4 |
| Completeness of stereo-specific assignmentsa [%] | |
| βCH2 | 38 (28/74) |
| Val and Leu isopropyl groups | 90 (19/21) |
| CYANA target function [Å2] | 0.30 ± 0.004 |
| Average r.m.s.d. to the meanCNS coordinates [Å] | |
| regular secondary structure elementsb, backbone heavy | 0.58±0.10 |
| regular secondary structure elements, all heavy atoms | 0.98±0.10 |
| residues 1–21, 34–102 backbone heavy atoms N, Cα, C′ | 0.72±0.12 |
| residues 1–21, 34–102 all heavy atoms | 1.17±0.12 |
| heavy atoms of molecular core (or best-defined SC)c | 0.72±0.11 |
| PROCHECK rawscored (φ and Ψ/all dihedral angles) | −0.09/−0.16 |
| PROCHECK Z-scoresd (φ and Ψ/all dihedral angles) | −0.04/−0.95 |
| MOLPROBITY raw score/Z-scoree | 16.64/−1.33 |
| Auto QF R/P/DP scores (%)f | 0.97/0.96/0.80 |
| Ramachandran plot summary ordered residue ranges: 7–47, 59–72, 76 – 82, 87 – 91 [%] | |
| most favored regions | 93.2 |
| additionally allowed regions | 6.7 |
| generously allowed regions | 0.1 |
| disallowed regions | 0 |
| Average number of distance constraints violations per CYANA conformer [A] | |
| 0.2–0.5 | 0.0 |
| > 0.5 | 0 |
| Average number of dihedral-angle constraint violations per CYANA conformer [degrees] | |
| > 10 | 0 |
Relative to pairs with non-degenerate chemical shifts.
Residues, 2–4, 8–10, 16–19, 37–42, 69–74 (β strands) 50–58, 80–100 (α-helices).
Includes 32 residues:4, 8, 10, 11, 13, 15, 18–20, 36–38, 43, 45, 46, 48–50, 53, 56, 69, 74, 79, 83, 85, 87, 88, 91,95, 96, 98, 99.
Scores defined in Ref.23.
Scores defined in Ref.24.
Scores defined in Ref.25.
Results and Discussion
The statistics summarized in Table I indicate that a high-quality NMR structure was obtained for the P. syringae PTH domain (Fig 1; PDB ID: 2jva). The structure exhibits a distinct α/β fold containing five β-strands with residues 2–4, 8–10, 16–19, 37–42, 69–74 (A-E), and two α-helices containing residues 50–58, 80–100 (I-II). β-strands C to E form the central anti-parallel sheet with topology C(↑), D(↓), E(↑). The β-strands A and B form a short β-hairpin structure (residues 1–10), which is in contact with both α-helices and the polypeptide segment connecting α-helix I and β-strand D. α-helix I itself is inserted between β-strands D and E. The large C-terminal α-helix II is attached to the β sheet on the same side of the β-hairpin. The flexible loop of residues 21–35 connects β-strands C and D, and contains the catalytically active GGQ tri-peptide segment (Fig. 1b).
Fig 1.
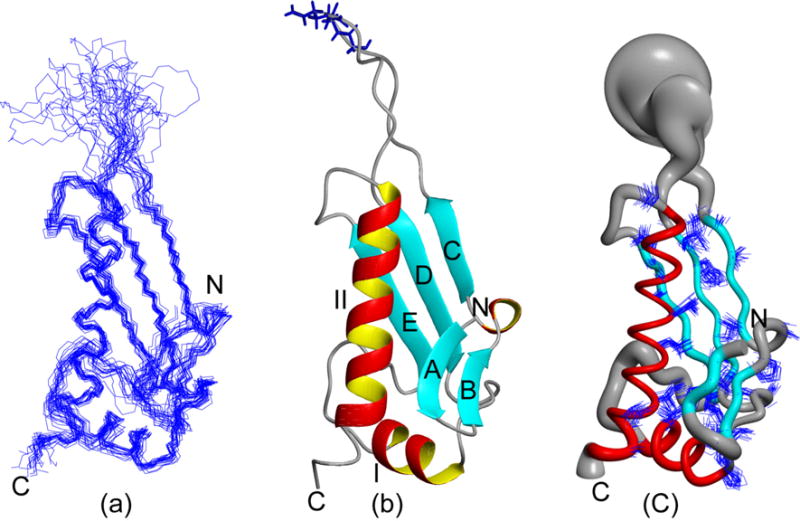
NMR structure of the P. syringae PTH domain (a): The 20 CYANA conformers with the lowest residual CYANA target function representing the NMR solution structure are shown after superposition for minimal r.m.s.d. of the backbone heavy atoms N, Cα and C′ atoms of regular secondary structure elements (Table 1). (b): Ribbon drawing of the CYANA conformer with the lowest residual target function value (Table I). The α-helices I to II are shown in red and yellow, β strands (A-E) are shown in cyan and other polypeptide segments are in grey, and the N- and C-terminal boundaries of the polypeptide segment are indicated as ‘N’ and ‘C’. Backbone and side chains of universally conserved GGQ (residues 26–28) motif are depicted in blue. (c): ‘Sausage’ representation of 20 superimposed conformers in the orientation of (a). For the presentation of the backbone a spline function was draw through the Cα positions and the thickness of the cylindrical rod is proportional to the mean of the global displacements of the 20 CYANA conformers calculated after superposition as in described in (a). The α-helices are shown in red and other polypeptide segments are displayed in grey. The 32 best-defined side chains (Table I) are also displayed. These figures were generated using the program MOLMOL.26
Fig-2 displays the structures of domains belonging to Pfam family PF00472 that have been solved so far. The sequence identity with the P. syringae PTH domain is 20% or less in all cases. In fact, a search for structurally similar proteins using the programs DALI20 (Z-score >4.0 as a cut-off criterion) and SSM21 identified only two of these structures as being structurally similar. Both programs identified the PTH domain of immature colon carcinoma transcript 1 protein from Mus musculus (MOUSE) (Fig. 2D; PDB ID: 1j26, gi:20306640, SwissProt/TrEMBL ID: ICT1_MOUSE, DALI Z-score 7.6, sequence identity of 23% with P. syringae PTH domain, r.m.s.d. of 3.7 A° with residues 6–26, 31–59 and 61–100 of P. syringae PTH domain, SSM Z-score 5.3, sequence identity of 17% with P. syringae PTH domain, r.m.s.d. of 1.89 A° with residues 15–19, 37–42, 49–58, 69–74 and 79–101 of P. syringae PTH domain), which is a thus far functionally uncharacterized protein. The comparison reveals that the P. syringae PTH domain has an additional short β-hairpin. DALI also identified the PTH domain of the polypeptide chain RF-2 from E. coli22 (PDB ID: 1gqe, gi: 146339, SwissProt/TrEMBL ID: RF2_ECOLI, Z-score 4.8, sequence identity of 16% with P. syringae PTH domain, rmsd of 3.9 A° with residues 2–5, 8–25, 30–43, 68–102, 103–108). The comparison reveals that the N-terminal β-hairpin and α-helix I of P. syringae PTH domain (Fig. 2A) are not present in this protein, but the overall similarity reveals that P. syringae PTH domain possess the fold of the PTH domain of class I peptide chain release factor.
Fig 2.
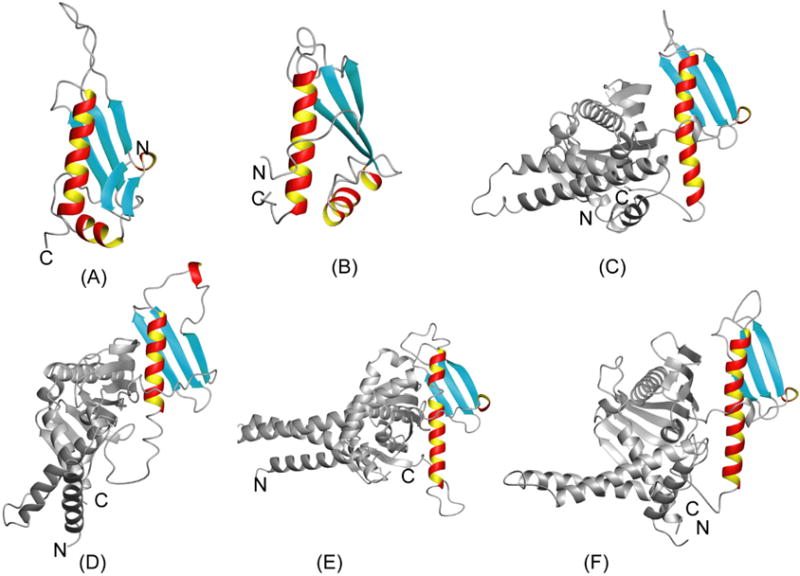
Ribbon diagrams of the six currently known structures belonging to PFam family PF00472. The PTH domains are shown in color and, for structures of full length proteins, the remainder of the protein chain is shown in grey. (A) P. syringae PTH domain (Fig. 1). (B) Immature colon carcinoma transcript 1 protein from Mus Musculus (mouse) (ICT1_MOUSE; PDB ID: 1j26, rmsd: 2.9 A°, sequence identity of 16% with P. syringae PTH domain). (C) Peptide chain RF-2 from E. coli (RF2_ECOLI; PDB ID: 1gqe, rmsd 3.8 A°, sequence identity of 13% with P. syringae PTH domain). (D) Peptide chain RF-1 from E.coli (RF1_ECOLI; PDB ID: 2b3t, rmsd 5.1 A°, sequence identity of 20% with P. syringae PTH domain). (E) Peptide chain RF-1 from Thermotoga maritima (RF1_THEMA; PDB ID: 1rq0, rmsd 4.9 A° sequence identity of 16% with P. syringae PTH domain). (F) Peptide chain RF-1 from Streptococcus mutans (RF1_STRMU; PDB ID: 1zbt, rmsd 4.19 A°, sequence identity of 15% with P. syringae PTH domain).
Taken together, the NMR structure of the P. syringae PTH domain significantly increases the ‘structural coverage’ of the large Pfam family PF00472 and finds 1395 proteins that are sufficiently related in sequence to build very reliable homology models (note that all those proteins belong to the same Pfam family and match at E-values <10–10) of members of this family. Moreover, the comparison of the solution structure of the P. syringae PTH domain with the domain structures in the ribosomal complex suggests that conformational changes upon formation of the complexes are moderate.
Acknowledgments
This work was supported by the National Institutes of Health (U54 GM074958-01) and the National Science Foundation (MCB 0416899 to T.S.).
References
- 1.Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, DeBoy RT, Durkin AS, Kolonay JF, Madupu R, Daugherty SC, Brinkac LM, Beanan MJ, Haft DH, Nelson WC, Davidsen TM, Zafar N, Zhou L, Liu J, Yuan Q, Khouri HM, Fedorova NB, Tran B, Russell D, Berry KJ, Utterback TR, Van Aken SE, Feldblyum TV, D’Ascenzo M, Deng WL, Ramos AR, Alfano JR, Cartinhour S, Chatterjee AK, Delaney TP, Lazarowitz SG, Martin GB, Schneider DJ, Tang X, Bender CL, White O, Fraser CM, Collmer A. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci USA. 2003;100:10181–10186. doi: 10.1073/pnas.1731982100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1- mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 4.Petry S, Broadersen DE, Murphy-IV FV, Dunham CM, Selmer M, Tarry JM, Kelley AC, Ramakrishnan V. Crystal structure of the ribosome in complex with release factor RF1 and RF2 bound to a cognate stop codon. Cell. 2005;123:1255–1266. doi: 10.1016/j.cell.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 5.Graille M, Heurgue-hamard V, Champ S, Mora L, Scrima N, Ulryck N, Tilbeurgh HV, Buckingham RH. J Biol Chem. 2005;20:917–927. doi: 10.1016/j.molcel.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 6.Liu G, Shen Y, Atreya HS, Parish D, Shao Y, Sukumaran DK, Xiao R, Yee A, Lemak A, Bhattacharya A, Acton TA, Arrowsmith CH, Montelione GT, Szyperski T. NMR data collection and analysis protocol for high-throughput protein structure determination. Proc Natl Acad Sci USA. 2005;102:10487–10492. doi: 10.1073/pnas.0504338102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acton TB, Gunsalus KS, Xiao R, Ma LC, Aramini JM, Baran MC, Chiang YW, Climent T, Cooper B, Denissova N, Douglas SM, Everett JK, Ho CK, Macapagal D, Paranji RK, Shastry R, Shih L-J, Swapna GVT, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT. Robot cloning and protein production platform of the Northeast Structural Genomics Consortium. Meth Enzymol. 2005;394:210–243. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Szyperski T. GFT NMR, a new approach to rapidly obtain precise high-dimensional NMR spectral information. J Am Chem Soc. 2003;125:1385–1393. doi: 10.1021/ja028197d. [DOI] [PubMed] [Google Scholar]
- 9.Atreya HS, Szyperski T. G-matrix Fourier transform NMR spectroscopy for complete protein resonance assignment. Proc Natl Acad Sci USA. 2004;101:9642–9647. doi: 10.1073/pnas.0403529101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen Y, Atreya HS, Liu G, Szyperski T. G-matrix Fourier transform NOESY based protocol for high-quality protein structure determination. J Am Chem Soc. 2005;127:9085–9099. doi: 10.1021/ja0501870. [DOI] [PubMed] [Google Scholar]
- 11.Delaglio F, Grzesiek G, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 12.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 13.Zimmerman DE, Kulikowski CA, Feng W, Tashiro M, Chien C-Y, Ríos CB, Moy FJ, Powers R, Montelione GT. Automated analysis of protein NMR assignments using methods from artificial intelligence. J Mol Biol. 1997;269:592–610. doi: 10.1006/jmbi.1997.1052. [DOI] [PubMed] [Google Scholar]
- 14.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program CYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 15.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Stereospecific Nuclear Magnetic Resonance Assignments of the Methyl Groups of Valine and Leucine in the DNA-Binding Domain of the 434 Repressor by Biosynthetically Directed Fractional 13C Labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 16.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shifts and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm CYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 18.Huang YJ, Moseley HN, Baran MC, Arrowsmith C, Powers R, Tejero R, Szyperski T, Montelione GT. An integrated platform for automated analysis of protein NMR structures. Meth Enzymol. 2005;394:111–141. doi: 10.1016/S0076-6879(05)94005-6. [DOI] [PubMed] [Google Scholar]
- 19.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 20.Holm L, Sander C. Mapping the Protein Universe. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 21.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 22.Vestergaard B, Van LB, Anderson GR, Nyborg J, Buckingham R, Kjeldgaard M. Bacterial polypeptide release factor RF2 is structurally distinct from Eukaryotic eRF1. Mol cell. 8:1375–1382. doi: 10.1016/s1097-2765(01)00415-4. [DOI] [PubMed] [Google Scholar]
- 23.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 24.Word JM, Bateman RC, Presley BK, Lovell SC, Richardson DC. Exploring steric constraints on protein mutations using MAGE/PROBE. Protein Sci. 2000;9:2251–2259. doi: 10.1110/ps.9.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang YJ, Powers R, Montelione GT. Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J Am Chem Soc. 2005;127:1665–1674. doi: 10.1021/ja047109h. [DOI] [PubMed] [Google Scholar]
- 26.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]