

Abstract
Purpose
The cone-rod homeobox (CRX) transcription factor is essential for photoreceptor gene expression, differentiation, and survival. Human CRX mutations can cause dominant retinopathies of varying onset and phenotype severity. In animal models, dominant frameshift Crx mutations introduce a premature termination codon (PTC), producing inactive truncated proteins that interfere with normal CRX function. Previously, a mutant mouse, TVRM65, was reported to carry a recessive late PTC mutation, Crx-L253X. More detailed phenotype analysis of the pathogenicity of Crx-L253X sheds new light on the variability of CRX-linked diseases.
Methods
Homozygous (L253X/X); heterozygous (L253X/+); Crx−/− and control C57BL/6J (WT) mice were analyzed at various ages for changes in retinal function (ERG), morphology (histology) and photoreceptor gene expression (qRT-PCR).
Results
At 1 month, L253X/X mice lack visual function, show greater reductions in retinal thickness, and distinct gene expression changes relative to Crx−/−, suggesting that the phenotype of L253X/X is more severe than Crx−/−. L253X/+ mice have reduced rod/cone function, but normal retinal morphology at all ages tested. qRT-PCR assays described a complex phenotype in which both developing and mature photoreceptors are unable to maintain proper gene expression. L253X mRNA/protein is overexpressed relative to normal Crx, suggesting a pathogenic mechanism similar to early PTC mutations. However, the overexpression is less pronounced, correlating with a relatively mild dominant phenotype.
Conclusions
The L253X mouse provides a valuable model for CRX-associated retinopathy. The pathogenicity of CRX frameshift mutations depends on the position of the PTC, which in turn determines the degree of mutant mRNA/protein overproduction.
Keywords: CRX mutations, animal models, inherited photoreceptor degeneration, cone-rod dystrophy, gene expression
The cone-rod homeobox protein (CRX) is a transcription factor (TF) that regulates expression of many photoreceptor genes essential for the development and maintenance of both rod and cone photoreceptors.1–4 Human CRX mutations are associated with various diseases, including retinitis pigmentosa (RP); cone-rod dystrophy (CoRD); and Leber congenital amaurosis (LCA).5,6 CRX-linked diseases are largely inherited in an autosomal dominant fashion or arise de novo, and vary widely in severity and age of onset.5,7–10 Since CRX mutations can be detected with early genetic testing, we need to be able to predict their effects and to define effective treatment and gene therapy regimens.7
Previous research has divided disease causing CRX mutations into four classes.11 Classes I and II are missense mutations that fall within or near the region coding for the DNA-binding homeodomain; class I mutations reduce the binding of CRX to its DNA targets.12 Previous work has shown by a variety of metrics that heterozygous mice carrying the class I R90W mutation suffer from a mild form of CoRD, similar to human patients with such mutations, while homozygotes exhibit an LCA-like condition.13 Class III and IV mutations represent frameshift mutations caused by insertions or deletions in the region coding for the transactivation domains of CRX.12 Class IV mutations, modeled by the RIP mouse, cause translation of a much longer peptide sequence due to a frameshift and extension of the open reading frame (ORF) into the 3′ untranslated region (3′UTR).14 In contrast to this extended peptide sequence, Class III mutations truncate the ORF of CRX with a premature termination codon (PTC) that results in a CRX protein with a shortened transactivation domain and leads to a severe dominant degenerative phenotype (LCA or CoRD).13 This phenotype has been modeled by the E168d2 mouse13 and Rdy (A182d1) cat15 (Fig. 1A). In vitro DNA binding and transactivation experiments determined that the E168d2 protein has a similar affinity for CRX target DNA sequences as normal CRX, but is unable to activate transcription on its own or in combination with other retinal TFs that normally synergize with CRX.12,13 Furthermore, when tested together with normal CRX, E168d2 protein interfered with normal CRX transactivation in a dominant-negative manner.13
Figure 1.

Class III Crx mutations introduce a PTC, resulting in accumulation of truncated CRX proteins and mutant mRNA with variable extended 3′UTR. (A) Schematic representation of normal and mutant Crx mRNA species, showing untranslated (UTR, gray/black lines) and protein-coding (open/striped boxes) with their relative sizes (bp), based on mouse (Normal, E168d213 and L253X) and cat (A182d1)15 models. The DNA-binding homeodomain (HD, dark strips) and the activation domain (AD, light strips) sequences12 are indicated. All three mutant mRNAs encode the complete HD, but ADs are truncated at different positions due to their PTC (indicated as the right-hand end of the protein coding box) induced by the mutation (marked by an asterisk). These PTCs also expand the 3′UTR to include different lengths of the original coding region (indicated by black line) in front of the normal 3′UTR (gray line). The position of the PTC relative to the normal stop codon determines the length of expanded 3′UTR: Early PTC mutants, E168d2 and A182d1 (Rdy cat mutation) produce a longer 3′UTR than the late PTC mutant L253X. Since 5′UTR and 3′UTR sequences of feline Crx mRNA are not available, these regions of the A182d1 transcript are indicated by dotted lines. (B) Immunoblot of CRX in the retinas of WT, L253X/+, and L253X/X mutants at 1-month old (mo) with GAPDH as a protein loading control. Black arrows indicate the running position for the normal and mutant (truncated) forms of CRX. (C) Quantification of relative amounts of normal and mutant CRX proteins in the indicated retinas at three ages (mean ± SEM, n ≥ 3; * P < 0.05, ** P < 0.01; 2-Way ANOVA with Tukey's multiple comparisons test). (D) ddPCR quantification of mutant (L253X) and normal Crx mRNA as percentage of total Crx. The specificity of the two allele-specific ddPCR assays was established using triplicate tail DNA samples (gray circles) from the indicated genotypes (DNA control). L253X/+ mRNA results are presented as percent of total Crx mRNA (mutant plus normal transcripts; mean ± SEM, n ≥ 3; **** P < 0.0001; unpaired t-test for mutant mRNA level relative to L253X/+ DNA control; n ≥ 3).
Class III PTC-causing CRX mutations also result in a novel untranslated region between the PTC and normal stop codon, which becomes a part of the mutant mRNA transcript's 3′UTR (Fig. 1A).11 In the E168d2 mouse and the Rdy (A182d1) cat, longitudinal studies showed gradual accumulation of mutant protein and mRNA.13,15 The mechanism for this accumulation is unknown, but was postulated to arise from decreased mutant RNA degradation,13 possibly due to the presence of cryptic stabilizing elements within the PTC-expanded mutant 3′UTR. Therefore, the position of the PTC (relative to the normal termination codon) could determine the stability, and thus abundance of mutant RNA.
Previously, another PTC mutant mouse, TVRM65 (Crx-L253X), was discovered in a chemical-induced mutagenesis screen conducted by The Jackson Laboratory16 (Bar Harbor, ME, USA; see Methods section for nomenclature specifics). The L253X mutation (Fig. 1A) occurs later (closer to the normal termination codon) than E168d2 and A182d1. L253X would be a candidate to model human late PTC class III mutations, such as L237I117 and Q256X18 associated with dominant CoRD. However, the initial histologic assessments suggested that the L253X mouse phenocopies the recessive null (Crx−/−) condition.16 Here, we describe more detailed phenotypic analysis of L253X mice, which demonstrates that this mouse models a new dominant but mild form of class III disease. Our results support the hypothesis that class III mutation pathogenicity depends on the position of the mutation-induced PTC (a late PTC produces a less severe phenotype than an early PTC), which correlates with the abundance of mutant protein products. These findings have implications for predicting human CRX disease severity and progression.
Methods
Nomenclature for the TVRM65 Mutation
There are several annotated transcripts for the murine Crx locus. The previously published amino acid change in the TVRM65 mouse was based on isoform 2 (NM001113330.1) that encodes a CRX protein with 323 amino acids.16,19 However, the major transcript in the retina is isoform 1 (NM_007770.4), which encodes a CRX protein homologous to human CRX, with 299 amino acids. Thus, here we refer to the TVRM65 mutation as p.L253X, based on isoform 1 numbering.
Mice
Crx-L253X (TVRM65) mice16 were obtained from The Jackson Laboratory (MGI ID: 4867395). Mice were mated and maintained with C57BL/6J (JAX Stock number 000664), and confirmed free of rd1 and rd8 mutations by PCR genotyping. Crx null mice (Crx−/−) were provided by Constance Cepko, Harvard University (Boston, MA, USA)20 and back-crossed onto C57BL/6J for more than 10 generations. All mice were housed in a barrier facility in the Division of Comparative Medicine of Washington University School of Medicine. All procedures involving the use of mice were approved by Washington University's Animal Care and Use Committee and followed the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
PCR genotyping, RNA purification and reverse transcription, and qRT-PCR were performed as previously described.13 Primer sets for genotyping and RT-PCR are listed in Supplementary Table S1. For qRT-PCR, all primers were tested for proper amplification efficiency prior to use. Relative gene expression was normalized to the retinal constitutively expressed genes Ubb, Tuba1B, and Gapdh. Data for 3 biologic replicates were then analyzed using the Delta Cq method in qPCR analysis software (QBase; Biogazelle, Ghent, Belgium). The results are presented by the heatmap.2 function of the R gplots package (v3.0.1).
Droplet Digital PCR (ddPCR)
Confirmatory genotyping and allelic expression data were generated using the droplet digital PCR system (BioRad, Hercules, CA, USA). A 20 μL ddPCR reaction mixture containing custom-designed normal and mutant primer/probe sets (see Supplementary Table S2) was prepared according to manufacturer's directions, droplets generated, and nanoreactions cycled on a thermal cycler (C1000 Touch; Bio-Rad Laboratories). The average number of allelic transcripts per biological replicate (n = 3) was then determined with a droplet reader and analytical software (QX 200 with QuantaSoft Analysis Pro; Bio-Rad Laboratories).
Transient Transfection Luciferase Reporter Assays
HEK293T cells (ATCC CRL-11286) were cultured in Dulbecco's modified Eagle medium with 10% fetal bovine serum and penicillin/streptomycin. Cells were transfected using a conventional CaCl and boric acid buffered saline method in 6-well plates as previously described.12,13 Experimental plasmids and transfection amounts included 2 μg BR130-Luc (a Rho promoter-luciferase reporter)12; 1 ng pRL-CMV (transfection normalization control); and protein expression vectors pED-NRL (100 ng), pcDNA3.1-hCRX, CRX1-254, and CRX-E168d2. Cells were harvested 48 hours posttransfection and assays performed using the dual-luciferase reporter assay system (Promega Corp., Madison, WI, USA).
ERG and Statistical Analyses
These were performed as previously described.13 Briefly, tests were performed on a visual electrodiagnostic system (UTAS-E3000 with EM for Windows; LKC Technologies, Inc., Gaithersburg, MD, USA) while mouse body temperature was maintained at 37°C ± 0.5°C with a heating pad controlled by a rectal temperature probe (FHC, Inc., Bowdoin, ME, USA). Pupils were dilated with 1.0% atropine sulfate (Bausch & Lomb, Tampa, FL, USA) and dilation and corneal hydration maintained during testing by positioning the platinum wire loop recording electrodes in a mixture of atropine and 1.25% hydroxypropyl methylcellulose (GONAK; Akorn, Inc., Buffalo Grove, IL, USA). Mice were tested without knowledge of genotype. Bilateral flash ERG responses were obtained; the set of recordings giving the higher amplitudes was correlated with genotype information for statistical analyses including 2-way ANOVA and post-hoc multiple comparison tests were performed using graphing software (GraphPad Prism; GraphPad Software, Inc., La Jolla, CA, USA).
Quantitative Western Blot Analysis
As previously described,13 experiments were performed using three biologic replicates (15 μg total protein extract from homogenates of two retinas). Membranes were probed with mouse monoclonal anti-CRX antibody M02 (1:200; Abnova Corp., Taipei City, Taiwan) and anti-GAPDH antibody (1:10,000, G9545; Sigma-Aldrich Corp., St. Louis, MO, USA), visualized with donkey anti-rabbit 680 nm and donkey anti-mouse 800 nm fluorescent dye (1:10,000 IRDyes; LI-COR, Inc., Lincoln, NE, USA), then imaged and quantified using commercial software (Odyssey Infrared Imager and ImageStudio 6; LI-COR, Inc.).
Histology, Morphometry, Immunohistochemistry, And Statistical Analyses
These assays were performed as previously described.13 Sagittal sections (4-μm) of paraffin-embedded eyecups, cut through the optic nerve head, were either stained with hematoxylin and eosin (H&E) or prepared for immunofluorescence staining. Slides containing sections were incubated overnight with primary antibody at 4°C (1:400 anti-Rhodopsin monoclonal RetP1; Sigma-Aldrich Corp.), washed, and incubated with secondary antibody (1:500 AlexaFluor 568 goat-anti-mouse; Invitrogen Corp., Carlsbad, CA, USA) for 2 hours at room temperature. Coverslips were mounted with mounting medium with DAPI (Vectashield Hardset; Vector Laboratories, Inc., Burlingame, CA, USA). All imaging was performed using a microscope and camera (DM5500B and DFC365FX; Leica Microsystems GmbH, Buffalo Grove, IL, USA).
Results
L253X Overproduces Mutant mRNA/Protein in Affected Retinas
A hallmark of class III CRX frameshift mutations is the overexpression of the mutant allele.13,15 To determine if L253X retinas accumulate mutant protein, we performed quantitative Western blots on total protein extracts from retinas of WT and heterozygous L253X/+ mice at postnatal day 10 (P10), 1 and 2 months old, and homozygous L253X/X mice at P10 only (because photoreceptors degenerate before later ages).16 Immunoblotting with an anti-CRX antibody revealed a truncated L253X protein that ran faster than the normal full-length CRX protein in samples from both heterozygous and homozygous mutant retinas (Fig. 1B). We quantified the total amount of CRX protein and of each isoform. Both L253X/+ and L235X/X displayed a 1.5- to 2.2-fold increase in total CRX protein compared to the WT retinas (Fig. 1C). The increases can be attributed to an accumulation of mutant protein (black bar), since normal CRX protein levels (gray bar) in L253X/+ retinas with one normal allele were roughly half of the total CRX protein present in WT retinas with two alleles.
To determine if the increased mutant protein corresponded with an increase in mutant mRNA, we quantified the relative allelic expression of L253X and normal Crx mRNA using droplet digital PCR (ddPCR). First, using mutant and WT mouse DNA, we developed a ddPCR assay that clearly distinguished the L253X and normal Crx sequences (Fig. 1D, DNA control). This assay was then used to quantify allele-specific mRNA species. At all ages tested, the mutant transcript was overrepresented, accounting for 57% of total Crx mRNA (Fig. 1D). Total Crx transcript levels quantified by conventional qRT-PCR (Supplementary Fig. S1) showed a statistically significant total Crx mRNA increase in 1-month L253X/+ retinas relative to WT, although levels were comparable to WT retinas during development at P10 and by 3 months.
Overall, L253X/+ retinas develop and maintain an elevated level of mutant mRNA that translates into an imbalance of the two protein forms, but this misregulation is not as severe as observed in other class III mutants. These results identify Crx-L253X as a bona fide class III mutation, but suggest that the position of the PTC determines the toxicity of the allele.
L253X/X Mice Have No Detectable Rod and Cone Function
Homozygous animals carrying class III or null Crx mutations lack measurable photoreceptor function.13,15,20 To confirm that L253X/X photoreceptors are functionally compromised, we quantified rod/cone light responses in L253X/X and WT control mice at 1 month using ERG. Even at the highest light intensity stimuli, L253X/X retinas displayed very little response (Figs. 2A–C). These results confirm that, like other class III models, homozygous L253X/X animals lack photoreceptor visual responses, resembling human LCA.
Figure 2.

L253X/X retinas lack rod and cone responses to light and degenerate earlier than Crx−/−. ERG was used to measure rod (dark-adapted) and cone (light-adapted) responses to various light stimuli in 1-mo L253X/X and WT mice. (A, B) Comparison of representative ERG traces at the highest light intensity: 0.89 log scot cd/m2 for dark-adapted responses, and 2.67 log scot cd/m2 for light-adapted responses. (C) Bar graphs compare mean A-wave and/or B-wave amplitude differences between the two genotypes at the highest light intensity tested (mean ± SEM, n ≥ 3; * P ≤ 0.05, *** P ≤ 0.0005, **** P < 0.0001; 2-way ANOVA with Sidak's multiple comparisons test). (D–F, H–J) H&E-stained paraffin-embedded sagittal retinal sections from the indicated mice at 1 and 3 mo. Images were taken approximately 500 μm from the ONH at a ×40 magnification (scale bar: 25 μm). (G, K) Morphometry measures at five set distances (in microns) on either side of the ONH (mean ± SEM, n ≥ 3; * P ≤ 0.05, ** P < 0.01, *** P ≤ 0.0003; 2-way ANOVA with Sidak's multiple comparisons test).
L253X/X Photoreceptors Degenerate Earlier Than Crx−/−
Other class III mutations show a greater impact on retina morphology and function than seen in Crx−/− mice.13,15,20 The original report noted that degeneration was more rapid in L253X/X than Crx−/− retinas, but suggested differences in mouse background might be a factor.16 To directly compare the impact of the L253X mutation with that of complete loss of Crx, we collected retinas from 1- and 3-month WT, L253X/X, and Crx−/− mice (all backcrossed 10 generations onto C57BL/6J) and assessed morphologic changes in H&E-stained sagittal sections through the optic nerve head (Figs. 2D–K). At 1 month, both L253X/X and Crx−/− retinas lacked photoreceptor outer segments (OS) and had thinner outer nuclear layers (ONL) than the WT control (Figs. 2E, 2F versus 2D). However, thinning was more pronounced in L253X/X than Crx−/−: L253X/X retina had only four to five rows of ONL nuclei, while Crx−/− maintained 7 to 8 rows at the same age (Fig. 2E versus 2F). Quantitative morphometry measures showed that the ONL of L253X/X was significantly thinner than that of Crx−/− at multiple positions (Fig. 2G, black versus solid gray). By 3 months, both models had similarly degenerated retinas with only 2–3 rows of ONL cells left (Fig. 2I versus 2J; 2K). Since both mutants showed normal ONL thickness at P7 through P10 (Supplementary Figs. S2A, S2B, and Tran et al.13), these results suggest that ONL degeneration occurs earlier or at a faster pace in L253X/X mice than in Crx−/−.
In addition to progressive ONL loss, L253X/X also underwent age-dependent thinning of the outer plexiform layer (OPL; Figs. 2E, 2I, Supplementary Figs. S2D, S2I, S2N, and Won et al.16). In contrast, nonphotoreceptor cell layers (e.g., inner nuclear layer, ganglion cell layer) were largely unchanged in L253X/X compared to WT controls at various ages (Supplementary Figs. S2C–Q). These results suggest that the L253X mutation mainly affects photoreceptor structural integrity and survival.
L235X/X Mice Exhibit Photoreceptor Gene Misregulation Distinct From Crx−/−
To further examine if the more severe morphologic phenotype of L253X/X retinas reflected greater changes in CRX target gene expression, we collected RNA from L253X/X and Crx−/− retinas at P10 during photoreceptor terminal differentiation prior to cell death.13 Transcripts of 14 known CRX-dependent genes were quantified by qRT-PCR and compared both with WT levels (Supplementary Fig. S2R) and between the two models (Fig. 3A). Although most of these genes were dysregulated in both mutants compared to WT mice (Supplementary Fig. S2R), half of them showed statistically significant expression differences between L253X/X and Crx−/− (Fig. 3A).13,21 Three of these, Gnat1, Grk1, and especially Rho differed markedly (>2-fold) between the two models (Fig. 3A). Such differences in essential photoreceptor gene expression likely contribute to the morphologic and phenotype differences between the two models.
Figure 3.
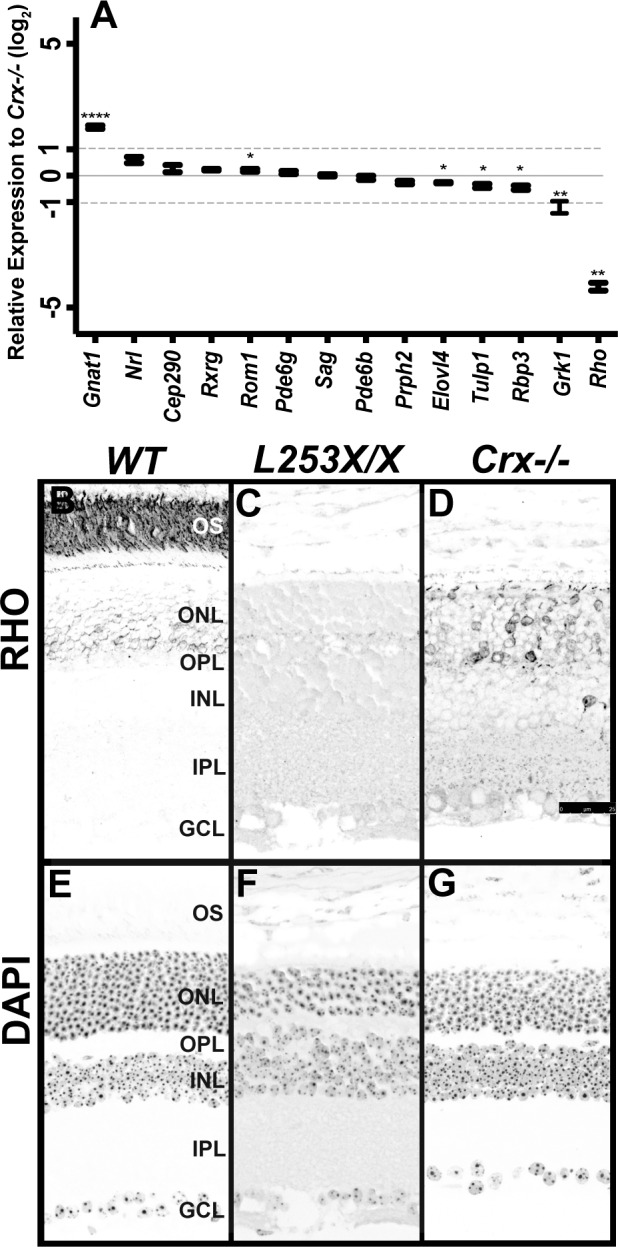
L253X/X has measurable gene expression differences relative to Crx−/−. (A) Expression at P10 of the indicated photoreceptor genes was measured by qRT-PCR and compared between L253X/X and Crx−/− at P10. The gray dashed lines mark 2-fold differences (±1 log2 relative expression) between the two genotypes (mean ± SEM, n ≥ 3; * P < 0.05, ** P < 0.01, **** P < 0.0001; unpaired t-test with Welch's Correction). (B–D) RHO immunostaining of retinal sections of the indicated mice at 1 mo revealed less RHO expression in L253X/X retinas than Crx−/− (signal in black, scale bar: 25 μm). (E–G) DAPI counterstaining of the above sections (signal in black).
To confirm qRT-PCR results at the protein level, we performed immunofluorescent staining for RHO on retinal sections of 1-month-old L253X/X, Crx−/− and WT control mice. RHO was chosen because it displayed the largest difference in the RNA level between L253X/X and Crx−/−. As expected, in WT retina, RHO was predominantly localized to the rod outer segments (OS) with faint staining seen in ONL cell bodies (Fig. 3B). In contrast, L253X/X retina displayed no detectable RHO immunostaining (Fig. 3C), while Crx−/− retina still had weak and spotty RHO signals localized to individual ONL cell bodies and inner segments (Fig. 3D). These results supported the RNA expression data and suggest that L253X mutant protein negatively affects transcription.
Overall, the comparison between L253X/X and Crx−/− indicates that L253X/X disease is more severe than that resulting from loss of Crx, supporting a dominant effect of the mutation.8,13 The more severe photoreceptor deficits in L253X/X thus presumably result from antimorphic activity of the truncated CRX protein.
Heterozygous L253X/+ Mice Show Mild Decreases in Rod and Cone Function
The L253X (TVRM65) mutation was originally reported to cause recessive retinal disease.16 However, if the mutant protein possesses antimorphic activity, we would hypothesize that one copy would produce a photoreceptor phenotype. To test this, we investigated photoreceptor function in L253X/+ retinas by assessing electrical responses to whole retina light stimulation. ERGs recorded at 1, 2, 3, and 5 months revealed L253X/+ mice exhibit mild but significant reductions in ERG amplitudes compared to WT mice (Fig. 4). At all ages tested, dark-adapted A-waves were reduced in L253X/+ mice, especially at high stimulus intensities (Figs. 4A–D, black line versus gray line). Dark-adapted B-waves of L253X/+ mice were comparable to WT at 1 month (Fig. 4E), but significantly reduced at 2, 3, and 5 months (Figs. 4F–H, black line versus gray line). The degree of A-wave or B-wave reductions appeared to be constant, suggesting that the defects were unlikely a result of progressive degeneration. Reduction in cone function was also observed: Light-adapted B-waves were significantly decreased in L253X/+ mice at 2, 3 and 5 months (Figs. 4J–L), but not at 1 month (Fig. 4I). These results suggest that L253X/+ mice have deficits in both rod and cone function, resembling a mild retinopathy.
Figure 4.

L253X/+ retinas exhibit reduced rod and cone light responses. ERG responses of L253X/+ mice (black lines) and WT (gray lines) were tested at four adult ages, and dark-adapted and light-adapted A- and B-wave amplitudes quantified: (A, E, I) 1 mo; (B, F, J) 2 mo; (C, G, K) 3 mo; and (D, H, L) 5 mo (mean ± SEM, n ≥ 3; * P < 0.05, **P < 0.01, *** P < 0.0006, **** P < 0.0001; 2-way ANOVA with Sidak's multiple comparisons test).
L253X/+ Retinas Do Not Degenerate
To test whether the functional defects correspond to morphologic changes, we measured retinal thickness in H&E stained retinal sections, comparing L253X/+ and L253X/X with WT mice at three ages (Fig. 5). Despite the progressive retinal degeneration seen in L253X/X mice (Figs. 5C, 5D, 5G, 5H, 5K, 5L; Supplementary Figs. S2C–Q), L253X/+ showed no degeneration or morphologic abnormalities compared to WT controls up to 3 months of age (Figs. 5B, 5D, 5F, 5H, 5J, 5L; Supplementary Figs. S2C–Q).
Figure 5.

L253X/+ retinas appear morphologically similar to WT up to 3 mo. H&E-stained retinal sections from L253X/+, L253X/X, and WT mice at the indicated ages: 1 mo (A–C); 2 mo (E–G); and 3 mo (I–K) were imaged at ×40 magnification (scale bar: 25 μm). Images were taken approximately 500 μm from the ONH, where differences in layer thickness are less significant. (D, H, L) ONL thickness was quantified by morphometry at five set distances (in microns) on either side of the ONH (mean ± SEM, n ≥ 3; * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001; 2-way ANOVA with Tukey's multiple comparisons test).
L253X/+ Retinas Display Dynamic Changes of Photoreceptor Gene Expression
To examine developmentally regulated photoreceptor gene expression in L253X/+ versus WT mice, we performed qRT-PCR analyses at two early ages: P10, when rods/cones are still terminally differentiating, and 1 month when rods/cones are fully mature in normal retinas. The genes tested were previously shown to dramatically change in expression during WT rod and cone development.22 Figure 6A shows expression changes of these genes in L253X/+ relative to WT mice at both ages. Gene expression conformed to three general trends in addition to the slight increase in total Crx expression (dashed black line) noted above: Gnat1, Rho, and Sop (solid black lines), markers of mature photoreceptors, were significantly decreased in L253X/+ at P10 but attained WT levels by 1 month. Rxrg (dotted gray line), which in WT retina loses expression over development,22 was elevated in L253X/+ at P10 but decreased closer to WT levels by 1 month. The other genes tested, which increase over development in WT retina, showed similar relative expression levels at both ages that did not vary considerably from WT (solid gray lines). Together, these gene expression trends suggest that L253X/+ retinas exhibit altered gene expression early in photoreceptor development, similar to other Crx mutant models.21
Figure 6.

L253X/+ retinas display dynamic changes in photoreceptor gene expression. qRT-PCR analyses of L253X/+ expression levels (relative to WT mice) of the indicated rod/cone genes are shown at P10 and 1 mo (mean ± SEM, n ≥ 3). Lines connecting expression levels of each gene at the two ages show the dynamic changes in expression, and reveal four different expression trends indicated here by line color and solidity, as described in Results. (B) Heatmap depicting qRT-PCR-detected expression changes of the listed photoreceptor genes in L253X/+ relative to WT at the indicated ages. Data are clustered based on gene expression patterns (log2 [mean fold-change], n ≥ 3). Colored gene names indicate expression in specific photoreceptor subtype(s). TF indicates transcription factors expressed by rods and/or cones that are essential for cell fate specification.
To determine if L253X/+ retinas maintain photoreceptor gene expression after differentiation, we used qRT-PCR to examine expression of 17 essential rod and cone genes in WT and L253X mutants at other adult ages. The results at 1, 2, 3 and 5 months (presented in a heatmap in Fig. 6B) suggest a complex phenotype. Genes coding for transcription factors Crx, Rxrg, Otx2, and Nrl were upregulated at many of the time points tested. The expression of essential rod and cone phototransduction genes showed a less clear pattern across ages, but in general all exhibit phases of reduced expression relative to WT.
Late PTC-Caused C-Terminal Truncation Reduces Transactivation Function of CRX
As an initial step to understand the molecular mechanism underlying misregulation of gene expression in L253X mutant retinas, we asked whether a short C-terminal truncation caused by a late PTC affects the ability of CRX to transactivate target gene expression. This was tested using transient transfection assays in HEK293T cells with a Rhodopsin (Rho) promoter-luciferase reporter (BR130-Luc).12 Vectors expressing either the full-length (normal) or truncated CRX1-254 were cotransfected with an NRL vector to measure their ability to transactivate the BR130-Luc reporter. CRX1-254 did show some dose-dependent ability to activate the Rho promoter, suggesting that it does bind DNA and interacts with NRL, but the maximum activity was greatly reduced compared to the normal CRX protein (∼30% of normal activity; Fig. 7).
Figure 7.
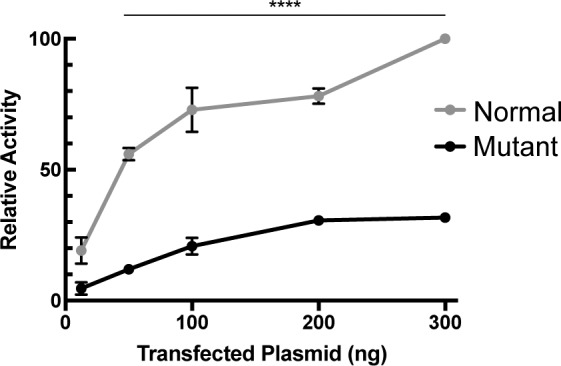
L253X retains minimal ability to transactivate rhodopsin promoter in the presence of NRL. Dual luciferase assays showing a dose-dependent increase in CRX transactivation activity of normal or mutant (CRX1-254) in the presence of constant amount of NRL (mean ± SEM, n = 3; **** P < 0.0001; 2-way ANOVA with Bonferroni's multiple comparisons test).
Discussion
L253X Mouse Provides a New Class III Model for Mild Dominant Cone-Rod Dystrophy
Although L253X (TVRM65) was originally identified as a recessive Crx mutation,16 several pieces of evidence suggest that L253X is antimorphic with dominant inheritance. First, L253X retinas overexpress the mutant gene product, a hallmark of class III mutations that amplifies the mutation's effects. Second, the photoreceptors in homozygous L253X/X mice degenerate earlier than Crx−/−, and show patterns of photoreceptor gene dysregulation that are distinct from Crx.−/− Thus, the presence of the mutant L253X protein causes more severe pathology than absence of CRX altogether, which is consistent with the antimorphic effect of other class III mutations11,13,15 Furthermore, heterozygous L253X/+ mice have early gene expression differences that largely resolve by 1 month, suggesting that full maturation may be delayed in these retinas. Although gene expression changes in adults were modest, rod and cone functional deficits were detectable as early as 1 month. Previous work on class III mutants has emphasized the effects that small changes in gene expression over a large number of genes can have on photoreceptor function.21 Collectively, these findings identify L253X as a class III Crx mutation with the phenotypes summarized in the Table.
Table.
Summary of L253X Related Phenotypes

Compared to previously reported class III mutations, pathogenicity of L253X is relatively mild. Unlike E168d2/+ photoreceptors, which lose their OS and undergo progressive degeneration,13 L253X/+ photoreceptors show normal morphology and do not degenerate. The ERG deficits do not worsen with age relative to C57BL/6J controls. The gene expression changes are also relatively modest with a multiphasic nature. This evolving expression pattern with increased levels of many TFs, but modest depletion of phototransduction cascade components, likely reflects an inability of the cells to maintain a proper homeostasis. This data would suggest that unknown feedback mechanisms are operational but unable to achieve the necessary precision. In adults, L253X/+ photoreceptors are alive and functional through 5 months, but it remains to be determined if the phenotype worsens at older ages or if the retina is more susceptible to environmental insults as reported with other class III mutants. Nevertheless, given the huge phenotypic variability of human CRX diseases, the L253X mouse provides a valuable animal model for understanding the pathogenesis of class III mutations and the underlying molecular mechanisms that determine phenotype severity, age of onset and disease progression.
The Molecular Mechanism(s) Underlying L253X Pathogenicity
The pathogenicity of class III mutations is generally determined by two factors, the activity and dose of the mutant protein. L253X protein contains an intact DNA binding domain but lacks a portion of the C-terminal activation domain (Fig. 1A). Thus, it likely retains the ability to bind DNA, but loses some aspect of transactivation function. Transient transfection assays showed that CRX1-254, a truncated CRX protein similar to L253X, retained only 30% of the normal CRX activity, very similar to the published reports.12,14 By comparison, the class III mutant protein E168d2 showed 25% to 30% of the transactivation activity under similar conditions.12,13 Previous reports have also shown CRX1–254 binds appropriate DNA sequence elements very similar to full-length CRX.14 It is conceivable that, as previously reported for E168d2,13 L253X protein may compete with normal CRX to bind to target genes, interfering with normal CRX function in heterozygous mice. However, transient transfection assays testing this hypothesis did not show this effect on the Rho-promoter luciferase reporter (data not shown). The antimorphic competition may not be limited to CRX, but could also affect other homeodomain transcription factors that bind to similar DNA targets, and as such transient cell transfection assays on a single reporter that lacks the native chromatin context and regulatory architecture may not be the proper experiment. Supporting this possibility, the retinas of L253X/X mutants, like E168d2/d2,13 showed more severe degeneration and biochemical defects than Crx−/− retinas. Other potential targets could include OTX2,23 which is upregulated in L253X retinas (Fig. 6B). Additional experiments are required to demonstrate dominant-negative activity of L253X on CRX and OTX2. Despite the similarity between L253X and E168d2, L253X/+ mice have a remarkably milder phenotype than E168d2/+; it is unlikely that the <10% difference in transactivation activity can account entirely for the significant difference between the two models. We therefore propose that the relative amounts of mutant and normal CRX protein must also play a role in phenotype severity.
L253X Allele-Specific Overproduction of Mutant mRNA and Protein Has Implications for the PTC Position Effect
Careful analysis of Crx mRNA and protein in L253X/+ and L253X/X retinas clearly shows an increased ratio of mutant/normal protein and mRNA. This finding supports the hypothesis that, like other class III models, L253X retinas accumulate mutant transcripts, leading to overproduction of the CRX mutant protein. Interestingly, however, the degree of mutant RNA accumulation in L253X/+ is lower than that of the other class III models carrying early PTC mutations. L253X mice only display a modest accumulation (57% mutant/43% normal), especially when compared to that observed in E168d2/+ mice at the same age (85% mutant/15% normal).13 Also, when considered with the quantification of total Crx mRNA, the L253X/+ must result in a downregulation of the normal allele as there is no aggregate overproduction of Crx. This suggests that the L253X mRNA is still “sensed” by the feedback regulatory mechanism, which functions normally in the mutant retina. Future studies could probe this feedback mechanism by comparing to the E168d2/+ retinas where although there is a gross increase in total Crx mRNA, the normal allele maintains normal expression levels.13 Both mouse models show overproduction of mutant protein relative to WT, but L253X accumulates at a slower rate than E168d2.
The mechanism for allele-specific overproduction of mutant Crx in class III models is not well understood, but likely stems from increased RNA stability due to the presence of the PTC in the last exon. This hypothesis further predicts that the position of the PTC influences the rate of mutant RNA accumulation. Indeed, L253X with a later PTC showed less RNA accumulation than the two early PTC alleles, E168d2 and A182d1 (Fig. 1). Thus, the earlier the PTC is, the more mutant product accumulates. These differences between early versus late PTCs may be attributed to the length of PTC-determined 3′UTR, within which multiple discrete elements could independently and additively contribute to mRNA hyperstability. Future identification of novel RNA regulatory elements shared among different PTC mutations may shed light on the mechanisms for mutant transcript hyperstability and phenotype variability in class III CRX disease. It is also notable that the accumulation of mutant protein in L253X retinas is more prominent than mutant RNA, raising the new possibility that altered protein turnover rate could also contribute to mutant protein accumulation.
In conclusion, our in-depth characterization of the Crx-L253X (TVRM65) mouse supports the classification of L253X as a dominant class III Crx mutation and provides a new animal model for understanding CRX-associated dominant retinopathies. The insights gained will hopefully lead to new treatment strategies for this complex disease.
Supplementary Material
Acknowledgments
The authors thank Connie Cepko for providing Crx−/− mice, GuangYi Ling, and Belinda Dana of the Immunology core for technical assistance in histology and immunohistochemistry assays, and Mingyan Yang for mouse genotyping and other technical assistance. The authors thank Brandon McKethan of Bio-Rad Laboratories for designing ddPCR assays to quantify mutant versus normal transcripts in L253X/+ retinas.
Supported by National Institutes of Health grants EY012543 and EY025272 (SC); EY013360 (Washington University [WU]) and EY002687 (WU, Department of Visual Science [WU-DOVS]); and unrestricted funds from Research to Prevent Blindness (WU-DOVS).
Disclosure: P.A. Ruzycki, None; C.D. Linne, None, A.K. Hennig, None; S. Chen, None
References
- 1. Freund CL, Gregory-Evans CY, Furukawa T,et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997; 91: 543– 553. [DOI] [PubMed] [Google Scholar]
- 2. Hennig AK, Peng GH, Chen S. . Regulation of photoreceptor gene expression by Crx-associated transcription factor network. Brain Res. 2008; 1192: 114– 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furukawa T, Morrow EM, Cepko CL. . Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997; 91: 531– 541. [DOI] [PubMed] [Google Scholar]
- 4. Swaroop A, Kim D, Forrest D. . Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci. 2010; 11: 563– 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivolta C, Berson EL, Dryja TP. . Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum Mutat. 2001; 18: 488– 498. [DOI] [PubMed] [Google Scholar]
- 6. Swain PK, Chen S, Wang Q-L,et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997; 19: 1329– 1336. [DOI] [PubMed] [Google Scholar]
- 7. Sohocki MM, Sullivan LS, Mintz-Hittner HA,et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998; 63: 1307– 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jacobson SG, Cideciyan AV, Huang Y,et al. Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Invest Ophthalmol Vis Sci. 1998; 39: 2417– 2426. [PubMed] [Google Scholar]
- 9. Koenekoop RK, Loyer M, Dembinska O, Beneish R. . Visual improvement in Leber congenital amaurosis and the CRX genotype. Ophthalmic Genet. 2002; 23: 49– 59. [DOI] [PubMed] [Google Scholar]
- 10. Nichols LL, Alur RP, Boobalan E,et al. Two novel CRX mutant proteins causing autosomal dominant leber congenital amaurosis interact differently with NRL. Hum Mutat. 2010; 31: 1472– 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran NM, Chen S. . Mechanisms of blindness: animal models provide insight into distinct CRX-associated retinopathies. Dev Dyn. 2014; 243: 1153– 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen S, Wang Q-L, Xu S,et al. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum Mol Genet. 2002; 11: 873– 884. [DOI] [PubMed] [Google Scholar]
- 13. Tran NM, Zhang A, Zhang X, Huecker JB, Hennig AK, Chen S. . Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS Genet. 2014; 10: e1004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roger JE, Hiriyanna A, Gotoh N,et al. OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. J Clin Invest. 2014; 124: 631– 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Occelli LM, Tran NM, Narfström K, Chen S, Petersen-Jones SM. . Retinal cell biology cat: a large animal model for CRX-associated Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2016; 57: 3780– 3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Won J, Shi LY, Hicks W,et al. Mouse model resources for vision research. J Ophthalmol. 2011; 2011: 1– 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang L, Xiao X, Li S,et al. CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun. 2012; 426: 498– 503. [DOI] [PubMed] [Google Scholar]
- 18. Lu Q-K, Zhao N, Lv Y-S,et al. A novel CRX mutation by whole-exome sequencing in an autosomal dominant cone-rod dystrophy pedigree. Int J Ophthalmol. 2015; 8: 1112– 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chau KY, Chen S, Zack DJ, Ono SJ. . Functional domains of the cone-rod homeobox (CRX) transcription factor. J Biol Chem. 2000; 275: 37264– 37270. [DOI] [PubMed] [Google Scholar]
- 20. Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. . Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999; 23: 466– 470. [DOI] [PubMed] [Google Scholar]
- 21. Ruzycki PA, Tran NM, Kefalov VJ, Kolesnikov AV, Chen S. . Graded gene expression changes determine phenotype severity in mouse models of CRX-associated retinopathies. Genome Biol. 2015; 16: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim JW, Yang HJ, Brooks MJ,et al. NRL-regulated transcriptome dynamics of developing rod photoreceptors. Cell Rep. 2016; 17: 2460– 2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Samuel A, Housset M, Fant B, Lamonerie T. . Otx2 ChIP-seq reveals unique and redundant functions in the mature mouse retina. PLoS One. 2014; 9: e89110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.